

Abstract
Advances in synthetic biology and metabolic engineering have enabled the construction of novel biological routes to valuable chemicals using suitable microbial hosts. Aldehydes serve as chemical feedstocks in the synthesis of rubbers, plastics, and other larger molecules. Microbial production of alkanes is dependent on the formation of a fatty aldehyde intermediate which is converted to an alkane by an aldehyde deformylating oxygenase (ADO). However, microbial hosts such as Escherichia coli are plagued by many highly active endogenous aldehyde reductases (ALRs) that convert aldehydes to alcohols, which greatly complicates strain engineering for aldehyde and alkane production. It has been shown that the endogenous ALR activity outcompetes the ADO enzyme for fatty aldehyde substrate. The large degree of ALR redundancy coupled with an incomplete database of ALRs represents a significant obstacle in engineering E. coli for either aldehyde or alkane production. In this study, we identified 44 ALR candidates encoded in the E. coli genome using bioinformatics tools, and undertook a comprehensive screening by measuring the ability of these enzymes to produce isobutanol. From the pool of 44 candidates, we found five new ALRs using this screening method (YahK, DkgA, GldA, YbbO, and YghA). Combined deletions of all 13 known ALRs resulted in a 90–99% reduction in endogenous ALR activity for a wide range of aldehyde substrates (C2–C12). Elucidation of the ALRs found in E. coli could guide one in reducing competing alcohol formation during alkane or aldehyde production.
Keywords: Biofuel, Synthetic biology, Metabolic engineering, Aldehyde, Alkane
1. Introduction
Concerns regarding the future availability of petroleum and its adverse environmental effects have generated interest in developing alternative sources for fuels and chemicals. For these reasons, the biological production of fuels and chemicals is viewed as a promising method for curbing reliance on petroleum. Advances in synthetic biology and metabolic engineering have enabled the construction of novel biological routes to valuable chemicals by rationally combining genes from various natural sources into a suitable microbial host (Rabinovitch-Deere et al., 2013; Peralta-Yahya et al., 2012; Geddes et al., 2011). Metabolic engineering approaches then allow tuning of the metabolism by eliminating or reducing competing pathways in the host to achieve high carbon flux toward the desired end product while also maintaining cell fitness (Rabinovitch-Deere et al., 2013; Peralta-Yahya et al., 2012; Geddes et al., 2011). The marriage of these techniques has enabled the biological production of a multitude of fuels and chemicals in user-friendly hosts such as Escherichia coli (Rabinovitch-Deere et al., 2013; Peralta-Yahya et al., 2012; Geddes et al., 2011; Zhang et al., 2012).
Aldehydes serve as chemical feedstocks in the synthesis of rubbers, plastics, and other larger molecules. Aldehydes such as butanal and isobutyraldehyde are high volume chemicals, each chemically produced from petroleum substrates in quantities greater than 1 million ton per year (Raff, 2013). Producing these compounds using microbes would be a renewable alternative to current methods. However, microbial hosts such as E. coli are plagued by many highly active endogenous aldehyde reductases (ALR) that convert aldehydes to alcohols (Rodriguez and Atsumi, 2012; Zhu et al., 2011), which greatly complicates strain engineering for aldehyde production. As a result, E. coli production of only acetaldehyde (Zhu et al., 2011) and isobutyraldehyde (Rodriguez and Atsumi, 2012) has been shown. In contrast, alcohol and organic acid production spanning C2–C18 has been demonstrated in E. coli and other microbes (Rabinovitch-Deere et al., 2013; Zhang et al., 2012; Atsumi et al., 2008; Dellomonaco et al., 2011; Schirmer et al., 2010; Ingram et al., 1998; Cintolesi et al., 2014; Liu et al., 2014).
The presence of ALRs also impairs microbial production of alkanes, which has been a prominent area of interest with the discovery of the aldehyde deformylating oxygenase (ADO) (Schirmer et al., 2010; Li et al., 2012; Warui et al., 2011). Biological alkane production has been shown in the range from C3 to C20 alkanes (Schirmer et al., 2010; Khara et al., 2013; Harger et al., 2013; Choi and Lee, 2013; Akhtar et al., 2013; Howard et al., 2013; Andre et al., 2013), but overall titers and productivities remain lower than those of the corresponding alcohols. This biological pathway is dependent on the formation of a fatty aldehyde intermediate, which can be converted to an alkane or an alcohol by ADO or ALR, respectively (Schirmer et al., 2010; Li et al., 2012). It has been shown that the endogenous ALR activity outcompetes the ADO enzyme for fatty aldehyde substrate (Schirmer et al., 2010; Howard et al., 2013). This large degree of ALR redundancy coupled with an incomplete database of ALRs (Rodriguez and Atsumi, 2012) represents a significant obstacle in engineering E. coli for either aldehyde and alkane production. Thus, a comprehensive elucidation and characterization of ALRs would facilitate targeted reduction of endogenous ALR activity for the production of specific aldehydes or alkanes.
In a previous study, we engineered a strain of E. coli (AL626) capable of producing isobutyraldehyde at titers up to 35 g L−1 (Rodriguez and Atsumi, 2012). This was accomplished by deleting six ALR genes (yqhD, adhP, eutG, yiaY, yjgB, and fucO) from the genome. However, this strain still had a notable amount of isobutanol formation (~10 g L−1) indicating that AL626 still contains enzymes with isobutyraldehyde reductase activity (Rodriguez and Atsumi, 2012). Owing to the high aldehyde to alcohol ratio exhibited by E. coli strain AL626, this strain can serve as an effective tool for in vivo screening of ALR activity by overexpressing ALR gene candidates in the presence of aldehyde and measuring the resulting aldehyde and/or alcohol formation.
In this study, we sought to complete four major objectives. First, using our previously engineered isobutyraldehyde producing strain, AL626, we aimed to undertake a comprehensive screening of candidate ALRs encoded in the E. coli genome. Second, any positive hits for isobutyraldehyde reductase activity should be confirmed in vitro along with the acquisition of a broad substrate profile (C2–C10) of the elucidated ALRs. Third, we aimed to delete any ALR gene(s) we uncover from AL626, creating a strain with the potential to have complete or near abolishment of endogenous ALR activity. Lastly, we sought to characterize this strain for its ability (or inability) to convert a wide range of aldehydes (C2–C12) to their corresponding alcohols as compared to a strain without ALR deletions. The comprehensive elucidation of the ALRs in the E. coli genome could guide one in reducing competing alcohol formation pathways and achieve improved alkane and aldehyde production from E. coli.
2. Methods
2.1. Reagents
All enzymes were purchased from New England Biolabs (Ipswich, MA). All synthetic oligonucleotides were ordered from Integrated DNA Technologies (Coralville, IA). DNA sequencing services were done by Davis Sequencing (Davis, CA). All chemicals for gas chromatography (GC) standards except for ethanol and 1-pentanol were purchased from Sigma-Aldrich (St. Louis, MO). 1-Pentanol was purchased from Acros Organics (Belgium). Ethanol (200 Proof) was purchased from VWR (Radnor, PA, Canada).
2.2. Plasmid and strain construction
All plasmids were constructed using sequence and ligation-independent cloning (SLIC) (Machado et al., 2012). Plasmids were verified by PCR, by digestion with restriction enzymes, and by sequencing. All oligonucleotides and plasmids are listed in Tables 1 and 2, respectively. To construct the plasmids listed in Table 2, the target gene(s) were amplified from the E. coli genome DNA with corresponding primers in Table 1 and pSA138 (Atsumi et al., 2008) vector was amplified with primers GR258 and GR259. The resulting fragments were purified and combined with SLIC (Machado et al., 2012; Rodriguez et al., 2014).
Table 1.
Oligonucleotides used in this study.
| Name | Sequence 5′–>3′ | Description |
|---|---|---|
| GR258 | TCTAGAGGCATCAAATAAAACGAAA | Forward pSA138 Vector |
| GR259 | GTGACCTTTCTCCTGCATGCTTATG | Reverse pSA138 Vector |
| GR260 | CATAAGCATGCAGGAGAAAGGTCACATGAAGATCAAAGCTGTTGGTGCAT | Forward yahK |
| GR261 | TTTCGTTTTATTTGATGCCTCTAGATCAGTCTGTTAGTGTGCGATTATCG | Reverse yahK |
| GR262 | CATAAGCATGCAGGAGAAAGGTCACATGAAAACGATGCTGGCAGCTTATT | Forward yphC |
| GR263 | TTTCGTTTTATTTGATGCCTCTAGATTAATCCGGGAAGTTAATCACAACT | Reverse yphC |
| GR264 | CATAAGCATGCAGGAGAAAGGTCACATGAAAAAGTTAGTAGCCACAGCAC | Forward ycjQ |
| GR265 | TTTCGTTTTATTTGATGCCTCTAGATTAAAACGTAACGCCCATTTTGATG | Reverse ycjQ |
| GR266 | CATAAGCATGCAGGAGAAAGGTCACATGAAAGCATTGACTTATCACGGCC | Forward ybdR |
| GR267 | TTTCGTTTTATTTGATGCCTCTAGATCATATTGTTCCCCCCGGCATCGCA | Reverse ybdR |
| GR268 | CATAAGCATGCAGGAGAAAGGTCACATGAAAAGCATATTAATTGAAAAAC | Forward rspB |
| GR269 | CTTTCGTTTTATTTGATGCCTCTAGATTATTCAGAAAAAGTGAGTAAGAC | Reverse rspB |
| GR270 | CATAAGCATGCAGGAGAAAGGTCACATGAAAAAGATACCTTTAGGCACAA | Forward ydjG |
| GR271 | CTTTCGTTTTATTTGATGCCTCTAGATTAACGCTCCAGGGCCTCTGCCAT | Reverse ydjG |
| GR272 | CATAAGCATGCAGGAGAAAGGTCACATGAAGCCGTCCGTTATCCTCTACA | Forward ghrB |
| GR273 | TTTCGTTTTATTTGATGCCTCTAGATTAGTCCGCGACGTGCGGATTCACA | Reverse ghrB |
| GR307 | CATAAGCATGCAGGAGAAAGGTCACATGTCTACGATGAATGTTTTAATTT | Forward yjjN |
| GR308 | TTTCGTTTTATTTGATGCCTCTAGATCAGAAAGTAATTACGCCTTTAATT | Reverse yjjN |
| GR309 | CATAAGCATGCAGGAGAAAGGTCACATGATCGTTTTAGTAACTGGAGCAA | Forward ydfG |
| GR310 | TTTCGTTTTATTTGATGCCTCTAGATTACTGACGGTGGACATTCAGTCCG | Reverse ydfG |
| GR311 | CATAAGCATGCAGGAGAAAGGTCACATGGCTATCCCTGCATTTGGTTTAG | Forward dkgB |
| GR312 | TTTCGTTTTATTTGATGCCTCTAGATTAATCCCATTCAGGAGCCAGACCT | Reverse dkgB |
| GR313 | CATAAGCATGCAGGAGAAAGGTCACATGAAAAATTCAAAAGCAATATTGC | Forward ydjJ |
| GR314 | TTTCGTTTTATTTGATGCCTCTAGATTAATCGCTAATTTTAATAACGCCT | Reverse ydjJ |
| GR315 | CATAAGCATGCAGGAGAAAGGTCACATGAAAGCACTGGCTCGGTTTGGCA | Forward ydjL |
| GR316 | TTTCGTTTTATTTGATGCCTCTAGATTATTCATCAAAGTCGTAAGTCATG | Reverse ydjL |
| GR317 | CATAAGCATGCAGGAGAAAGGTCACATGCAGGCGTTACTTTTAGAACAGC | Forward yhdH |
| GR318 | TTTCGTTTTATTTGATGCCTCTAGATTAGTTAACCTTCACCAGCGTGCGA | Reverse yhdH |
| GR319 | CATAAGCATGCAGGAGAAAGGTCACATGACTCATAAAGCAACGGAGATCC | Forward ybbO |
| GR320 | TTTCGTTTTATTTGATGCCTCTAGATCACCCCTGCAATATTTTGTCCATC | Reverse ybbO |
| GR321 | CATAAGCATGCAGGAGAAAGGTCACATGGACCGCATTATTCAATCACCGG | Forward gldA |
| GR322 | TTTCGTTTTATTTGATGCCTCTAGATTATTCCCACTCTTGCAGGAAACGC | Reverse gldA |
| GR491 | GCATGCAGGAGAAAGGTCACATGGCTAATCCAACCGTTATTAAGCTACAGG | Forward dkgA |
| GR492 | TTTTATTTGATGCCTCTAGATTAGCCGCCGAACTGGTCAGG | Reverse dkgA |
| GR493 | GCATGCAGGAGAAAGGTCACATGCAACAAAAAATGATTCAATTTAGTGGCG | Forward yeaE |
| GR494 | TTTTATTTGATGCCTCTAGATCACACCATATCCAGCGCAGTTTTTC | Reverse yeaE |
| GR495 | GCATGCAGGAGAAAGGTCACATGGTCTGGTTAGCGAATCCCGAAC | Forward yghZ |
| GR496 | TTTTATTTGATGCCTCTAGATCATTTATCGGAAGACGCCTGCC | Reverse yghZ |
| GR497 | GCATGCAGGAGAAAGGTCACATGCAATATCACCGTATACCCCACAGTTC | Forward tas |
| GR498 | TTTTATTTGATGCCTCTAGATTATGGTGCCGGATAAGTATAAACCTGATGC | Reverse tas |
| GR499 | GCATGCAGGAGAAAGGTCACATGAAAACGGGATCTGAGTTTCATGTCG | Forward ygbJ |
| GR500 | TTTTATTTGATGCCTCTAGATCATGATTTCGCTCCCGGTAGAGTG | Reverse ygbJ |
| GR501 | GCATGCAGGAGAAAGGTCACATGGATATCATCTTTTATCACCCAACGTTCG | Forward ghrA |
| GR502 | TTTTATTTGATGCCTCTAGATTAGTAGCCGCGTGCGCG | Reverse ghrA |
| GR511 | GCATGCAGGAGAAAGGTCACGTGAAAAGTGCAATGACAAGCTCTCC | Forward ycjS |
| GR512 | TTTTATTTGATGCCTCTAGATCATAATTCCACACGCGTCCCTG | Reverse ycjS |
| GR513 | GCATGCAGGAGAAAGGTCACGTGAAAAAATTACGTATCGGCGTAGTGG | Forward yceM |
| GR514 | TTTTATTTGATGCCTCTAGATTATTCACTCATCGCATCGCGC | Reverse yceM |
| GR515 | GCATGCAGGAGAAAGGTCACGTGGAAAGGTTTGATGCCATTATTATAGGC | Forward yhiN |
| GR516 | TTTTATTTGATGCCTCTAGATCAGGACGACTTTGCTGCAATC | Reverse yhiN |
| GR517 | GCATGCAGGAGAAAGGTCACATGGTCATCAACTGCGCCTTTATTG | Forward yhhX |
| GR518 | TTTTATTTGATGCCTCTAGATTACTTAGCGAGAGTTACTGTGGAGGGAG | Reverse yhhX |
| GR519 | GCATGCAGGAGAAAGGTCACATGAGCAGCAATACATTTACTCTCGGTAC | Forward ydbC |
| GR520 | TTTTATTTGATGCCTCTAGATTATTCTCGCGAAATACCATCCAACGTAG | Reverse ydbC |
| GR521 | GCATGCAGGAGAAAGGTCACATGATACGTTTCGCTGTGATTGGTACG | Forward ygjR |
| GR522 | TTTTATTTGATGCCTCTAGATTATAGTTTTACGCTATCTGCCGGAAAAATC | Reverse ygjR |
| GR523 | GCATGCAGGAGAAAGGTCACATGGCTATAGCACTTGTGACTGGTGG | Forward ygfF |
| GR524 | TTTTATTTGATGCCTCTAGATTATTTCCCGCCCGCCAAATC | Reverse ygfF |
| GR525 | GCATGCAGGAGAAAGGTCACATGGCACAGGTTGCGATTATTACC | Forward yohF |
| GR526 | TTTTATTTGATGCCTCTAGACTATTCTGGGTTGAACTGTGGATTCG | Reverse yohF |
| GR617 | GCATGCAGGAGAAAGGTCACATGCATTACCAGCCAAAACAAGATTTAC | Forward yciK |
| GR618 | TTTTATTTGATGCCTCTAGATCATTGGGAAATTCCTGGTTTACGG | Reverse yciK |
| GR619 | GCATGCAGGAGAAAGGTCACATGAATGTCAATTTCTTTGTCACCTGTATTGG | Forward ykgE |
| GR620 | TTTTATTTGATGCCTCTAGATCAGCGGCTCATCAACACTTCAG | Reverse ykgE |
| GR621 | GCATGCAGGAGAAAGGTCACATGTCAATCGAATCTCTCAATGCGTTC | Forward ygcW |
| GR622 | TTTTATTTGATGCCTCTAGATTAGCGCACTAAATAACCGCCATCAAC | Reverse ygcW |
| GR623 | GCATGCAGGAGAAAGGTCACATGGGTAAACTCACGGGCAAGACA | Forward upcA |
| GR624 | TTTTATTTGATGCCTCTAGATCAGATACCGACGCTAACCGTCTCC | Reverse upcA |
| GR625 | GCATGCAGGAGAAAGGTCACATGCAATACAACCCCTTAGGAAAAACCG | Forward yajO |
| GR626 | TTTTATTTGATGCCTCTAGATTATTTAAATCCTACGACAGGATGCGG | Reverse yajO |
| GR627 | GCATGCAGGAGAAAGGTCACATGTTTAATTCTGACAACCTGAGACTCGAC | Forward hdhA |
| GR628 | TTTTATTTGATGCCTCTAGATTAATTGAGCTCCTGTACCCCACCA | Reverse hdhA |
| GR629 | GCATGCAGGAGAAAGGTCACATGGTTCAGCGTATTACTATTGCGCC | Forward ydhF |
| GR630 | TTTTATTTGATGCCTCTAGATTACGGTACGTCGTACCCCAGTGC | Reverse ydhF |
| GR631 | GCATGCAGGAGAAAGGTCACATGTCTCATTTAAAAGACCCGACCACG | Forward yghA |
| GR632 | TTTTATTTGATGCCTCTAGATTAACCTAAATGCTCGCCGCCG | Reverse yghA |
| GR655 | GCATGCAGGAGAAAGGTCACATGGAAAGTGGTCATCGCTTTGATG | Forward ybiC |
| GR656 | TTTTATTTGATGCCTCTAGATTAGCTGGCTAACTGCTGACAGAAAG | Reverse ybiC |
| GR657 | GCATGCAGGAGAAAGGTCACATGAAAGCGTTATCCAAACTGAAAG | Forward tdh |
| GR658 | TTTTATTTGATGCCTCTAGATTAATCCCAGCTCAGAATAACTTTCC | Reverse tdh |
| GR659 | GCATGCAGGAGAAAGGTCACATGAAAACCAAAGTTGCTGCTATTT | Forward yggP |
| GR660 | TTTTATTTGATGCCTCTAGATCATTGCGCGGCCTCCC | Reverse yggP |
| GR661 | GCATGCAGGAGAAAGGTCACATGAAGCGTTACACACCTGACTTTC | Forward yqiB |
| GR662 | TTTTATTTGATGCCTCTAGACTAATAAACCGGAATCGCCATCGCT | Reverse yqiB |
| GR663 | GCATGCAGGAGAAAGGTCACATGAAATCACGTGCTGCCGTTGCAT | Forward frmA |
| GR664 | TTTTATTTGATGCCTCTAGATCAGTAACGAATTACGGTTCGAATG | Reverse frmA |
| GR665 | GCATGCAGGAGAAAGGTCACATGATTAATTATGGCGTTGTTGGTG | Forward yjhC |
| GR666 | TTTTATTTGATGCCTCTAGATTACATTACTGATGTATGTTTAATG | Reverse yjhC |
| GR667 | GCATGCAGGAGAAAGGTCACATGAGCGACAACATCCGTGTTGGGT | Forward ydgJ |
| GR668 | TTTTATTTGATGCCTCTAGATCATGCAAGGCACAAAGTCG | Reverse ydgJ |
Table 2.
Strains and plasmids used in this study.
| E. coli strain | Genotype | Reference |
|---|---|---|
| BW25113 | rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLC78 | (Datsenko and Wanner, 2000) |
| JCL16 | BW25113/F′ [traD 36, proAB+ lacIq ZΔM15 Tn10(tetr)] | (Atsumi et al., 2008) |
| JCL260 | Same as JCL16 but with ΔadhE Δfrd-ldhA Δpta Δp3B Δfnr | (Atsumi et al., 2008) |
| AL626 | Same as JCL260 but with ΔyqhD ΔadhP ΔeutG ΔyiaY ΔyjgB ΔfucO | (Rodriguez and Atsumi, 2012) |
| AL1728 | Same as AL626 but with ΔeutE ΔyahK ΔyqhE ΔgldA ΔybbO ΔyghA | This work |
| AL1550 | JCL260 harboring pSA129 | This work |
| AL1827 | AL1728 harboring pSA129 | This work |
| Plasmid name | ||
| pGR03 | p15A ori; CmR; PLlacO1: alsS-ilvCD | (Rodriguez and Atsumi, 2012) |
| pSA138 | ColE1 ori; AmpR; PLlacO1: kivd-yqhD | (Atsumi et al., 2008) |
| pSA129 | ColE1 ori; AmpR; PLlacO1: kivd | (Atsumi et al., 2010) |
| pZE12-luc | ColE1 ori; AmpR; PLlacO1: luc | (Lutz and Bujard, 1997) |
| pAL337 | ColE1 ori; AmpR; PLlacO1: kivd-yahk | This work |
| pAL338 | ColE1 ori; AmpR; PLlacO1: kivd-yphC | This work |
| pAL339 | ColE1 ori; AmpR; PLlacO1: kivd-ycjQ | This work |
| pAL340 | ColE1 ori; AmpR; PLlacO1: kivd-ybdR | This work |
| pAL341 | ColE1 ori; AmpR; PLlacO1: kivd-rspB | This work |
| pAL342 | ColE1 ori; AmpR; PLlacO1: kivd-ydjG | This work |
| pAL343 | ColE1 ori; AmpR; PLlacO1: kivd-ghrB | This work |
| pAL358 | ColE1 ori; AmpR; PLlacO1: kivd-yjjN | This work |
| pAL359 | ColE1 ori; AmpR; PLlacO1: kivd-ydfG | This work |
| pAL360 | ColE1 ori; AmpR; PLlacO1: kivd-dkgB | This work |
| pAL361 | ColE1 ori; AmpR; PLlacO1: kivd-ydjJ | This work |
| pAL362 | ColE1 ori; AmpR; PLlacO1: kivd-ydjL | This work |
| pAL363 | ColE1 ori; AmpR; PLlacO1: kivd-yhdH | This work |
| pAL364 | ColE1 ori; AmpR; PLlacO1: kivd-ybbO | This work |
| pAL365 | ColE1 ori; AmpR; PLlacO1: kivd-gldA | This work |
| pAL504 | ColE1 ori; AmpR; PLlacO1: kivd-dkgA | This work |
| pAL505 | ColE1 ori; AmpR; PLlacO1: kivd-yeaE | This work |
| pAL506 | ColE1 ori; AmpR; PLlacO1: kivd-yghZ | This work |
| pAL507 | ColE1 ori; AmpR; PLlacO1: kivd-tas | This work |
| pAL508 | ColE1 ori; AmpR; PLlacO1: kivd-ygbJ | This work |
| pAL509 | ColE1 ori; AmpR; PLlacO1: kivd-ghrA | This work |
| pAL514 | ColE1 ori; AmpR; PLlacO1: kivd-ycjS | This work |
| pAL515 | ColE1 ori; AmpR; PLlacO1: kivd-yceM | This work |
| pAL516 | ColE1 ori; AmpR; PLlacO1: kivd-yhiN | This work |
| pAL517 | ColE1 ori; AmpR; PLlacO1: kivd-yhhX | This work |
| pAL518 | ColE1 ori; AmpR; PLlacO1: kivd-ydbC | This work |
| pAL519 | ColE1 ori; AmpR; PLlacO1: kivd-ygjR | This work |
| pAL520 | ColE1 ori; AmpR; PLlacO1: kivd-ygfF | This work |
| pAL521 | ColE1 ori; AmpR; PLlacO1: kivd-yohF | This work |
| pAL553 | ColE1 ori; AmpR; PLlacO1: kivd-yciK | This work |
| pAL554 | ColE1 ori; AmpR; PLlacO1: kivd-ykgE | This work |
| pAL555 | ColE1 ori; AmpR; PLlacO1: kivd-ygcW | This work |
| pAL556 | ColE1 ori; AmpR; PLlacO1: kivd-upcA | This work |
| pAL557 | ColE1 ori; AmpR; PLlacO1: kivd-yajO | This work |
| pAL558 | ColE1 ori; AmpR; PLlacO1: kivd-hdhA | This work |
| pAL559 | ColE1 ori; AmpR; PLlacO1: kivd-ydhF | This work |
| pAL560 | ColE1 ori; AmpR; PLlacO1: kivd-yghA | This work |
| pAL580 | ColE1 ori; AmpR; PLlacO1: kivd-ybiC | This work |
| pAL581 | ColE1 ori; AmpR; PLlacO1: kivd-thd | This work |
| pAL582 | ColE1 ori; AmpR; PLlacO1: kivd-yggP | This work |
| pAL583 | ColE1 ori; AmpR; PLlacO1: kivd-yqiB | This work |
| pAL584 | ColE1 ori; AmpR; PLlacO1: kivd-frmA | This work |
| pAL585 | ColE1 ori; AmpR; PLlacO1: kivd-yjhC | This work |
| pAL586 | ColE1 ori; AmpR; PLlacO1: kivd-ydgJ | This work |
2.3. Aldehyde reductase activity assay
The strains were grown to OD600 of ~0.4 in 5 mL Luria Broth (LB) medium at 37 °C, followed by addition of 1 mM isopropyl-β-D-thio-galactoside (IPTG). Protein overexpression was performed at 30 °C for 2 h. Then 1.8 mL of cells were centrifuged at 17,000g for 10 min, resuspended in 300 μL BugBuster Protein Extraction Reagent (Novagen, San Diego, CA, USA), and incubated at room temperature for 20 min for cell lysis. The samples were centrifuged 16,000g for 20 min at 4 °C. Supernatants were taken for enzyme assays. ADH activities were measured by following the reduction of aldehyde with NADH or NADPH at 340 nm at 37 °C using a Synergy H1 Hybrid Plate Reader (BioTek Instruments, Inc., Winooski, VT). The enzyme assay was performed in a 96-well format as a 200 μL reaction. The reaction mixture was made up of 86 μL of H2O, 10 μL of 1 M MOPS (pH 7.0), 4 μL of 10 mM NAD(P)H in 10 mM Tris–HCl (pH 7.0), 50 μL of 100 mM aldehyde, and 50 μL diluted enzyme (10 μL cell lysate in 40 μL phosphate buffer (pH 7.5)). One unit of activity is defined as the oxidation of 1 μmol of NAD(P)H per minute per mg protein. Protein concentrations were measured using Advanced Protein Assay Reagent (Cytoskeleton Inc., Denver, CO). Bovine Serum Albumin (NEB) was used to prepare a standard curve.
2.4. Culture conditions
Overnight cultures were grown in 5 mL LB containing appropriate antibiotics. Antibiotic concentrations were as follows: kanamycin (50 μg/mL), chloramphenicol (40 μg/mL), ampicillin (250 μg/ mL), and tetracycline (20 μg/mL). Production was carried out with modified M9 medium as used in previous studies (Rodriguez and Atsumi, 2012; Atsumi et al., 2008; Rodriguez et al., 2014) (hereby referred to as M9P medium): 33.7 mM Na2HPO4, 22 mM KH2PO4, 8.55 mM NaCl, 9.35 mM NH4Cl, 1 mM MgSO4, 0.1 mM CaCl2, 5 g L−1 yeast extract, 50 g L−1 glucose, and 1000-fold dilution of A5 trace metal mix (2.86 g H3BO3, 1.81 g MnCl2 · 4H2O, 0.222 g ZnSO4 · 7H2O, 0.39 g Na2MoO4 · 2H2O, 0.079 g CuSO4 · 5H2O, 49.4 mg Co(NO3)2 · 6H2O per liter water). Optical densities (OD) were measured at 600 nm with a Plate Reader (BioTek Instruments, Inc., Winooski, VT).
2.5. Isobutyraldehyde production for ALR candidate screening
Overnight cultures were inoculated 1% in 5 mL M9P in 15 mL screw-cap culture tubes. Cells were grown to an OD600 of ~ 0.4 at 37°C in a rotary shaker (250 rpm), followed by addition of 1 mM IPTG. Production was performed at 37 °C in a rotary shaker (250 rpm) for 24 h. Screw-cap tubes were tightly sealed to prevent evaporation of products. 1.5 mL of culture was taken for analysis every 24 h. 1.5 mL of the cultures was centrifuged at 17,000g for 3 min; then 1 mL of the supernatants was transferred to 2 mL GC vials for GC analysis.
2.6. Bioinformatics and literature search for candidate ALRs
To acquire the list of ALR candidate screened in this study, we first searched the E. coli genome database EcoCyc (Keseler et al., 2013) for genes annotated as oxidoreductase, reductase, or dehydrogenase which also bind NAD(P)H or FAD. Literature availability on any of these genes, especially regarding the enzyme activity, was used to determine likely and unlike candidates. For example, the methyl-glyoxal reductase class of enzymes (DkgA (Ko et al., 2005; Habrych et al., 2002; Yum et al., 1999; Jeudy et al., 2006), DkgB (Ko et al., 2005; Di Luccio et al., 2006), YeaE (Ko et al., 2005), YdjG (Di Luccio et al., 2006), YghZ (Ko et al., 2005; Totir et al., 2012; Grant et al., 2003)) are known to reduce methylglyoxal, but have not been characterized with primary aliphatic aldehydes. Overexpression of ucpA is known to increase furan tolerance in E. coli and potentially act on a range of other substrates (Wang et al., 2012). The glyoxylate reductases GhrA and GhrB reduce the beta-keto group to yield a beta-hydroxy acid (Nunez et al., 2001). Though unlikely to act on primary aldehydes, these two were selected for completion.
Most of the NAD(P)H-binding oxidoreductases in E. coli have not been characterized in the literature. In this case, protein BLAST searching against non-Escherichia taxids was used to obtain information regarding conserved domains, family of the enzyme, and substrate preference of any homolog(s) characterized in other organisms (Supplementary Table S1). Specifically, BLAST searches that resulted in similarity (>80% sequence identity) to the short chain dehydrogenase family and alcohol dehydrogenase family were considered strong candidates (YahK, YgfF, YbbO, YohF, RspB, YbdR, YcjQ, YdjJ, YdjL, YphC, UcpA, YciK, YgcW, and YghA) (Supplementary Table S1). BLAST searches that identified enzymes as aldo/keto reductase or other families were also screened (YdbC, YdhF, Tas, YajO, YjhC, YkgE, GldA, Tdh, YbiC, YggP, YjjN, HdhA, and FrmA). In some cases, BLAST searching yielded little information about the enzyme, only identifying the candidate as NAD(P)H-binding oxidoreductases (YceM, YqiB, YhiN, YhhX, YhdH, YcjS, YgjR, and YdgJ). Such candidates were also included for the sake of completion.
The known ALRs (YqhD, AdhP, EutG, YiaY, YjgB, FucO, and BetA) were also blasted against the E. coli genome (Supplementary Table S1). Oxidoreductases that yielded an Expect value score less than ~E–20 (corresponding to a score= ~90 bits) were also screened, if not already selected for screening. This score threshold was chosen because it closely matched protein family and cofactor usage targeted in this study, and thus helped to narrow down the pool of candidates to test.
2.7. Deletion of all proven aldehyde reductases in E. coli
All gene deletions were carried out by the method developed by Datsenko and Wanner (2000). The deleted fragments were verified by PCR and sequencing. The method was repeated to delete all candidate genes. After each deletion, we have verified the deletions of the target genes and other previously deleted genes by PCR and sequencing.
All strains used in this study are listed in Table 2. During construction of AL1728, we found that AL626 and AL1728 had likely acquired multiple copies of the yahK gene. These genes were verifiably deleted from the target sites, but detected by PCR to still be present in another location. This rendered it difficult to construct a strain with a complete set of both the ALR gene deletions and those for competing pathways from AL626. This could have come about from the multiple iterations of P1 transduction (Thomason et al., 2007) used to create AL626 and its parent JCL260.
2.8. Ketoacid substrate feeding experiments
Overnight cultures were inoculated 1% in 5 mL M9P in 15 mL screw-cap culture tubes. Cells were grown to an OD600 of ~0.4 at 37°C in a rotary shaker (250 rpm), followed by addition of 1 mM IPTG. The cultures were incubated for 1 h after induction at 30 °C. Then metabolites of interest were added to the cultures. Production was performed at 30 °C in a rotary shaker (250 rpm) for 24 h. Screw-cap tubes were tightly sealed to prevent evaporation of products.
2.9. C10 and C12 aldehyde feeding experiments
Overnight cultures were inoculated 1% in 20 mL M9P in shake flasks. Cells were grown to an OD600 of ~0.4 at 37 °C in a rotary shaker (250 rpm). Then 10 or 15 mL of culture was added to a 250 mL screw cap flask, and 500 mg L−1 of C10 or C12 aldehyde was added to the cultures. In the case of nonane bilayer experiments, 2 mL of nonane was also added along with aldehyde. Cultures were incubates at 30 °C in a rotary shaker (250 rpm) for 24 or 48 h. Screw-cap tubes were tightly sealed to prevent evaporation of products.
2.10. Gas chromatography sample preparation
For ALR candidate screening and keto acid feeding, 1.5 mL of the cultures were centrifuged at 17,000g for 3 min; then 1 mL of the supernatants was transferred to 2 mL vials for gas chromatography (GC) analysis.
For straight chain aldehyde feeding (C10 and C12), an equal volume of ethyl acetate (Sigma) as culture was added to extract products. These samples were mixed for 10 min in a rotary shaker (250 rpm) at 30 °C. Then each sample was centrifuged at 2750g for 5 min, and 1 mL of the ethyl acetate layer was transferred into a vial GC analysis. For nonane bilayer experiments, the entire culture and nonane layer was centrifuged at 2750g for 5 min. Then1 mL of both the nonane layer and culture layer was taken for analysis.
2.11. GC analysis
Concentrations of all products were analyzed by GC equipped with a flame ionization detector (FID). The GC system is a Shimadzu GC-2010 with an AOC-20 S autosampler and AOC-20i Auto Injector. The column used was a DB-Wax capillary column (30 m length, 0.32-mm diameter, 0.50-μm film thickness) from Agilent Technologies. GC oven temperature was initially held at 40 °C for 3 min, then increased at a rate of 45 °C min until 230 °C and held for 4 min. Injector temperature was held at 225 °C and an FID detector was held at 330 °C. Injection volume was 0.5 μL, injected at a 15:1 split ratio. Helium was used as the carrier gas. Retention times from samples were compared with external standards. Standard curves were prepared by diluting pure aldehyde or alcohol into water at concentrations of 0.01, 0.1, and 1 g L−1. 100 mg L−1 of 1-pentanol was added to all samples and external standards as an internal standard.
In the case of C10 and C12 aldehydes and alcohols, the column used was a DB-FAPP capillary column (30 m length, 0.32-mm diameter, 0.50-μm film thickness) from Agilent Technologies. GC oven temperature was initially held at 40 °C for 3 min, then increased at a rate of 45 °C min until 250 °C and held for 10 min. Injector temperature was held at 225 °C and the FID detector was held at 330 °C. Injection volume was 1 μL, injected at a 15:1 split ratio. Helium was used as the carrier gas. Retention times from samples were compared with external standards. Standard curves were prepared by diluting purely the chemical into ethyl acetate at concentrations of 0.01, 0.1, and 1 g L−1. 100 mg L−1 of dodecane was added to samples and external standards as an internal standard.
3. Results and discussion
3.1. Screening aldehyde reductase candidates in E. coli
Due to the high aldehyde to alcohol ratio exhibited by E. coli strain AL626 (Rodriguez and Atsumi, 2012) (Table 2), this strain can serve as an effective tool for screening ALR activity by overexpressing candidate ALRs and measuring the resulting alcohol formation. An initial, larger pool of candidates (~70) was selected based on two initial requirements: Annotation of domains with 1. NAD(P)H/FAD dependency and 2. oxidoreductase activity. The initial pool of genes was further condensed by a BLAST (Altschul et al., 1990) analysis of those with high similarity to dehydrogenase-type enzymes (more details are in Section 2.6). This yielded 44 candidate ALRs genes encoded in the E. coli genome. We then undertook a comprehensive in vivo screening of the 44 candidate ALRs by measuring the ability of these enzymes to produce isobutanol from E. coli strain AL626 also expressing the isobutyraldehyde pathway (alsS (Bacillus subtilis), ilvCD (E. coli), kivd (Lactococcus lactis)) (Rodriguez and Atsumi, 2012) (Fig. 1a).
Fig. 1.

In vivo screening for ALR activity by isobutanol production. (a) The isobutanol pathway (AlsS, IlvCD, Kdc, and ALR). (b) Screening of 44 candidate ALRs from E. coli by measuring isobutanol production in strain AL626 which has greatly reduced ALR activity. Strain AL626 also expressing YqhD was used as a positive control and AL626 without any ALR was used as a negative control (N.C). Asterisks (*) indicates high ALR activity. Strains were grown in M9P media, then induced at OD600 ~ 0.4 with 1 mM IPTG and allowed to produce for 24 h at 37 °C in a rotary shaker (250 rpm). Error bars indicate SD (n=3).
The candidate ALR genes were cloned onto a high copy plasmid (~50 copies per cell) downstream of kivd (De la Plaza et al., 2004) (Table 2). The well-known ALR, YqhD (Rodriguez and Atsumi, 2012; Sulzenbacher et al., 2004; Perez et al., 2008; Atsumi et al., 2010; Miller et al., 2009; Jarboe, 2011; Lee et al., 2010), was used as a positive control while a plasmid with only kivd was used as a negative control. The isobutyraldehyde and isobutanol formation was then measured from each of the 44 strains (Fig. 1b).
Five of the 44 enzymes tested (~10%) produced high amounts (>0.5 g L−1 ) of isobutanol: YahK, YbbO, GldA, DkgA, and YghA (Fig. 1b). During the course of this study, the YahK enzyme was also shown by Pick et al., with extensive kinetic data, to have activity for a multitude of substrates (Pick et al., 2013). Interestingly, the dkgA (also called yqhE) gene is coded directly downstream of the well-known ALR gene, yqhD. DkgA is known to act on methylglyoxal, glyceraldehyde, valeraldehyde, benzaldehdye, and 2,5-diketo-D-gluconate (Ko et al., 2005; Jeudy et al., 2006). dkgA and yqhD are transcribed from the same promoter (Turner et al., 2011), but dkgA may also have its own promoter as well (Lee et al., 2010). Several studies using YqhD for microbial production of chemicals have been published (Atsumi et al., 2010; Jarboe, 2011; Tang et al., 2009; Clomburg and Gonzalez, 2011; Lan and Liao, 2012; Atsumi et al., 2009), yet DkgA has seen few applications. Of the other ALR enzymes identified in our study, GldA is known to act on 1,2-propanedial in addition to glycerol (Gonzalez et al., 2008). Little is known about the two enzymes, YbbO and YghA, besides that they are both annotated as oxidoreductases that have a Rossmann-fold domain (Keseler et al., 2013). The sequence similarity of these five ALRs is quite different from that of other known ALRs in E. coli (YqhD, AdhP, EutG, YiaY, YjgB, and FucO) (Rodriguez and Atsumi, 2012; Dellomonaco et al., 2011).
3.2. Activity profile of aldehyde reductases
With five positive hits for in vivo aldehyde reductase activity, we further confirmed this activity in vitro (Fig. 2). We tested the five ALRs for activity toward a spectrum of C2–C10 aldehydes: acetaldehyde, isobutyraldehyde, hexanal, octanal, and decanal. Enzymes were also tested for cofactor specificity (NADH and NADPH), and all five appear to be strictly NADPH dependent with the substrates tested.
Fig. 2.

In vitro substrate profile of ALRs. ALR enzyme activity from E. coli AL626 cell lysates expressing each ALR: (a) YahK, (b) DkgA, (c) GldA, (d) YbbO, (e) YghA, and (f) no ALR. One unit of activity is defined as the oxidation of 1 μM NADPH per minute per mg protein. Error bars indicate SD (n=3).
Both this work and the previously published work (Pick et al., 2013) confirm that YahK is strictly NAPDH dependent and has broad ALR activity (Fig. 2a). While acetaldehyde, isobutyraldehyde, and hexanal are known substrates for YahK (Pick et al., 2013), this study adds octanal and decanal to the list of accepted substrates (Fig. 2a). YahK showed activity with all substrates tested, including activity toward acetaldehyde and octanal (~0.5 μg/min/mg protein), and highest activity with isobutyraldehyde (1.2 μg/min/mg protein).
DkgA showed activity toward the medium chain substrates isobutyraldehyde and hexanal (~0.95 μg/min/mg protein) and less with octanal (0.23 μg/min/mg protein) (Fig. 2b). No activity with acetaldehyde or decanal was detected. Since DkgA has no acetaldehyde activity, it may serve as a good candidate for medium-chain alcohol production.
The other three enzymes GldA, YbbO, and YghA showed about 10-fold lower activity for any of the tested substrates than DkgA and YahK. GldA has been shown to use NAD+ for dehydrogenation of glycerol or 1,2-propanediol (Tang et al., 1979), but we did not detect any NADH dependent reductase activity with the substrates tested. GldA only had significant activity with isobutyraldehyde (0.12 μg/ min/mg protein), perhaps due to similar size compared to glycerol (Fig. 2c). YbbO showed broad substrate activity toward all the C2–C10 aldehydes with the highest activity toward hexanal and octanal (0.13 μg/min/mg protein) (Fig. 2d). YghA, on the other hand, showed activity toward isobutyraldehyde and decanal (0.05 μg/min/mg protein) and no activity toward hexanal and octanal (Fig. 2e).
3.3. Reduction in short and medium chain aldehyde reductase activity in E. coli
Now that the five candidates were shown to have ALR activity in vivo and in vitro, we were interested in the phenotype of an E. coli strain lacking all known ALRs. We deleted these five ALR genes from the E. coli AL626. All 13 targeted ALR genes (adhE, yqhD, adhP, eutG, yiaY, yjgB, fucO, betA, yahK, dkgA (yqhE), ybbO, gldA, and yghA) were deleted from the isobutanol production strain JCL260 (parent strain of AL626, Table 2), in which all major competing pathways have been deleted, creating AL1728. AL1728 showed ~60% reduction in isobutanol formation after 24 h as compared to AL626 (Fig. 3).
Fig. 3.

Comparison of isobutyraldehyde production from strain AL626 and AL1728. (a) Isobutyraldehyde pathway. (b) Isobutanol and isobutyraldehyde production from AL626 and AL1728 expressing the isobutyraldehyde pathway. AL626 also expressing yqhD was used as a positive control. (c) Isobutanol formation from AL626 and AL1728 (from (b)). Strains were grown in M9P media, then induced at OD600 ~ 0.4 with 1 mM IPTG and allowed to produce for 24 h at 37 °C in a rotary shaker (250 rpm). Error bars indicate SD (n=3).
Next, we tested the ability of this strain to convert a wide range of aldehydes to alcohols, and compared it to JCL260. It has been shown that 2-ketoacids from branched chain amino acid biosynthesis can be decarboxylated by a ketoacid decarboxylase (Kdc) to form corresponding aldehydes (Atsumi et al., 2008; De la Plaza et al., 2004) (Fig. 4a). These aldehydes are subsequently converted to alcohols by endogenous ALRs in E. coli (Rodriguez and Atsumi, 2012; Zhu et al., 2011; Atsumi et al., 2010). Thus, we first tested the ability of AL1728 to convert 2-ketoacid-derived aldehydes to alcohols as compared to JCL260. We expressed kivd in JCL260 and AL1728 (strains AL1550 and AL1827 respectively (Table 2)), and separately fed the cultures 8 g L−1 of each ketoacid (2-oxobutyrate, 2-oxovalerate, 2-oxoisovalerate, 3-methyl-2-oxovalerate, 4-methyl-2-oxovalerate, or phenylpyruvate). The growth of AL1827 was similar with that of AL1550. We then measured the resulting aldehyde and alcohol after 24 h of incubation (Fig. 4). With AL1550, the majority of ketoacids were ultimately converted to alcohols by endogenous ALR activity (Fig. 4b) to efficiencies between 89% (2-methyl-1-butanol) and 99% (1-butanol). In contrast, only 2–15% alcohol conversion occurred in AL1827, with the majority of product remaining as aldehydes (Fig. 4c). These data suggest that we have successfully identified and removed nearly all branched chain ALR activity from E. coli. That there was also a decrease in propanal and butanal as straight chain products from AL1827 versus AL1550 suggests that the majority of straight chain ALR activity has been removed as well.
Fig. 4.

Comparison of ketoacid-based aldehyde and alcohol formation from the parent strain and ALR deleted strain. (a) Ketoacid based alcohol formation by Kdc and ALR. (b, c) Measurement of individual alcohol formation from ketoacids (2-ketobutyrate, 2-ketovalerate, 2-ketoisovalerate, 2-keto-3-methylvalerate, 2-keto-4-methylvalerate, phenylpyruvate) by AL1550 (b) and AL1827 (c). Strains were grown in M9P, then induced at OD600 ~0.4 with 1 mM IPTG and allowed to express for 1 h. After 1 h, 8 g L−1 ketoacid was added to the cultures and the strains were allowed to produce for 24 h at 37 °C in a rotary shaker (250 rpm). Error bars indicate SD (n=3).
3.4. Reduction in acetaldehyde reductase activity in E. coli
Ethanol can be a major byproduct in the biological production of fuels and chemicals. Although AdhE represents a major source of ethanol formation in E. coli during fermentation (Membrillo-Hernandez and Lin, 1999), complete elimination of ethanol formation is difficult (Zhu et al., 2011). To test the degree of ethanol formation in AL1827 as compared to AL1550, we fed 8 g L−1 pyruvate into the media (Fig. 5). Pyruvate is decarboxylated to acetaldehyde which can then be converted to ethanol by endogenous ALRs (Fig. 5a). AL1550 generated 842 mg L−1 of ethanol after 24 h, while AL1827 generated only 1.3 mg L−1 ethanol (Fig. 5b). This represents 99.8% less ethanol produced than AL1550, implying that we have achieved near abolishment of acetaldehyde reductase activity in E. coli. Thus, the 13 ALRs (or a combination there of) deleted in AL1827 likely encompass the vast majority of acetaldehyde reductase activity in E. coli. It is also important to note that additional ALR activity could still exist, but may not have been expressed by the genome under the conditions used in this study.
Fig. 5.

Comparison of ethanol formation from strain AL1550 and AL1827 by decarboxylation of pyruvate. (a) Biological route to ethanol from pyruvate. Pyruvate is decarboxylated to acetaldehyde by the ketoacid decarboxylase, Kdc (L. lactis), followed by conversion of acetaldehyde to ethanol by endogenous ALR activity. Feeding pyruvate into the media increases flux toward acetaldehyde. (b) Measurement of extracellular ethanol formation from E. coli strain AL1550 and AL1827 (Table 2) expressing kivd (L. lactis) after 24 h with 8 g L−1 pyruvate added to the media. Strains were grown in M9P media, then induced at OD600 ~ 0.4 with 1 mM IPTG and allowed to express for 1 h. After 1 h, 8 g L−1 pyruvate was added to the cultures and the strains were allowed to produce for 24 h at 37 °C in a rotary shaker (250 rpm). Error bars indicate SD (n=3).
3.5. Reduced long chain aldehyde reductase activity in E. coli
Since E. coli strain AL1827 showed almost no ALR activity toward short or medium chain substrates after 24 h, we hypothesized it might also show reduced long-chain aldehyde reductase activity.
Thus, we tested AL1827 for its ability to convert long chain aldehydes (C10 and C12) to alcohols as compared to AL1550 (Fig. 6). We fed 500 mg L−1 of each aldehyde and observed the concentration of aldehyde and alcohol after 24 h (Fig. 6b and c). AL1550 was able to convert roughly 90% of C10 and C12 aldehyde after 24 h, while AL1827 showed 90:10 ratio of C10 aldehyde:alcohol and about 74:26 ratio of C12 aldehyde:alcohol. This low long chain alcohol phenotype is highly desirable for alkane production and may be useful for in vivo screening of ADO mutants since aldehydes remain in the culture with low alcohol conversion.
Fig. 6.

Comparison of C10 and C12 alcohol formation from strain AL1550 and AL1827 by aldehyde feeding. (a) Fatty aldehydes are converted to alcohols by endogenous ALRs. (b, c) Measurement of individual C10 and C12 aldehyde conversion to alcohol by E. coli strain AL1550 (b) and AL1827 (c) after 24 h with 500 mg L−1 of individual aldehydes added to the media. (d, e) Time course of C10 aldehyde conversion to alcohol in AL1550 (d) and AL1827 (e). Strains were grown in 15 mL M9P media. At OD600 ~ 0.4, 500 mg L−1 aldehyde was added to the cultures and the strains were incubated for 24 h at 30 °C in a rotary shaker (250 rpm). Error bars indicate SD (n=3).
We observed a ~90% decrease of aldehyde presence in AL1827 cultures during the course of 24 h (Fig. 6c). This trend can also be seen in an 8 h time course with 500 mg L−1 C10 aldehyde and either with AL1550 or AL1827 (Fig. 6d and e). However, while aldehyde decreased in AL1827 cultures, little corresponding alcohol was detected. Another possibility is that the aldehyde was converted to an acid, but only a small amount of C10 or C12 acid was observed. Thus, the destination of C10 and C12 aldehydes in AL1827 cultures is unknown. Whatever the final product, its conversion is likely a less benign product than alcohols, since we also observed toxicity with both C10 and C12 aldehydes in AL1827 cultures.
When engineering E. coli for the production of toxic products, bilayer systems have been previously incorporated to extract the toxic products in situ (Rodriguez et al., 2014; Connor et al., 2010). Thus, to reduce the unknown fate of the long chain aldehydes as well as the toxicity, we incorporated a nonane bilayer with C10 aldehyde substrate and measured conversion after 48 h (Fig. 7). AL1550 still displayed a high conversion rate of C10 aldehyde to alcohol, whereas AL1827 still showed little alcohol formation. In addition, the nonane layer protected the aldehyde from uptake or unknown degradation in AL1827, which retained 490 mg L−1 C10 aldehyde after 48 h. As demonstrated here and in previous studies, both bilayer systems (Rodriguez et al., 2014; Connor et al., 2010) as well as gas stripping systems (Rodriguez and Atsumi, 2012; Zhu et al., 2011) can be applied successfully to alleviate the toxic effects of aldehydes and other products in E. coli.
Fig. 7.
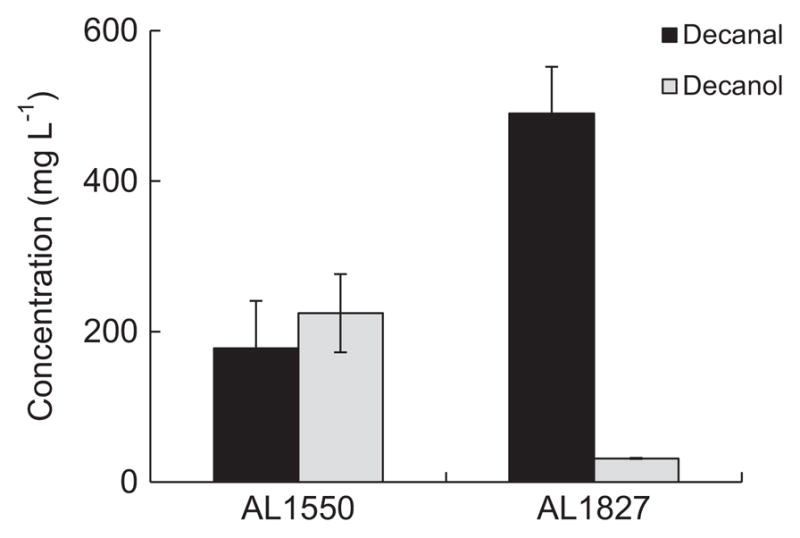
Comparison of C10 aldehyde conversion using a nonane bilayer from strain AL1550 and AL1827. Strains were grown in M9P media. At OD600 ~ 0.4, 500 mg L−1 aldehyde was added to the 10 mL of culture along with 2 mL nonane in a 250 mL screw cap flask, and the strains were incubated for 48 h at 30 °C in a rotary shaker (250 rpm). Error bars indicate SD (n=3).
4. Conclusion
In this study, we make available the information necessary to reduce short, medium, and long chain alcohol formation which could lead to increased productivities of aldehydes and alkanes from an engineered E. coli. We elucidated five additional aldehyde reductases coded on the E. coli genome, bringing the total amount of ALRs to 13 (AdhE, YqhD, AdhP, EutG, YiaY, YjgB, BetA, FucO, YahK, DkgA, YbbO, YghA, and GldA). We also demonstrated that an E. coli strain lacking these genes leads to near abolishment of endogenous ALR activity. Strains with reduced ALR activity such as the ones used in this study can serve as a valuable tool for rapid and convenient in vivo screening of ALRs and their substrate profile. While we attempted to remove all 13 aldehyde reductases with proven activity from the E. coli’s genome for the sake of completion, such an exhaustive effort would be unnecessary for engineering useful production strains. Targeting certain ALRs for their specific endogenous expression, activity, and substrate profile could reduce the necessary gene deletions to 3–5 genes.
For future efforts in alkane production from E. coli, the activity of ADO remains a limiting factor. Several studies have been conducted to improve alkane production, yet overall productivities remain low (Schirmer et al., 2010; Khara et al., 2013; Harger et al., 2013; Choi and Lee 2013; Akhtar et al., 2013; Howard et al., 2013; Andre et al., 2013). Better enzymes need to be engineered through either rational or directed evolution approaches, as well as improving the recharge rate of the diiron enzyme system.
Supplementary Material
Acknowledgments
This work was supported by University of California-Davis startup fund and the Hellman Fellowship to S.A. G.M.R. was supported by a US National Institutes of Health Biotechnology Training Grant Fellowship (T32-GM008799) and a Sloan Fellowship. We thank Eric Walters and Yohei Tashiro for experimental assistance.
Appendix A. Supporting information
Supplementary data associated with this paper can be found in the online version at http://dx.doi.org/10.1016/j.ymben.2014.07.012.
References
- Akhtar MK, Turner NJ, Jones PR. Carboxylic acid reductase is a versatile enzyme for the conversion of fatty acids into fuels and chemical commodities. Proc Natl Acad Sci USA. 2013;110:87–92. doi: 10.1073/pnas.1216516110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andre C, Kim SW, Yu XH, Shanklin J. Fusing catalase to an alkane-producing enzyme maintains enzymatic activity by converting the inhibitory byproduct H2O2 to the cosubstrate O2. Proc Natl Acad Sci USA. 2013;110:3191–3196. doi: 10.1073/pnas.1218769110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451:86–89. doi: 10.1038/nature06450. [DOI] [PubMed] [Google Scholar]
- Atsumi S, et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng. 2008;10:305–311. doi: 10.1016/j.ymben.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Atsumi S, Higashide W, Liao JC. Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol. 2009;27:1177–1180. doi: 10.1038/nbt.1586. [DOI] [PubMed] [Google Scholar]
- Atsumi S, et al. Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl Microbiol Biotechnol. 2010;85:651–657. doi: 10.1007/s00253-009-2085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Lee SY. Microbial production of short-chain alkanes. Nature. 2013;502:571–574. doi: 10.1038/nature12536. [DOI] [PubMed] [Google Scholar]
- Cintolesi A, Clomburg JM, Gonzalez R. In silico assessment of the metabolic capabilities of an engineered functional reversal of the beta-oxidation cycle for the synthesis of longer-chain (C>/=4) products. Metab Eng. 2014;23:100–115. doi: 10.1016/j.ymben.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Clomburg JM, Gonzalez R. Metabolic engineering of Escherichia coli for the production of 1,2-propanediol from glycerol. Biotechnol Bioeng. 2011;108:867–879. doi: 10.1002/bit.22993. [DOI] [PubMed] [Google Scholar]
- Connor MR, Cann AF, Liao JC. 3-Methyl-1-butanol production in Escherichia coli: random mutagenesis and two-phase fermentation. Appl Microbiol Biotechnol. 2010;86:1155–1164. doi: 10.1007/s00253-009-2401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellomonaco C, Clomburg JM, Miller EN, Gonzalez R. Engineered reversal of the beta-oxidation cycle for the synthesis of fuels and chemicals. Nature. 2011;476:355–359. doi: 10.1038/nature10333. [DOI] [PubMed] [Google Scholar]
- De la Plaza M, Fernandez de Palencia P, Pelaez C, Requena T. Biochemical and molecular characterization of alpha-ketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acids by Lactococcus lactis. FEMS Microbiol Lett. 2004;238:367–374. doi: 10.1016/j.femsle.2004.07.057. [DOI] [PubMed] [Google Scholar]
- Di Luccio E, Elling RA, Wilson DK. Identification of a novel NADH-specific aldo-keto reductase using sequence and structural homologies. Biochem J. 2006;400:105–114. doi: 10.1042/BJ20060660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes CC, et al. Simplified process for ethanol production from sugarcane bagasse using hydrolysate-resistant Escherichia coli strain MM160. Bioresour Technol. 2011;102:2702–2711. doi: 10.1016/j.biortech.2010.10.143. [DOI] [PubMed] [Google Scholar]
- Geddes CC, Nieves IU, Ingram LO. Advances in ethanol production. Curr Opin Biotechnol. 2011;22:312–319. doi: 10.1016/j.copbio.2011.04.012. [DOI] [PubMed] [Google Scholar]
- Gonzalez R, Murarka A, Dharmadi Y, Yazdani SS. A new model for the anaerobic fermentation of glycerol in enteric bacteria: trunk and auxiliary pathways in Escherichia coli. Metab Eng. 2008;10:234–245. doi: 10.1016/j.ymben.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Grant AW, Steel G, Waugh H, Ellis EM. A novel aldo-keto reductase from Escherichia coli can increase resistance to methylglyoxal toxicity. FEMS Microbiol Lett. 2003;218:93–99. doi: 10.1111/j.1574-6968.2003.tb11503.x. [DOI] [PubMed] [Google Scholar]
- Habrych M, Rodriguez S, Stewart JD. Purification and identification of an Escherichia coli beta-keto ester reductase as 2,5-diketo-D-gluconate reductase YqhE. Biotechnol Prog. 2002;18:257–261. doi: 10.1021/bp0101841. [DOI] [PubMed] [Google Scholar]
- Harger M, et al. Expanding the product profile of a microbial alkane biosynthetic pathway. ACS Synth Biol. 2013;2:59–62. doi: 10.1021/sb300061x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard TP, et al. Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli. Proc Natl Acad Sci USA. 2013;110:7636–7641. doi: 10.1073/pnas.1215966110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram LO, et al. Metabolic engineering of bacteria for ethanol production. Biotechnol Bioeng. 1998;58:204–214. doi: 10.1002/(sici)1097-0290(19980420)58:2/3<204::aid-bit13>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Jarboe LR. YqhD: a broad-substrate range aldehyde reductase with various applications in production of biorenewable fuels and chemicals. Appl Microbiol Biotechnol. 2011;89:249–257. doi: 10.1007/s00253-010-2912-9. [DOI] [PubMed] [Google Scholar]
- Jeudy S, Monchois V, Maza C, Claverie JM, Abergel C. Crystal structure of Escherichia coli DkgA, a broad-specificity aldo-keto reductase. Proteins. 2006;62:302–307. doi: 10.1002/prot.20710. [DOI] [PubMed] [Google Scholar]
- Keseler IM, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41:D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khara B, et al. Production of propane and other short-chain alkanes by structure-based engineering of ligand specificity in aldehyde-deformylating oxygenase. ChemBioChem. 2013;14:1204–1208. doi: 10.1002/cbic.201300307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, et al. Conversion of methylglyoxal to acetol by Escherichia coli aldo-keto reductases. J Bacteriol. 2005;187:5782–5789. doi: 10.1128/JB.187.16.5782-5789.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan EI, Liao JC. ATP drives direct photosynthetic production of 1-butanol in cyanobacteria. Proc Natl Acad Sci USA. 2012;109:6018–6023. doi: 10.1073/pnas.1200074109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, et al. Transcriptional activation of the aldehyde reductase YqhD by YqhC and its implication in glyoxal metabolism of Escherichia coli K-12. J Bacteriol. 2010;192:4205–4214. doi: 10.1128/JB.01127-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, et al. Evidence for only oxygenative cleavage of aldehydes to alk(a/e)nes and formate by cyanobacterial aldehyde decarbonylases. Biochemistry. 2012;51:7908–7916. doi: 10.1021/bi300912n. [DOI] [PubMed] [Google Scholar]
- Liu R, et al. Metabolic engineering of fatty acyl-ACP reductase-dependent pathway to improve fatty alcohol production in Escherichia coli. Metab Eng. 2014;22:10–21. doi: 10.1016/j.ymben.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado HB, Dekishima Y, Luo H, Lan EI, Liao JC. A selection platform for carbon chain elongation using the CoA-dependent pathway to produce linear higher alcohols. Metab Eng. 2012;14:504–511. doi: 10.1016/j.ymben.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Membrillo-Hernandez J, Lin EC. Regulation of expression of the adhE gene, encoding ethanol oxidoreductase in Escherichia coli: transcription from a downstream promoter and regulation by Fnr and RpoS. J Bacteriol. 1999;181:7571–7579. doi: 10.1128/jb.181.24.7571-7579.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EN, et al. Silencing of NADPH-dependent oxidoreductase genes (yqhD and dkgA) in furfural-resistant ethanologenic Escherichia coli. Appl Environ Microbiol. 2009;75:4315–4323. doi: 10.1128/AEM.00567-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez MF, Pellicer MT, Badia J, Aguilar J, Baldoma L. Biochemical characterization of the 2-ketoacid reductases encoded by ycdW and yiaE genes in Escherichia coli. Biochem J. 2001;354:707–715. doi: 10.1042/0264-6021:3540707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. Microbial engineering for the production of advanced biofuels. Nature. 2012;488:320–328. doi: 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- Perez JM, Arenas FA, Pradenas GA, Sandoval JM, Vasquez CC. Escherichia coli YqhD exhibits aldehyde reductase activity and protects from the harmful effect of lipid peroxidation-derived aldehydes. J Biol Chem. 2008;283:7346–7353. doi: 10.1074/jbc.M708846200. [DOI] [PubMed] [Google Scholar]
- Pick A, Ruhmann B, Schmid J, Sieber V. Novel CAD-like enzymes from Escherichia coli K-12 as additional tools in chemical production. Appl Microbiol Biotechnol. 2013;97:5815–5824. doi: 10.1007/s00253-012-4474-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch-Deere CA, Oliver JW, Rodriguez GM, Atsumi S. Synthetic biology and metabolic engineering approaches to produce biofuels. Chem Rev. 2013;113:4611–4632. doi: 10.1021/cr300361t. [DOI] [PubMed] [Google Scholar]
- Raff DK, Butanals Ullman’s Encyclopedia of Industrial Chemistry 2013 [Google Scholar]
- Rodriguez GM, Atsumi S. Escherichia coli by removing aldehyde reductase activity. Microb Cell Fact. 2012;11:90. doi: 10.1186/1475-2859-11-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Tashiro Y, Atsumi S. Expanding ester biosynthesis in Escherichia coli. Nat Chem Biol. 2014;10:259–265. doi: 10.1038/nchembio.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer A, Rude MA, Li X, Popova E, del Cardayre SB. Microbial biosynthesis of alkanes. Science. 2010;329:559–562. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]
- Sulzenbacher G, et al. Crystal structure of E. coli alcohol dehydrogenase YqhD: evidence of a covalently modified NADP coenzyme. J Mol Biol. 2004;342:489–502. doi: 10.1016/j.jmb.2004.07.034. [DOI] [PubMed] [Google Scholar]
- Tang CT, Ruch FE, Lin ECC. Purification and properties of a nicotinamide adenine dinucleotide-linked dehydrogenase that serves an Escherichia coli mutant for glycerol catabolism. J Bacteriol. 1979;140:182–187. doi: 10.1128/jb.140.1.182-187.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Tan Y, Zhu H, Zhao K, Shen W. Microbial conversion of glycerol to 1,3-propanediol by an engineered strain of Escherichia coli. Appl Environ Microbiol. 2009;75:1628–1634. doi: 10.1128/AEM.02376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. Curr Protoc Mol Biol. 2007;1(17):1–8. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
- Totir M, et al. Macro-to-micro structural proteomics: native source proteins for high-throughput crystallization. Plos One. 2012;7:e32498. doi: 10.1371/journal.pone.0032498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PC, et al. Escherichia coli genes yqhD and dkgA that are involved in furfural tolerance. J Ind Microbiol Biotechnol. 2011;38:431–439. doi: 10.1007/s10295-010-0787-5. [DOI] [PubMed] [Google Scholar]
- Wang X, Miller EN, Yomano LP, Shanmugam KT, Ingram LO. Increased furan tolerance in Escherichia coli due to a cryptic ucpA gene. Appl Environ Microbiol. 2012;78:2452–2455. doi: 10.1128/AEM.07783-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warui DM, et al. Detection of formate, rather than carbon monoxide, as the stoichiometric coproduct in conversion of fatty aldehydes to alkanes by a cyanobacterial aldehyde decarbonylase. J Am Chem Soc. 2011;133:3316–3319. doi: 10.1021/ja111607x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum DY, Lee BY, Pan JG. Identification of the yqhE and yafB genes encoding two 2, 5-diketo-D-gluconate reductases in Escherichia coli. Appl Environ Microbiol. 1999;65:3341–3346. doi: 10.1128/aem.65.8.3341-3346.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, et al. Enhancing fatty acid production by the expression of the regulatory transcription factor FadR. Metab Eng. 2012;14:653–660. doi: 10.1016/j.ymben.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Zhu H, Gonzalez R, Bobik TA. Coproduction of acetaldehyde and hydrogen during glucose fermentation by Escherichia coli. Appl Environ Microbiol. 2011;77:6441–6450. doi: 10.1128/AEM.05358-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.