

Abstract
The advent of next-generation sequencing (NGS) technology has plummeted the cost of whole genome sequencing, which has provided a long list of putative drug targets for a variety of diseases ranging from infectious diseases to cancers. The majority of these drug targets are still awaiting high-quality small molecule ligands to validate their therapeutic potential and track their druggability. Screening compound libraries based on privileged scaffolds is an efficient strategy to identify potential ligands to distinct biological targets. 7H-Pyrrolo[3,2-f]quinazoline (PQZ) is a potential privileged heterocyclic scaffold with diverse pharmacological properties. A number of biological targets have been identified for different derivatives of PQZ. This review summarized the synthetic strategies to access the chemical space associated with PQZ and discussed their unique biological profiles.
1. Introduction
High throughput screening (HTS) has become one of the primary strategies to discover novel chemical modulators of various proteins involved in different disease states.1 Often these proteins are discovered by genomic and epigenomic studies enabled by next-generation sequencing technology.2, 3 When the protein targets are not well-characterized for rational drug design, HTS is perhaps the only avenue to identify the small molecule modulators. One of the keys to a successful HTS campaign is to have ready access to high-quality chemical library. Hundreds of thousands of small molecule compounds have been collected in the screening libraries in the public domain. Additional millions of compounds are in the possession of private sector in the large pharmaceutical companies. These libraries have been screened numerous times against multiple targets and pathways in the past two decades. As new biological targets are evolved from genomic studies, these libraries will continue to provide starting points to identify potential chemical modulators. Because many of the newly unveiled targets fall into the category of “undruggable” targets like transcription factors and structural proteins,4–6 additional library members with unique structural and biological features will be in critical need. In this context, chemical library members from privileged chemical scaffolds will be highly desirable.
As initially put by Evans,7 a privileged chemical structure is defined as a chemical scaffold with distinct and specific biological activities towards multiple unrelated biological targets. For example, benzodiazepine is a classical privileged scaffold. Prominent members with benzodiazepine scaffold include widely used sedative drugs diazepam (Figure 1) and nitrazepam.8 These drugs bind γ-aminobutyric acid (GABAA)-receptors to potentiate the inhibitory neurotransmitter GABA’s action.9 Other members with benzodiazepine scaffold have been shown to be cholecystokinin (CCK) receptor antagonists7, 10 (e.g. L-364,718) and apoptosis-inducing agents11, 12 (e.g. Bz-423) (Figure 1). Spirooxindole is another widely studied privileged scaffold that is present in both synthetic and naturally occurring products with drug-like properties. These include MDM2 inhibitor MI-77301,13 muscarinic M1 and 5-HT2 receptor modulator pteropodine,14 and cell cycle inhibitor spirotryprostatin A (Figure 1).15, 16 Chemical libraries from privileged scaffolds thus represents rich source of chemical ligands to various biological targets. There have been excellent reviews to cover the array of privileged scaffolds.17, 18 In this review, we will discuss the chemistry and pharmacological activities of 7H-pyrrolo[3,2-f]quinazoline-1,3-diamine (PQZ). We conclude that PQZ is emerging as a potential privileged scaffold.
Figure 1.

Selected structures with privileged benzodiazepine or spirooxindole.
2. Chemical Synthesis of PQZs
The synthesis of the tricyclic PQZs, 7H-pyrrolo[3,2-f]quinazoline-1,3-diamine (2) in particular, has attracted much of chemists’ attention due to their wide range of pharmacological activities. For example, compound 2 potently inhibited human dihydrofolate reductase (DHFR) with Ki = 1 nM.19 One core issue of the synthetic pathways is to construct the unique tricyclic skeleton. The other focus of the syntheses is to efficiently and regioselectively introduce different substitutions to different positions on the scaffold. Therefore, diverse methods have been employed to synthesize functionalized PQZs. The first method to prepare the tricyclic ring system in 2 was reported by Ledig (Scheme 1)20 and this method remains the method of choice with some variations. In his synthesis, 5-aminoindole hydrochloride (1) was chosen as the starting material to condense with sodium dicyanamide under reflux in 1-octanol for 13 h to directly afford PQZ 2.
Scheme 1.
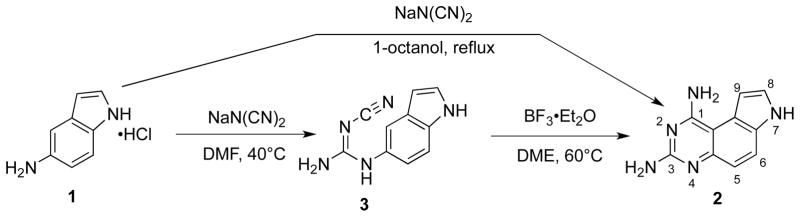
Synthesis of PQZ 2.
To avoid the long-time heating and consequent decomposition of intermediate 3 in Ledig’s one-pot procedure, Jones et al isolated intermediate 3 and synthesized 2 through a two-step process as shown in Scheme 1.21 With the assistance of boron trifluoride (BF3) as a Lewis acid, the cyclization condition was much milder than Ledig’s method. Importantly, the overall yield of 2 was higher and formation of the alternative linear isomer was efficiently suppressed. All of these were beneficial for product isolation and purification. We later found that the currently commercially available 5-aminoindole had to be converted into its corresponding hydrochloride in order to obtain high yield of 2.22
The differential pKas of the three nitrogen atoms (N1, N3 and N7) is a key factor for regioselective substitution reactions on PQZ.22 To predict the pKa difference, Chen et al optimized the structure of 2 using quantum mechanics calculation at HF/6-31G** level of theory and calculated the Mulliken atomic charges.23 The results suggested that the order of pKa is N7<N1≤N3. Therefore, it was predicted that N7–H could be selectively deprotonated for substitution. This hypothesis was consistent with experimental results. For example, regioselective N7 alkylation24 and acylation22 were realized with benzylic halides and acylating reagents (e.g. acid anhydrides and N-hydroxysuccinimide esters) under basic condition to afford 4 and 5, respectively (Scheme 2). The minor pKa difference between N1 and N3 was exploited to convert 5 to N1-acylated analogs 6 with a strong base like NaH in DMF.22
Scheme 2.

Regioselectively synthesis of mono-N-substituted PQZs 4, 5, 6 and 9.
To achieve regioselective N3-acylation of 2, Chen et al took advantage of the relatively high nucleophilicity of N3.22 Therefore, the N1-amino group was temporarily protected as –OH by acidic hydrolysis25 to give compound 7. Nucleophilic acylation of 7 selectively generated 8. Then, with the aid of BOP reagent,26, 27 N1 amine was regenerated using an SNAr displacement reaction between ammonia (NH3/MeOH) and 8 to afford N3-acylated analogs 9.
To illustrate the generality in exploiting the differential pKas of the three nitrogen atoms (N1, N3 and N7), Chen et al further demonstrated the synthesis of all the possible combinations of bisacetylated PQZs (Scheme 3).22 For example, as N7-H in 6a and 8a could be selectively deprotonated by NaH in DMF to react with acetic anhydride to provide bisacetylated PQZs 10 and 11. Rearrangement of acetyl group from N7 to N1 in 11 gave N1, N3-biacetylated derivative 12. Finally, N1,N3,N7-triacetylated compound 13 could be obtained by heating a mixture of 2 and excess acetic anhydride in DMF at 100 °C.22
Scheme 3.
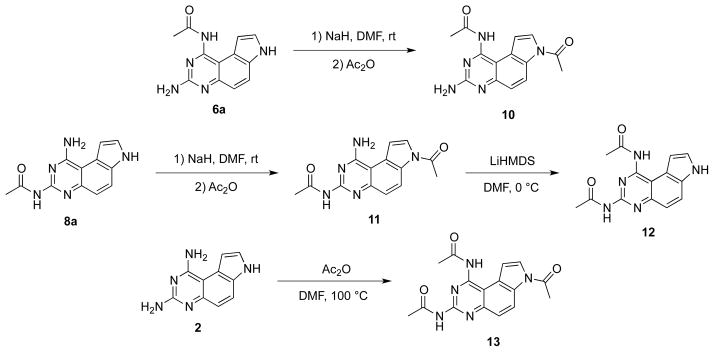
Synthesis of bis- and tri-acetylated PQZs 10, 11, 12 and 13.
Ahn et al reported a synthesis of N7-(isopropyl)benzylpyrrolo[3,2-f]quinazolines 17 and 18, which were substituted by different amino groups at C1 and C3, respectively (Scheme 4).24 In this synthesis, 5-nitroindole (14) was used as the starting material, which was N-alkylated with 4-isopropylbenzyl chloride followed by catalytic hydrogenation to afford substituted 5-aminoindole 15. Compound 16 was obtained by chlorination of corresponding hydroxyl intermediate to be obtained by cyclization between 15 and trichloromethyl isocyanate. Based on the differential leaving group ability of the chlorine atoms, different amino groups were sequentially introduced at position 1 and 3.
Scheme 4.

Synthesis of PQZs 17 and 18.
Kuyper and co-workers synthesized a series of 8-alkyl and 7,8-dialkyl-1,3-diaminopyrrolo[3,2-f]quinazolines (Scheme 5).19 These syntheses were based on the original method developed by Ledig, but using different substituted 5-aminoindoles. The key intermediates, 5-nitroindole derivatives 26, were obtained by two approaches. In the first one, an SNAr substitution of 4-fluoronitrobenzene (19) or reductive amination of 4-nitroaniline (20) was employed to prepare para-substituted nitro anilines 21. After iodination of 21 with ICl, an alkynyl moiety was introduced to the ortho-position of the amino group by palladium-catalyzed Sonogashira coupling to yield 22. Conversion of 22 into 26 was catalyzed by CuI in DMF under reflux. In the alternative method to prepare 26, the nitro group was introduced after construction of the indole ring in 25. Finally, the target compounds 28 were prepared from intermediates 27 under the same conditions as reported by Ledig or Jones.20, 21
Scheme 5.

Synthesis of 8-substituted PQZs 28.
A series of 3-phenyl-pyrroloquinazolinones with platelet aggregation inhibitory activity were discovered by Ferlin et al (Scheme 6).28 In their synthesis of the target compounds, condensation between 31 and substituted 5-aminoindoles 29 afforded 32, which were cyclized at high temperature (250 °C) to give the final products 33. In an analogous synthesis of tetracyclic pyrimido[4,5-c]carbazoles 36, montmorillonite K-10 clay was used as a Lewis acid to catalyze the cyclization of N-indole-N′-ethoxycarbonylamidine compounds 35 (Scheme 7).29
Scheme 6.
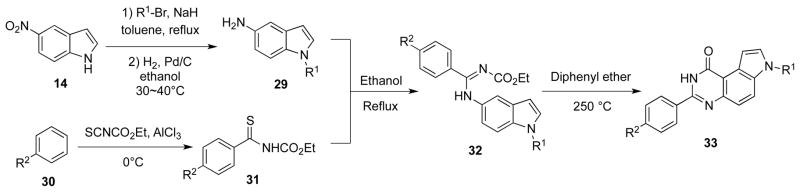
Synthesis of 3-phenyl-pyrroloquinazolinones 33.
Scheme 7.
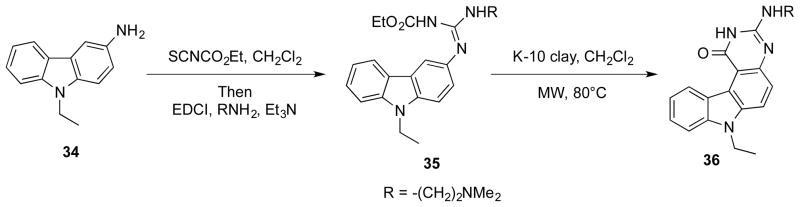
Synthesis of pyrimido[4,5-c]carbazole 36.
Pyrrolo[3,2-f]quinazolinone was a motif in indolo[2,3-a]pyrimido[5,4-c]carbazoles 42 (Scheme 8), which showed potent inhibitory activity against renal cancer cells. These compounds were synthesized by a key C-C bond ring-closure reaction of 41. This oxidation step was effected by photocyclization with iodine under continuous UV irradiation.30
Scheme 8.

Synthesis of indolo[2,3-a]pyrimido[5,4-c]carbazoles 42.
3. Pharmacological activities of PQZs
A wide range of biological activities have been described for various substituted and fused PQZs. The biochemical targets behind these bioactivities were better understood for some than the others. We divided this section into the biochemical targets that were modulated by different PQZs. Representative members from each class are presented to illustrate their potential medical applications.
3.1 PQZs as dihydrofolate reductase (DHFR) inhibitors
DHFR is an enzyme that catalyzes the reduction of dihydrofolate to tetrahydrofolate in the presence of NADPH as the cofactor.31 In proliferating cells, this DHFR-catalyzed reaction is the sole source to tetrahydrofolate, which is necessary for the one-carbon transfer reactions to synthesize purine, thymidine and some amino acids. Inhibition of DHFR will lead to blockage of DNA and protein synthesis, both of which are critical for the proliferating cells. Therefore, DHFR inhibitors exhibit activity against various cancers, bacterial infections and parasitic diseases. A large number of PQZs have been evaluated as inhibitors of DHFR from various species.19 In fact, the original design of 2 was to mimic the known structural elements required for DHFR inhibition.19, 20 These structure-activity relationship (SAR) studies showed that a large hydrophobic group appended to N7 was optimal for DHFR inhibition.19, 32 Therefore, the majority of structural analogs of 2 in the literature possess an N7-alkyl group.19, 20
3.1.1 Antimicrobial agents
Castaldo et al reported a series of PQZs DHFR inhibitors against Candida infections.33 Among these compounds, 43 and 44 (Figure 2) showed the most potent inhibitory activity against 40 clinical Candida isolates, with mean minimal inhibitory concentrations (MICs) of 0.64 μg/mL and 1.39 μg/mL, respectively. In comparison with the treatment with these agents alone, the combination of 44 and sulfamethoxazole (SMX), a structural analog of para-aminobenzoic acid (PABA) to suppress the synthesis of folic acid, resulted in synergistic effect in inhibiting the growth of susceptible Candida isolates. This synergistic effect was due to the sequential suppression of dihydropteroate synthetase (DHPS) and DHFR, both of which are key enzymes in the synthesis of tetrahydrofolic acid.
Figure 2.

Representative PQZs as DHFR inhibitors.
Kuyper et al designed and synthesized 7,8-dialkyl-1,3-diaminopyrrolo[3,2-f]quinazolines as novel DHFR inhibitors.19 These compounds were designed based on the X-ray crystal structure of Candida albicans DHFR in complex with a PQZ derivative.34 SAR studies indicated that alkyl substitutions at N7 were indispensable to retain potent DHFR inhibitory activity. This was consistent with the binding mode that the N7 substituent pointed toward a large hydrophobic pocket in DHFR.19 In addition, they found that the inhibitory activity on fungal DHFR increased significantly with compounds having C8 substituted with a small group. As one of the most potent inhibitors among these congeners, 45 (Figure 2) not only exhibited potent inhibitory potency against C. albicans and human DHFR (Kis 30 pM and 0.3 pM respectively), but also efficiently protected the mice from infections caused by Pneumocystis carinii and Candida albicans.19
3.1.2 Anti-cancer agents
McCormack et al evaluated a series of PQZs as potential anticancer agents.35 A benzyl group attached to N7 was crucial to retain excellent inhibitory potency, while the non-substituted or methylated derivatives were almost three orders of magnitude less potent. N7-Heteroaromatic substituents such as picolyl groups conferred a comparable activity to the N7-benzyl groups. Consistent with the known roles of DHFR in cancer cells, the potent DHFR inhibitor 46 (Figure 2) significantly suppressed the growth of L1210 leukemia cell line (IC50 40 nM). This was similar to the inhibitory effect of methotrexate, a widely used anticancer DHFR inhibitor.
In addition to the mentioned antimicrobial activity (see section 3.1.1), compound 45 also showed potent inhibitory activity against the growth of HCT-8 colon cancer cells (IC50 0.74 nM) and many other human cancer cell lines.19 Importantly, 45 did not seem to be a substrate for human P-glycoprotein because it was equipotent in the parental and multiple drug resistance (MDR) cell lines such as KBV-1, MCF-7/ADR and P388/ADR.19 Furthermore, 45 significantly extended the survival time of the mice with intraperitoneal implants of P388 and P388/ADR tumors in vivo.
Compound 45 showed excellent pharmacokinetic profile in male beagle dogs.36 After intravenous and oral administration, the corresponding mean plasma half-lives (t1/2) were 2.8 ± 0.66 and 4.0 ± 0.76 h. The maximum concentration (Cmax = 0.17 ± 0.039 μg/mL) was reached in about 1.8 h after oral administration. In comparison with another DHFR inhibitor piritrexim (PTX), 45 exhibited more extensive distribution in tissues, with a volume of distribution (Vd) about 9.3 L/g. The absolute bioavailability in beagle dogs was 49 ± 16%. Moreover, the pharmacokinetic profile of 45 was not significantly affected by co-administration of calcium leucovorin that is often used to prevent the adverse effect of DHFR inhibitors in the clinic. These preclinical data suggested that 45 was a good candidate for further clinical development.
3.2 PQZs as protease-activated receptor (PAR) antagonists
Thrombin is a plasma trypsin-like serine protease and promotes platelet activation and aggregation.37 The cellular effect of thrombin is partially mediated by G protein-coupled protease-activated receptors (PARs).38 Activation of PARs is initiated by the cleavage of the receptor’s N-terminal exodomain by thrombin. Then the resulting new N-terminus, acting as a tethered ligand, binds to an extracellular loop of the receptor and induces transmembrane signaling.38, 39 Among the four identified PARs (PAR1, PAR2, PAR3 and PAR4), PAR1 is critical for modulation of thrombin signalling in vascular endothelial cells.40 Moreover, PAR1, expressed on the surface of human platelets, shows higher affinity to thrombin than PAR4. This higher affinity translates to activation of human platelets by lower concentrations of thrombin.41, 42
Since inhibition of platelet aggregation could be a promising treatment strategy for thrombosis and restenosis,43 there has been strong interest in developing PAR antagonists. Researchers at then Schering-Plough initiated a HTS campaign to identify non-peptidic PAR1 antagonists.24 From this screening, PQZ 47 (Figure 3) was identified as a hit with an IC50 of 300 nM. A series of related PQZs were designed and synthesized to investigate their SARs. It was found that modifications including alkylation, acylation and sulfonylation at N7 other than benzylation significantly decreased binding activity to PAR1. Modifications at N1 and N3 generated compounds with variable potency. Small alkyl groups at N3 were favoured while substitution at N1 was generally detrimental. In the absence of a crystal structure of the complex between PAR1 and PQZs, these SAR results were rationalized by the later-developed 3D-QSAR (three-dimensional quantitative structure-activity relationship) models.44
Figure 3.

Representative PQZs as PAR1 antagonists.
Among the synthesized PQZs as PAR antagonists, N3-cyclopropylated compounds 48 (SCH79797) and 49 (SCH203099) were the most potent analogs (Figure 3).24 Both of the compounds significantly blocked the binding between PAR1 on the platelet membranes and a PAR1 peptidic agonist ha-TRAP [Ala-Phe(p-F)-Arg-Cha-HArg-Tyr-NH2]45 with IC50 values of 56 and 52 nM, respectively. Although none of these PAR antagonists were evaluated for DHFR inhibition, 47 would be predicted to be a sub-nM to low nM DHFR inhibitor based on known SAR,19, 32, 35 suggesting its preferential selectivity towards DHFR. However, it is well-accepted that primary amino groups at N1 and N3 are required for optimal DHFR inhibition. This is rationalized by the crystal structure of a PQZ in complex with DHFR where extensive hydrogen-bond networks are involved with the primary amino groups.46 Importantly, additional pockets do not exist in DHFR to tolerate substitutions at N1 and N3.46 By this reasoning, compounds 48 and 49 would not be expected to be potent DHFR inhibitors, suggesting that different substitutions in PQZ can impart different target selectivity. This feature is often the case for privileged scaffolds.
When tested in functional assays, compounds 48 and 49 blocked platelet aggregation induced either by ha-TRAP or thrombin.24 Further biochemical and pharmacological characterization of 48 suggested that it presented high PAR subtype selectivity for PAR1 by using subtype-selective agonists.24, 47 For example, it did not inhibit platelet aggregation induced by a PAR4-selective agonist (GYPGQV).24 When the PAR selectivity profile was evaluated in hCASMC (human coronary artery smooth muscle cells), 48 substantially inhibited the transient increase of cytosolic free Ca2+ concentration ([Ca2+]i) elicited by thrombin (3 nM) or TFLLRNPNDK-NH2 (30 μM), a PAR-1-selective agonist. On the other hand, it could not suppress the increase of [Ca2+]i induced by a PAR2-selective agonist, SLIGKV-NH2 (10 μM).47
In addition to its role in blood clotting, thrombin is also an important factor to mediate myocardial ischemia/reperfusion (I/R) injury.48 Functional inhibition of thrombin could result in a significant decrease in infarct size following I/R.49 Based on these findings, PAR1 was hypothesized as a potential target for I/R.50 Before or during ischemia, treatment with 48 introduced an immediate reduction of infarct size and decrease of myocardial necrosis.50 If given after ischemia, 48 could also decrease adverse left ventricular (LV) remodelling and preserve LV function through late stage antifibrotic events.51 Compound 48 down-regulated the downstream signalling activities of PAR1, which is a crucial initiator of tissue stiffening in cardiac fibroblasts after I/R injury. Therefore, PAR1 inhibitors could be a potential therapy to both myocardial I/R injury and postinfarction cardiomyopathy.
During a study on the fused pyrroloheterocycles, Ferlin et al reported new phenyl pyrroloquinolinones as potential antimitotic agents.52 They also discovered that a series of 3-phenyl-pyrroloquinazolinones displayed platelet aggregation inhibitory activity.28 Compound 50 (Figure 3) exhibited the most potent antagonistic activity towards the thrombin-induced platelet aggregation among these derivatives. The comparison between 50 and other analogs showed that both alkyl groups at pyrrolic nitrogen and a methoxy group on the appendant phenyl ring attenuated inhibitory activity. Besides blocking platelet activation, 50 also affected PAR agonist-induced increase of cytosolic [Ca2+] in platelets. Although these biological readouts would be consistent with antagonism of PAR, direct PAR binding data were not established. In fact, its inhibitory effect on platelet aggregation induced by PAR-independent collagen would suggest alternative mechanism of action than PAR inhibition.28 Overall, the SAR of PQZs as PAR antagonists is distinctly different from that of DHFR inhibition.
3.3 PQZs as protein tyrosine phosphatase 1B (PTP1B) inhibitors
PTP1B is the first protein tyrosine phosphatase to be cloned. Functional studies of PTP1B provided convincing evidence that it is a negative regulator of insulin sensitivity.53, 54 Therefore, pharmacological inhibitors of PTP1B would represent a promising strategy to develop therapeutics for type 2 diabetes (T2D).55 Roche scientists screened their internal collection of compounds for small molecule PTP1B inhibitors to yield a series of substituted PQZs (Figure 4).56–58 A large number of PQZs, most of which contained an aryl substitution at C6, were prepared to evaluate their PTP1B inhibitory activity. No detailed SAR studies have been published, but the IC50 for these compounds ranged from 1.09–91.79 μM,58 which is ~1000 fold less potent than the prototypical PQZ DHFR inhibitors. Two representative compounds 51 and 52 are shown in Figure 4. NMR studies showed that these compounds do not directly bind to the phosphate binding pocket, suggesting a unique mechanism of action.56
Figure 4.

Representative PQZs as PTP1B inhibitors.
3.4 Miscellaneous targets
Guan et al followed up a series of antimalarial PQZs initially prepared by Ledig20 and identified WR227825 (Figure 5), which exhibited potent antimalarial activity both in vitro and in vivo, but with high host toxicity.25 To reduce the host toxicity, different types of substitution were introduced to the amino groups at C1 and C3, such as alkyl and acyl groups. One such modified compound 53 (Figure 5) not only showed excellent antimalarial activity against various clones of Plasmodium falciparum in vitro, but also good efficacy in mice and non-human primates. Compound 53 could be rapidly deacetylated in plasma to generate WR227825 and therefore was considered as a prodrug.59 Although it has not been established if WR227825 inhibits pfDHFR biochemically, its structure would predict it was a potent pfDHFR inhibitor and its antimalarial activity likely arose from pfDHFR inhibition.
Figure 5.

Representative PQZs with miscellaneous pharmacological activities.
The safety profile of 53 improved significantly compared to that of WR227825.60 In the adult rat P. berghei infection model, the therapeutic index of 53 was 80, while that of WR227825 was only 10. The metabolism of 53 to WR227825 included three steps of sequential hydrolysis of acetamide.59 Since the metabolic intermediates had similar activity to WR227825, the therapeutic potency of 53 was not affected by such transformations in vivo. In fact, the multiple-step metabolic process was the major reason for its decreased host toxicity to allow gradual release of toxic WR227825.59
Electrostatic complementarity is a crucial force for binding interactions between small molecule ligands and receptors.61–63 Chen et al designed and synthesized a series of mono-N-acylated PQZs to modulate the electrostatics of PQZ ligands in a different way from the other reported PQZ modifications.22 These regioselectively mono-N-acylated compounds were evaluated in a phenotypic anti-proliferative assay with two triple negative breast cancer cell lines, MDA-MB-231 and MDA-MB-468. It was found that N7-acylated derivatives dramatically decreased their solubility in DMSO or aqueous buffers, which precluded detailed biological evaluations. On the other hand, the N1- and N3-acylated analogs displayed reasonable solubility and they presented variable anti-proliferative activity. In general, the N1-acylated compounds were less potent than N3-acylated ones. Within the series of N3-acylated PQZs, N3-arylacyl substitution was favoured.22 Among these compounds, 54 exhibited superior activity in both of the two cell lines (Figure 5). Importantly, 54 was not toxic to normal human mammary epithelial cells. The anti-proliferative activity of 54 was not due to human DHFR inhibition and its biological target(s) to mediate its selective toxicity in cancer cells remain to be identified.22
In addition to the diverse biological activities mentioned above, other bioactivities have also been reported for PQZs (Table 1). For example, PQZ 55 was reported to inhibit the replication of herpes simplex virus 1 (HSV-1) with IC50 of 15 μM (Figure 5).64 PQZ 56 was shown to weakly (IC50 ~100 μM) inhibit serum paraoxonase (PON), a calcium-dependent esterase to hydrolyze organophosphate (Figure 5).65 Other more extensively fused PQZs have also been shown to present different biological activities. Tetracyclic pyrimido[4,5-c]carbazoles 36 (Scheme 7) showed moderate potency to inhibit growth of leukemia cell line HL60.29 Through replacement of the lactam ring in the prototypical kinase inhibitor staurosporine (57) with a bioisosteric pyrimidone ring, indolo[2,3-a]pyrimido[5,4-c]carbazole 58 was obtained as a novel anti-cancer agent to inhibit proliferation of various cancer cell lines in the low μM concentration range (Figure 5).30 Finally, compound 2 is currently collected in the National Institutes of Health (NIH) molecular library and has been screened more 600 times in different assays,66 and it was found to be active as an inhibitor of dual specificity tyrosine-phosphorylation-regulated kinase 1A (Dyrk1A), DNA repair proteins Rad52 and Rad54 (Table 1). These screening results still await further validation. It is known that compound 2 is brightly fluorescent with excitation at 340 nm and emission at 450 nm35 and appropriate controls should be included during validation to eliminate fluorescent interference. Interestingly, a survey of screenings performed at NIH suggested that compound 2 does not seem to be a good scaffold for kinases although aminoquinazolines are present in many potent kinase inhibitors.67–70 Among the 22 screening assays deposited on PubChem against different kinases, only Dyrk1A was weakly inhibited with an IC50 of 5.51 μM.66
Table 1.
Miscellaneous bioactivities of PQZs.
4. Conclusions and perspectives
Since the initial design and synthesis of PQZ in the 1970s, a large number of PQZs has been prepared from different medicinal chemistry programs. Although PQZs were originally designed as DHFR inhibitors, many new biological activities have been described for the PQZs. These new discoveries would not be possible without the aid of HTS assays. As the PQZs will be continued to be assayed in new screening programs, other bioactivities will undoubtedly be uncovered. These results clearly show that PQZ is emerging as a potential privileged scaffold. Different synthetic strategies have been developed to differentially functionalize the PQZ nucleus, allowing us to access the previously untouched chemical space to fuel further discovery of novel bioactivities. As discussed above, many of the bioactivities associated with PQZs were from phenotypic screening assays without knowing their exact biochemical targets. Identification of their targets and understanding their selectivity profiles will be critical in delivering high quality chemical probes to interrogate the biology and/or therapeutic candidates for various diseases. The recent development in chemical proteomics will definitively contribute to our better understanding of these latter aspects.71, 72 Further biological studies with compounds derived from PQZ will likely provide novel chemical tools to modulate various targets and potential therapeutics for different diseases.
Acknowledgments
We thank the members of Xiao laboratory for helpful discussions. The presented work from our group was partially supported by Oregon Health & Science University, National Institutes of Health (R01GM087305) and Susan G. Komen for the Cure.
References
- 1.Kodadek T. Nat Chem Biol. 2010;6:162–165. doi: 10.1038/nchembio.303. [DOI] [PubMed] [Google Scholar]
- 2.Mwenifumbo JC, Marra MA. Nat Rev Genet. 2013;14:321–332. doi: 10.1038/nrg3445. [DOI] [PubMed] [Google Scholar]
- 3.Fricke WF, Rasko DA. Nat Rev Genet. 2014;15:49–55. doi: 10.1038/nrg3624. [DOI] [PubMed] [Google Scholar]
- 4.Network TCGA. Nature. 2012;490:61–70. [Google Scholar]
- 5.Darnell JE. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 6.Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Curr Cancer Drug Targets. 2010;10:384–391. doi: 10.2174/156800910791208535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 8.Sternbach LH. J Med Chem. 1979;22:1–7. doi: 10.1021/jm00187a001. [DOI] [PubMed] [Google Scholar]
- 9.Sigel E. Curr Top Med Chem. 2002;2:833–839. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- 10.Chang RS, Lotti VJ. Proc Natl Acad Sci U S A. 1986;83:4923–4926. doi: 10.1073/pnas.83.13.4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blatt NB, Bednarski JJ, Warner RE, Leonetti F, Johnson KM, Boitano A, Yung R, Richardson BC, Johnson KJ, Ellman JA, Opipari AW, Jr, Glick GD. J Clin Invest. 2002;110:1123–1132. doi: 10.1172/JCI16029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boitano A, Ellman JA, Glick GD, Opipari AW., Jr Cancer Res. 2003;63:6870–6876. [PubMed] [Google Scholar]
- 13.Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, Stuckey JA, Meagher JL, Bai L, Liu L, Hoffman-Luca CG, Lu J, Shangary S, Yu S, Bernard D, Aguilar A, Dos-Santos O, Besret L, Guerif S, Pannier P, Gorge-Bernat D, Debussche L. Cancer Res. 2014;74:5855–5865. doi: 10.1158/0008-5472.CAN-14-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang TH, Matsumoto K, Tohda M, Murakami Y, Takayama H, Kitajima M, Aimi N, Watanabe H. Eur J Pharmacol. 2002;444:39–45. doi: 10.1016/s0014-2999(02)01608-4. [DOI] [PubMed] [Google Scholar]
- 15.Cui CB, Kakeya H, Osada H. Tetrahedron. 1996;52:12651–12666. [Google Scholar]
- 16.Edmondson S, Danishefsky SJ, Sepp-Lorenzino L, Rosen N. J Am Chem Soc. 1999;121:2147–2155. [Google Scholar]
- 17.Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drewry DH, Macarron R. Curr Opin Chem Biol. 2010;14:289–298. doi: 10.1016/j.cbpa.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 19.Kuyper LF, Baccanari DP, Jones ML, Hunter RN, Tansik RL, Joyner SS, Boytos CM, Rudolph SK, Knick V, Wilson HR, Caddell JM, Friedman HS, Comley JCW, Stables JN. J Med Chem. 1996;39:892–903. doi: 10.1021/jm9505122. [DOI] [PubMed] [Google Scholar]
- 20.Ledig KW. 4118561. US. 1978
- 21.Jones ML, Kuyper LF, Styles VL, Caddell JM. J Heterocycl Chem. 1994;31:1681–1683. [Google Scholar]
- 22.Chen J, Kassenbrock A, Li BX, Xiao X. MedChemComm. 2013;4:1275–1282. doi: 10.1039/C3MD00134B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mulliken RS. J Chem Phys. 1955;23:1833–1840. [Google Scholar]
- 24.Ahn HS, Arik L, Boykow G, Burnett DA, Caplen MA, Czarniecki M, Domalski MS, Foster C, Manna M, Stamford AW, Wu Y. Bioorg Med Chem Lett. 1999;9:2073–2078. doi: 10.1016/s0960-894x(99)00339-x. [DOI] [PubMed] [Google Scholar]
- 25.Guan J, Zhang Q, O’Neil M, Obaldia N, Ager A, Gerena L, Lin AJ. Antimicrob Agents Chemother. 2005;49:4928–4933. doi: 10.1128/AAC.49.12.4928-4933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan ZK, Wacharasindhu S, Binnun E, Mansour T. Org Lett. 2006;8:2425–2428. doi: 10.1021/ol060815y. [DOI] [PubMed] [Google Scholar]
- 27.Wan ZK, Wacharasindhu S, Levins CG, Lin M, Tabei K, Mansour TS. J Org Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 28.Ferlin MG, Borgo C, Deana R. Eur J Med Chem. 2012;48:275–283. doi: 10.1016/j.ejmech.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 29.Debray J, Zeghida W, Baldeyrou B, Mahieu C, Lansiaux A, Demeunynck M. Bioorg Med Chem Lett. 2010;20:4244–4247. doi: 10.1016/j.bmcl.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 30.Pierce LT, Cahill MM, Winfield HJ, McCarthy FO. Eur J Med Chem. 2012;56:292–300. doi: 10.1016/j.ejmech.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Schnell JR, Dyson HJ, Wright PE. Annu Rev Biophys Biomol Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 32.Chen JC, Qian L, Shen Y, Chen LM, Zheng KC. Chin J Chem. 2006;24:1531–1537. [Google Scholar]
- 33.Castaldo RA, Gump DW, McCormack JJ. Antimicrob Agents Chemother. 1979;15:81–86. doi: 10.1128/aac.15.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baccanari DP, Tansik RL, Joyner SS, Fling ME, Smith PL, Freisheim JH. J Biol Chem. 1989;264:1100–1107. [PubMed] [Google Scholar]
- 35.McCormack JJ, Allen BA, Ledig KW, Hillcoat BL. Biochem Pharmacol. 1979;28:3227–3229. doi: 10.1016/0006-2952(79)90066-2. [DOI] [PubMed] [Google Scholar]
- 36.Studenberg SD, Woolley JL, Deangelis DV, Wargin WA. Biopharm Drug Disposition. 1997;18:433–442. doi: 10.1002/(sici)1099-081x(199707)18:5<433::aid-bdd32>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 37.Stassen JM, Arnout J, Deckmyn H. Curr Med Chem. 2004;11:2245–2260. doi: 10.2174/0929867043364603. [DOI] [PubMed] [Google Scholar]
- 38.Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, Weis WI, Coughlin SR, Kobilka BK. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vu TKH, Hung DT, Wheaton VI, Coughlin SR. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 40.Coughlin SR. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 41.Covic L, Gresser AL, Kuliopulos A. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- 42.Shapiro MJ, Weiss EJ, Faruqi TR, Coughlin SR. J Biol Chem. 2000;275:25216–25221. doi: 10.1074/jbc.M004589200. [DOI] [PubMed] [Google Scholar]
- 43.Baykal D, Schmedtje JF, Jr, Runge MS. Am J Cardiol. 1995;75:82B–87B. doi: 10.1016/0002-9149(95)80019-o. [DOI] [PubMed] [Google Scholar]
- 44.Dixit A, Kashaw SK, Gaur S, Saxena AK. Biorg Med Chem. 2004;12:3591–3598. doi: 10.1016/j.bmc.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 45.Feng DM, Veber DF, Connolly TM, Condra C, Tang MJ, Nutt RF. J Med Chem. 1995;38:4125–4130. doi: 10.1021/jm00020a029. [DOI] [PubMed] [Google Scholar]
- 46.Whitlow M, Howard AJ, Stewart D, Hardman KD, Kuyper LF, Baccanari DP, Fling ME, Tansik RL. J Biol Chem. 1997;272:30289–30298. doi: 10.1074/jbc.272.48.30289. [DOI] [PubMed] [Google Scholar]
- 47.Ahn HS, Foster C, Boykow G, Stamford A, Manna M, Graziano M. Biochem Pharmacol. 2000;60:1425–1434. doi: 10.1016/s0006-2952(00)00460-3. [DOI] [PubMed] [Google Scholar]
- 48.Raivio P, Lassila R, Petäjä J. Ann Thorac Surg. 2009;88:318–325. doi: 10.1016/j.athoracsur.2008.12.097. [DOI] [PubMed] [Google Scholar]
- 49.Chong AJ, Pohlman TH, Hampton CR, Shimamoto A, Mackman N, Verrier ED. Ann Thorac Surg. 2003;75:S649–S655. doi: 10.1016/s0003-4975(02)04691-x. [DOI] [PubMed] [Google Scholar]
- 50.Strande JL, Hsu A, Su J, Fu X, Gross GJ, Baker JE. Basic Res Cardiol. 2007;102:350–358. doi: 10.1007/s00395-007-0653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, Strande JL. J Cardiovasc Pharmacol Ther. 2013;18:460–475. doi: 10.1177/1074248413485434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gasparotto V, Castagliuolo I, Ferlin MG. J Med Chem. 2007;50:5509–5513. doi: 10.1021/jm070534b. [DOI] [PubMed] [Google Scholar]
- 53.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 54.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Combs AP. J Med Chem. 2010;53:2333–2344. doi: 10.1021/jm901090b. [DOI] [PubMed] [Google Scholar]
- 56.Cheung AWH, Banner B, Bose J, Kim K, Li S, Marcopulos N, Orzechowski L, Sergi JA, Thakkar KC, Wang BB, Yun W, Zwingelstein C, Berthel S, Olivier AR. Bioorg Med Chem Lett. 2012;22:7518–7522. doi: 10.1016/j.bmcl.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 57.Berthel SJ, Cheung AW-H, Kim K, Thakkar KC, Yun W. 2004101568. WO. 2004
- 58.Berthel SJ, Thakkar KC. US 7226915 B2. US Pat. 2004
- 59.Li Q, Kozar MP, Shearer TW, Xie LH, Lin AJ, Smith KS, Si Y, Anova L, Zhang J, Milhous WK, Skillman DR. Antimicrob Agents Chemother. 2007;51:2898–2904. doi: 10.1128/AAC.00932-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie LH, Li Q, Lin AJ, Smith K, Zhang J, Skillman DS. Antimicrob Agents Chemother. 2006;50:1649–1655. doi: 10.1128/AAC.50.5.1649-1655.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao X, Antony S, Pommier Y, Cushman M. J Med Chem. 2005;48:3231–3238. doi: 10.1021/jm050017y. [DOI] [PubMed] [Google Scholar]
- 62.Muzet N, Guillot B, Jelsch C, Howard E, Lecomte C. Proc Natl Acad Sci. 2003;100:8742–8747. doi: 10.1073/pnas.1432955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kangas E, Tidor B. J Phys Chem B. 2001;105:880–888. [Google Scholar]
- 64.Dicker IB, Blasecki JW, Seetharam S. Antiviral Res. 1995;28:213–224. doi: 10.1016/0166-3542(95)00049-r. [DOI] [PubMed] [Google Scholar]
- 65.Aviram M, Rosenblat M, Bisgaier CL, Newton RS, Primo-Parmo SL, La Du BN. J Clin Invest. 1998;101:1581–1590. doi: 10.1172/JCI1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. [Accessed Jun 26, 2014];The PubChem Project. http://pubchem.ncbi.nlm.nih.gov.
- 67.DiMauro EF, Newcomb J, Nunes JJ, Bemis JE, Boucher C, Buchanan JL, Buckner WH, Cee VJ, Chai L, Deak HL, Epstein LF, Faust T, Gallant P, Geuns-Meyer SD, Gore A, Gu Y, Henkle B, Hodous BL, Hsieh F, Huang X, Kim JL, Lee JH, Martin MW, Masse CE, McGowan DC, Metz D, Mohn D, Morgenstern KA, Oliveira-dos-Santos A, Patel VF, Powers D, Rose PE, Schneider S, Tomlinson SA, Tudor YY, Turci SM, Welcher AA, White RD, Zhao H, Zhu L, Zhu X. J Med Chem. 2006;49:5671–5686. doi: 10.1021/jm0605482. [DOI] [PubMed] [Google Scholar]
- 68.Morphy R. J Med Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- 69.Ren L, Ahrendt KA, Grina J, Laird ER, Buckmelter AJ, Hansen JD, Newhouse B, Moreno D, Wenglowsky S, Dinkel V, Gloor SL, Hastings G, Rana S, Rasor K, Risom T, Sturgis HL, Voegtli WC, Mathieu S. Bioorg Med Chem Lett. 2012;22:3387–3391. doi: 10.1016/j.bmcl.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 70.Vasbinder MM, Aquila B, Augustin M, Chen H, Cheung T, Cook D, Drew L, Fauber BP, Glossop S, Grondine M, Hennessy E, Johannes J, Lee S, Lyne P, Mortl M, Omer C, Palakurthi S, Pontz T, Read J, Sha L, Shen M, Steinbacher S, Wang H, Wu A, Ye M. J Med Chem. 2013;56:1996–2015. doi: 10.1021/jm301658d. [DOI] [PubMed] [Google Scholar]
- 71.Burdine L, Kodadek T. Chem Biol. 2004;11:593–597. doi: 10.1016/j.chembiol.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 72.Schenone M, Dancik V, Wagner BK, Clemons PA. Nat Chem Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]