

Abstract
Muscle stem cell (satellite cell) activation post muscle injury is a transient and critical step in muscle regeneration. It is regulated by physiological cues, signaling molecules, and epigenetic regulatory factors. The mechanisms that coherently turn on the complex activation process shortly after trauma are just beginning to be illuminated. In this review, we will discuss the current knowledge of satellite cell activation regulation.
Keywords: Satellite cells, Quiescent stem cells, Epigenetics, Non-coding RNA, DNA methylation, Growth factors
Introduction
Skeletal muscle is voluntarily controlled striated muscle tissue that produces locomotion, postural behavior, and breathing. It is also the largest insulin-stimulated glucose utilization tissue in the body [1]. As the most abundant tissue in the human body, on average, it accounts for 40–50 % of an adult male’s and 30–40 % of an adult female’s body weight. Maintenance of muscle mass is not only critical for precise movements, but also important for optimal metabolic homeostasis. Unfortunately, due to the function and location of skeletal muscle, it is susceptible to the damage caused by overstretching, straining, trauma, everyday wear and tear, and several degenerative muscle disorders. These damages can be repaired through muscle regeneration mediated by muscle stem cells. Satellite cells represent a major group of muscle stem cells. Initially identified by Mauro [2] in 1961, satellite cells are located between the sarcolemma and the basal lamina of myofibers. These cells usually remain quiescent with a large nuclear-to-cytoplasmic ratio and a low number of mitochondria [3]. In response to exercise and injury, quiescent satellite cells are activated to enter the cell cycle, proliferate, and eventually exit at G1, fusing to form terminally differentiated multinucleated myofibers.
In addition to satellite cells, several other types of muscle-resident adult stem cells have recently been found [3]. These stem cells are also capable of muscle lineage differentiation and their activation also represents an important part of muscle regeneration, although the regulatory mechanism remains largely unknown.
There are many sophisticated reviews on satellite cells and muscle regeneration [3–12]. Here, we summarize the current literature on regulation of satellite cells and other muscle-resident stem cell activation.
Satellite cells and satellite cell activation
Although multiple types of stem cells with muscle lineage differentiation potential have been identified [13], satellite cells are the major contributor to the remarkable regenerative capabilities of skeletal muscle. Satellite cells were initially discovered by Alex Mauro more than 50 years ago using electron microscopy, as mononucleated cells located at the periphery of muscle fibers [2]. Mauro suggested that satellite cells “might be pertinent to the vexing problem of skeletal muscle regeneration” [2]. Indeed, later experiments revealed that satellite cells were able to give rise to terminally differentiated multinucleated myotubes through cell fusion to regenerate damaged myofibers [14–20].
During embryonic development, satellite cells emerge together with the muscle in which they reside and share the same origin as muscle. Satellite cells from the trunk and limb muscles originate from the dermomyotome, while the majority of the satellite cells of the craniofacial muscles are derived from the head mesoderm [4, 21–24]. The number of satellite cells reaches a peak at the neonatal stage, accounting for about 30–35 % of the total myofiber nuclei. The number decreases to ~2–7 % of the total myofiber nuclei in adulthood [25–29].
Satellite cells are activated and are more proliferative during the neonatal period to support the rapid gain in muscle mass [25, 30–32]. In contrast to the situation in neonates, the majority of satellite cells are mitotically quiescent in adults, remaining at the G0 stage, although the mechanism by which active satellite cells become quiescent after the burst of postnatal muscle mass growth is not clear yet.
The quiescent satellite cells reside in a unique niche in intact muscles [5]. They are located closely juxtaposed between the sarcolemma of muscle fibers and the basal lamina that surrounds the fiber [2]. These cells display specific gene expression profiles compared to actively proliferating satellite cells. Pax7, Pax3, M-cadherin, Syndecan-4, CD34, α7-Integrin, and CXCR4 [33–35] dominantly express in quiescent satellite cells (Fig. 1), and MyoD expression is absent in quiescent satellite cells [36, 37]. They can be quickly activated to re-enter the cell cycle and proliferate in response to extrinsic signals, a process referred to as satellite cell activation.
Fig. 1.
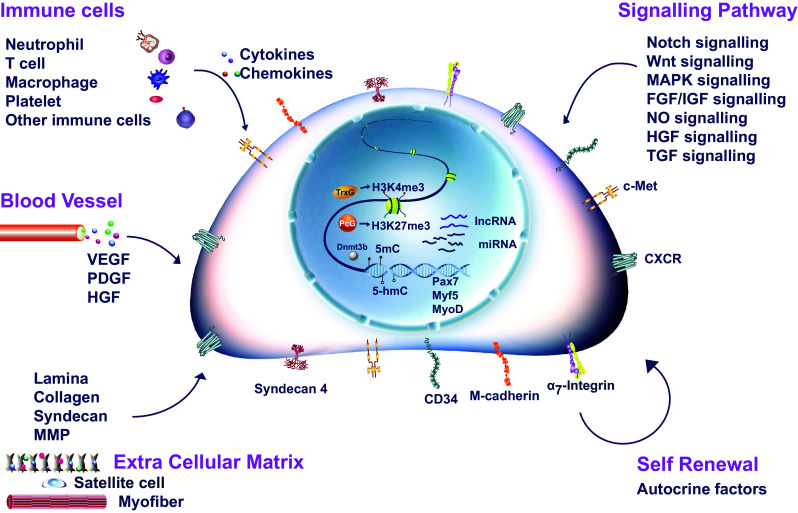
Regulation of satellite cell activation. Satellite cell specific surface markers were indicated on the cell membrane. Key transcription factors involved in satellite cell activation were indicated in the nuclei of satellite cell. Key signaling pathways regulating satellite cell activation were listed. Major microenvironment components including extra cellular matrix and neighboring cell types (immune cells and blood vessels) affecting satellite cell activation were illustrated
The morphology of activated satellite cells is different from that of quiescent satellite cells. Quiescent satellite cells are usually spindle-shaped with little cytoplasm and few organelles [38, 39], whereas activated satellite cells are larger with an expanded cytoplasm and more organelles [39, 40]. The earliest marker for activated satellite cells is phosphorylated p38, followed by MyoD [41–43]. Although detected in the majority of quiescent satellite cells, Myf5 is highly upregulated on activation [44, 45]. During G1 phase, shortly after exit from quiescence, MyoD does not promote differentiation, but instead directly regulates the expression of Cdc6, a gene involved in rendering chromatin accessible for DNA replication, allowing cell cycle entry [43, 46].
MyoD plays multiple roles at different stages of muscle differentiation in a context-dependent manner. For example, MyoD expressed in quiescent cells has been reported to inhibit cell proliferation in a differentiation-independent way [47]. In myoblasts, MyoD can also inhibit cell cycle entry by the induction of p21 and upregulation of apoptotic genes [48, 49]. After the first cell division, the proliferative cells enter the cell cycle and continue to divide every 10 h [50].
Upon stimulation, such as muscle damage, exercise, or pathogenic conditions, the satellite cells start to proliferate and give rise to a myogenic precursor cell population called myoblasts. Myoblasts can go through several rounds of amplification, then exit the cell cycle and fuse to each other or to the existing myofibers to form terminally differentiated myofibers. Some of the progeny of the activated satellite cells can restore the pool of quiescent stem cells by asymmetric self-renewal [51]. ERK signaling regulates the reversible quiescence of a subpopulation of satellite cells through ERK signaling inhibitor Spry1 [52]. Depletion of Spry1 in satellite cells increases the number of cells committed to apoptosis and reduced the number of quiescent satellite cells after muscle injury reparation, suggesting that Spy1 is required for reversible quiescence [52]. Activation of satellite cells is the critical step in the initiation of muscle regeneration. It is subjected to multiple layers of tight regulation. Physiological cues, signaling molecules, and epigenetic regulators are all involved in the orchestration of the orderly activation of satellite cells upon stimulation.
Physiological cues to induce satellite cell activation
Disruption of muscle fibers and basal lamina
The intact myofiber sarcolemma and the basal lamina provide an important niche to maintain the quiescent state of satellite cells [11]. They are destroyed in muscle injury, leading to disruption of the protective niche for quiescent satellite cells. Damages to the basal lamina destroy the collagen–laminin network, where satellite cells anchor themselves through α7/β1 integrins [53]. The mobilization of satellite cells contributes to their activation. Furthermore, destruction of the sarcolemma and the basal lamina allows an influx of calcium and the release of hepatocyte growth factor (HGF) from the extra cellular matrix (ECM) to directly activate the unprotected satellite cells [54–58].
Muscle damage also generates a large amount of destructed muscle fibers. Selective induction of fiber damage using bupivacaine, without disruption of the basal lamina and other cell types, results in elevated satellite cell activation and proliferation [59], suggesting that the damaged fibers provide signals that activate quiescent satellite cells. Indeed, these dying fibers produce nitric oxide (NO) that further activates HGF and downstream signaling to induce satellite cell activation [58]. Therefore, both disruption of the protective niche and the factors released by dying fibers contribute to the activation of quiescent satellite cells.
Infiltration of immune cells
Muscle injury, stretching, overuse, and degenerative muscle diseases induce the infiltration of large amounts of immune cells, initiated by early neutrophil invasion and followed by macrophage infiltration [60–62]. Chemokines, cytokines, and growth factors produced by macrophages, together with fibroblasts, attract more circulating inflammatory cells, including T cells and B cells. Cytokines, growth factors, and chemokines secreted by these inflammatory cells, such as IL-6 and IFNγ, can promote satellite cell activation and proliferation [50, 63–65]. Inhibition of inflammation by non-steroidal anti-inflammatory drugs in humans reduces the number of activated satellite cells, thus slowing down muscle regeneration [66–68]. Immune cells and the inflammation reaction followed by immune cell infiltration provide a microenvironment for satellite cell activation and proliferation (Fig. 1).
Blood vessels
Upon muscle injury, blood vessels are also severely disrupted. The vascular endothelial cells of damaged vessels release growth factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and HGF [13]. These factors promote satellite cell activation and proliferation [11]. Vascularization and angiogenesis are essential steps in muscle regeneration. Many growth factors enriched at the injury site to promote vessel restoration can also promote satellite cell proliferation [69, 70]. The restored blood vessels then signal the activated satellite cells to return to the quiescent state [71]. Communication between satellite cells and blood vessels can, thus, regulate the dynamic cycle of satellite cell activation and quiescence maintenance.
Signals to activate satellite cells
Upon muscle injury, a combination of signals is generated by damaged myofibers, blood vessels, and immune cells to wake up the quiescent satellite cells. The activated satellite cells also signal back to the environment to orchestrate orderly muscle regeneration (Fig. 1).
HGF
HGF is a mesenchyme-derived heparin-binding glycoprotein that regulates cell proliferation, cell survival, cell motility, and morphogenesis [72]. HGF can bind to the c-Met receptor to regulate cell growth, cell motility, morphogenesis, and organ regeneration by activating a tyrosine kinase signaling cascade [73]. Mice deficient in HGF or its receptor, c-Met, lack all the muscle groups derived from migratory hypaxial precursor cells [57, 74]. The application of exogenous HGF to somites induces ectopic delamination of myogenic precursor cells into the lateral plate mesoderm [75, 76]. These results suggest that HGF is essential in inducing the migration of myogenic precursor cells in embryonic myogenesis.
Satellite cells express both HGF and c-Met. Muscle cells and non-muscle cells in close proximity produce and secret HGF, which is sequestered in the ECM around intact muscle fibers [39, 54, 58]. Upon muscle damage, HGF is released from the ECM, promoting the entry of quiescent satellite cells into cell cycle [54–56]. Satellite cells also express HGF; therefore, the activation of satellite cells by HGF can undergo both paracrine and autocrine [56].
The presence of HGF-bound c-Met has been considered to be the initial step in satellite cell activation [40, 55]. Recently, Rodgers et al. showed that injury-related systemic signals could induce the quiescent satellite cells to transit from G0 to GAlert. The GAlert cells are primed for activation. HGF, c-Met, and the downstream mTORC1 signaling are required for this transition [77]. The addition of HGF to satellite cells cultured with single fibers induces prominent satellite cell activation [40, 56, 78], further indicating the pivotal role of HGF in this process. HGF could respond to weak signals induced by injury and prime the quiescent satellite cells for the “alert” stage. If the injury continues and the induced systemic signals cross the threshold, the alerted satellite cells will be quickly activated and muscle injury will be efficiently repaired.
The molecular mechanism by which HGF further activates satellite cells has not been fully elucidated. It may activate the downstream tyrosine kinase signaling pathway to alter the expression levels of cell cycle-related genes. The expression levels of c-fos and c-jun, the early genes of tyrosine kinase signaling, are rapidly upregulated in satellite cells 3–6 h after muscle injury. This is considered to be the immediate response to HGF-c-Met-mediated signaling. Receptor-bound HGF can also increase Twist expression [79] which further activates EMT (epithelial mesenchymal transition) [80]. Meanwhile HGF-mediated downregulation of the protein levels of p27kip1 in a p21Cip1/Waf1-independent manner in satellite cells [79], helps the cells to overcome cell cycle blockage. HGF can activate satellite cells rapidly after trauma by promoting EMT to release them from their quiescent prone niche and removing cell cycle blockages.
NO
NO is a freely diffusible small messenger capable of pleiotropic cellular functions, such as survival, stress resistance, and neurotransmission [81]. NO is produced in skeletal muscle through reactions catalyzed by nitric oxide synthase (NOS). Within 6 h post-injury, NOS mRNA levels are significantly increased in both damaged muscle fibers and the infiltrating macrophages, therefore elevating the NO levels at the injury site [82–84]. In iNOS(−/−) mice, satellite cells fail to proliferate and differentiate after injury [85], suggesting that NO is required for normal muscle reparation after injury.
NO plays multiple roles during the muscle regeneration process. At the early stage of muscle damage, it promotes macrophages to lyse damaged muscle cells in a reactive oxygen species (ROS)-independent manner to protect cells from further ROS damage [86], and stimulates the release of HGF, together with other growth factors and cytokines to activate satellite cells [87]. At the second stage of muscle regeneration, NO inhibits neutrophil-mediated lysis of muscle cells and reduces ROS generated from prolonged inflammation, protecting the activated satellite cells from ROS stress and apoptosis [82].
NO activates satellite cells not only by facilitating the release of HGF, but also by antagonizing the inhibitory effects of TGF-β on satellite cells. The administration of L-NAME, an NOS inhibitor, at the injury site in rat muscle leads to abnormally elevated TGF-β level that induces fibrosis [88].
IGF and FGFs
Insulin-like growth factor (IGF) is a circulating hormone critical for development and regeneration of almost every organ [89]. IGF signaling is initiated by binding of IGF to the IGF receptor (IGFR) to activate its tyrosine kinase activity and autophosphorylation, which in turn phosphorylates insulin receptor substrate 1 (IRS-1). Phosphorylated IRS-1 recruits the regulatory subunit of PI3K and activates it. Activated PI3K phosphorylates Akt, which then activates mTOR and p70S6 kinase to turn on the IGF-PI3K/Akt-mTOR-S6K axis of signaling pathway. This signaling process has been shown to be important for muscle mass maintenance [90]. Six IGF binding proteins, named IGFBP1-6, bind IGF in the extracellular fluid and the circulation to further regulate IGF activities [91]. The expression of IGF and all six IGFBPs has been detected in regenerating skeletal muscle [92], suggesting their roles in muscle wound healing.
Muscle damage induces the expression of alternative splicing isoforms of IGF named mechano-growth factor (MGF) and IGF-IEa [93]. MGF is only expressed in the damaged muscle and its expression is correlated with the activation of quiescent satellite cells [94]. IGF-IEa is expressed later than MGF during muscle regeneration, correlating with myoblast proliferation and differentiation [95, 96]. MGF elevates the activity of superoxide dismutase, the enzyme required for decreasing the level of ROS [97], thus protecting the satellite cells from ROS-induced damage. IGFBP6 is an IGF sequester, which increases the expression levels of IGF isoforms. However, its expression level is dramatically decreased at the early stage of muscle regeneration to allow more IGF available to activate satellite cells and promote their proliferation [98]. IGF-IR heterozygous mice display decreased the levels of MyoD expression and satellite cell activation [99], further confirming the importance of IGF in the satellite cell activation process.
The mechanism of IGF-mediated satellite cell activation has not been fully elucidated but may involve the upregulation of Myf5 expression upon injury. After muscle injury, an influx of calcium triggers calcineurin and calmodulin kinase through calcium binding to calmodulin, to activate Myf5 expression. IGF can activate Myf5 through the calcium-mediated activation pathway [100]. In addition, it could also activate Myf5 expression through PI3K/Akt and ERK signaling pathways [100, 101]. It can also activate expression of cyclin D2 to promote entry to cell cycle and cell proliferation through MEK/ERK and PI3K/AKT signaling pathways in C2C12 cells. IGF2 expression levels could be regulated by N-cadherin signaling through activation of p38α and β [102]. An immunoglobulin superfamily member Cdo can cooperate with the scaffold protein JLP to increase the level of active p38α and β, thus increasing the expression level of IGF2 during the myoblast differentiation process [103]. The similar mechanism may also contribute to the activation of satellite cells by IGF. Another immunoglobulin superfamily member, the receptor for advanced glycation end-products (RAGE) activated by HMGB1, also plays an important role in satellite cell activation. RAGE is transiently expressed in satellite cells located in injured muscles and represses Pax7 expression through activation of p38-MAPK signaling [104–107]. Deletion of RAGE in muscles leads to increased satellite cell number and Pax7 expression level [106], suggesting that RAGE is required for proper timing of muscle regeneration. RAGE may also be able to regulate satellite cell activation by repressing Pax7 expression.
Fibroblast growth factors (FGFs) belong to the family of heparin-binding circulating mitogens that regulate cell survival, proliferation, migration, differentiation, and morphogenesis. The expression of many FGF family members has been detected in skeletal muscle [108]. Among them, FGF6 has been shown to be expressed predominantly in myogenic cells, and its expression level is upregulated during muscle regeneration. In FGF6−/− mice, the number of activated satellite cells is significantly reduced and the size of quiescent satellite cells pool remains constant, suggesting that FGF6 is essential for satellite cell activation and proliferation [109]. Consistent with the results from gene knockout experiments, the addition of exogenous FGF1, 2, 4, 6, or 9 stimulates satellite cell proliferation significantly in vitro. The stimulating effects of FGF2, 4, 6, and 9 are further enhanced in combination with HGF [110, 111].
FGFs activate the downstream signaling pathways through binding to FGF receptor 1–4 (FGFR1-4), which are transmembrane tyrosine receptors. All four FGFRs are detected in satellite cells. Among them, FGFR1 and 4 are the most prominent ones [110]. The expression level of FGFR1 is significantly upregulated shortly after muscle injury, correlating with the activation of satellite cells [110]. Overexpression of FGFR1 facilitates the proliferation of cultured myoblasts and represses myoblast differentiation [112]. It may also be able to promote the proliferation of activated satellite cells.
The binding of FGF to FGFR leads to dimerization and autophosphorylation of the receptor, followed by activation of Ras signaling pathways. Overexpression of constitutively activated Ras can bypass FGF, promoting myoblast proliferation [113]. FGF can also activate MKK-ERK signaling cascade, facilitating the transition from G1 to S phase in myoblasts, thus increasing their proliferation [114]. FGF can also induce the activation of satellite cells by enhancing the G1-S transition through ERK signaling pathway.
Similar to IGFs, FGF can also regulate calcium-mediated signaling. IGF enhances intracellular calcium intake in CD34+ satellite cells, as indicated by lighting up cells with the calcium-sensitive fluorescent dye X-Rhod-1. The elevated intracellular calcium level induced by FGF2 triggers nuclear translocation of NFATc3 and NFATc2, which facilitates MyoD expression in satellite cells leading to their activation. The effects of IGF on the calcium intake of satellite cells and satellite cell activation are antagonized by the blockage of the TRPC ion channel [115].
Notch
Notch signaling is one of the major regulatory pathways in cell fate determination. Notch is a family of transmembrane receptors containing four members, Notch1–4. After binding to its ligands (Delta-like 1, Delta-like 4, Jagged 1, and Jagged 2), Notch undergoes protease cleavage to free its intracellular domain, NICD, a transcription coactivator that facilitates RBP-J kappa (Rbpj, the Notch target transcription factor) -mediated transcription activation [116, 117]. In embryonic muscle development, Notch signaling is required for myogenic cell fate commitment and muscle stem cell maintenance. Notch ligand Delta 1 (Dll1) null mutant mice displayed hypotrophy due to premature differentiation of satellite cells [118]. Consistent with it, muscle-specific depletion of Rbpj leads to loss of myogenic stem cells due to increasing differentiation during embryogenesis [119]. Muscle-specific overexpression of NICD increases Pax7+ muscle stem cell numbers and maintains these cells in an undifferentiated state [120].
Notch1, 2, 3, Notch/Rbpj targets Hey1, HeyL, and Hes1 are all expressed robustly in quiescent satellite cells. Disruption of Notch signaling by Rbpj depletion in satellite cells leads to precocious differentiation and depletion of satellite cells [121]. All these genetic data support that Notch signaling is essential for maintenance of satellite cell quiescence, and it should be downregulated in satellite cell activation. Indeed, endogenous Notch signaling is dramatically reduced in activated satellite cells isolated from regenerating muscle, compared with quiescent satellite cells [121–123]. This reduction in activity occurs within 20 h of an injury [122]. The first mitosis of activated satellite cells after injury occurs at about the same time (18-24 h) [50]. The correlation of the timing of Notch signaling downregulation and the cell cycle entrance of quiescent satellite cells indicates that these two events may be tightly coupled.
Notch signaling has also been reported to be required for satellite cell proliferation. An abnormally high numbers of satellite cells are found in Notch3 knockout mice [124]. Activation of Notch signaling in cultured satellite cells also promotes proliferation of these cells [125, 126]. These seemingly contradictory results can be explained by the presence of two waves of Notch signaling during the activation of satellite cells. The first wave maintains the quiescent state of the satellite cells, which is inactivated upon cell activation. The second wave is turned on at the proliferative stage of the activated satellite cells. The changes in Notch signaling levels during muscle regeneration support this notion. Notch signaling level decreases almost immediately after muscle injury when satellite cells are activated [122], and increases again 4–5 days post-injury [127].
Most of the current observations about the function of Notch in satellite cell activation are correlations. Whether Notch is just a passive downstream responder of satellite cell activation or actively causes the activation is still under debating. To clarify this issue, continuous monitoring of Notch signaling level during the satellite cell activation process and additional well-controlled manipulations of Notch signaling at various stages of satellite cell activation are needed.
Wnt
Wnt proteins are soluble signaling molecules regulating multiple cellular processes, including cell fate determination, stem cell proliferation, cell polarity, morphology, and tumorigenesis. The canonical Wnt signaling cascade is turned on by the binding of Wnt ligand to transmembrane receptors Frizzled and low-density lipoprotein receptor-related protein co-receptor (LRP). Ligand binding stimulates the phosphorylation of Dishevelled (Dsh) and inactivates GSK3β, triggering the stabilization of the common downstream Wnt effecter β-catenin. When Wnt signaling is off, a destruction complex composed of GSK3β, Adenomatous polyposis coli (APC), and Axin2 associates with β-catenin to drive its ubiquitination and degradation. When Wnt signaling is on, β-catenin accumulates due to the disassembly of the destruction complex and is translocated into nucleus to serve as a coactivator for TCF/LEF transcription factors to activate the expression of target genes [128].
Wnt signaling is upregulated upon muscle injury. There is increased TCF reporter activity in myogenic cells two days post-muscle injury [125]. Exogenous Wnt1, 3a, and 5a promote satellite cell proliferation, whereas Wnt4 and 6 inhibit it [3]. β-catenin can promote satellite cell self-renewal and prevent immediate satellite cell differentiation [125, 129, 130]. Consistent with the notion that Wnt promotes satellite cell proliferation, nuclear localization of β-catenin has been detected only in activated satellite cells and myoblasts, but not in differentiating muscle cells (myogenin+). Perplexingly, other observations seem to oppose the pro-proliferative functions of Wnt. When β-catenin expression level is reduced by RNAi in satellite cells, more activated Pax7+MyoD+ satellite cells are observed, whereas constitutive expression of β-catenin leads to downregulation of MyoD [130], suggesting that β-catenin inhibits satellite cell activation. In aged mice, elevated serum Wnt level inhibits the proliferation of satellite cells and directs their fate toward the fibrogenic lineage. Furthermore, injection of Wnt inhibitors Dkk1 and sFRP3 in aged mice leads to reduction of fibrogenic lineage differentiation [131, 132]. These seemingly contradictory results could be due to different systems used in the experiments and potentially altered Wnt signaling pathways in aged animals.
In addition to the canonical Wnt pathway, Wnt can also activate Rho/Rac and JNK signaling through crosstalk mediated via Dsh, to regulate cell polarity [133]. This non-canonical Wnt signaling functions in satellite cells. Wnt7a stimulates symmetrical division of satellite cells to expand the activated satellite cell pool via activation of planar cell polarity (PCP) signaling. The satellite cell number increases dramatically upon Wnt7a overexpression, whereas the depletion of Wnt7a leads to a marked decrease of satellite cell number [134]. The expansion of the activated satellite cell pool induces the expression of fibronectin, which further modifies the satellite cell niche and stimulates Wnt7a-Frizzled 7-PCP signaling to form a feedback loop during muscle regeneration [135].
Both canonical and non-canonical Wnt are involved in the regulation of satellite cell activation and proliferation. Experiments, that can pinpoint Wnt functions at a particular cell stage, i.e., the initial stage of satellite cell activation, activated satellite cell stage, myoblast stage etc., will help to elucidate the functions of Wnt signaling during satellite cell activation.
TGF-β
The transforming growth factor beta (TGF-β) superfamily comprises many secreted factors essential in nearly every aspect of cellular behavior. It is grouped into subfamilies based on sequence homology, including the TGF-β, activin, glial cell line derived neurotrophic factor (GDNF), growth/differentiation factor (GDF), and bone morphogenetic protein (BMP) subfamilies. TGF-β ligands bind to type I and II serine/threonine kinase cell surface receptors. Upon ligand binding, the type II receptor phosphorylates the type I receptor to activate it. The activated type I receptor subsequently phosphorylates receptor-regulated Smads (R-Smads), which forms a heterodimer with common mediator Smad (co-Smad) to mediate its nuclear translocation and transcription activation [136].
In general, TGF-β signaling plays a negative role in the regulation of myogenesis by repressing the expression of MyoD and myogenin [137–140]. It is highly expressed in quiescent satellite cells and repressing cell cycle progression [98]. Myostatin is a TGF-β family member expressed specifically in muscle tissue to prevent muscle growth and differentiation [141]. Myostatin maintains satellite cell quiescence and represses satellite cell self-renewal by inducing p21CIP expression [142]. Myostatin represses the expression of MyoD to prevent satellite cells from activation [143]. It can also increase Pax7 expression level through ERK signaling pathway to help maintaining satellite cell quiescence [144]. Many growth factors, including IGF and FGF, as described above, antagonize the inhibitory effects of TGF-β to activate satellite cells during muscle regeneration, although the mechanisms remain to be defined.
TGF-β level is elevated in circulation with aging, preventing satellite cells from entering into cell cycle by inducing the expression of cell cycle inhibitors such as p15, p16, p21, and p27 [145]. The injection of an antibody against TGF-β at the injury site of aged mice rejuvenates satellite cells by promoting their activation [145].
Extracellular matrix signals
Satellite cells have close contact with ECM. In addition to intracellular responses to signaling molecules, remodeling of ECM has been shown to be a critical step in satellite cell activation. Quiescent satellite cells express ECM components such as versican, fibrillin-2, and glypicans. These ECM proteins bind HGF and other growth factors to lower their effective concentration around satellite cells. Once trauma occurs in a muscle, ECM is damaged and the trapped growth factors are released to activate the quiescent satellite cells (Fig. 1).
Syndecans
Heparin sulfate proteoglycans (HSPGs) are composed of two or three linear polysaccharides covalently attached to various proteins at the cell surface and in ECM. They act as adhesion proteins and receptors for many growth factors, morphogens, and adhesion proteins [146]. Transmembrane syndecans are one of the major forms of membrane HSPGs. Satellite cells specifically express syndecan-3 and -4 [147], which serve as low affinity FGF receptors [148]. Satellite cell activation and proliferation is abolished in syndecan-4 knockout mice due to loss of MAPK signaling [149], showing syndecan-4 to be required for satellite cell activation. Syndecan-4 can also form a co-receptor complex with Frizzled-7 to facilitate Wnt7-mediated non-canonical Wnt signaling to regulate satellite cell activation [135]. In contrast, satellite cell activation is accelerated in syndecan-3 knockout mice, although the cells are arrested in S phase [149]. Syndecan-3 and -4 may work together to regulate the coherent chain of reactions during satellite cell activation and proliferation.
MMPs
Matrix metalloproteinases (MMPs) belong to a family of zinc-dependent endopeptidases capable of degrading ECM proteins. In skeletal muscle, MMPs are released at the reparation site by damaged myofibers or immune cells recruited by muscle injury [150]. MMP2 is produced by satellite cells and damaged myofibers [151–153], while MMP9 is generated by infiltrating leukocytes and macrophages [153]. NO induces the increase of the enzymatic activity and expression levels of MMP2 and MMP9. Both MMP2 and 9 cleave collagens, laminin, and other ECM components to release HGF from the sequestered sites to activate satellite cells [154, 155].
Inhibitors of TIMPs (metalloproteinases) are also involved in satellite cell activation. TIMP3 is highly expressed in quiescent satellite cells and downregulated in activated satellite cells [98]. Consequently, the activity of MMP2 is elevated in activated satellite cells [151–153]. In addition to MMP2, TIMP3 can also repress the activity of TNF-α converting enzyme to block TNF-α mediated p38 signaling pathway activation and further inhibit satellite cell activation [156].
Pax7 and Pax3 in satellite cell activation
Pax7 and Pax3 are satellite cell markers. Pax3 is expressed in satellite cells lying in most trunk and forelimb, but not hindlimb muscles [157], whereas Pax7 is expressed in satellite cells resident in all muscles [158]. The expression of Pax7, but not Pax3, prevents satellite cells from undergoing apoptosis [159]. Heterogeneity has been demonstrated in satellite cells. Some satellite cells express high levels of Pax7 (Pax7High), whereas ~3 % of satellite cells express low levels (Pax7Low) [98]. The Pax7High cells take longer to enter the cell cycle after satellite cell activation [11]. Whether the Pax7High and Pax7Low satellite cells are activated in distinct patterns remains unclear.
Pax3 and Pax7 can control the expression of Myf5 and MyoD [160], but how Pax7/3 activates MyoD upon muscle injury, in particular, the transcriptional regulatory mechanism involved, is not known. Recent work on embryonal rhabdomyosarcomas has shown that the p38 MAPK signaling pathway mediated by the receptor for advanced glycation end-products could downregulate Pax7. Pax7 may reduce MyoD level by promoting its degradation in rhabdomyosarcoma [161]. Whether the similar degradation of MyoD controlled by Pax7 occurs in satellite cells remains to be examined. Pax7 has been shown to be able to activate the transcription of MyoD and Myf5 during embryonic development and in primary myoblasts [157, 160]. Whether Pax7-mediated MyoD degradation also occurs in satellite cells, in particular at the transition from a quiescent to an activated cell, requires further study. In addition, the relationship between Pax7’s role as a transcriptional activator of MyoD, and Myf5 expression and its function as a factor promoting MyoD degradation, are of interest and should be further explored.
Inhibitor of DNA binding (Id) proteins are negative regulators of MyoD [162]. Pax7 can also regulate the expression of Id proteins [163]. Sine oculis homeobox (Six) homeodomain transcription factors are also involved in satellite cell activation by regulating MyoD expression [164]. Six1 activates MyoD expression by directly binding the MyoD promoter [165]. MyoD expression is diminished by conditional depletion of Six1 in Pax7-expressing satellite cells [166].
Satellite heterogeneity and satellite activation
Satellite cells are heterogeneous cell populations. They have various developmental origins, fiber associations, and expression profiles [10]. For example, the expression level of Pax7 varies dramatically in distinct subgroups of satellite cells. Pax7High cells display reduced metabolic activity and delayed entrance into cell cycle upon stimulation, whereas Pax7Low cells are more primed be activated [12]. A small proportion of quiescent satellite cells do not express CD34 and Myf5 [167], although the physiological significance of this has not been elucidated. The heterogeneity of satellite cells could affect the dynamics of satellite cell activation.
Myoblasts expressing desmin, MyoD, and myogenin have been observed within 12 h post-injury, when satellite cell activation is incomplete and proliferation has not yet been initiated [168]. The presence of the apparently differentiated myoblast population before satellite cell activation and proliferation could suggest the existence of two distinct satellite cell populations, the normal quiescent satellite cells and another group of more ready to differentiate [168]. Each population might be activated through different signaling pathways and display distinct kinetics. Identification of markers for each satellite cell subpopulation and development of methods for the specific isolation of each subpopulation will contribute to elucidate the specific activation mechanism governing each subpopulation. The accumulation of genomewide profiling at the single cell level will further deepen our understanding of the activation of heterogeneous satellite cells.
Epigenetic regulation in satellite cell activation
Histone modifications
Changes in histone modifications on Pax7, MyoD, Myf5, and other MRF genes occur during muscle regeneration. The Polycomb group (PcG) and Ttrithorax group (TrxG) epigenetic regulators play important roles in satellite cell activation and differentiation. PcG is composed of PRC1 and PRC2 complexes. KMT6, Ezh1, and Ezh2 are subunits of these two complexes with lysine methyltransferase activities. MLL1, MLL2, MLL3, MLL4, SET1a, and SET1b are subunits of TrxG complex with lysine methyltransferase activities. PcG marks chromatin with repressive H3K27me3 modification through its lysine methyltransferase activities whereas TrxG establishes the permissive H3K4me3 modification on chromatin [169, 170]. The transition from the transcriptionally permissive H3K4me3 modification to the repressive H3K27me3 modification is critical in cell fate determination [170]. This transition is induced by Ezh2 on the Pax7 gene upon satellite cell activation and during the subsequent proliferation stages, switching off Pax7 expression in a p38-MAPK-dependent manner [171]. The genes involved in cell cycle progression are also enriched for the permissive H3K4me3 modification in activated satellite cells [172].
Pax7 also participates in the establishment of the epigenetic pattern in satellite cell activation. In activated satellite cells and myoblasts derived from satellite cells, Pax7 recruits the Wdr5-Ash2L-MLL2 histone methyltransferase complex that methylates the H3K4 site to establish a permissive H3K4me3 modification on Myf5 [173].
A genomewide analysis of H3K4me3 and H3K27me3 profiles in purified quiescent satellite cells and activated satellite cells revealed a general lack of H3K27me3 repressive marker and the presence of H3K4me3 permissive marker on a large number of genes in quiescent satellite cells. Many non-myogenic-specific genes are labeled by the bivalent histone marker in quiescent satellite cells, which are replaced by the repressive H3K27me3 marker in activated satellite cells. The changes in the histone modification status in activated satellite cells compared with quiescent satellite cells may be due to the significant increase in the PcG subunit Ezh2 [123].
Sirt1, a member of the NAD+-dependent protein deacetylase family, is expressed in both quiescent and activated satellite cells. The reduction of Sirt1 level leads to premature differentiation [174], whereas its overexpression promotes satellite cell proliferation [175]. Sirt1 deacetylates MyoD and MEF2D to regulate their activities [176, 177]. Sirt1 interacts with Notch signaling pathway components Hes1 and Hey2 interfering with Notch activity [178]. This may also contribute to control of satellite cell activation (Fig. 1). Sirt1 can also serve as a nutrient sensor. Sirt1 interacts with ATG7 to activate autophagy through AMPK pathway. The activation of autophagic flux helps meet the high bioenergetic demands of satellite cell activation [179].
DNA methylation in satellite cell activation
DNA methylation is an important aspect of epigenetic regulation. More than three decades ago, it was demonstrated that inhibition of DNA methylation transdifferentiated fibroblasts to muscle lineage [180]. In satellite cells, Dnmt-3b, a member of the DNA methyltransferase family, is recruited to CpG islands in Notch1 promoter in an Ezh2 binding-dependent manner to mediate increased methylation of Notch1 promoter under the control of TNF-α and NF-κB. Hyper-DNA methylation of Notch1 promoter mediated by TNF-α and NF-κB leads to a reduction of satellite cell self-renewal and proliferation [181]. The application of sulforaphane, a DNA methyltransferase inhibitor, to isolated satellite cells results in downregulation of the myostatin signaling pathway, which may promote satellite cell proliferation and differentiation [182].
The importance of DNA methylation in regulating stem cell functions has only begun to be unraveled in recent years and the data accumulated so far are limited. The current observations hint that DNA methylation might be an additional aspect to regulate satellite cell activation, but the direct link between satellite cell activation and DNA methylation is still missing. More systematic investigations are required to clarify the function of DNA methylation in this process.
Non-coding RNA
Non-coding RNA (ncRNA) has been shown in the last decade to act as key regulators of gene expression. NcRNA makes up the majority of transcription products of the eukaryotic genome. It can be divided into structural and regulatory RNAs. Ribosomal RNA (rRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and tRNA are grouped as structural RNAs whose functions have been thoroughly studied for decades. Small regulatory RNA comprises microRNAs (miRNAs), piwi-interacting RNAs (piRNAs), and small interfering RNAs (siRNAs). Regulatory RNA molecules longer than 200 nucleotides are characterized as long non-coding RNA (lncRNA) [183]. In this review, we focus on the functions of miRNA and lncRNA in the process of satellite cell activation.
Micro RNA
The myomiR family is a group of miRNAs specifically expressed in muscle, including miR-1a, miR-133, miR-206, miR-208, miR-486, and miR-499. The myomiR family is involved in regulating muscle differentiation and is capable of transdifferentiating fibroblasts to muscle lineage cells [184]. miR-1 expression is upregulated during muscle regeneration. It directly represses Pax7 expression to promote satellite cell proliferation and differentiation [185].
In addition to myomiRs, there are several other miRNAs involved in satellite cell activation. Both miR-31 and the mRNA of Myf5 are sequestered in ribonucleoprotein (RNP) granules, where miR-31 interacts with the 3’ UTR of Myf5 to suppress its translation in quiescent satellite cells [44, 186]. Upon satellite cell activation, Myf5 mRNA is released by downregulation of miR-31 and breakdown of RNP granules to rapidly switch on Myf5 protein translation [44, 186]. MiR-489 is enriched in quiescent satellite cells, and downregulated in activated satellite cells. It represses the expression of oncogene Dek to prevent quiescent satellite cells from entering cell cycle [187]. MiR-181 is upregulated during muscle regeneration. It targets Hox-A11, a negative regulator of MyoD, to promote satellite cell activation and differentiation [188]. MiR-206 is highly upregulated in activated satellite cells and myoblasts. It is also highly expressed in Duchenne muscles probably due to an intensified activation of satellite cells [186, 189–192]. Upregulation of miR-206 represses expression of Pax7, Notch3, IGFBP5, and Hmgb3 to promote satellite cell differentiation [193]. Downregulation of miR-125b occurs after injury to relieve IGF2 repression and promote subsequent satellite cell activation [194]. MiR-221/222 can also regulate satellite cell activation by promoting cell cycle progression [195, 196]. MiR-1192 can inhibit the translation of HMGB1, which is highly expressed in satellite cells and myoblasts, inducing the expression of myogenic factors such as MyoD and myogenin [197, 198].
A global downregulation of miRNAs has been observed in human satellite cells during the transition from quiescence to activation, in particular miR-106b, miR-25, miR-29c, and miR-320c [199]. The functions of these miRNAs remain to be elucidated.
LncRNA
LncRNA can be grouped into cis-acting RNA and trans-acting RNA. Cis-acting RNA works in proximity to its transcription sites, whereas trans-acting RNA works at distinct loci. Both cis- and trans-acting lncRNA can recruit chromatin remodeling factors to alter the local or overall chromatin status to regulate transcription [200]. As the majority of the studies on lncRNA have been performed in C2C12 myoblast cell line, little is known about its functions in satellite cells.
The majority of known lncRNA molecules in myoblasts, such as Malat1, linc-MD1, SRA, Neat1, and YAM, are upregulated upon myoblast differentiation and are required for normal differentiation [201–207]. Linc-MD1 is located 13 kb upstream of pre-miR133b. Two microRNAs, miR133b and miR206, are located in the gene body of linc-MD1. Linc-MD1 fine-tunes the differentiation timing of myoblasts by sponging miR133b and miR206 to antagonize their repression on MAML1 and MEF2C [207]. The RNA-binding protein HuR binds linc-MD1 to facilitate the accumulation of linc-MD1 in cells and reinforce its sponge activity [201]. H19 is transcribed from the Igf2 locus and is highly expressed in adult muscle [208–210]. It interacts with PRC2 to repress the expression of Igf2, therefore inhibiting proliferation [211, 212]. H19 has several let-7 binding sites and serves as a microRNA let-7 sponge to relieve the inhibitory effects of let-7 on Igfbp2, an inhibitor of the IGF signaling pathway [213]. In myoblasts, H19 downregulates IGF signaling pathway to repress proliferation and may also play a similarly negative role in IGF signaling in satellite cells. To investigate the functions of lncRNA in satellite cell activation, expression profiles and functional assays should be carried out in quiescent and activated satellite cells.
The functions of ncRNA are just beginning to be realized. The specific functions of ncRNA during satellite cell activation, in particular, whether it can directly drive satellite cell activation, require further investigation.
Conclusion
The activation of quiescent satellite cells is orchestrated by physiological cues, signaling pathways, and epigenetic regulators. We are just beginning to unravel how this process is regulated. Many questions remain unanswered, especially with regard to transcription and epigenetic regulation. Satellite cell activation is an asynchronized and transient process in vivo. Following the live cell activation process by high-resolution imaging and other new techniques will reveal more information about the process in vivo. The identification of more key genetic mutants affecting satellite cell activation will also help to reveal the missing links in the regulatory network, while genomewide analysis of the binding profile of epigenetic regulators will further deepen our understanding.
Many factors have been found to exhibit dramatic changes when satellite cells are activated. However, whether these changes are the causes of satellite cell activation or the consequences of it remains to be identified. Genetic mutations and careful characterization of the order of events during the satellite cell activation process will shed light on this. Another important question that remains to be explored is the link between uncontrolled satellite cell activation and cancer, especially non-alveolar rhabdomyosarcoma. Further exploration of this question will reveal more targets for drug development to treat rhabdomyosarcoma.
Acknowledgments
This work was sponsored by grants from the Ministry of Science and Technology of China (2014CB964700 to P.H.), the ‘‘Strategic Priority Research Program’’ of the Chinese Academy of Sciences (XDA01010204 to P. H.), the National Natural Science Foundation of China (31171420 to P. H.), One Hundred Talents Program of Chinese Academy of Sciences 2012OHTP02 to P. H., Science and Technology Commission of Shanghai Municipality (12PJ1409700, 13JC1406500 to P. H.), and CAS-CSIRO Research Program brant No. GJHZ1504.
References
- 1.Stump CS, et al. The metabolic syndrome: role of skeletal muscle metabolism. Ann Med. 2006;38(6):389–402. doi: 10.1080/07853890600888413. [DOI] [PubMed] [Google Scholar]
- 2.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93(1):23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Relaix F, Marcelle C. Muscle stem cells. Curr Opin Cell Biol. 2009;21(6):748–753. doi: 10.1016/j.ceb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Cosgrove BD, et al. A home away from home: challenges and opportunities in engineering in vitro muscle satellite cell niches. Differentiation Res Biol Divers. 2009;78(2–3):185–194. doi: 10.1016/j.diff.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aziz A, Sebastian S, Dilworth FJ. The origin and fate of muscle satellite cells. Stem cell reviews. 2012;8(2):609–622. doi: 10.1007/s12015-012-9352-0. [DOI] [PubMed] [Google Scholar]
- 7.Pannerec A, Marazzi G, Sassoon D. Stem cells in the hood: the skeletal muscle niche. Trends Mol Med. 2012;18(10):599–606. doi: 10.1016/j.molmed.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Pallafacchina G, Blaauw B, Schiaffino S. Role of satellite cells in muscle growth and maintenance of muscle mass. Nutr Metab Cardiovasc Dis. 2013;23(Suppl 1):S12–S18. doi: 10.1016/j.numecd.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Yusuf F, Brand-Saberi B. Myogenesis and muscle regeneration. Histochem Cell Biol. 2012;138(2):187–199. doi: 10.1007/s00418-012-0972-x. [DOI] [PubMed] [Google Scholar]
- 10.Biressi S, Rando TA. Heterogeneity in the muscle satellite cell population. Semin Cell Dev Biol. 2010;21(8):845–854. doi: 10.1016/j.semcdb.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montarras D, L’Honore A, Buckingham M. Lying low but ready for action: the quiescent muscle satellite cell. FEBS J. 2013;280(17):4036–4050. doi: 10.1111/febs.12372. [DOI] [PubMed] [Google Scholar]
- 12.Brack AS, Rando TA. Tissue-specific stem cells: lessons from the skeletal muscle satellite cell. Cell Stem Cell. 2012;10(5):504–514. doi: 10.1016/j.stem.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ceafalan LC, Popescu BO, Hinescu ME. Cellular players in skeletal muscle regeneration. BioMed Res Int. 2014;2014:957014. doi: 10.1155/2014/957014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bischoff R. Regeneration of single skeletal muscle fibers in vitro. Anat Rec. 1975;182(2):215–235. doi: 10.1002/ar.1091820207. [DOI] [PubMed] [Google Scholar]
- 15.Konigsberg IR. Clonal analysis of myogenesis. Science. 1963;140(3573):1273–1284. doi: 10.1126/science.140.3573.1273. [DOI] [PubMed] [Google Scholar]
- 16.Konigsberg UR, Lipton BH, Konigsberg IR. The regenerative response of single mature muscle fibers isolated in vitro. Dev Biol. 1975;45(2):260–275. doi: 10.1016/0012-1606(75)90065-2. [DOI] [PubMed] [Google Scholar]
- 17.Moss FP, Leblond CP. Nature of dividing nuclei in skeletal muscle of growing rats. J Cell Biol. 1970;44(2):459–462. doi: 10.1083/jcb.44.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reznik M. Thymidine-3H uptake by satellite cells of regenerating skeletal muscle. J Cell Biol. 1969;40(2):568–571. doi: 10.1083/jcb.40.2.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snow MH. Myogenic cell formation in regenerating rat skeletal muscle injured by mincing. II. An autoradiographic study. Anat Rec. 1977;188(2):201–217. doi: 10.1002/ar.1091880206. [DOI] [PubMed] [Google Scholar]
- 20.Yaffe D. Cellular aspects of muscle differentiation in vitro. Curr Top Dev Biol. 1969;4:37–77. doi: 10.1016/s0070-2153(08)60480-9. [DOI] [PubMed] [Google Scholar]
- 21.Hutcheson DA, et al. Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for beta-catenin. Genes Dev. 2009;23(8):997–1013. doi: 10.1101/gad.1769009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gros J, et al. A common somitic origin for embryonic muscle progenitors and satellite cells. Nature. 2005;435(7044):954–958. doi: 10.1038/nature03572. [DOI] [PubMed] [Google Scholar]
- 23.Relaix F, et al. A Pax3/Pax7-dependent population of skeletal muscle progenitor cells. Nature. 2005;435(7044):948–953. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 24.Harel I, et al. Distinct origins and genetic programs of head muscle satellite cells. Dev Cell. 2009;16(6):822–832. doi: 10.1016/j.devcel.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allbrook DB, Han MF, Hellmuth AE. Population of muscle satellite cells in relation to age and mitotic activity. Pathology. 1971;3(3):223–243. doi: 10.3109/00313027109073739. [DOI] [PubMed] [Google Scholar]
- 26.Schmalbruch H, Hellhammer U. The number of satellite cells in normal human muscle. Anat Rec. 1976;185(3):279–287. doi: 10.1002/ar.1091850303. [DOI] [PubMed] [Google Scholar]
- 27.Lindstrom M, Thornell LE. New multiple labelling method for improved satellite cell identification in human muscle: application to a cohort of power-lifters and sedentary men. Histochem Cell Biol. 2009;132(2):141–157. doi: 10.1007/s00418-009-0606-0. [DOI] [PubMed] [Google Scholar]
- 28.Sajko S, et al. Frequency of M-cadherin-stained satellite cells declines in human muscles during aging. J Histochem Cytochem. 2004;52(2):179–185. doi: 10.1177/002215540405200205. [DOI] [PubMed] [Google Scholar]
- 29.Bareja A, Billin AN. Satellite cell therapy—from mice to men. Skelet Muscle. 2013;3(1):2. doi: 10.1186/2044-5040-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schultz E. Satellite cell proliferative compartments in growing skeletal muscles. Dev Biol. 1996;175(1):84–94. doi: 10.1006/dbio.1996.0097. [DOI] [PubMed] [Google Scholar]
- 31.Campion DR, et al. Changes in the satellite cell population during postnatal growth of pig skeletal muscle. J Anim Sci. 1981;52(5):1014–1018. doi: 10.2527/jas1981.5251014x. [DOI] [PubMed] [Google Scholar]
- 32.Mesires NT, Doumit ME. Satellite cell proliferation and differentiation during postnatal growth of porcine skeletal muscle. Am J Physiol Cell Physiol. 2002;282(4):C899–C906. doi: 10.1152/ajpcell.00341.2001. [DOI] [PubMed] [Google Scholar]
- 33.Kuang S, et al. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172(1):103–113. doi: 10.1083/jcb.200508001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seale P, et al. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102(6):777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 35.Alfaro LA, et al. CD34 promotes satellite cell motility and entry into proliferation to facilitate efficient skeletal muscle regeneration. Stem Cells. 2011;29(12):2030–2041. doi: 10.1002/stem.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornelison DD, Wold BJ. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol. 1997;191(2):270–283. doi: 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- 37.Zammit PS, et al. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166(3):347–357. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schultz E, McCormick KM. Skeletal muscle satellite cells. Rev Physiol Biochem Pharmacol. 1994;123:213–257. doi: 10.1007/BFb0030904. [DOI] [PubMed] [Google Scholar]
- 39.Wozniak AC, et al. Signaling satellite-cell activation in skeletal muscle: markers, models, stretch, and potential alternate pathways. Muscle Nerve. 2005;31(3):283–300. doi: 10.1002/mus.20263. [DOI] [PubMed] [Google Scholar]
- 40.Anderson JE. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol Biol Cell. 2000;11(5):1859–1874. doi: 10.1091/mbc.11.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kondoh K, Sunadome K, Nishida E. Notch signaling suppresses p38 MAPK activity via induction of MKP-1 in myogenesis. J Biol Chem. 2007;282(5):3058–3065. doi: 10.1074/jbc.M607630200. [DOI] [PubMed] [Google Scholar]
- 42.Jones NC, et al. The p38alpha/beta MAPK functions as a molecular switch to activate the quiescent satellite cell. J Cell Biol. 2005;169(1):105–116. doi: 10.1083/jcb.200408066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang K, Sha J, Harter ML. Activation of Cdc6 by MyoD is associated with the expansion of quiescent myogenic satellite cells. J Cell Biol. 2010;188(1):39–48. doi: 10.1083/jcb.200904144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crist CG, Montarras D, Buckingham M. Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell. 2012;11(1):118–126. doi: 10.1016/j.stem.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 45.Gayraud-Morel B, et al. Myf5 haploinsufficiency reveals distinct cell fate potentials for adult skeletal muscle stem cells. J Cell Sci. 2012;125(Pt 7):1738–1749. doi: 10.1242/jcs.097006. [DOI] [PubMed] [Google Scholar]
- 46.Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123(1):13–24. doi: 10.1016/j.cell.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 47.Sorrentino V, et al. Cell proliferation inhibited by MyoD1 independently of myogenic differentiation. Nature. 1990;345(6278):813–815. doi: 10.1038/345813a0. [DOI] [PubMed] [Google Scholar]
- 48.Halevy O, et al. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267(5200):1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- 49.Asakura A, et al. Increased survival of muscle stem cells lacking the MyoD gene after transplantation into regenerating skeletal muscle. Proc Natl Acad Sci USA. 2007;104(42):16552–16557. doi: 10.1073/pnas.0708145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocheteau P, et al. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148(1–2):112–125. doi: 10.1016/j.cell.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 51.Kuang S, et al. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129(5):999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shea KL, et al. Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell. 2010;6(2):117–129. doi: 10.1016/j.stem.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanes JR. The basement membrane/basal lamina of skeletal muscle. J Biol Chem. 2003;278(15):12601–12604. doi: 10.1074/jbc.R200027200. [DOI] [PubMed] [Google Scholar]
- 54.Allen RE, et al. Hepatocyte growth factor activates quiescent skeletal muscle satellite cells in vitro. J Cell Physiol. 1995;165(2):307–312. doi: 10.1002/jcp.1041650211. [DOI] [PubMed] [Google Scholar]
- 55.Tatsumi R, et al. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev Biol. 1998;194(1):114–128. doi: 10.1006/dbio.1997.8803. [DOI] [PubMed] [Google Scholar]
- 56.Sheehan SM, et al. HGF is an autocrine growth factor for skeletal muscle satellite cells in vitro. Muscle Nerve. 2000;23(2):239–245. doi: 10.1002/(sici)1097-4598(200002)23:2<239::aid-mus15>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 57.Maina F, et al. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell. 1996;87(3):531–542. doi: 10.1016/s0092-8674(00)81372-0. [DOI] [PubMed] [Google Scholar]
- 58.Wozniak AC, Anderson JE. Nitric oxide-dependence of satellite stem cell activation and quiescence on normal skeletal muscle fibers. Dev Dyn. 2007;236(1):240–250. doi: 10.1002/dvdy.21012. [DOI] [PubMed] [Google Scholar]
- 59.Bischoff R. Interaction between satellite cells and skeletal muscle fibers. Development. 1990;109(4):943–952. doi: 10.1242/dev.109.4.943. [DOI] [PubMed] [Google Scholar]
- 60.Toumi H, F’Guyer S, Best TM. The role of neutrophils in injury and repair following muscle stretch. J Anat. 2006;208(4):459–470. doi: 10.1111/j.1469-7580.2006.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Orimo S, et al. Analysis of inflammatory cells and complement C3 in bupivacaine-induced myonecrosis. Muscle Nerve. 1991;14(6):515–520. doi: 10.1002/mus.880140605. [DOI] [PubMed] [Google Scholar]
- 62.Fielding RA. Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am J Physiol. 1993;265(1 Pt 2):R166–R172. doi: 10.1152/ajpregu.1993.265.1.R166. [DOI] [PubMed] [Google Scholar]
- 63.Kharraz Y, et al. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm. 2013;2013:491497. doi: 10.1155/2013/491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saclier M, et al. Differentially activated macrophages orchestrate myogenic precursor cell fate during human skeletal muscle regeneration. Stem Cells. 2013;31(2):384–396. doi: 10.1002/stem.1288. [DOI] [PubMed] [Google Scholar]
- 65.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 2010;298(5):R1173–R1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mackey AL. The influence of anti-inflammatory medication on exercise-induced myogenic precursor cell responses in humans. J Appl Physiol (1985) 2007;103(2):425–431. doi: 10.1152/japplphysiol.00157.2007. [DOI] [PubMed] [Google Scholar]
- 67.Mikkelsen UR, et al. Local NSAID infusion does not affect protein synthesis and gene expression in human muscle after eccentric exercise. Scand J Med Sci Sports. 2011;21(5):630–644. doi: 10.1111/j.1600-0838.2010.01170.x. [DOI] [PubMed] [Google Scholar]
- 68.Ziltener JL, Leal S, Fournier PE. Non-steroidal anti-inflammatory drugs for athletes: an update. Ann Phys Rehabil Med. 2010;53(4):278–282. doi: 10.1016/j.rehab.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 69.Ziche M, Morbidelli L. Nitric oxide and angiogenesis. J Neurooncol. 2000;50(1–2):139–148. doi: 10.1023/a:1006431309841. [DOI] [PubMed] [Google Scholar]
- 70.Anderson JE, Wozniak AC. Satellite cell activation on fibers: modeling events in vivo—an invited review. Can J Physiol Pharmacol. 2004;82(5):300–310. doi: 10.1139/y04-020. [DOI] [PubMed] [Google Scholar]
- 71.Abou-Khalil R, et al. Autocrine and paracrine angiopoietin 1/Tie-2 signaling promotes muscle satellite cell self-renewal. Cell Stem Cell. 2009;5(3):298–309. doi: 10.1016/j.stem.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miyazawa K, et al. Proteolytic activation of hepatocyte growth factor in response to tissue injury. J Biol Chem. 1994;269(12):8966–8970. [PubMed] [Google Scholar]
- 73.Zarnegar R, Michalopoulos GK. The many faces of hepatocyte growth factor: from hepatopoiesis to hematopoiesis. J Cell Biol. 1995;129(5):1177–1180. doi: 10.1083/jcb.129.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bladt F, et al. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376(6543):768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- 75.Brand-Saberi B, et al. Scatter factor/hepatocyte growth factor (SF/HGF) induces emigration of myogenic cells at interlimb level in vivo. Dev Biol. 1996;179(1):303–308. doi: 10.1006/dbio.1996.0260. [DOI] [PubMed] [Google Scholar]
- 76.Heymann S, et al. Regulation and function of SF/HGF during migration of limb muscle precursor cells in chicken. Dev Biol. 1996;180(2):566–578. doi: 10.1006/dbio.1996.0329. [DOI] [PubMed] [Google Scholar]
- 77.Rodgers JT, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510(7505):393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tatsumi R, et al. Mechanical stretch induces activation of skeletal muscle satellite cells in vitro. Exp Cell Res. 2001;267(1):107–114. doi: 10.1006/excr.2001.5252. [DOI] [PubMed] [Google Scholar]
- 79.Leshem Y, et al. Hepatocyte growth factor (HGF) inhibits skeletal muscle cell differentiation: a role for the bHLH protein twist and the cdk inhibitor p27. J Cell Physiol. 2000;184(1):101–109. doi: 10.1002/(SICI)1097-4652(200007)184:1<101::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 80.Khan MA, et al. Twist: a molecular target in cancer therapeutics. Tumour Biol. 2013;34(5):2497–2506. doi: 10.1007/s13277-013-1002-x. [DOI] [PubMed] [Google Scholar]
- 81.Filippin LI, et al. Nitric oxide and repair of skeletal muscle injury. Nitric Oxide. 2009;21(3–4):157–163. doi: 10.1016/j.niox.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 82.Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81(1):209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 83.Rubinstein I, et al. Involvement of nitric oxide system in experimental muscle crush injury. J Clin Invest. 1998;101(6):1325–1333. doi: 10.1172/JCI810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Park CS, et al. Neuronal isoform of nitric oxide synthase is expressed at low levels in human retina. Cell Mol Neurobiol. 1996;16(4):499–515. doi: 10.1007/BF02150230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rigamonti E, et al. Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J Immunol. 2013;190(4):1767–1777. doi: 10.4049/jimmunol.1202903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tidball JG. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288(2):R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- 87.Ganea E, et al. Matrix metalloproteinases: useful and deleterious. Biochem Soc Trans. 2007;35(Pt 4):689–691. doi: 10.1042/BST0350689. [DOI] [PubMed] [Google Scholar]
- 88.Darmani H, et al. Expression of nitric oxide synthase and transforming growth factor-beta in crush-injured tendon and synovium. Mediators Inflamm. 2004;13(5–6):299–305. doi: 10.1155/S0962935104000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Serra C, et al. The role of GH and IGF-I in mediating anabolic effects of testosterone on androgen-responsive muscle. Endocrinology. 2011;152(1):193–206. doi: 10.1210/en.2010-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sandri M, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Song YH, et al. The therapeutic potential of IGF-I in skeletal muscle repair. Trends in endocrinology and metabolism: TEM. 2013;24(6):310–319. doi: 10.1016/j.tem.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamaguchi M, et al. The CLOCK 3111T/C SNP is associated with morning gastric motility in healthy young women. Physiol Behav. 2012;107(1):87–91. doi: 10.1016/j.physbeh.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 93.Hill M, Wernig A, Goldspink G. Muscle satellite (stem) cell activation during local tissue injury and repair. J Anat. 2003;203(1):89–99. doi: 10.1046/j.1469-7580.2003.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Owino V, Yang SY, Goldspink G. Age-related loss of skeletal muscle function and the inability to express the autocrine form of insulin-like growth factor-1 (MGF) in response to mechanical overload. FEBS Lett. 2001;505(2):259–263. doi: 10.1016/s0014-5793(01)02825-3. [DOI] [PubMed] [Google Scholar]
- 95.Hill M, Goldspink G. Expression and splicing of the insulin-like growth factor gene in rodent muscle is associated with muscle satellite (stem) cell activation following local tissue damage. J Physiol (London) 2003;549(2):409–418. doi: 10.1113/jphysiol.2002.035832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang SY, Goldspink GE. Different roles of the IGF-I Ec peptide (MGF) and mature IGF-I in myoblast proliferation and differentiation. FEBS Lett. 2002;522(1–3):156–160. doi: 10.1016/s0014-5793(02)02918-6. [DOI] [PubMed] [Google Scholar]
- 97.Dobrowolny G, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168(2):193–199. doi: 10.1083/jcb.200407021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pallafacchina G, et al. An adult tissue-specific stem cell in its niche: a gene profiling analysis of in vivo quiescent and activated muscle satellite cells. Stem Cell Res. 2010;4(2):77–91. doi: 10.1016/j.scr.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 99.Dong Y, et al. Interactions between p-Akt and Smad3 in injured muscles initiate myogenesis or fibrogenesis. Am J Physiol Endocrinol Metab. 2013;305(3):E367–E375. doi: 10.1152/ajpendo.00644.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perez-Ruiz A, Gnocchi VF, Zammit PS. Control of Myf5 activation in adult skeletal myonuclei requires ERK signalling. Cell Signal. 2007;19:1671–1680. doi: 10.1016/j.cellsig.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 101.Elia D, et al. Sonic hedgehog promotes proliferation and differentiation of adult muscle cells: involvement of MAPK/EIK and PI3 K/Akt pathways. Biochimica Et Biophysica Acta -Mol Cell Res. 2007;1773(9):1438–1446. doi: 10.1016/j.bbamcr.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 102.Lovett FA, et al. Convergence of Igf2 expression and adhesion signalling via RhoA and p38 MAPK enhances myogenic differentiation. J Cell Sci. 2006;119(23):4828–4840. doi: 10.1242/jcs.03278. [DOI] [PubMed] [Google Scholar]
- 103.Takaesu G, et al. Activation of p38 alpha/beta MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J Cell Biol. 2006;175(3):383–388. doi: 10.1083/jcb.200608031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Riuzzi F, Sorci G, Donato R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J Biol Chem. 2006;281(12):8242–8253. doi: 10.1074/jbc.M509436200. [DOI] [PubMed] [Google Scholar]
- 105.Riuzzi F, Sorci G, Donato R. RAGE expression in rhabdomyosarcoma cells results in myogenic differentiation and reduced proliferation, migration, invasiveness, and tumor growth. Am J Pathol. 2007;171(3):947–961. doi: 10.2353/ajpath.2007.070049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Riuzzi F, et al. HMGB1-RAGE regulates muscle satellite cell homeostasis through p38-MAPK- and myogenin-dependent repression of Pax7 transcription. J Cell Sci. 2012;125(Pt 6):1440–1454. doi: 10.1242/jcs.092163. [DOI] [PubMed] [Google Scholar]
- 107.Sorci G, et al. Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol Cell Biol. 2004;24(11):4880–4894. doi: 10.1128/MCB.24.11.4880-4894.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kastner S, et al. Gene expression patterns of the fibroblast growth factors and their receptors during myogenesis of rat satellite cells. J Histochem Cytochem. 2000;48(8):1079–1096. doi: 10.1177/002215540004800805. [DOI] [PubMed] [Google Scholar]
- 109.Floss T, Arnold HH, Braun T. A role for FGF-6 in skeletal muscle regeneration. Genes Dev. 1997;11(16):2040–2051. doi: 10.1101/gad.11.16.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sheehan SM, Allen RE. Skeletal muscle satellite cell proliferation in response to members of the fibroblast growth factor family and hepatocyte growth factor. J Cell Physiol. 1999;181(3):499–506. doi: 10.1002/(SICI)1097-4652(199912)181:3<499::AID-JCP14>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 111.Yablonka-Reuveni Z, Seger R, Rivera AJ. Fibroblast growth factor promotes recruitment of skeletal muscle satellite cells in young and old rats. J Histochem Cytochem. 1999;47(1):23–42. doi: 10.1177/002215549904700104. [DOI] [PubMed] [Google Scholar]
- 112.Scata KA, et al. FGF receptor availability regulates skeletal myogenesis. Exp Cell Res. 1999;250(1):10–21. doi: 10.1006/excr.1999.4506. [DOI] [PubMed] [Google Scholar]
- 113.Fedorov YV, Rosenthal RS, Olwin BB. Oncogenic Ras-induced proliferation requires autocrine fibroblast growth factor 2 signaling in skeletal muscle cells. J Cell Biol. 2001;152(6):1301–1305. doi: 10.1083/jcb.152.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jones NC, et al. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol. 2001;186(1):104–115. doi: 10.1002/1097-4652(200101)186:1<104::AID-JCP1015>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 115.Liu Y, Schneider MF. FGF2 activates TRPC and Ca(2 +) signaling leading to satellite cell activation. Front Physiol. 2014;5:38. doi: 10.3389/fphys.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Louvi A, Artavanis-Tsakonas S. Notch and disease: a growing field. Semin Cell Dev Biol. 2012;23(4):473–480. doi: 10.1016/j.semcdb.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Castel D, et al. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013;27(9):1059–1071. doi: 10.1101/gad.211912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schuster-Gossler K, Cordes R, Gossler A. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc Natl Acad Sci USA. 2007;104(2):537–542. doi: 10.1073/pnas.0608281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vasyutina E, et al. RBP-J (Rbpsuh) is essential to maintain muscle progenitor cells and to generate satellite cells. Proc Natl Acad Sci USA. 2007;104(11):4443–4448. doi: 10.1073/pnas.0610647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mourikis P, et al. Cell-autonomous Notch activity maintains the temporal specification potential of skeletal muscle stem cells. Development. 2012;139(24):4536–4548. doi: 10.1242/dev.084756. [DOI] [PubMed] [Google Scholar]
- 121.Bjornson CR, et al. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30(2):232–242. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mourikis P, et al. A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells. 2012;30(2):243–252. doi: 10.1002/stem.775. [DOI] [PubMed] [Google Scholar]
- 123.Liu L, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4(1):189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Delfini MC, et al. Delta 1-activated notch inhibits muscle differentiation without affecting Myf5 and Pax3 expression in chick limb myogenesis. Development. 2000;127(23):5213–5224. doi: 10.1242/dev.127.23.5213. [DOI] [PubMed] [Google Scholar]
- 125.Brack AS, et al. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2(1):50–59. doi: 10.1016/j.stem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 126.Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. 2002;3(3):397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- 127.Mourikis P, Tajbakhsh S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev Biol. 2014;14:2. doi: 10.1186/1471-213X-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 129.Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45 + adult stem cells during muscle regeneration. Cell. 2003;113(7):841–852. doi: 10.1016/s0092-8674(03)00437-9. [DOI] [PubMed] [Google Scholar]
- 130.Perez-Ruiz A, et al. beta-Catenin promotes self-renewal of skeletal-muscle satellite cells. J Cell Sci. 2008;121(Pt 9):1373–1382. doi: 10.1242/jcs.024885. [DOI] [PubMed] [Google Scholar]
- 131.Brack AS, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317(5839):807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 132.Naito AT, et al. Complement C1q activates canonical Wnt signaling and promotes aging-related phenotypes. Cell. 2012;149(6):1298–1313. doi: 10.1016/j.cell.2012.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Simons M, Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008;42:517–540. doi: 10.1146/annurev.genet.42.110807.091432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Le Grand F, et al. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009;4(6):535–547. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bentzinger CF, et al. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell. 2013;12(1):75–87. doi: 10.1016/j.stem.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley interdisciplinary reviews. Dev Biol. 2013;2(1):47–63. doi: 10.1002/wdev.86. [DOI] [PubMed] [Google Scholar]
- 137.Vaidya TB, et al. Fibroblast growth factor and transforming growth factor beta repress transcription of the myogenic regulatory gene MyoD1. Mol Cell Biol. 1989;9(8):3576–3579. doi: 10.1128/mcb.9.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Martin JF, Li L, Olson EN. Repression of myogenin function by TGF-beta 1 is targeted at the basic helix-loop-helix motif and is independent of E2A products. J Biol Chem. 1992;267(16):10956–10960. [PubMed] [Google Scholar]
- 139.Li X, McFarland DC, Velleman SG. Transforming growth factor-beta1-induced satellite cell apoptosis in chickens is associated with beta1 integrin-mediated focal adhesion kinase activation. Poult Sci. 2009;88(8):1725–1734. doi: 10.3382/ps.2008-00534. [DOI] [PubMed] [Google Scholar]
- 140.Kollias HD, McDermott JC. Transforming growth factor-beta and myostatin signaling in skeletal muscle. J Appl Physiol (1985) 2008;104(3):579–587. doi: 10.1152/japplphysiol.01091.2007. [DOI] [PubMed] [Google Scholar]
- 141.Joulia-Ekaza D, Cabello G. The myostatin gene: physiology and pharmacological relevance. Curr Opin Pharmacol. 2007;7(3):310–315. doi: 10.1016/j.coph.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 142.McCroskery S, et al. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162(6):1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Amthor H, et al. The regulation and action of myostatin as a negative regulator of muscle development during avian embryogenesis. Dev Biol. 2002;251(2):241–257. doi: 10.1006/dbio.2002.0812. [DOI] [PubMed] [Google Scholar]
- 144.McFarlane C, et al. Myostatin signals through Pax7 to regulate satellite cell self-renewal. Exp Cell Res. 2008;314(2):317–329. doi: 10.1016/j.yexcr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 145.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454(7203):528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iozzo RV. Matrix proteoglycans: from molecular design to cellular function. Annu Rev Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 147.Cornelison DD, et al. Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev Biol. 2001;239(1):79–94. doi: 10.1006/dbio.2001.0416. [DOI] [PubMed] [Google Scholar]
- 148.Quinlan JG, et al. Radiation inhibition of mdx mouse muscle regeneration: dose and age factors. Muscle Nerve. 1995;18(2):201–206. doi: 10.1002/mus.880180209. [DOI] [PubMed] [Google Scholar]
- 149.Cornelison DD, et al. Essential and separable roles for syndecan-3 and syndecan-4 in skeletal muscle development and regeneration. Genes Dev. 2004;18(18):2231–2236. doi: 10.1101/gad.1214204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Carmeli E, et al. Matrix metalloproteinases and skeletal muscle: a brief review. Muscle Nerve. 2004;29(2):191–197. doi: 10.1002/mus.10529. [DOI] [PubMed] [Google Scholar]
- 151.Ferre PJ, et al. Longitudinal analysis of gene expression in porcine skeletal muscle after post-injection local injury. Pharm Res. 2007;24(8):1480–1489. doi: 10.1007/s11095-007-9266-8. [DOI] [PubMed] [Google Scholar]
- 152.Fukushima K, et al. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (CXMDJ) BMC Musculoskelet Disord. 2007;8:54. doi: 10.1186/1471-2474-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kherif S, et al. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: a study in experimentally injured and mdx muscles. Dev Biol. 1999;205(1):158–170. doi: 10.1006/dbio.1998.9107. [DOI] [PubMed] [Google Scholar]
- 154.Yamada M, et al. Matrix metalloproteinase-2 mediates stretch-induced activation of skeletal muscle satellite cells in a nitric oxide-dependent manner. Int J Biochem Cell Biol. 2008;40(10):2183–2191. doi: 10.1016/j.biocel.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 155.Yamada M, et al. Matrix metalloproteinases are involved in mechanical stretch-induced activation of skeletal muscle satellite cells. Muscle Nerve. 2006;34(3):313–319. doi: 10.1002/mus.20601. [DOI] [PubMed] [Google Scholar]
- 156.Liu H, et al. TIMP3: a physiological regulator of adult myogenesis. J Cell Sci. 2010;123(Pt 17):2914–2921. doi: 10.1242/jcs.057620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Relaix F, et al. Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol. 2006;172(1):91–102. doi: 10.1083/jcb.200508044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sambasivan R, et al. Distinct regulatory cascades govern extraocular and pharyngeal arch muscle progenitor cell fates. Dev Cell. 2009;16(6):810–821. doi: 10.1016/j.devcel.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 159.Zanou N, Gailly P. Skeletal muscle hypertrophy and regeneration: interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell Mol Life Sci. 2013;70(21):4117–4130. doi: 10.1007/s00018-013-1330-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hu P, et al. Codependent activators direct myoblast-specific MyoD transcription. Dev Cell. 2008;15(4):534–546. doi: 10.1016/j.devcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Riuzzi F, et al. RAGE signaling deficiency in rhabdomyosarcoma cells causes upregulation of PAX7 and uncontrolled proliferation. J Cell Sci. 2014;127(Pt 8):1699–1711. doi: 10.1242/jcs.136259. [DOI] [PubMed] [Google Scholar]
- 162.Boldrin L, et al. Mature adult dystrophic mouse muscle environment does not impede efficient engrafted satellite cell regeneration and self-renewal. Stem Cells. 2009;27(10):2478–2487. doi: 10.1002/stem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kumar D, et al. Id3 is a direct transcriptional target of Pax7 in quiescent satellite cells. Mol Biol Cell. 2009;20(14):3170–3177. doi: 10.1091/mbc.E08-12-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yajima H, et al. Six family genes control the proliferation and differentiation of muscle satellite cells. Exp Cell Res. 2010;316(17):2932–2944. doi: 10.1016/j.yexcr.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 165.Lee H, Habas R, Abate-Shen C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science. 2004;304(5677):1675–1678. doi: 10.1126/science.1098096. [DOI] [PubMed] [Google Scholar]
- 166.Liu Y, et al. Six1 regulates MyoD expression in adult muscle progenitor cells. PLoS One. 2013;8(6):e67762. doi: 10.1371/journal.pone.0067762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Beauchamp JR, et al. Expression of CD34 and Myf5 defines the majority of quiescent adult skeletal muscle satellite cells. J Cell Biol. 2000;151(6):1221–1234. doi: 10.1083/jcb.151.6.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rantanen J, et al. Satellite cell proliferation and the expression of myogenin and desmin in regenerating skeletal muscle: evidence for two different populations of satellite cells. Lab Invest. 1995;72(3):341–347. [PubMed] [Google Scholar]
- 169.Hansen KH, et al. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10(11):1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 170.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 171.Palacios D, et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7(4):455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sebastian S, et al. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc Natl Acad Sci USA. 2009;106(12):4719–4724. doi: 10.1073/pnas.0807136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.McKinnell IW, et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008;10(1):77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fulco M, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14(5):661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rathbone CR, Booth FW, Lees SJ. FoxO3a preferentially induces p27Kip1 expression while impairing muscle precursor cell-cycle progression. Muscle Nerve. 2008;37(1):84–89. doi: 10.1002/mus.20897. [DOI] [PubMed] [Google Scholar]
- 176.Fulco M, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12(1):51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 177.Zhao X, et al. Regulation of MEF2 by histone deacetylase 4- and SIRT1 deacetylase-mediated lysine modifications. Mol Cell Biol. 2005;25(19):8456–8464. doi: 10.1128/MCB.25.19.8456-8464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Guarani V, et al. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473(7346):234–238. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tang AH, Rando TA. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014;33(23):2782–2797. doi: 10.15252/embj.201488278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Taylor SM, Jones PA. Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell. 1979;17(4):771–779. doi: 10.1016/0092-8674(79)90317-9. [DOI] [PubMed] [Google Scholar]
- 181.Acharyya S, et al. TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One. 2010;5(8):e12479. doi: 10.1371/journal.pone.0012479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fan H, et al. Sulforaphane causes a major epigenetic repression of myostatin in porcine satellite cells. Epigenetics : Off J DNA Methylation Soc. 2012;7(12):1379–1390. doi: 10.4161/epi.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Erriquez D, Perini G, Ferlini A. Non-coding RNAs in muscle dystrophies. Int J Mol Sci. 2013;14(10):19681–19704. doi: 10.3390/ijms141019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kirby TJ, McCarthy JJ. MicroRNAs in skeletal muscle biology and exercise adaptation. Free Radic Biol Med. 2013;64:95–105. doi: 10.1016/j.freeradbiomed.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen JF, et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol. 2010;190(5):867–879. doi: 10.1083/jcb.200911036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cacchiarelli D, et al. miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 2011;12(2):136–141. doi: 10.1038/embor.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cheung TH, et al. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature. 2012;482(7386):524–528. doi: 10.1038/nature10834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Naguibneva I, et al. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol. 2006;8(3):278–284. doi: 10.1038/ncb1373. [DOI] [PubMed] [Google Scholar]
- 189.van Rooij E, Liu N, Olson EN. MicroRNAs flex their muscles. Trends in genetics : TIG. 2008;24(4):159–166. doi: 10.1016/j.tig.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 190.Chen JF, Callis TE, Wang DZ. microRNAs and muscle disorders. J Cell Sci. 2009;122(Pt 1):13–20. doi: 10.1242/jcs.041723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Eisenberg I, Alexander MS, Kunkel LM. miRNAS in normal and diseased skeletal muscle. J Cell Mol Med. 2009;13(1):2–11. doi: 10.1111/j.1582-4934.2008.00524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ge Y, Chen J. MicroRNAs in skeletal myogenesis. Cell Cycle. 2011;10(3):441–448. doi: 10.4161/cc.10.3.14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu N, et al. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Invest. 2012;122(6):2054–2065. doi: 10.1172/JCI62656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ge Y, Sun Y, Chen J. IGF-II is regulated by microRNA-125b in skeletal myogenesis. J Cell Biol. 2011;192(1):69–81. doi: 10.1083/jcb.201007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cardinali B, et al. Microrna-221 and microrna-222 modulate differentiation and maturation of skeletal muscle cells. PLoS One. 2009;4(10):e7607. doi: 10.1371/journal.pone.0007607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Maciotta S, et al. Hmgb3 is regulated by microRNA-206 during muscle regeneration. PLoS One. 2012;7(8):e43464. doi: 10.1371/journal.pone.0043464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Dormoy-Raclet V. HuR and miR-1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat Commun. 2013;4:2388. doi: 10.1038/ncomms3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Riuzzi F, et al. HMGB1-RAGE regulates muscle satellite cell homeostasis through p38-MAPK- and myogenin-dependent repression of Pax7 transcription. J Cell Sci. 2012;125(6):1440–1454. doi: 10.1242/jcs.092163. [DOI] [PubMed] [Google Scholar]
- 199.Koning M, et al. A global downregulation of microRNAs occurs in human quiescent satellite cells during myogenesis. Differentiation. 2012;84(4):314–321. doi: 10.1016/j.diff.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 200.Neguembor MV, Jothi M, Gabellini D. Long noncoding RNAs, emerging players in muscle differentiation and disease. Skelet Muscle. 2014;4(1):8. doi: 10.1186/2044-5040-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Legnini I, et al. A feedforward regulatory loop between HuR and the long noncoding RNA linc-MD1 controls early phases of myogenesis. Mol Cell. 2014;53(3):506–514. doi: 10.1016/j.molcel.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Watts R, et al. Myostatin-induced inhibition of the long noncoding RNA Malat1 is associated with decreased myogenesis. Am J Physiol Cell Physiol. 2013;304(10):C995–C1001. doi: 10.1152/ajpcell.00392.2012. [DOI] [PubMed] [Google Scholar]
- 203.Sunwoo H, et al. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009;19(3):347–359. doi: 10.1101/gr.087775.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Caretti G, et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell. 2006;11(4):547–560. doi: 10.1016/j.devcel.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 205.Hube F, et al. Steroid receptor RNA activator protein binds to and counteracts SRA RNA-mediated activation of MyoD and muscle differentiation. Nucleic Acids Res. 2011;39(2):513–525. doi: 10.1093/nar/gkq833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lu L, et al. Genome-wide survey by ChIP-seq reveals YY1 regulation of lincRNAs in skeletal myogenesis. EMBO J. 2013;32(19):2575–2588. doi: 10.1038/emboj.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Twayana S, et al. Biogenesis and function of non-coding RNAs in muscle differentiation and in Duchenne muscular dystrophy. Biochem Soc Trans. 2013;41(4):844–849. doi: 10.1042/BST20120353. [DOI] [PubMed] [Google Scholar]
- 208.Brannan CI, et al. The product of the H19 gene may function as an RNA. Mol Cell Biol. 1990;10(1):28–36. doi: 10.1128/mcb.10.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gabory A, Jammes H, Dandolo L. The H19 locus: role of an imprinted non-coding RNA in growth and development. BioEssays News Rev Mol Cell Dev Biol. 2010;32(6):473–480. doi: 10.1002/bies.200900170. [DOI] [PubMed] [Google Scholar]
- 210.Onyango P, Feinberg AP. A nucleolar protein, H19 opposite tumor suppressor (HOTS), is a tumor growth inhibitor encoded by a human imprinted H19 antisense transcript. Proc Natl Acad Sci USA. 2011;108(40):16759–16764. doi: 10.1073/pnas.1110904108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ripoche MA, et al. Deletion of the H19 transcription unit reveals the existence of a putative imprinting control element. Genes Dev. 1997;11(12):1596–1604. doi: 10.1101/gad.11.12.1596. [DOI] [PubMed] [Google Scholar]
- 212.Forne T, et al. Loss of the maternal H19 gene induces changes in Igf2 methylation in both cis and trans. Proc Natl Acad Sci USA. 1997;94(19):10243–10248. doi: 10.1073/pnas.94.19.10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Li Z, et al. An HMGA2-IGF2BP2 axis regulates myoblast proliferation and myogenesis. Dev Cell. 2012;23(6):1176–1188. doi: 10.1016/j.devcel.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]