

Abstract
Mutations in FGF14, which encodes intracellular fibroblast growth factor 14 (iFGF14), have been linked to spinocerebellar ataxia (SCA27). In addition, mice lacking Fgf14 (Fgf14−/−) exhibit an ataxia phenotype resembling SCA27, accompanied by marked changes in the excitability of cerebellar granule and Purkinje neurons. It is not known, however, whether these phenotypes result from defects in neuronal development or if they reflect a physiological requirement for iFGF14 in the adult cerebellum. Here, we demonstrate that the acute and selective Fgf14-targeted short hairpin RNA (shRNA)-mediated in vivo “knock-down” of iFGF14 in adult Purkinje neurons attenuates spontaneous and evoked action potential firing without measurably affecting the expression or localization of voltage-gated Na+ (Nav) channels at Purkinje neuron axon initial segments. The selective shRNA-mediated in vivo “knock-down” of iFGF14 in adult Purkinje neurons also impairs motor coordination and balance. Repetitive firing can be restored in Fgf14-targeted shRNA-expressing Purkinje neurons, as well as in Fgf14−/− Purkinje neurons, by prior membrane hyperpolarization, suggesting that the iFGF14-mediated regulation of the excitability of mature Purkinje neurons depends on membrane potential. Further experiments revealed that the loss of iFGF14 results in a marked hyperpolarizing shift in the voltage dependence of steady-state inactivation of the Nav currents in adult Purkinje neurons. We also show here that expressing iFGF14 selectively in adult Fgf14−/− Purkinje neurons rescues spontaneous firing and improves motor performance. Together, these results demonstrate that iFGF14 is required for spontaneous and evoked action potential firing in adult Purkinje neurons, thereby controlling the output of these cells and the regulation of motor coordination and balance.
Keywords: channel inactivation, FGF14, fibroblast growth factor homologous factor 4 (FHF4), intrinsic excitability, spinocerebellar ataxia 27, voltage-gated sodium (Nav) channels
Introduction
The intracellular fibroblast growth factors 11-14 (iFGFs 11-14) are differentially expressed throughout the developing and adult nervous systems (Smallwood et al., 1996, Hartung et al., 1997; Wang et al., 2000). Despite sequence and structural homology to prototypical FGFs, the iFGFs are not secreted and do not activate FGF receptors (Smallwood et al., 1996; Olsen et al., 2003; Goldfarb, 2005; Ornitz and Itoh, 2001) and, as a result, are also referred to as fibroblast growth factor homologous factors. The first insight into the possible functional role(s) of the iFGFs was provided with the demonstration that iFGF12 interacts with the voltage-gated pore forming (α) subunit Na+ (Nav) channel, Nav 1.9 (Liu et al., 2001). Numerous subsequent studies revealed the generality of this observation for iFGF12-14 and various Nav α-subunits, Nav1.1-Nav.1.9 (Liu et al., 2003; Wittmack et al., 2004; Lou et al., 2005; Wang et al., 2011a). It has also been demonstrated that the iFGFs bind directly to Nav α-subunit C-terminal domains through the common iFGF core domain and that alternative splicing at the iFGF N termini does not affect binding (Liu et al., 2003; Wittmack et al., 2004; Lou et al., 2005; Goldfarb et al., 2007; Laezza et al., 2007, 2009; Goetz et al., 2009; Wang et al., 2011b, 2012). In heterologous cells, iFGF coexpression affects the amplitudes and the time- and voltage-dependent properties of Nav α-subunit-encoded currents (Liu et al., 2003; Wittmack et al., 2004; Lou et al., 2005; Goetz et al., 2009; Laezza et al., 2009; Wang et al., 2011b).
Evidence for the in vivo association of iFGFs with neuronal Nav channels was provided with the demonstration of iFGF13 expression at nodes of Ranvier in sensory fibers of the dorsal root (Wittmack et al., 2004) and iFGF14 expression at the axon initial segments (AIS) of hippocampal pyramidal neurons (Goldfarb et al., 2007; Laezza et al., 2007), structures that contain high densities of Nav channels. iFGF14 is also concentrated at the AIS in Purkinje, granule, stellate, and basket cells in the cerebellum (Xiao et al., 2013). Mutations in FGF14 have been linked to spinocerebellar ataxia 27 (SCA27), an autosomal-dominant disorder characterized by gait and movement disorders, nystagmus, and cognitive impairment (van Swieten et al., 2003, Dalski et al., 2005, Brusse et al., 2006, Misceo et al., 2009; Coebergh et al., 2014). In addition, Fgf14−/− mice exhibit paroxysmal dyskinesia and movement and spatial learning deficits (Wang et al., 2002; Wozniak et al., 2007), phenotypes that resemble those seen in SCA27. Electrophysiological studies have demonstrated attenuated spontaneous and evoked firing in Fgf14−/− cerebellar granule and Purkinje neurons (Goldfarb et al., 2007, Shakkottai et al., 2009), as well as reduced postsynaptic potentials at granule-Purkinje cell synapses (Yan et al., 2013). The physiological significance and the functional implications of these findings are unclear, however, because Fgf14−/− mice lack iFGF14 throughout development and no causal links between the observed change in granule and/or Purkinje neuron firing and the observed motor impairment have been established. The experiments here were undertaken to test the hypotheses that iFGF14 is required for the regulation of excitability in mature Purkinje neurons, the sole output neurons of the cerebellum (Apps and Garwicz, 2005), and that the acute loss of iFGF14 in these cells results in balance and motor deficits. Additional experiments were designed to test the hypothesis that introducing iFGF expression in adult Fgf14−/− Purkinje neurons can rescue spontaneous and evoked firing in these cells and improve motor performance.
Materials and Methods
Animals.
All experiments involving animals were performed in accordance with the guidelines published in the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and all protocols were approved by the Washington University Animal Studies Committee. The (Fgf14−/−) mice, harboring a targeted disruption of the Fgf14 locus, have been described previously (Wang et al., 2002). For experiments, Fgf14−/− mice were generated by crossing Fgf14+/− heterozygotes congenic in the C57BL/6 background and genotypes were confirmed by PCR screening (Wang et al., 2002; Xiao et al., 2013). Adult male and female wild-type (WT) and Fgf14−/− C57BL/6 mice of varying ages (as indicated) were used in the various experiments completed here.
Plasmids.
The mouse-Fgf14A-myc (mFgf14-myc) plasmid was generated as described previously (Laezza et al., 2009). Lentiviral constructs (pLKO.1) containing short hairpin RNAs (shRNAs) targeting Fgf14 were obtained from the RNAi Consortium (TRC) through the Genome Institute at Washington University. The following (seven) Fgf14-targeted shRNA sequences were screened in vitro, as detailed below: 5′-GCCATTGGAAGTTGCCATGTA-3′, 5′-GCAATAATGAATGGAGGCAAA-3′, 5′-CCTTGCATGATGTTGGTGAAA-3′, 5′-CCAAGGATGACAGCACCAATT-3′, 5′-GCAAGTTT AAAGAGTCTGTTT-3′,5′-ACCAGGTTATATTGCAGGCAA-3′, and 5′-GCAAGGCTACTACTTGCA GAT-3′. An shRNA, 5′-CAACAAGATGAAGAGCACCAA-3′, which targets a variant of green fluorescent protein (GFP), was also obtained for use as the control, nontargeted shRNA.
Each shRNA 21-nucleotide sense sequence was synthesized (IDT) into the corresponding 97-nucleotide miRNA-adapted shRNA oligonucleotide, containing sense and antisense sequences, linked by a (19 nucleotide) hairpin loop. Forward and reverse strands were annealed and cloned into a miR30 context in the 3′-untranslated region (3′-UTR) of tdTomato (see Fig. 1A) in the pPRIME vector (Stegmeier et al., 2005). The entire tdTomato-shRNA cassette was then cloned into a modified adeno-associated virus (AAV) transfer plasmid AAV-CAG containing the CAG (chicken β-actin with CMV enhancer) promoter generated from AAV-CAG-IRES-GFP (pTR-UF-12.1; Klein et al., 2002) by removal of the IRES-GFP cassette. In preliminary experiments, several lentiviral and AAV vectors with various promoter combinations were generated and compared for efficient and selective in situ transduction of Purkinje neurons, as assayed by fluorescent reporter protein expression (Bosch et al., 2014). Of the viruses tested, AAV1 with a CAG promoter exhibited the highest efficiency and specificity for Purkinje neurons. For in vivo knock-down experiments, the 21-mer sense sequence 5′-GCAAGTTTAAAGAGTCTGTTT-3′ was used.
Figure 1.

Generation and validation of Fgf14-targeted shRNA. A, Schematic representation of the AAV transfer vector with an shRNA embedded in a miRNA (miR30) context in the 3′-UTR of tdTomato. Expression is driven by the chicken β-actin (with a CMV enhancer) promoter (CAG). ITR, Inverted terminal repeat; pA, polyadenylation signal. B, Mouse Fgf14-myc was expressed in CHL cells with increasing amounts of either the nontargeted or the Fgf14-targeted shRNA plasmid. Western blot analyses using an anti-myc antibody revealed that the Fgf14-targeted, but not the nontargeted, shRNA reduced iFGF14 protein expression in a dose-dependent manner. Blots were also probed with an anti-actin antibody to verify equal sample loading. C, Wild-type mice were injected into the cerebellum with the nontargeted shRNA-targeted (top) or the Fgf14-targeted (bottom) shRNA-expressing AAV1. Parasaggital sections were cut and stained with anti-Ankyrin G (blue) and anti-FGF14 (green) antibodies. In A, Representative low-magnification images of nontargeted shRNA- and Fgf14-targeted shRNA-transduced cells are shown. Viral-transduced neurons were identified by tdTomato expression and cell type was determined by morphology and location. Purkinje neurons, for example, were readily identified by large somata and extensively branched dendritic trees extending into the molecular layer; TdTomato expression in Purkinje neurons is robust. Arrowheads indicate the few tdTomato expressing (virally transduced) granule neurons. Scale bars, 25 μm. m, Molecular layer; p, Purkinje layer; g, granule layer. Overall anti-FGF14 labeling in the granule layer is similar in sections prepared after injections of the nontargeted and Fgf14-targeted shRNA-expressing AAV1.
To create AAV-FGF14-IRES-tdTomato, the GFP in AAV-CAG-IRES-GFP was replaced with tdTomato. Human FGF14B amplified by PCR from pQBI-hFGF14B-GFP (Lou et al., 2005) was inserted into AAV-IRES-tdTomato upstream of the IRES. To generate AAV-CAG-GFP, GFP was amplified by PCR from pQBI-fC2 (Quantum Biotechnology) and inserted into AAV-CAG-IRES-GFP in place of IRES-GFP. All plasmids were verified by sequencing.
Cell culture and shRNA screening.
All reagents were purchased from Sigma unless otherwise noted. Chinese hamster lung (CHL) cells (McEwen et al., 2004) were maintained in RPMI-1640 supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, at 37°C in a 95% air/5% CO2 incubator. Cells were transfected at 80–90% confluence using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions.
For shRNA screening, CHL cells were seeded into 6-well tissue culture dishes and cotransfected with 4 μg of the mFgf14-myc plasmid and increasing amounts (0.2–2 μg) of the Fgf14-targeted shRNA or the nontargeted shRNA plasmid. Approximately 24 h later, cells were washed with PBS and lysed in 20 mm Tris-HCl, 150 mm NaCl, and 1% NP-40 containing protease inhibitors (Protease Inhibitor Cocktail Set III; Millipore). Cell extracts were collected, incubated for 15 min at 4°C, and centrifuged at 3000 rpm for 10 min at 4°C to remove insoluble materials. For Western blot analyses, 50 mm TCEP (Thermo Fisher) was added to each cell lysate and 50 μg of each protein lysate was fractionated by polyacrylamide (4–15% gradient) gel electrophoresis.
After fractionation, proteins were transferred to Immobilon-P or Immobilon-FL membranes (Millipore) for 1.5 h at 4°C and blocked with 3% (v/v) nonfat dry milk in PBS-containing 0.1% Tween 20 (PBST) or Odyssey blocking buffer (LI-COR Biosciences) at room temperature (22–24°C). Membranes were probed for 2 h at 22–24°C with a mouse monoclonal anti-myc (1:1000; Santa Cruz Biotechnology) and/or a rabbit monoclonal anti-β-actin (1:1000; LI-COR) antibody diluted in 0.5% (v/v) nonfat dry milk in PBST or Odyssey blocking buffer. After washing with PBST, membranes were incubated with goat anti-mouse-horseradish peroxidase (1:5000; Santa Cruz Biotechnology) or goat anti-mouse-IR800 (1:10,000; LI-COR) and goat anti-rabbit-IR680 (1:20,000; LI-COR) for 1 h at 22–24°C. Signals were detected with the SuperSignal Fempto chemiluminescent substrate (Pierce) or the Odyssey Infrared Imaging System (LI-COR).
AAV vector production.
AAV1 vectors were prepared by the Hope Center Viral Vectors Core at Washington University, as described previously (Zolotukhin et al., 2002). Briefly, HEK-293 cells, maintained in Dulbecco's modified Eagle's medium supplemented with 5% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin at 37°C in a 95% air/5% CO2 incubator, were plated at 30–40% confluence in CellSTACS (Corning). Approximately 24 h later, cells were cotransfected with 0.6 mg of the AAV transfer plasmid containing the construct of interest (e.g., FGF14) and 1.8 mg of helper plasmid (pXYZ1 for AAV1) using the calcium phosphate precipitation method (Zolotukhin et al., 2002). The transfection medium was removed after 6 h and the cells were incubated at 37°C for 3 d. For harvesting, cells were lysed by (three) freeze/thaw cycles and the cell lysates collected and treated with 50 U/ml Benzonaze, followed by iodixanol gradient centrifugation. The iodixanol gradient fraction was further purified on a HiTrap Q column and concentrated (Zolotukhin et al., 2002). Vector titers, determined by Dot blot assay, were as follows: Fgf14-targeted shRNA = 1.4 × 1013 vg/ml (viral genomes/ml); nontargeted shRNA = 5 × 1012 vg/ml; GFP = 7.1 × 1012 vg/ml; hFGF14B-IRES-tdTomato = 5 × 1012 vg/ml.
Stereotaxic virus injections.
Juvenile (3–4 weeks old) and adult (2–4 months old) male and female WT and Fgf14−/− C57BL/6 mice were anesthetized by intraperitoneal injection of 1 ml/kg ketamine/xylene (30 mg/ml and 4 mg/ml, respectively) mixture and secured in a stereotaxic head holder; body temperature was maintained at 37°C with a feedback-controlled heating pad. For acute FGF knock-down experiments, 1–2 μl of virus was injected into the Purkinje neuron layer of lobule VI (coordinates: midline 1–2 mm anterior to interparietal-occipital suture, or ∼5–6 mm caudal to bregma, and 0.35 mm below the pial surface) using a glass pipette (18–25 μm) connected to a Picospritzer (Parker Hannifin). For behavioral experiments, a total of 6 μl of virus was injected in two midline locations (1–2 mm anterior and 2 mm posterior to the interparietal–occipital suture and both 0.35 mm below the pial surface) to target lobules VI and VII with anterior–posterior spreading to lobules IV/V and/or VIII and lateral spreading into the lateral cerebellar hemispheres. Injection site and spread were determined during microscopic imaging for electrophysiology and/or immunostaining. The iFGF14- and GFP-expressing viruses were mixed at a ratio of 5:1 (iFGF14:GFP) before injection.
Preparation of acute cerebellar slices.
Slices were prepared from the cerebella of 8- to 10-week-old WT and Fgf14−/− mice using standard procedures (Davie et al., 2006). Briefly, mice were deeply anesthetized with 1.25% Avertin and perfused transcardially with ice-cold cutting solution containing the following (in mm): sucrose, 240; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 25; CaCl2, 0.5; and MgCl2 7, saturated with 95% O2/5% CO2. Brains were rapidly removed and placed in ice-cold cutting solution. Parasagittal slices (350 μm) were cut on a Leica VT1000 S vibrating blade microtome and incubated in a holding chamber with oxygenated artificial CSF (ACSF) containing the following (in mm): NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 25; CaCl2, 2; MgCl2, 1; and dextrose, 25 (∼310 mosmol l−1), saturated with 95% O2/5% CO2, at 33°C for 25 min and then at 22–23°C for at least 35 min before transfer to the recording chamber.
Electrophysiological recordings.
Electrophysiological experiments were performed blind to experimental group on sections prepared from WT and Fgf14−/− animals and from virus-injected WT animals 4–5 weeks after injection(s). Whole-cell current-clamp recordings were obtained at 33 ± 1°C from WT and Fgf14−/− Purkinje neurons and from visually identified tdTomato- or GFP-expressing Purkinje neurons using differential interference contrast optics with infrared illumination. Current-clamp (and voltage-clamp) protocols were controlled and data were collected using a Multiclamp 700B patch-clamp amplifier interfaced with a Digidata 1332 acquisition system and the pCLAMP 9 software (Molecular Devices) to a Gateway computer. Slices were perfused continually with ACSF saturated with 95% O2/5% CO2. In all current-clamp experiments, recording pipettes contained the following (in mm): KMeSO4 120, KCl 20, HEPES 10, EGTA 0.2, NaCl 8, Mg-ATP 4, Tris-GTP 0.3, and phosphocreatine 14, pH 7.25, ∼300 mosmol/L. Tip potentials were zeroed before membrane–pipette seals were formed; pipette capacitances and series resistances were compensated using the PClamp software. After a whole-cell recording was established, spontaneous firing was recorded for 1 min. Input resistances were calculated from voltage deflections recorded in response to 100 pA hyperpolarizing current injections. Signals were acquired at 100 kHz and filtered at 10 kHz before digitization and storage. Action potential trains, elicited in response to (250 ms) depolarizing current injections of variable amplitudes, were also recorded. The effects of the membrane potential on the firing properties of Purkinje neurons were examined by evoking single action potentials and action potential trains in response to brief (5 ms) and prolonged (250 ms) depolarizing current injections from hyperpolarized membrane potentials.
Voltage-clamp recordings were obtained from WT and Fgf14−/− Purkinje neurons in acute cerebellar slices at 33 ± 1°C. Recording pipettes contained the following (in mm): CsCl 110, TEA-Cl 30, CaCl2 1, MgCl2 2, EGTA 10, Mg-ATP 4, Tris-GTP 0.3 and 10 HEPES, pH 7.25 ∼310 mosmol/L). After formation of a gigaseal, the control ACSF superfusing the slice was changed to a low sodium bath solution containing the following (in mm): NaCl 50, TEA-Cl 75, KCl 2.5, 1.25 NaH2PO4 1.25, NaHCO3 25, CaCl2 2, MgCl2 1, CdCl2 0.1, and dextrose 25, ∼310 mosmol/L, also saturated with 95% O2/5% CO2. Series resistances were compensated by ≥75%; voltage errors resulting from the uncompensated series resistances were always ≤4 mV and were not corrected. Using the strategy of Milescu et al. (2010) to minimize space-clamp errors and to enable the successful measurement of fast transient Nav currents in neurons in acute brain slices, voltage-clamp protocols were designed, tested, and optimized.
To determine the voltage dependences of steady-state inactivation of the Nav currents in WT and Fgf14−/− Purkinje neurons, a three-step protocol (Milescu et al., 2010) was used. To inactivate axial Nav currents, a brief (3 ms) depolarizing voltage step to 0 mV (determined optimal for Purkinje neurons) was first delivered from the holding potential of −70 mV. This was followed by a brief (2.5 ms) hyperpolarizing step to various conditioning potentials (ranging from −110 mV to −15 mV in 5 mV increments) to allow recovery (Milescu et al., 2010) and, finally, by a second (3 ms) depolarizing voltage step to −20 mV to evoke (and measure) current through available somatic Nav channels. Before analysis of steady-state inactivation of the transient Nav current in each cell, the component of the current that does not inactivate—that is, the persistent Nav current—was digitally subtracted from the transient portion of the current. The voltage-clamp paradigm and a representative set of subtracted voltage-clamp records, with selected voltage steps shown and color-coded with the corresponding current traces, are presented in Figure 8A. Current amplitudes at each test potential in each cell were measured and normalized to the current evoked (in the same cell) from the most hyperpolarized test potential (of −110 mV). Mean ± SEM normalized current amplitudes (INa/INa,max) were then plotted as a function of the conditioning membrane potential (Vm) and fitted using the Boltzmann, INa/INa,max = 1/(1 + e[(Vh − Vm)/k]), where Vh (also called V1/2i) is the membrane potential at half-maximal inactivation and k is the slope.
Figure 8.

iFGF14 shifts the voltage dependence of steady-state inactivation of the Nav currents in cerebellar Purkinje neurons. Whole-cell voltage-clamp recordings were obtained at 33 ± 1°C from WT and Fgf14−/− Purkinje neurons in acute cerebellar slices as described in Materials and Methods. A, Representative recordings of Nav currents (left) evoked in a WT cerebellar Purkinje neuron from various conditioning voltages; the voltage-clamp paradigm is illustrated above the records and the currents are shown in the color of the corresponding voltage step. Note that the persistent currents were digitally subtracted (see Materials and Methods) and only the inactivating, transient components of the currents are shown. The transient Nav currents evoked at 0 mV were measured and normalized (in the same cell) to the current evoked from the most hyperpolarized test potential (of −110 mV). Mean ± SEM normalized transient Nav current amplitudes in WT and Fgf14−/− Purkinje neurons were then plotted (right) as a function of the conditioning membrane potential and fitted using a Boltzmann equation (see Materials and Methods). The transient Nav current is inactivated at a significantly (p < 0.001) more hyperpolarized membrane potential in Fgf14−/− (V1/2 = −71.1 ± 0.9 mV, slope = 11.3 ± 1.0; n = 10), than in WT (V1/2 = −58.1 ± 0.9 mV, slope = 10.3 ± 1.0; n = 11) Purkinje neurons. B, Representative recordings of the transient and persistent Nav currents (left) evoked in a WT Purkinje neuron at various test potentials; the voltage-clamp paradigm is illustrated above the records and the currents are shown in the color of the corresponding voltage step; the raw, unsubtracted current records used to quantify the transient and persistent compoents of the Nav currents are shown in the inset. The transient and persistent Nav conductances for the currents evoked at each test potential were determined and normalized (in the same cell) to the maximal transient and persistent Nav conductances and mean ± SEM normalized transient and persistent Nav conductances in WT and Fgf14−/− Purkinje neurons were plotted (right) as a function of the test potential. Although well described by single Boltzmann functions (see Materials and Methods), the voltage dependences of activation of both the transient and the persistent components of the Nav currents (right) are similar in WT and Fgf14−/− Purkinje neurons.
To determine the voltage dependences of activation of the transient and persistent components of the Nav currents in WT and Fgf14−/− Purkinje neurons, a three-step protocol was also used. In this case, cells were first briefly (3 ms) depolarized to −40 mV from a holding potential of −80 mV. After a 1 ms step to −55 mV, transient and persistent Nav currents evoked in response to voltage steps to various test potentials (ranging from −70 to 0 mV in 5 mV increments) were recorded. The voltage–clamp paradigm and a representative set of records, with selected voltage steps shown and color-coded with the corresponding current traces, are presented in Figure 8B. The transient and persistent Nav current amplitudes at each test potential in each cell were measured; persistent currents were measured at the end of the test voltage step (1 ms after the peak) and the transient currents were measured as the difference between the peak and the persistent inward Nav currents. The transient and persistent Nav conductances (GNa) were then calculated and normalized to their respective maximal (GNa,max) conductance values determined (in the same cell) at the most depolarized test potential (0 mV). Mean ± SEM normalized transient and persistent Nav conductances (GNa/GNa,max) were then plotted as a function of the test potential (Vm) and fitted using the Boltzmann, GNa/GNa,max = 1 + e[(Va − Vm)/k], where Va is the membrane potential of half-maximal activation and k is the slope factor.
Whole-cell current-clamp and voltage-clamp data were compiled and analyzed using ClampFit (Molecular Devices), Microsoft Excel, and Prism (GraphPad) software.
Immunostaining.
Mice were deeply anesthetized with 1 ml/kg intraperitoneal ketamine/xylazine mixture and perfused transcardially with 0.9% NaCl, followed by freshly prepared, ice-cold 1% formaldehyde (Goldin et al., 2000) in 0.1 m PBS, pH 7.4. Brains were removed and postfixed for 1 h in the same fixative at 4°C, followed by overnight cryoprotection in 30% sucrose in 0.1 m PBS at 4°C. Brains were embedded in OCT and frozen; sagittal (40 μm) cryostat sections of the cerebellum were collected into PBS at 22–23°C and stored at 4°C until processed for staining.
Free-floating sections were rinsed twice in 0.01 m PBS and permeabilized for 30 min in PBS with 0.1% Triton X-100 (v/v). Sections were incubated with blocking solution (PBS plus 10% goat serum) for 1 h, followed by staining overnight with primary antibodies diluted in PBS with 0.1% Triton X-100 and 0.1% bovine serum albumin. After washing with PBS, sections were incubated with appropriate goat secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor 647 (1:400; Life Technologies) diluted in PBS for 1 h. Sections were again washed with PBS, mounted on positively charged slides, and coated with a drop of Vectashield Hardset (Vector Laboratories). After placing coverslips on the slides, the sections were allowed to dry overnight at 4°C. The following primary antibodies were used: mouse monoclonal anti-iFGF14 (1:1000; NeuroMab), mouse monoclonal anti-Ankyrin G clone N106/36 (1:1000; NeuroMab), and mouse monoclonal anti-pan Nav α-subunit (1:1000; Sigma).
Image acquisition and analysis.
Image acquisition using a confocal laser scanning microscope (Olympus Fluoview-500) and 60× oil-immersion objective and image analyses were performed with the experimenter blinded to experimental group. Laser intensity, gain, and pinhole were kept constant between experimental groups. Sequential acquisition of multiple channels was used, and z-stacks were collected at 0.5 μm steps. Image stacks were converted into maximum intensity z-projections using ImageJ and were analyzed separately with ImageJ or MATLAB (Mathworks): ImageJ was used to generate line scan graphs and MATLAB was used to generate total integrated intensity graphs. For ImageJ analysis, three channel TIFF files were analyzed by drawing line profiles along the AIS starting at the soma and using anti-Ankyrin G staining as the marker of AIS location. Fluorescence intensity plot profiles for each channel were obtained and imported into Excel for averaging among experimental groups. Alternatively, single 2D RGB TIFF files were imported into MATLAB and analyzed using previously described methods (Grubb and Burrone, 2010). Briefly, a line profile was drawn along each axon starting at the soma and continuing past the AIS. The start and end of each AIS was determined by the proximal and distal points, respectively, at which the normalized and smoothed anti-Ankyrin G fluorescence intensity declined to 0.33 of the maximal value. Fluorescence intensities obtained from these line profiles were smoothed using a 40-point sliding mean (Grubb and Burrone, 2010) and normalized to the maximal and minimal values in the same AIS; normalized values are reported. Integrated fluorescence intensity along each AIS was determined as the area under the curve between the start and end of the AIS.
Balance beam.
Motor coordination was evaluated with the experimenter blinded to experimental group by assessing the ability of mice to traverse an 80 cm texturized (5 mm) cylindrical metal beam (shRNA experiments) or an 80 cm texturized (12 mm) square Plexiglas beam (rescue experiments) to reach an enclosed black Plexiglas (20 cm × 20 cm × 20 cm) escape box (Carter et al., 2001). Each mouse was tested for its ability to cross the beam placed horizontally 50 cm above a table from a clear Plexiglas (15 cm × 15 cm) start platform to the escape box. Baseline performance was assessed at 2–4 months of age. After intracerebellar AAV1 injections, mice were allowed to recover for 4–5 weeks and then reevaluated. Animals were videotaped during testing and scoring was performed offline. Time to traverse the beam and number of hindlimb foot slips were determined for each mouse before and after AAV1 injections. Scoring of performance on the balance beam was performed as previously described by Heng et al. (2007). Falls from the beam were scored as follows: the first fall was scored as a single foot slip and the second fall was scored as a fail. Fails were scored as a maximum time of 20 s and the maximum number of foot slips. The maximum number of foot slips observed in each experiment (10 for the shRNA experiment and 16 for the rescue experiment) was used as the maximum foot-slip score for a mouse that failed. Hindlimb dragging behavior, in which the mice wrapped their hind feet around the beam, pressed their bodies against the beam, and used their forelimbs to pull their bodies across the beam, was scored as the fraction of the beam over which that mouse dragged multiplied by a factor of 10 for the shRNA experiments or 16 for the rescue experiments, such that a mouse that dragged >100% of the beam was scored as having made the maximum number of foot slips (i.e., 10 or 16 foot slips, respectively).
Trials took place over 8 d (Heng et al., 2007). On day 1, naive mice were habituated to the start platform and escape box for several minutes. Mice were then allowed to traverse the beam three times, once as a group with cage mates and twice individually, before the test cross. On each subsequent day, each mouse crossed the beam once. Testing of mice after intracerebellar injections was performed in the same way, although the practice trials on day 1 were omitted. The largest improvement in performance was observed over the first 3 d and these were considered training days. Performance typically plateaued during the last 5 d and these trials were analyzed separately.
Statistical analyses.
All results are presented as means ± SEM. Statistical analyses were performed using the (unpaired) Student's t test, one-/two-way ANOVA, or the Kolmogorov–Smirnov test, as noted in the text and figure legends.
Results
Acute “knock-down” of iFGF14 in mature Purkinje neurons in vivo
To identify shRNA sequences that reduce iFGF14 expression, seven Fgf14-targeted shRNAs were screened in CHL fibroblasts coexpressing FGF14-myc (see Materials and Methods). Western blot analysis revealed that three of the seven shRNAs reduced iFGF14 to a similar extent (data not shown). One (5′-GCAAGTTTAAAGAGTCTGTTT-3′) of these validated Fgf14-targeted shRNA sequences and a control, nontargeted shRNA sequence (targeting GFP; see Materials and Methods) were then cloned (individually) in an miR-30 context into the 3′-UTR of the tdTomato reporter gene (Stegmeier et al., 2005) and inserted into an adeno-associated virus (AAV) transfer vector containing a CAG (chicken β-actin with CMV enhancer) promoter (Fig. 1A) determined in preliminary experiments to provide robust and selective AAV serotype1 (AAV1)-mediated expression of transgenes in adult cerebellar Purkinje neurons (see Materials and Methods). To confirm that the Fgf14-targeted shRNA was effective in reducing iFGF14 expression in the miR-30 context, FGF14-myc was coexpressed with increasing amounts of the plasmid expressing the Fgf14-targeted shRNA or the nontargeted shRNA in CHL cells (Fig. 1B). Western blot analyses revealed that iFGF14 was robustly expressed in cells coexpressing the nontargeted shRNA, but was decreased in a dose-dependent manner, in cells coexpressing the Fgf14-targeted shRNA (Fig. 1B). AAV1 viruses expressing the Fgf14-targeted or the nontargeted shRNA driven by the CAG promoter were generated (see Materials and Methods) for transduction of mature Purkinje neurons in vivo.
Acute “knock-down” of iFGF14 expression in mature Purkinje neurons in vivo was examined following stereotaxic injection of the Fgf14-targeted, or the nontargeted, shRNA-expressing AAV1 into mature WT mouse cerebellum. Cerebellar sections were stained using anti-Ankyrin G and anti-iFGF14 specific antibodies 4–8 weeks after the injections. Regions of the cerebellum injected with virus were identified by tdTomato expression and AIS regions were identified by anti-Ankyrin G immunolabeling (Kordeli et al., 1995). As illustrated in Figure 1C, tdTomato expression was readily detected in large numbers of Purkinje neurons in the cerebellum of animals transduced with either the nontargeted or the Fgf14-targeted shRNA-expressing AAV1. In marked contrast, tdTomato expression was seen in only a very small number of granule cells and was undetectable in stellate and basket cells in the molecular layer (Fig. 1C). Analyses of anti-Ankyrin G immunofluorescence intensities at Purkinje neuron AIS revealed that Ankyrin G expression was indistinguishable in nontargeted and Fgf14-targeted shRNA-transduced Purkinje neurons (Fig. 2A,B,D), suggesting that the organization of Purkinje neuron AIS is not affected by the acute in vivo “knock-down” of iFGF14.
Figure 2.

Ankyrin G expression at the axon initial segment is robust in Purkinje neurons transduced with the Fgf14-targeted shRNA. A, Representative images of nontargeted and Fgf14-targeted shRNA-transduced Purkinje neurons identified by tdTomato fluorescence (red) and stained with anti-Ankyrin G (blue) and anti-iFGF14 (green) specific antibodies. In each panel, the arrowheads indicate the AIS. Scale bars, 5 μm. B, Representative line scans of anti-Ankyrin G and anti-iFGF14 immunofluorescence intensities along the AIS of a nontargeted (top) and an Fgf14-targeted (bottom) shRNA-transduced Purkinje neuron. Vertical (red) dotted lines indicate the start and end of each AIS. C, Mean ± SEM anti-iFGF14 (top) and anti-Ankyrin G (bottom) immunofluorescence intensities along the AIS of Purkinje neurons transduced with either the nontargeted (n = 29 AIS, 2 animals) or the Fgf14-targeted (n = 26 AIS, 2 animals) shRNA. D, Mean ± SEM anti-iFGF14 (top) integrated immunofluorescence intensity was reduced significantly (≠p < 0.0001; Student's t test) in Fgf14-targeted shRNA-expressing (n = 26) compared with nontargeted shRNA-expressing (n = 29) Purkinje neurons, whereas mean ± SEM anti-ankyrin G integrated immunofluorescence intensities were not measurably affected.
Similar to WT animals (Xiao et al., 2013), anti-iFGF14 immunolabeling was detected at the AIS of neurons throughout the cerebellar cortex, including in stellate, basket, granule, and Purkinje neurons, of mice injected with the nontargeted shRNA (Figs. 1C, 2A). Anti-iFGF14 immunostaining was particularly intense in the granule layer (Fig. 1C), consistent with the robust expression of iFGF14 in granule neurons (Xiao et al., 2013). In Purkinje neurons expressing the nontargeted shRNA, anti-iFGF14 labeling is evident in a proximal to distal gradient at the AIS (Fig. 2A–C), similar to WT Purkinje neurons (Xiao et al., 2013). In sections from mice injected with the Fgf14-targeted shRNA-expressing AAV1, however, little to no anti-iFGF14 labeling was detected at the AIS of transduced Purkinje neurons (Fig. 2A–C). Quantification of the total (integrated) fluorescence intensity (see Materials and Methods) along Purkinje neuron AIS further revealed that anti-iFGF14 labeling was significantly (‡p < 0.0001) lower in Purkinje neurons expressing the Fgf14-targeted shRNA, compared with cells expressing the nontargeted shRNA (Fig. 2D). In contrast, anti-iFGF14 labeling in the molecular and granule cell layers was indistinguishable in mice injected with the nontargeted and the Fgf14-targeted shRNA-expressing viruses (Fig. 1C).
Acute “knock-down” of iFGF14 attenuates spontaneous and evoked firing in Purkinje neurons
Whole-cell current-clamp recordings were obtained from td-Tomato-positive Purkinje neurons (Fig. 3A) in acute cerebellar slices prepared from mice 3–4 weeks after stereotaxic intracerebellar injections of either the nontargeted shRNA- or the Fgf14-targeted shRNA-expressing AAV1. Similar to WT Purkinje neurons (Häusser and Clark, 1997), spontaneous firing (Fig. 3B) was observed in the vast majority (26 of 30) of td-Tomato-positive Purkinje neurons expressing the nontargeted shRNA (Fig. 3D). In marked contrast, most (40 of 46) of the td-Tomato-positive Purkinje neurons expressing the Fgf14-targeted shRNA were silent (Fig. 3C,D). Spontaneous firing was observed in only six (of the 46) Fgf14-targeted shRNA-expressing Purkinje neurons (Fig. 3D). Additional experiments revealed that the firing properties of noninfected Purkinje neurons in Fgf14-targeted shRNA transduced cerebella were indistinguishable from WT Purkinje neurons.
Figure 3.

Spontaneous and evoked firing is attenuated in Purkinje neurons transduced with the Fgf14-targeted shRNA. A, Representative fluorescence images of acute cerebellar slices prepared from a wild-type mouse injected with tdTomato-shRNA AAV1. Low-magnification image (left) shows widespread infectivity of an entire lobule of the cerebellum and the high-magnification image (right) reveals infected Purkinje neurons. B, C, Representative recordings of spontaneous activity in a nontargeted shRNA-transduced Purkinje neuron (B) and a quiescent Fgf14-targeted shRNA-transduced Purkinje neuron (C). D, Percentages of nontargeted (n = 30) and Fgf14-targeted (n = 46) shRNA-transduced Purkinje neurons that fired spontaneously or were silent. The vast majority (>85%) of the nontargeted shRNA-transduced cells were spontaneously active, whereas most (>85%) of the Fgf14-targeted shRNA-transduced Purkinje neurons were silent. E, F, Representative recordings from nontargeted (E) and Fgf14-targeted (F) shRNA-transduced Purkinje neurons before and during a prolonged (250 ms) 1000 pA depolarizing current injection. G, Mean ± SEM numbers of action potentials evoked in nontargeted (n = 28) and Fgf14-targeted (n = 46) shRNA transduced Purkinje neurons are plotted as a function of the amplitude of the injected current. H, Inverse cumulative frequency plots of spike numbers (elicited by 250 ms 1000 pA depolarizing current injections) in nontargeted (n = 28) and Fgf14-targeted (n = 46) shRNA-transduced Purkinje neurons. The frequency distribution in Fgf14-targeted shRNA-transduced neurons is shifted significantly (p < 0.0001, Kolmogorov–Smirnov test) to the left compared with nontargeted shRNA-transduced cells.
Analyses of current-clamp records obtained during prolonged (250 ms) depolarizing current injections (Fig. 3E,F) revealed that, similar to recordings from WT Purkinje neurons (Kalume et al., 2007, Shakkottai et al., 2009), the numbers of action potentials evoked in nontargeted shRNA-transduced Purkinje neurons increased as a function of the amplitude of the injected current (Fig. 3G). Importantly, action potentials could also be evoked in response to depolarizing current injections in Purkinje neurons expressing the Fgf14-targeted shRNA (Fig. 3F), although firing was not sustained (Fig. 3G). Rather, prolonged depolarizing current injections in these cells elicited only one or a few action potentials independent of the amplitude of the injected current (Fig. 3G).
As illustrated in the inverse cumulative frequency plots (Fig. 3H), the numbers of spikes evoked in response to 1 nA depolarizing current injections in Fgf14-targeted shRNA-expressing Purkinje neurons is shifted significantly (≠p < 0.0001) to the left (fewer spikes) compared with the distribution in cells expressing the nontargeted shRNA. In response to a prolonged (250 ms) 1 nA depolarizing current injection, for example, ∼70% of the Purkinje neurons transduced with the nontargeted shRNA-expressing AAV1 fired >20 action potentials (in 250 ms), whereas <10% of the Fgf14-targeted shRNA-expressing Purkinje neurons fired >20 action potentials (Fig. 3H). Acute “knock-down” of iFGF14, therefore, markedly reduces spontaneous and evoked repetitive firing in mature Purkinje neurons (see Discussion).
Acute “knock-down” of iFGF14 in Purkinje neurons impairs motor performance
To determine the functional consequences of the loss of iFGF14 in mature Purkinje neurons, motor performance was assessed using the balance beam task (Carter et al., 2001, Heng et al., 2007). Baseline motor performance was evaluated in 2- to 3-month-old naive animals (before virus injections). After acclimation and training (see Materials and Methods), the time to traverse a 5 mm cylindrical beam and the number of hindlimb placement errors (foot slips) made during the crossing were recorded for each animal. Stereotaxic injections of the nontargeted shRNA- or the Fgf14-targeted shRNA-expressing AAV1 were then made into each animal and motor performance was evaluated again 4–5 weeks later (Fig. 4A). As illustrated in the bright-field and fluorescence images in Figure 4B, widespread infectivity in the cerebellum was evident after two intracerebellar injections (see Materials and Methods).
Figure 4.

Motor performance and coordination are impaired in mice expressing the Fgf14-targeted shRNA in Purkinje neurons. A, Motor performance and coordination were evaluated in the balance beam test (see Materials and Methods) before and 4 weeks after intracerebellar injections of the nontargeted (n = 11) or the Fgf14-targeted (n = 9) shRNA-expressing AAV1. B, Representative bright-field (top) and fluorescence (bottom) images of the posterior part of a brain dissected from a wild-type mouse 4 weeks after 2 stereotaxic injections of tdTomato-shRNA AAV1 into the cerebellum. Left, Dorsal views; right, midsagittal views. C, D, The time to traverse a 5 mm beam (C), as well as the number of hindlimb placement errors (foot slips) during crossing (D), were evaluated before and after intracerebellar injections of the nontargeted (n = 11) or the Fgf14-targeted (n = 9) shRNA AAV1. Times to traverse the beam and number of foot slips were indistinguishable before and after injection of the nontargeted shRNA. The mean ± SEM time to cross was significantly (**p < 0.01, two-way ANOVA) longer and the mean ± SEM number of foot slips was significantly (**p < 0.01, two-way ANOVA) higher, however, in the animals injected with the Fgf14-targeted shRNA AAV1 whether compared with the same animals before injection or to the animals injected with the nontargeted shRNA AAV1.
The mean ± SEM time to traverse the beam (Fig. 4C) and the mean ± SEM number of foot slips (Fig. 4D) were indistinguishable in animals before and after intracerebellar injections of the nontargeted shRNA-expressing AAV1. Motor performance and coordination (Fig. 4C,D), however, were markedly impaired in mice injected with the Fgf14-targeted shRNA-expressing virus. The mean ± SEM time to traverse the beam, for example, was significantly (**p < 0.01) longer in mice after injection of the Fgf14-targeted shRNA-expressing virus compared with before the injections or compared with the control (Fig. 4C). Similarly, the mean ± SEM number of foot slips was significantly (**p < 0.01) higher in mice after injection of the Fgf14-targeted shRNA (Fig. 4D). The acute “knock-down” of iFGF14 in mature Purkinje neurons, therefore, markedly impairs motor performance and coordination (see Discussion).
Nav α-subunit expression and localization are unaffected by acute “knock-down” of iFGF14
To determine whether the acute loss of iFGF14 in mature Purkinje neurons affects the expression and/or localization of Nav α-subunits, cerebellar sections prepared from mice injected with the nontargeted shRNA- or the Fgf14-targeted shRNA-expressing AAV1 were stained with anti-Ankyrin G and pan anti-Nav α-subunit specific antibodies. Similar to WT Purkinje neurons (Buttermore et al., 2012; Xiao et al., 2013), robust anti-Nav α-subunit immunostaining was observed at the AIS of Purkinje neurons transduced with the nontargeted shRNA- or the Fgf14-targeted shRNA-expressing AAV1 (Fig. 5A). Quantification of total anti-Nav α-subunit immunofluorescence intensity along the AIS revealed that Nav α-subunit expression is indistinguishable in Purkinje neurons transduced with the Fgf14-targeted, compared with the nontargeted, shRNA AAV1 (Fig. 5B–D), suggesting that Nav α-subunit expression in mature Purkinje neurons is unaffected by the acute “knock-down” of iFGF14 (see Discussion).
Figure 5.

Anti-Nav α-subunit immunofluorescence intensity and localization at Purkinje neuron AIS are unaffected by Fgf14-targeted shRNA. After injections of the nontargeted (top) or the Fgf14-targeted (bottom) shRNA AAV1, parasagittal sections were cut and stained with anti-Ankyrin G (blue) and anti-panNav α-subunit- (green) specific antibodies. A, Representative images of nontargeted and Fgf14-targeted shRNA-transduced Purkinje neurons, identified by tdTomato fluorescence (red); in each panel, arrowheads indicate AIS regions and scale bars are 5 μm. B, Representative line scans of anti-Ankyrin G and anti-panNav immunofluorescence intensities along the AIS of a nontargeted (top) and an Fgf14-targeted (bottom) shRNA-transduced Purkinje neuron. Vertical (red) dotted lines indicate the starts and ends of the AIS. C, Mean ± SEM immunofluorescence intensities of anti-pan Nav (top) and anti-Ankyrin G (bottom) along the AIS of Purkinje neurons transduced with either the nontargeted (n = 51 AIS, 2 animals) or the Fgf14-targeted (n = 60 AIS, 2 animals) shRNA. D, Mean ± SEM integrated immunofluorescence intensity of anti-panNav (top) and anti-Ankyrin G (bottom) staining along the AIS of Purkinje neurons transduced with the nontargeted (n = 51 AIS, 2 animals) or the Fgf14-targeted (n = 60 AIS, 2 animals) shRNA. Mean ± SEM anti-Ankyrin G and anti-Nav α-subunit labeling intensities are indistinguishable (Student's t test) in adult Purkinje neurons transduced with the nontargeted and the Fgf14-targeted shRNAs.
Hyperpolarization rescues firing in Fgf14-targeted shRNA-expressing Purkinje neurons
The lack of effect of the Fgf14-targeted shRNA on Nav α-subunit immunolabeling at Purkinje neuron AIS (Fig. 5) suggests that the observed reduction in excitability (Fig. 3) reflects the loss of iFGF14-mediated regulation of the biophysical properties of the Nav channels (Liu et al., 2003; Lou et al., 2005; Goldfarb et al., 2007; Laezza et al., 2009) underlying action potential generation rather than changes in the number and/or localization of these channels. In previous studies, we found that expression of iFGF14 in Neuro2A cells resulted in a marked depolarizing shift in the voltage dependence of Nav current inactivation (Lou et al., 2005), suggesting that the marked reduction in the excitability of mature cerebellar Purkinje neurons with the acute “knock-down” of iFGF14 results from a hyperpolarizing shift in Nav current inactivation. To explore this hypothesis, the effects of prior membrane hyperpolarization on spontaneous and evoked firing were examined (Fig. 6). Hyperpolarizing current injections (of 500 pA) reduced (hyperpolarized) the membrane potentials of Purkinje neurons transduced with either the nontargeted (Fig. 6A) or the Fgf14-targeted (Fig. 6B) shRNA-expressing AAV1. Injection of the hyperpolarizing current silenced the vast majority (24 of 25) of the Purkinje neurons expressing the nontargeted shRNA (Fig. 6A). In addition, the mean ± SEM membrane potentials measured (with the 500 pA hyperpolarizing current applied) in Purkinje neurons expressing the nontargeted shRNA (−64.9 ± 0.8 mV; n = 25) and the Fgf14-targeted shRNA (−63.4 ± 0.6 mV; n = 42) were indistinguishable. The input resistances of Purkinje neurons expressing the nontargeted shRNA (24.8 ± 2.0 MΩ; n = 24) and the Fgf14-targeted shRNA (27.5 ± 1.6 MΩ; n = 41) were also indistinguishable.
Figure 6.
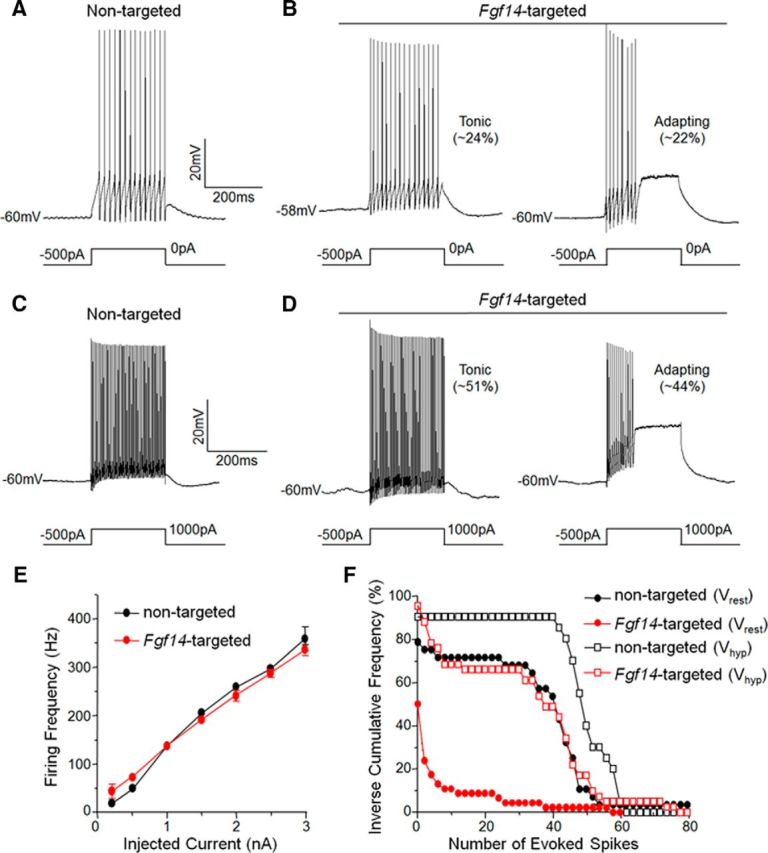
Membrane hyperpolarization rescues repetitive firing in Purkinje neurons transduced with Fgf14-targeted shRNA. A–D, Representative evoked firing recorded in nontargeted (A,C) and Fgf14-targeted (B,D) shRNA-transduced Purkinje neurons in response to membrane depolarization from a hyperpolarized membrane potential. Removal of a (500 pA) hyperpolarizing current injection resulted in spontaneous firing in most (20 of 25) of the nontargeted shRNA-transduced Purkinje neurons (A) and in 19 of 41 Fgf14-targeted shRNA-transduced Purkinje neurons: 10 of these cells fired tonically and 9 adapted; the remaining (12) cells were silent. Depolarizing (1000 pA) current injections (250 ms) after membrane hyperpolarization elicited high-frequency repetitive firing in most (23 of 25) Purkinje neurons expressing the nontargeted shRNA, as well as in most (39 of 41) Purkinje neurons expressing the Fgf14-targeted shRNA (D). Mean ± SEM firing frequencies in nontargeted and Fgf14-targeted shRNA-transduced Purkinje neurons in response to (250 ms) depolarizing current injections of varying amplitudes presented after membrane hyperpolarization are plotted in E. Firing rates were indistinguishable (two-way ANOVA) in nontargeted and Fgf14-targeted shRNA-transduced Purkinje neurons. F, Inverse cumulative frequency plots of numbers of action potentials evoked by 250 ms, 1000 pA depolarizing current injections after membrane hyperpolarization (Vhyp) in nontargeted and Fgf14-targeted shRNA-transduced Purkinje neurons are plotted. Histograms of spike numbers elicited from rest (Vrest) are replotted from Figure 3H for comparison purposes. Membrane hyperpolarization significantly (p < 0.0001) shifted the frequency distributions to the right (more spikes) in both nontargeted shRNA and Fgf14-targeted shRNA-transduced Purkinje neurons. In addition, the distribution of spikes evoked from a hyperpolarized potential in Fgf14-targeted shRNA-transduced Purkinje neurons is not significantly different from the distribution of spikes evoked from rest in Purkinje neurons expressing the nontargeted shRNA.
Removal of the hyperpolarizing current resulted in repetitive firing in most (20 of 25) of the nontargeted shRNA-expressing Purkinje neurons (Fig. 6A). Interestingly, removal of the applied hyperpolarizing current also resulted in repetitive firing in 19 of the 41 Fgf14-targeted shRNA-transduced Purkinje neurons: of these, 10 fired tonically and 9 fired at least one action potential and then adapted (Fig. 6B). The remaining (22 of 41) Fgf14-targeted shRNA-expressing Purkinje neurons were electrically silent. These results are in marked contrast to the quiescent phenotype of Fgf14-targeted shRNA-transduced Purkinje neurons (Fig. 3C) in the absence of hyperpolarizing current injection and suggest that the electrophysiological effects of the acute “knock-down” of iFGF14 depend on membrane potential (see Discussion).
To explore the effects of membrane hyperpolarization on evoked firing in Purkinje neurons (250 ms), depolarizing current injections of varying amplitudes were delivered from the hyperpolarized membrane potential. Analyses of the evoked responses revealed high-frequency repetitive firing in most (23 of 25) Purkinje neurons transduced with the nontargeted shRNA-expressing AAV1 (Fig. 6C). High-frequency firing was also observed in the vast majority (39 of 41) of Purkinje neurons expressing the Fgf14-targeted shRNA: 21 of these cells fired tonically and 18 cells adapted (Fig. 6D). These results are in striking contrast to the inability of Fgf14-targeted shRNA-expressing Purkinje neurons to sustain evoked repetitive firing in response to depolarizing current injections from rest (Fig. 3F–H). Analyses of the rates of repetitive firing, evoked from a hyperpolarized membrane potential, in tonically firing nontargeted shRNA- and Fgf14-targeted shRNA-expressing Purkinje neurons further revealed that the frequency increased as a function of the amplitude of the injected current (Fig. 6E) and that the mean ± SEM firing frequencies (Fig. 6E) of the nontargeted shRNA- and Fgf14-targeted shRNA-expressing Purkinje neurons that fired tonically in response to depolarizing current injections from hyperpolarized membrane potentials were indistinguishable.
Comparison of inverse cumulative frequency plots of the numbers of spikes evoked in response to 1 nA depolarizing current injections from a hyperpolarized potential revealed that membrane hyperpolarization significantly (p < 0.0001) shifted the distributions to the right (more spikes) in both nontargeted shRNA- and Fgf14-targeted shRNA-expressing Purkinje neurons, compared with the distributions of spike numbers elicited from rest (Fig. 6F). In response to a 1 nA depolarizing current injection from rest, for example, <10% of the Purkinje neurons transduced with the Fgf14-targeted shRNA virus fired >20 action potentials (Fig. 6F). In contrast, ∼65% of these cells fired >20 action potentials when the depolarizing current was injected from a hyperpolarized membrane potential (Fig. 6F). Interestingly, the distribution of the numbers of spikes evoked from a hyperpolarized potential in Purkinje neurons transduced with Fgf14-targeted shRNA virus is indistinguishable from the distribution of spikes evoked from rest in Purkinje neurons transduced with nontargeted shRNA virus (Fig. 6F). The effects of acute “knock-down” of iFGF14 on spontaneous and evoked repetitive firing in mature Purkinje neurons, therefore, are steeply dependent on the membrane potential (see Discussion).
Hyperpolarization also rescues repetitive firing in Fgf14−/− Purkinje neurons
Spontaneous and evoked firing are reduced markedly in Fgf14−/− Purkinje neurons (Shakkottai et al., 2009). Subsequent experiments were aimed at determining whether membrane hyperpolarization can also rescue repetitive firing in adult Fgf14−/− Purkinje neurons. Similar to nontargeted shRNA-expressing WT cells (Fig. 6), membrane hyperpolarization electrically silenced most (30 of 32) WT Purkinje neurons (Fig. 7). The mean ± SEM membrane potentials measured (with the 500 pA hyperpolarizing current injection) in WT and Fgf14−/− Purkinje neurons were identical: the mean ± SEM membrane potentials measured were −64.4 ± 0.6 mV (n = 30) and −64.4 ± 0.6 mV (n = 18) in WT and Fgf14−/− Purkinje neurons, respectively. The input resistances of WT (28.6 ± 1.3 MΩ; n = 30) and Fgf14−/− (28.9 ± 1.7 MΩ; n = 22 neurons) were also indistinguishable. Removal of the hyperpolarizing current resulted in spontaneous firing in most (24 of 32; 75%) WT neurons and in 11 of 23 (50%) Fgf14−/− neurons (Fig. 7).
Figure 7.

Membrane hyperpolarization rescues repetitive firing in adult Fgf14−/− Purkinje neurons. A–D, Representative recordings from WT and Fgf14−/− Purkinje neurons obtained using the protocols described in the legend to Figure 5. Removal of the (500 pA) hyperpolarizing current resulted in repetitive firing in most (24 of 32) of the WT neurons (A) and in 11 of the 23 Fgf14−/− neurons (B). Depolarizing (1000 pA) current injections, presented from hyperpolarized membrane potentials, resulted in repetitive firing in most (29 of 32) WT (C) and (21 of 23) Fgf14−/− (D) Purkinje neurons. E, Mean ± SEM firing frequencies in WT and Fgf14−/− Purkinje neurons in response to prolonged (250 ms) depolarizing current injections of varying amplitudes. Firing rates increased as a function of the amplitudes of the depolarizing current injections and were indistinguishable (two-way ANOVA) in WT (n = 32) and Fgf14−/− (n = 23) Purkinje neurons.
In addition, and similar to the results obtained in the experiments performed on Fgf14-targeted shRNA-transduced Purkinje neurons (Fig. 6), depolarizing current injections presented from a hyperpolarized membrane potential resulted in repetitive firing in the vast majority of WT (29 of 32) and Fgf14−/− (21 of 23) Purkinje neurons (Fig. 7C,D). Analyses of repetitive firing rates recorded in response to prolonged depolarizing current injections from a hyperpolarized membrane potential further revealed that firing frequencies increased as a function of the amplitude of the injected current (Fig. 7E). In addition, the mean ± SEM firing frequencies in WT (n = 32) and Fgf14−/− (n = 23) Purkinje neurons were indistinguishable (Fig. 7E). Similar to the findings with acute “knock-down” of iFGF14 in mature Purkinje neurons (Fig. 6), therefore, the functional effects of the germline-targeted disruption of Fgf14 on spontaneous and evoked repetitive firing in mature Purkinje neurons can be modified by hyperpolarizing the membrane potential.
Loss of iFGF14 affects voltage-dependent Nav current inactivation in Purkinje neurons
The observations that the functional consequences of the acute “knock-down” of iFGF14 and of the germline-targeted disruption of Fgf14 on spontaneous and evoked repetitive firing in mature Purkinje neurons can be modified in situ by hyperpolarizing the membrane potential suggested an effect of iFGF14 on the voltage-dependent properties of the Nav currents in these cells, as has been demonstrated previously in other cells (Liu et al., 2003; Wittmack et al., 2004; Lou et al., 2005; Goldfarb et al., 2007; Goetz et al., 2009; Laezza et al., 2009; Dover et al., 2010; Wang et al., 2011b). To test this hypothesis, voltage-clamp recordings were obtained from WT and Fgf14−/− Purkinje neurons in acute cerebellar slices, as described in Materials and Methods, using voltage-clamp protocols similar to those described by Milescu et al. (2010) designed to minimize space-clamp errors and to enable the reliable measurements of fast transient Nav currents in neurons in acute brain slices.
To determine the voltage dependences of steady-state inactivation of the transient Nav currents in WT and Fgf14−/− Purkinje neurons, a three-step protocol (see Materials and Methods) was used and is illustrated in Figure 8A. Current amplitudes at each test potential in each cell were measured and normalized to the current evoked in the same cell from −110 mV. Analysis of the mean ± SEM normalized transient Nav current amplitudes plotted as a function of the conditioning membrane potential (Fig. 8A) revealed that the voltage at which 50% of the Nav channels are inactivated (V1/2) in Fgf14−/− (V1/2 = −71.1 ± 0.9 mV; n = 10) Purkinje neurons is significantly (p < 0.001) more hyperpolarized in Fgf14−/− than in WT Purkinje neurons, with V1/2 values of −71.1 ± 0.9 mV (n = 10) and −58.1 ± 0.9 mV (n = 11) in Fgf14−/− and WT Purkinje neurons, respectively (Fig. 8A). The slopes of the fits to the steady-state inactivation data (Fig. 8A) in WT and Fgf14−/− Purkinje neurons, in contrast, are not significantly different [slopes = 10.3 ± 1.0 (n = 11) and 11.3 ± 1.0 (n = 10) in WT and Fgf14−/− Purkinje neurons, respectively].
A voltage-clamp protocol was also designed (see Materials and Methods) to allow examination of the voltage dependences of activation of the transient and persistent components of the Nav currents in WT and Fgf14−/− Purkinje neurons and is illustrated in Figure 8B. Analyses of the mean ± SEM normalized transient and persistent Nav conductances measured in WT (n = 7) and Fgf14−/− (n = 7) Purkinje neurons plotted as a function of test potential revealed that, in contrast to steady-state inactivation (Fig. 8A), the voltage dependences of activation (Fig. 8B) of the transient Nav currents in WT and Fgf14−/− Purkinje neurons are not significantly different: the V1/2 (and slope) values determined for the transient Nav currents in Fgf14−/− and WT Purkinje neurons were −33.8 ± 1.3 mV (7.7 ± 1.2) and −35.7 ± 0.6 mV (5.7 ± 0.5), respectively. The voltage dependences of activation of the persistent Nav currents in WT (V1/2 = −33.1 ± 1.1 mV; slope = 11.9 ± 1.4) and Fgf14−/− (V1/2 = −37.8 ± 2.1 mV; slope = 10.5 ± 1.4) Purkinje neurons are also not significantly different (Fig. 8B).
Expression of iFGF14 in Fgf14−/− Purkinje neurons rescues spontaneous firing and improves motor performance
To test the hypothesis that the acute expression of iFGF14 in adult Fgf14−/− Purkinje neurons might restore spontaneous firing, GFP-expressing AAV1, alone or in combination with hFGF14B-expressing AAV1 (Fig. 9), was injected into Fgf14−/− mouse cerebellum. Approximately 4–8 weeks after the injections, cerebellar sections were cut and stained with anti-iFGF14 and anti-Ankyrin G specific antibodies. No anti-iFGF14 immunolabeling was detected in Fgf14−/− Purkinje neurons transduced with the GFP virus (Fig. 9B,C). Robust anti-iFGF14 immunostaining at the Purkinje neuron AIS, identified by colabeling with the AIS marker Ankyrin G, however, was detected in cells transduced with the iFGF14B- and GFP-expressing viruses (Fig. 9B,C). Examination of images at low magnification revealed that anti-iFGF14 labeling was also present at the AIS of cells in the molecular, but not the granule, layer (Fig. 9B). Whole-cell current-clamp recordings obtained from Purkinje neurons in acute cerebellar slices from mice 3–4 weeks after stereotaxic intracerebellar AAV1 injections revealed that most (31 of 33; >90%) Fgf14−/− Purkinje neurons transduced with the GFP virus were electrically silent; spontaneous firing was observed in only two (of 33) Fgf14−/− cells (Fig. 10A,C). Spontaneous firing was observed, however, in the majority (39 of 57; 70%) of Fgf14−/− Purkinje neurons cotransduced with the iFGF14B- and GFP-expressing viruses (Fig. 10B,C).
Figure 9.
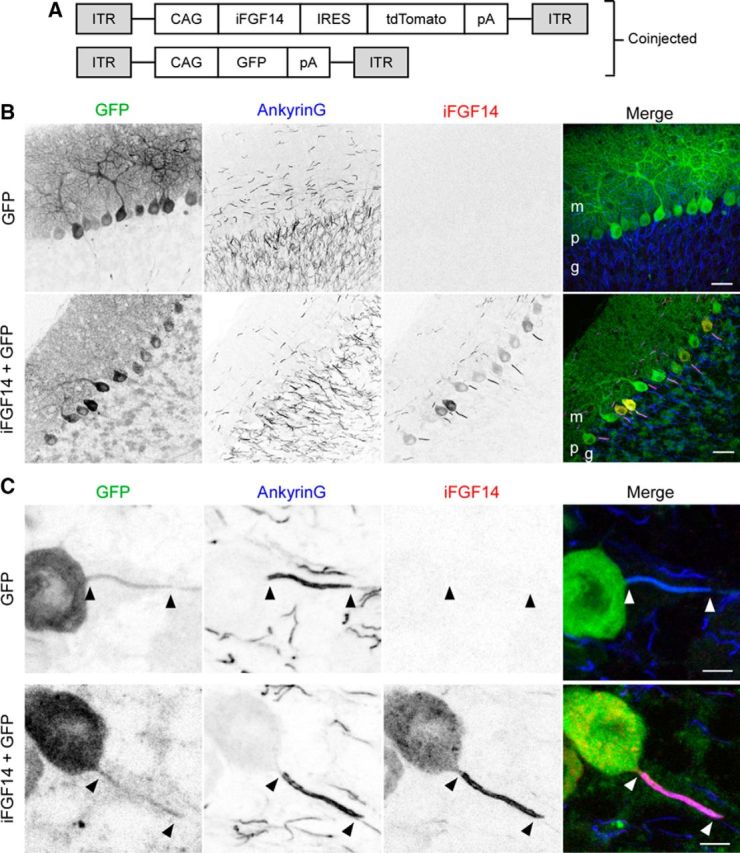
Viral-mediated expression of iFGF14 in adult Fgf14−/− Purkinje neurons. A, Schematic of the AAV transfer plasmid (see Materials and Methods) chicken β actin with CMV enhancer promoter (CAG). IRES, Internal ribosome entry site; ITR, inverted terminal repeat; pA, polyadenylation signal. B, C, Representative low-magnification (B) and high magnification (C) images of parasagital sections of adult Fgf14−/− mouse cerebellum injected with AAV1 expressing GFP alone (top panels) or in combination with the AAV1 expressing iFGF14 (bottom panels). Scale bars: B, 25 μm; C, 5 μm. Anti-iFGF14 and anti-Ankyrin G immunostaining are shown in red and blue, respectively. Transduced Purkinje neurons were identified by GFP fluorescence.
Figure 10.

Acute expression of iFGF14 in Fgf14−/− Purkinje neurons rescues spontaneous firing and improves motor coordination in Fgf14−/− mice. Representative recordings of a quiescent GFP-transduced Fgf14−/− Purkinje neuron (A) and of spontaneous activity in an Fgf14−/− Purkinje neuron cotransduced with the iFGF14 and GFP viruses (B). C, Percentages of GFP (n = 33) and GFP + iFGF14 (n = 57) transduced Purkinje neurons that fired spontaneously or were silent. The vast majority (>93%) of the GFP-transduced Fgf14−/− Purkinje neurons were silent, whereas most (>68%) of the Fgf14−/− Purkinje neurons transduced with iFGF14 were spontaneously active. D, Representative bright-field (top) and fluorescence (bottom) images of the posterior part of a brain dissected from a wild-type mouse 4 weeks after 2 stereotaxic injections of GFP virus into the cerebellum. Left, Dorsal views; right, midsagittal views. The mean ± SEM times to traverse a 12 mm beam (E) and numbers of hindlimb foot slips (F) in Fgf14−/− mice (n = 8) before and after intracerebellar injections of the GFP virus are not significantly different. In the Fgf14−/− mice injected with both the iFGF14 and GFP viruses (n = 9), however, the mean ± SEM time to cross the beam (E) was significantly (**p < 0.001) faster and the mean ± SEM number of foot slips (F) was significantly (**p < 0.01) lower after the injections compared with the same animals before the injections. The measured values are also significantly (**p < 0.01) different from the Fgf14−/− mice injected with the GFP virus.
To determine the functional consequences of the electrophysiological “rescue” of Fgf14−/− Purkinje neurons, balance beam performance was examined in Fgf14−/− animals at 2–4 months of age before virus injections and again 4–5 weeks after the injections. As illustrated in the bright-field and fluorescence images in Figure 10D, widespread infectivity in the cerebellum was evident after intracerebellar injections of the GFP-expressing and/or iFGF14B-expressing AAV1. Motor performance in Fgf14−/− mice was evaluated as the ability to cross a 12 mm rectangular balance beam; the larger diameter beams were used because the Fgf14−/− mice could not reliably traverse the 5 mm cylindrical beam that was used in the “knock-down” experiments (see Fig. 4). The mean ± SEM time to traverse the 12 mm beam (Fig. 10) and the mean ± SEM numbers of hindlimb foot slips (Fig. 10F) were indistinguishable in animals examined before and after injection of the GFP virus.
Motor performance and coordination (Fig. 10E,F), however, were markedly improved in the Fgf14−/− mice coinjected with the iFGF14- and GFP-expressing viruses. Fgf14−/− mice injected with the iFGF14 and GFP viruses, for example, took significantly (**p < 0.01) less time to traverse the beam after the injection compared with the same mice before the injection or with Fgf14−/− mice injected with only the GFP-expressing virus (Fig. 10E). The mean ± SEM number of foot slips was also significantly (**p < 0.01) lower in mice after coinjection of the iFGF14 and GFP viruses compared with before the injection or with the Fgf14−/− mice injected with the GFP-expressing virus (Fig. 10F). The in vivo rescue of iFGF14 expression in adult Fgf14−/− Purkinje neurons, therefore, improves motor coordination.
Discussion
The results of the experiments here demonstrate that the acute, Fgf14-shRNA-mediated, “knock-down” of iFGF14 eliminates spontaneous firing in the vast majority (>85%) of mature cerebellar Purkinje neurons, results that are remarkably similar to previous findings in Fgf14−/− Purkinje neurons in in vitro slices prepared from 25- to 30-d-old animals (Shakkottai et al., 2009). These findings demonstrate a physiological role of iFGF14 in mature Purkinje neurons that is independent for any developmental function(s) of iFGF14. Further electrophysiological experiments revealed that membrane hyperpolarization rescues high-frequency firing in adult Purkinje neurons with acute, Fgf14-shRNA-mediated “knock-down” of iFGF14 expression, as well as in adult Fgf14−/− Purkinje neurons. Consistent with a critical physiological role for iFGF14 in the functioning of mature cerebellar Purkinje neurons, additional experiments here demonstrate that viral-mediated rescue of iFGF14 expression restores spontaneous and evoked firing in Fgf14−/− Purkinje neurons and improves motor coordination and balance in Fgf14−/− animals. These studies provide evidence that the lack of iFGF14 throughout development does not result in irreversible changes in the properties of cerebellar Purkinje neurons.
iFGF14 regulates the voltage dependence of Nav channel availability in mature Purkinje neurons
The initial finding that iFGF12 binds to the C-terminal tails of Nav α-subunits (Liu et al., 2001, 2002) suggested possible roles for the iFGFs in determining the expression, biophysical properties, and/or subcellular distributions of Nav channels. Consistent with this hypothesis, subsequent studies in heterologous expression systems demonstrated effects of iFGF coexpression on Nav current amplitudes, as well as on the voltage dependences of Nav current activation and inactivation (Liu et al., 2003; Wittmack et al., 2004; Lou et al., 2005; Laezza et al., 2009; Wang et al., 2011b; Goldfarb, 2012). Comparison of the results obtained in various studies, however, also revealed that the functional effects of the individual iFGFs vary with the iFGF splice variant and the Nav α-subunit coexpressed, as well as with the cellular expression environment (Wittmack et al., 2004; Lou et al., 2005; Laezza et al., 2007, 2009; Wang et al., 2011b), making extrapolation to the likely “physiological roles” of the iFGFs in the regulation of native Nav channels difficult if not impossible. Indeed, in HEK-293 cells, as well as in Neuro 2A cells, coexpression of either iFGF14A or iFGF14B shifted the voltage dependences of activation and inactivation of Nav1.1 or Nav1.2 α-subunit-encoded Nav currents, but only iFGF14B measurably affected the amplitudes of the Nav currents in isolated hippocampal neurons (Lou et al., 2005, Laezza et al., 2007). In addition, although iFGF14B attenuated Nav α-subunit-encoded currents in HEK-293 cells, expression of iFGF14B in hippocampal pyramidal neurons markedly increased Nav current amplitudes (Lou et al., 2005, Laezza et al., 2007).
The results of the immunohistochemical analyses here revealed no significant differences in the distribution or the intensity of anti-pan Nav α-subunit labeling in nontargeted shRNA- and Fgf14-shRNA-expressing cerebellar Purkinje neurons. The current-clamp experiments here, however, revealed that the attenuation of spontaneous and evoked firing in Purkinje neurons with the acute Fgf14-shRNA-mediated “knock-down” of iFGF14, as well as in Fgf14−/− Purkinje neurons, is restored by prior membrane hyperpolarization, suggesting that iFGF14 affects the voltage-dependent properties of the Nav currents in these cells. Voltage-clamp recordings designed to explore this hypothesis indeed revealed that the V1/2 (−71.1 ± 1.0 mV) determined for steady-state inactivation of the transient component of the Nav currents in Fgf14−/− Purkinje neurons is significantly (p < 0.001) more hyperpolarized than the V1/2 (−58.1 ± 1.0 mV) measured in WT Purkinje neurons (Fig. 8A), whereas no differences in the voltage dependences of activation of either the transient or the persistent components of the Nav currents in WT and Fgf14−/− Purkinje neurons (Fig. 8B) were observed. Similar results were reported recently by Yan et al. (2014) in studies conducted on cerebellar Purkinje neurons isolated from postnatal day 6–8 animals and maintained for 12–14 d in vitro. These investigators, for example, reported that shRNA-mediated knock-down of iFGF14 expression resulted in an ∼10 mV hyperpolarizing shift in the V1/2 for steady-state inactivation of the transient component of the Nav current without measurably affecting the voltage dependence of current activation (Yan et al., 2014). Although the magnitude (∼10 mV) of the shift was similar to that (∼13 mV) observed here, the absolute V1/2 values measured in the absence (∼−60 mV) and presence (∼−50 mV) of iFGF14 were much more depolarized than the V1/2 values of −71 mV (Fgf14−/−) and −58 mV (WT) determined here. These apparent discrepancies may reflect differences in the maturity of the cells studied and/or in the experimental preparations (acute slices vs isolated cells maintained in tissue culture) used. Additional experiments will be needed to determine the contributing factors. Interestingly, it has also been reported that the in vitro shRNA-mediated knock-down of iFGF14 expression in immature Purkinje neurons maintained in culture accelerates the rate of Nav current inactivation and reduces the resurgent Nav current, effects that are also expected to affect firing rates (Yan et al., 2014). In vitro shRNA-mediated knock-down of iFGF14 expression has also been shown to reduce voltage-gated Ca2+ currents in cerebellar granule neurons (Yan et al., 2013). It has not been possible, however, to reliably voltage-clamp and measure Ca2+ currents or resurgent Nav currents in mature Purkinje neurons in acute cerebellar slices. Alternative experiments, perhaps coupled with modeling approaches, therefore, will need to be exploited to determine the functional importance of iFGF14-mediated modulation of Ca2+ currents and resurgent Nav currents in mature cerebellar Purkinje neurons.
Together, the results presented here suggest a model (Fig. 11) in which iFGF14 regulates the voltage dependence of Nav channel inactivation in mature cerebellar Purkinje neurons. In the presence of iFGF14, most Nav channels are closed (green) and available to open at the resting potential, whereas in the absence of iFGF14, most Nav channels are inactivated (red). In the absence of iFGF14, low Nav channel availability precludes spontaneous firing (Fig. 11A). Membrane hyperpolarization increases the proportion of closed Nav channels available to open (green), allowing for “rescue” of repetitive firing despite the loss of iFGF14 (Fig. 11B). The similarity in the distributions of spike numbers in nontargeted shRNA-expressing neurons at rest and iFGF14-deficient neurons at hyperpolarized potentials (Fig. 6F) suggests, however, that Nav channel availability in iFGF14 deficient neurons at hyperpolarized membrane potentials overlaps with Nav channel availability in control neurons at rest (Fig. 11C). This model could be further adapted if experimental evidence emerges suggesting additional effects of iFGF14 on Nav channel kinetics and/or on resurgent Nav currents in mature Purkinje neurons.
Figure 11.
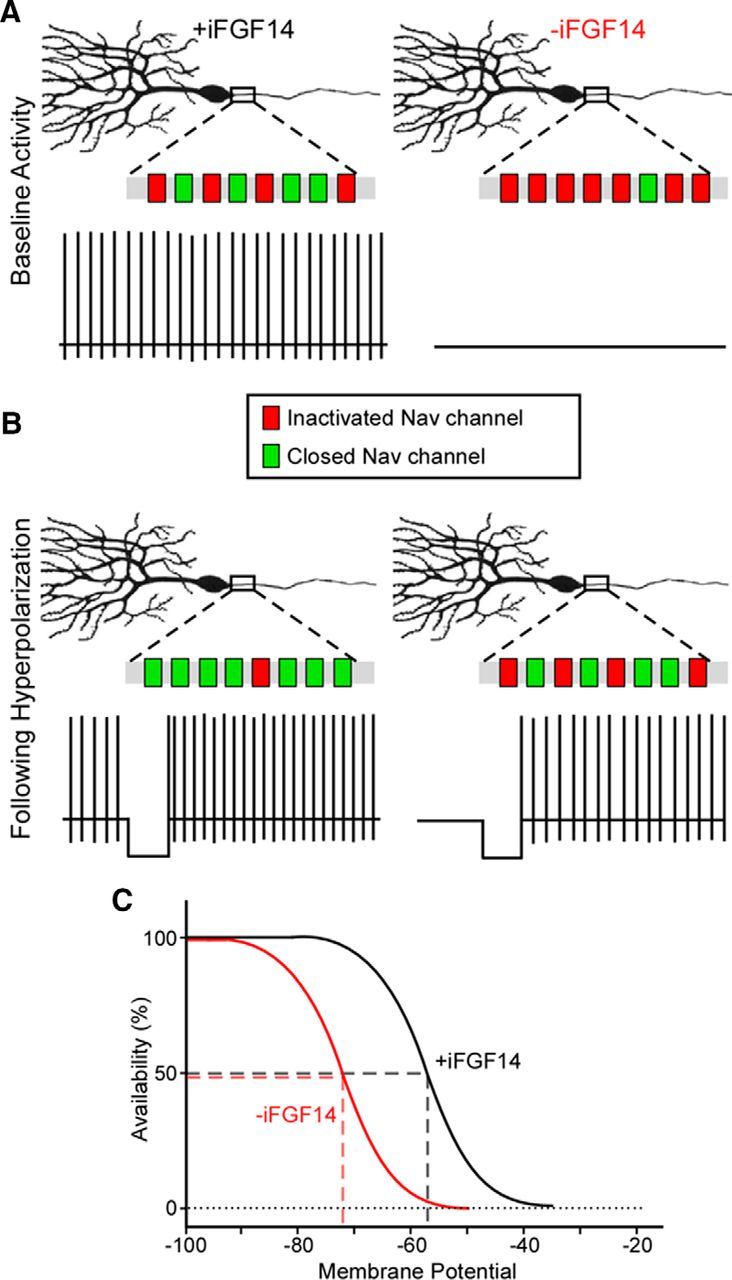
Schematic illustration of the modulatory effect of iFGF14 on Nav channel availability and repetitive firing in Purkinje neurons. A, Nav channel availability at rest is high in WT Purkinje neurons, which express iFGF14, and low in iFGF14 deficient Purkinje neurons. In addition, WT Purkinje neurons are spontaneously active, whereas most iFGF14-deficient Purkinje neurons are quiescent. B, Hyperpolarization of the membrane potential increases Nav channel availability in both WT and iFGF14-deficient Purkinje neurons and, importantly, “rescues” repetitive firing in the iFGF14-deficient cells. The total numbers of Nav channels are unchanged; only the numbers of closed channels that are available to open are affected by the membrane hyperpolarization. In addition, the availability of Nav channels is similar in iFGF14-deficient Purkinje neurons at hyperpolarized membrane potentials and in control Purkinje neurons at rest. C, In the presence of iFGF14, Nav channel availability in Purkinje neurons is shifted markedly to the right (depolarized).
It has also been reported that excitability is attenuated in cerebellar granule neurons from animals lacking iFGF14 (Fgf14−/−) or both iFGF12 and iFGF14 (Fgf12−/−/Fgf14−/−) (Goldfarb et al., 2007). In addition, voltage-clamp recordings from Fgf12−/−/Fgf14−/− granule neurons in in vitro slices and from isolated granule neurons maintained in culture also revealed a marked (∼−13–14 mV) hyperpolarizing shift V1/2 for Nav current inactivation (Goldfarb et al., 2007; Dover et al., 2010; Goldfarb, 2012). Interestingly, this shift and the absolute V1/2 values measured in WT and Fgf12−/−/Fgf14−/− cerebellar granule neurons are very similar to those determined here for Nav current inactivation in mature Fgf14−/− and WT Purkinje neurons in in vitro slices. It will be of interest to determine the molecular bases of the observed differences in the voltage dependences of inactivation of the Nav currents measured in immature Purkinje neurons maintained in culture (Yan et al., 2014) and in mature Purkinje neurons in acute in vitro slices.
The electrophysiological studies here also revealed heterogeneity in the voltage-dependent modulation of repetitive firing in Purkinje neurons lacking iFGF14. Spontaneous firing after the removal of membrane hyperpolarization, for example, was observed in only ∼1/2 of the Fgf14-targeted shRNA-expressing (Fig. 6B) and Fgf14−/− (Fig. 7B) Purkinje neurons. The repetitive firing patterns recorded in response to depolarizing current injections from a hyperpolarized membrane potential were also variable in that 1/2 of the cells fired tonically and the others adapted (Figs. 6D, 7D). This phenotypic heterogeneity likely reflects differences in the functional impact of iFGF14-mediated modulation of molecularly distinct Nav channels and/or in the repertoire of other ion channels expressed in individual Purkinje neurons. Functional and variable iFGF14-mediated effects on non-Nav channels could also contribute to observed differences in Purkinje neuron firing properties. Additional experiments focused on identifying the underlying cellular and molecular mechanisms will be of interest.
iFGF14-mediated regulation of Purkinje neuron firing is required for motor coordination
Previous electrophysiological studies on Fgf14−/− mice revealed marked effects of the loss of iFGF14 on the excitability of cerebellar granule and Purkinje neurons (Goldfarb et al., 2007; Shakkottai et al., 2009). Although these observations clearly suggested a critical role(s) for reduced neuronal excitability in the cerebellum in producing the motor deficits evident in Fgf14−/− mice (Goldfarb et al., 2007; Shakkottai et al., 2009), the causal link(s) between the reduced excitability of these (cerebellar granule and Purkinje) neurons and the motor deficits observed was not established. The results presented here demonstrate that the acute and selective loss of iFGF14 in mature Purkinje neurons markedly reduces the excitability of these cells and impairs motor coordination and balance. Importantly, further experiments revealed that viral-mediated acute and selective rescue of iFGF14 expression in adult Fgf14−/− Purkinje neurons restores spontaneous and evoked firing and, in addition, improves motor coordination and balance in adult Fgf14−/−animals. These combined results demonstrate a link between the expression of iFGF14 in Purkinje neurons, the excitability of these cells, and the regulation of motor coordination and balance. Nevertheless, additional experiments using approaches similar to those developed and exploited here are needed to determine the functional effects of the acute and selective loss of iFGF14 in mature cerebellar granule neurons.
Physiological and pathophysiological implications
The results presented here clearly demonstrate a critical physiological role for iFGF14 in the regulation of the excitability of Purkinje neurons through modulation of Nav channel availability (Fig. 11) and that the loss of this interaction has dramatic effects on Purkinje neuron excitability, cerebellar function, motor coordination, and balance. It is unclear, however, whether iFGF14 should be considered an obligatory accessory subunit of Nav channels in Purkinje (and other) neurons and/or if iFGF14-Nav α-subunit interactions are dynamically regulated. It is clear, however, that if intracellular events regulate the interactions between iFGF14 and Nav channels, these would be expected to affect Nav channel inactivation/availability and therefore to affect the excitability of Purkinje neurons. Interestingly, it has been demonstrated recently that the interaction between iFGF14 and Nav channel α-subunits is modulated by pharmacological inhibition of glycogen synthase kinase-3 (Shavkunov et al., 2013), revealing a potential signaling pathway to modulate iFGF14-Nav channel interactions in situ. Further studies are needed to determine the physiological relevance of these observations in Purkinje and other neurons. Delineation of cell signaling pathways that modulate (attenuate or enhance) iFGF14-Nav channel interactions could provide insights into the development of novel therapeutic strategies to treat disorders of excitability in the cerebellum and, indeed, throughout the nervous system.
Footnotes
This work was supported by the National Institutes of Health (NIH Grant R01NS065761 to D.M.O. and J.M.N. and NIH Training Grants T32 GM007200 and T32 HL007275 to M.K.B.). The monoclonal anti-iFGF14 and anti-Ankyrin G antibodies were developed by and obtained from the UC Davis/NIH NeuroMab facility, which is supported by NIH Grant U24NS050606 and maintained by the University of California at Davis. All interfering RNAs were obtained from the Washington University RNAi Core, which supported by the Genome Institute and the Children's Discovery Institute (Grant CDI-LI-2010-94) at Washington University. All viruses were generated in the Hope Center Viral Vectors Core, which is supported by a Neuroscience Blueprint Core grant (Grant P30 NS057105). We thank A. Burkhalter for assistance with and training in stereotaxic mouse surgery, M. Grubb for providing us with MATLAB scripts for analyses of immunohistochemical data, M. Heng for advice on balance beam setup and scoring, and L. Li for technical assistance.
The authors declare no competing financial interests.
References
- Apps R, Garwicz M. Anatomical and physiological foundations of cerebellar information processing. Nat Rev Neurosci. 2005;6:297–311. doi: 10.1038/nrn1646. [DOI] [PubMed] [Google Scholar]
- Bosch MK, Nerbonne JM, Ornitz DM. Dual transgene expression in murine cerebellar Purkinje neurons by viral transduction in vitro. PLoS One. 2014;9:e104062. doi: 10.1371/journal.pone.0104062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusse E, de Koning I, Maat-Kievit A, Oostra BA, Heutink P, van Swieten JC. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): A new phenotype. Mov Disord. 2006;21:396–401. doi: 10.1002/mds.20708. [DOI] [PubMed] [Google Scholar]
- Buttermore ED, Piochon C, Wallace ML, Philpot BD, Hansel C, Bhat MA. Pinceau organization in the cerebellum requires distinct functions of neurofascin in Purkinje and basket neurons during postnatal development. J Neurosci. 2012;32:4724–4742. doi: 10.1523/JNEUROSCI.5602-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Morton J, Dunnett SB. Motor coordination and balance in rodents. Curr Protoc Neurosci. 2001 doi: 10.1002/0471142301.ns0812s15. Chapter 8:Unit 8.12. [DOI] [PubMed] [Google Scholar]
- Coebergh JA, Fransen van de Putte DE, Snoeck IN, Ruivenkamp C, van Haeringen A, Smit LM. A new variable phenotype in spinocerebellar ataxia 27 (SCA27) caused by a deletion in the FGF14 gene. Eur J Paediatr Neurol. 2013;18:413–415. doi: 10.1016/j.ejpn.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Dalski A, Atici J, Kreuz FR, Hellenbroich Y, Schwinger E, Zühlke C. Mutation analysis in the fibroblast growth factor 14 gene: frameshift mutation and polymorphisms in patients with inherited ataxias. Eur J Hum Genet. 2005;13:118–120. doi: 10.1038/sj.ejhg.5201286. [DOI] [PubMed] [Google Scholar]
- Davie JT, Kole MH, Letzkus JJ, Rancz EA, Spruston N, Stuart GJ, Häusser M. Dendritic patch-clamp recording. Nat Protoc. 2006;1:1235–1247. doi: 10.1038/nprot.2006.164. [DOI] [PubMed] [Google Scholar]
- Dover K, Solinas S, D'Angelo E, Goldfarb M. Long-term inactivation particle for voltage-gated sodium channels. J Physiol. 2010;588:3695–3711. doi: 10.1113/jphysiol.2010.192559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz R, Dover K, Laezza F, Shtraizent N, Huang X, Tchetchik D, Eliseenkova AV, Xu CF, Neubert TA, Ornitz DM, Goldfarb M, Mohammadi M. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J Biol Chem. 2009;284:17883–17896. doi: 10.1074/jbc.M109.001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 2005;16:215–220. doi: 10.1016/j.cytogfr.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M. Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell Mol Life Sci. 2012;69:1067–1076. doi: 10.1007/s00018-011-0832-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, Giza J, Tchetchik D, Kelley K, Vega A, Matthews G, Rossi P, Ornitz DM, D'Angelo E. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 2007;55:449–463. doi: 10.1016/j.neuron.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/S0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung H, Feldman B, Lovec H, Coulier F, Birnbaum D, Goldfarb M. Murine FGF-12 and FGF-13 expression in embryonic nervous system, connective tissue and heart. Mech Dev. 1997;64:31–39. doi: 10.1016/S0925-4773(97)00042-7. [DOI] [PubMed] [Google Scholar]
- Häusser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/S0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington's disease. J Neurosci. 2007;27:8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci. 2007;27:11065–11074. doi: 10.1523/JNEUROSCI.2162-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, King MA, Hamby ME, Meyer EM. Dopaminergic cell loss induced by human A30P alpha-synuclein gene transfer to the rat substantia nigra. Hum Gene Ther. 2002;13:605–612. doi: 10.1089/10430340252837206. [DOI] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V. Ankyrin G. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270:2352–2359. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci. 2009;42:90–101. doi: 10.1016/j.mcn.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CJ, Dib-Hajj SD, Waxman SG. Fibroblast growth factor homologous factor 1B binds to the C terminus of the tetrodotoxin-resistant sodium channel rNav1.9a (NaN) J Biol Chem. 2001;276:18925–18933. doi: 10.1074/jbc.M101606200. [DOI] [PubMed] [Google Scholar]
- Liu CJ, Dib-Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem. 2002;278:1029–1036. doi: 10.1074/jbc.M207074200. [DOI] [PubMed] [Google Scholar]
- Lou JY, Laezza F, Gerber BR, Xiao M, Yamada KA, Hartmann H, Craig AM, Nerbonne JM, Ornitz DM. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol. 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL. Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem. 2004;279:16044–16049. doi: 10.1074/jbc.M400856200. [DOI] [PubMed] [Google Scholar]
- Milescu LS, Bean BP, Smith JC. Isolation of somatic Na+ currents by selective inactivation of axonal channels with a voltage prepulse. J Neurosci. 2010;30:7740–7748. doi: 10.1523/JNEUROSCI.6136-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misceo D, Fannemel M, Barøy T, Roberto R, Tvedt B, Jaeger T, Bryn V, Strømme P, Frengen E. SCA27 caused by a chromosome translocation: further delineation of the phenotype. Neurogenetics. 2009;10:371–374. doi: 10.1007/s10048-009-0197-x. [DOI] [PubMed] [Google Scholar]
- Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, Mohammadi M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J Biol Chem. 2003;278:34226–34236. doi: 10.1074/jbc.M303183200. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:REVIEWS3005. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, Xiao M, Xu L, Wong M, Nerbonne JM, Ornitz DM, Yamada KA. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33:81–88. doi: 10.1016/j.nbd.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shavkunov AS, Wildburger NC, Nenov MN, James TF, Buzhdygan TP, Panova-Elektronova NI, Green TA, Veselenak RL, Bourne N, Laezza F. The fibroblast growth factor 14.voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3) J Biol Chem. 2013;288:19370–19385. doi: 10.1074/jbc.M112.445924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood PM, Munoz-Sanjuan I, Tong P, Macke JP, Hendry SH, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc Natl Acad Sci U S A. 1996;93:9850–9857. doi: 10.1073/pnas.93.18.9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, Heutink P. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected] Am J Hum Genet. 2003;72:191–199. doi: 10.1086/345488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Hennessey JA, Kirkton RD, Wang C, Graham V, Puranam RS, Rosenberg PB, Bursac N, Pitt GS. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res. 2011a;109:775–782. doi: 10.1161/CIRCRESAHA.111.247957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang C, Hoch EG, Pitt GS. Identification of novel interaction sites that determine specificity between Fibroblast Homologous Factors and voltage-gated sodium channels. J Biol Chem. 2011b;286:24253–24263. doi: 10.1074/jbc.M111.245803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chung BC, Yan H, Lee SY, Pitt GS. Crystal structure of the ternary complex of a Nav C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Cell Structure. 2012;20:1167–1172. doi: 10.1016/j.str.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, McEwen DG, Ornitz DM. Subcellular and developmental expression of alternatively spliced forms of fibroblast growth factor 14. Mech Dev. 2000;90:283–287. doi: 10.1016/S0925-4773(99)00241-5. [DOI] [PubMed] [Google Scholar]
- Wang Q, Bardgett ME, Wong M, Wozniak DF, Lou J, McNeil BD, Chen C, Nardi A, Reid DC, Yamada K, Ornitz DM. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35:25–38. doi: 10.1016/S0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- Wittmack EK, Rush AM, Craner MJ, Goldfarb M, Waxman SG, Dib-Hajj SD. Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J Neurosci. 2004;24:6765–6775. doi: 10.1523/JNEUROSCI.1628-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Xiao M, Xu L, Yamada KA, Ornitz DM. Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol Dis. 2007;26:14–26. doi: 10.1016/j.nbd.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Bosch MK, Nerbonne JM, Ornitz DM. FGF14 localization and organization of the axon initial segment. Mol Cell Neurosci. 2013;56:393–403. doi: 10.1016/j.mcn.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bao L, Yang L, Wu Q, Li S. Roles of intracellular fibroblast growth factors in neural development and functions. Sci China Life Sci. 2012;55:1038–1044. doi: 10.1007/s11427-012-4412-x. [DOI] [PubMed] [Google Scholar]
- Yan H, Pablo JL, Pitt GS. FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Reports. 2013;4:66–75. doi: 10.1016/j.celrep.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Pablo JL, Wang C, Pitt GS. FGF14 modulates resrugent sodium current in mouse cerebellar Purkinje neuron. eLife. 2014;3:e04193. doi: 10.7554/eLife.04193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/S1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]