

Abstract
The soybean cyst nematode (SCN) resistance locus Rhg1 is a tandem repeat of a 31.2 kb unit of the soybean genome. Each 31.2-kb unit contains four genes. One allele of Rhg1, Rhg1-b, is responsible for protecting most US soybean production from SCN. Whole-genome sequencing was performed, and PCR assays were developed to investigate allelic variation in sequence and copy number of the Rhg1 locus across a population of soybean germplasm accessions. Four distinct sequences of the 31.2-kb repeat unit were identified, and some Rhg1 alleles carry up to three different types of repeat unit. The total number of copies of the repeat varies from 1 to 10 per haploid genome. Both copy number and sequence of the repeat correlate with the resistance phenotype, and the Rhg1 locus shows strong signatures of selection. Significant linkage disequilibrium in the genome outside the boundaries of the repeat allowed the Rhg1 genotype to be inferred using high-density single nucleotide polymorphism genotyping of 15 996 accessions. Over 860 germplasm accessions were found likely to possess Rhg1 alleles. The regions surrounding the repeat show indications of non-neutral evolution and high genetic variability in populations from different geographic locations, but without evidence of fixation of the resistant genotype. A compelling explanation of these results is that balancing selection is in operation at Rhg1.
Keywords: copy number variation, resistance gene, Rhg1, selection, soybean
Introduction
Genetic variation encompasses a wide range of distinct types of DNA sequence polymorphism, from single nucleotide variants (SNVs) to insertions, deletions and copy number variation of DNA segments (CNV) ranging in size from a few base pairs to entire chromosomes (Sebat et al. 2004; Conrad et al. 2006; Redon et al. 2006). CNVs influence gene expression, cause disorders such as human disease, are involved in adaptation during evolutionary process and drive phenotypic diversity in a wide range of organisms (McCarroll et al. 2006; Nguyen et al. 2006; Repping et al. 2006; Stranger et al. 2007). There is increasing evidence of high levels of CNV in plant genomes (Swanson-Wagner et al. 2010; Cao et al. 2011; Zheng et al. 2011; McHale et al. 2012; Hanikenne et al. 2013; Iovene et al. 2013), and evidence is emerging that CNVs mediate a number of valuable crop traits (Sutton et al. 2007; Cook et al. 2012; Dı′az et al. 2012). Questions such as the origin of these structural variations, as well as their contributions to both evolutionary adaptation and phenotypic traits, remain unresolved.
Soybean [Glycine max (L.) Merr.] is the world's most cultivated legume. It has provided on average 57% of oilseed production as well as 68% of protein meal consumption worldwide since 2000 (http://www.soystats.com). In addition, soybean has been used to provide industrial resources such as biodiesel and plastics. The total value of the US soybean crop was more than $42 billion in 2013 and doubled in the 5 years up to 2013. It has been estimated that the loss of soybean production caused by soybean cyst nematode (SCN, Heterodera glycines Ichinohe), the most damaging pest of soybean in the USA in yield loss terms, was equivalent to 4–6% of the total production from 2006 to 2010 (http://aes.missouri.edu/delta/research/soyloss.stm). SCN has spread to most soybean producing areas worldwide, and genetic resistance is a key component for its control (Niblack et al. 2006).
Soybean cyst resistance is a quantitative trait, and the Rhg1 locus on soybean chromosome 18 was found to confer the strongest and most useful SCN resistance of any known quantitative trait locus (Concibido et al. 2004; Kim et al. 2010a). Rhg1 has been successfully introgressed into high-yielding germplasm in the USA. Approximately 95% of the commercially cultivated, SCN-resistant soybean cultivars in the northcentral USA utilize the Rhg1-b allele, originally derived from the soybean germplasm collection accession PI 88788, as the main gene for resistance (Cregan et al. 1999). The Rhg1 alleles in the genomes of Fayette (a cultivar derived from PI 88788) and Peking (also known as PI 548402) are CNV loci carrying 10 and 3 tandemly replicated copies of a 31.2-kb segment of the genome, in nose-to-tail orientation (Cook et al. 2012). The sequence of the 31.2-kb repeated segment encodes four intact genes. None of the genes in the repeat resemble a typical plant resistance gene, which contains a nucleotide-binding site–leucine-rich repeat (NBS-LRR) domain (McHale et al. 2006). However, SCN resistance can be conferred on a susceptible plant by increasing the expression levels of three of the genes at the locus. Thus, enhanced expression of multiple genes (analogous to that caused by CNV of the genes) is capable of conferring resistance (Cook et al. 2012).
Investigation of more germplasm accessions revealed variation in both the number of copies of the repeat and the sequence of the individual repeat units. Different sequences in the repeat units are present even in the same homozygous genotype (Cook et al. 2014). Thus, this locus consists of several genes per unit, and several units per locus, where the individual units and genes have different DNA sequences and occur in different permutations within the same organism. As the tools of molecular biology provide limited opportunities to investigate such a complex system, here we use the tools of genomics, phylogenetics and population genetics to probe the structure and evolution of this locus.
In the study below, we examine diversity at the Rhg1 locus across 106 G. max and Glycine soja germplasm accessions using a genomic qPCR assay, validated with whole-genome sequencing (WGS) assays. We identify and quantify duplication events at the Rhg1 locus and find a wide distribution of copy number. We interpret the order of the individual units of the multicopy versions of the locus and use these sequences to perform evolutionary analysis on the individual repeats. Using our data for 106 resistant accessions, WGS on selected accessions and large-scale single nucleotide polymorphism (SNP) data obtained by Infinium genotyping of the entire soybean germplasm collection, we investigate signatures of selection at the Rhg1 locus. We discuss the implications of evolution at this locus for both soybean population genetics and future breeding approaches.
Materials and methods
Soybean germplasm
The term ‘accession’ is used here to mean a genetic line registered and stored by the United States Department of Agriculture (USDA) soybean germplasm collection (Urbana, IL, USA). We use the term plant introduction (PI) as it is used by this collection, to designate a soybean or Glycine soja line originating outside the USA that is registered in the USA and stored by USDA. Some cultivars, especially those bred in the USA, are not PIs but are still USDA germplasm accessions. All soybean germplasm used in this study, including PIs and soybean cultivars with resistance to SCN, was obtained from the USDA soybean germplasm collection. Based on two independent studies (Diers et al. 1997; Chen et al. 2006), SCN-resistant germplasm with resistance to at least one of SCN types 1, 2, 3, 5 and 14 was chosen as ‘SCN-resistant accessions’. A total of 106 SCN-resistant accessions (102 PIs collected in diverse geographic regions and four US cultivars) that were available as of June 2013 were obtained (Table S1, Supporting information). Plants were grown in a growth chamber set at a photocycle of 18/6 h (day/night), 23/20 °C (day/night) and 50% relative humidity for about 10 days. Young leaf tissue was collected from two individuals for each line and kept individually at −80 °C for genomic DNA isolation.
Quantitative PCR validation of copy number variation
Genomic DNA extraction was performed as described in a previous study [‘Fosmid library construction’ section in ‘Supplementary Materials’ in Cook et al. (2012)]. A pool of two plants of each germplasm accession was used for the DNA extraction. The presence of the junction between two adjacent copies of the 31.2-kb repeat described by Cook et al. (2012) was first investigated using tandem repeat site-specific primers that span the junction between two repeat units and thus only produce a product if the 31.2-kb unit is repeated at least twice (Primer IDs 1 and 2 in Table S4, Supporting information). Having identified lines that contain more than one copy of the 31.2-kb unit, we then investigated copy number. Genomic qPCR (quantitative polymerase chain reaction on the genomic DNA from the locus) was performed on the genomic DNA samples described above using the Brilliant II QPCR Master Mix with Low ROX kit (Agilent Technologies) and the Mx3000P QPCR system (Agilent Technologies). Relative quantification using the ΔΔCT measurement method (Livak & Schmittgen 2001) was used to measure copy number relative to the soybean reference genome, from the Williams 82 line. Amplification efficiencies of all DNA preparations from all samples were determined by 10× dilution series. A heat-shock protein gene (hsp; Li et al. 2009) was used as an endogenous control for all assays. A sequence of Glyma18g02590, one of the Rhg1 genes in the duplicated region, was chosen for primer design (Primer IDs 3 and 4 in Table S4, Supporting information) using PrimerQuestSM (Integrated DNA Technologies) based on the reference genome and fosmid clone sequences from Cook et al. 2012. CT values for technical replicate(s) of both of the internal control and target genes were obtained from the same plate and run at all times. A minimum of four technical replicates were prepared to generate ΔCT values. 95% confidence intervals were calculated to give error bars, and the copy number was assigned to the nearest integer value.
Whole-genome shotgun sequencing
DNA extracted as above was treated with RNase (Roche Applied Science) by incubating in 25 μg/mL RNase at 4 °C overnight. We performed whole-genome shotgun sequencing of nine germplasm accessions using Illumina technology. 1.5 μg of genomic DNA was sequenced using the Illumina HiSeq 2500 instrument with 150- or 155-bp paired-end sequencing at the University of Illinois Biotechnology Center. A total of nine DNA sequencing libraries were prepared with the Illumina TruSeq DNA Sample Preparation kit (Illumina). The libraries were loaded into lanes and sequenced using version one of the Illumina TruSeq SBS sequencing Rapid kit. A total of 41–48 Gb of reads having average quality scores 30 or higher was produced from each lane (Table S5, Supporting information). Our data from the accession LD09-15087a were previously published (Cook et al. 2012). Illumina whole-genome raw read data sets obtained from independent studies were as follows: 41 SoyNAM founder line genotypes (courtesy of Perry Cregan, unpublished) and 31 accessions from Lam et al. 2010; including W06 (SRR064619), LD00-2817P (Schmitz et al. 2013) and PI 437654, PI 90763, PI 89772, PI 548402, PI 548316 and PI 209332 (Cook et al. 2014). These data were variable in quality and coverage. A total of 89 accessions were analysed, 26 of which contained the Rhg1 resistance repeat. Of these 89 data sets, 22 met the 8× coverage cut-off within the Rhg1 repeat itself; these were used for assembly and analysis of the repeat. Just 18 met the 8× cut-off across the 1.5-Mbp region around Rhg1; this set was used for the population and evolutionary analysis around the repeat that uses whole-genome sequence data. Of the 41 SoyNAM data sets, only three (PI 427136, PI 518751 and LD00-3309) were used for variant calling, as other data sets had substantial gaps in coverage.
Confirmation of copy number variation by read depth
To confirm copy number variation within the tandemly duplicated region of the Rhg1 allele, reads from each sample were aligned to the glyma 1.1 version of the soybean genome assembly. novoalign (version 3.00.03; http://www.novocraft.com) with paired-end options was used to align the reads to the reference genome. Only unique alignment locations were allowed for reads. The number of reads aligned to the repeat unit reference sequence was counted from a BAM file using samtools (version 0.1.18). Copy number, C, for a repeat unit (31.2-kb) was calculated by the average of six measurements (± standard deviation) where C = rw−1. Here r represents the total number of reads aligning to the repeat locus, and ω represents the total number of reads aligning to one of six 31.2-kb reference sequence windows immediately outside the repeated region, as described in the ‘Whole-genome shotgun sequencing and read depth in duplicated region’ section of ‘Supplementary Materials’ for fig. 2b in Cook et al. (2012).
Fig 2.
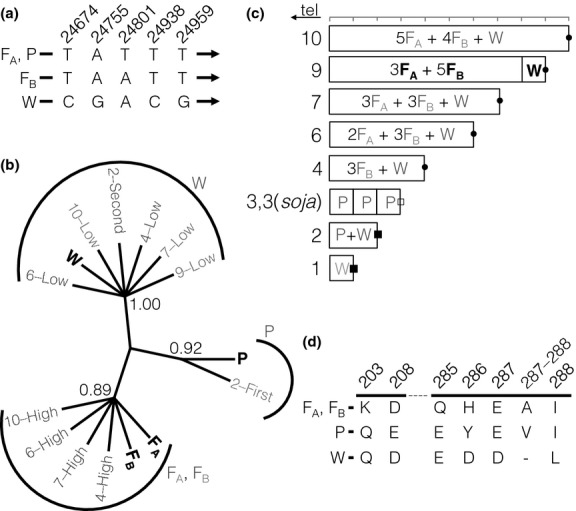
Sequence of the Rhg1 repeat units. (a) Five examples of sequence variants used for reconstruction of repeat units in an intergenic region between Glyma18g02610 and Glyma18g02620 are displayed. Three patterns in these five nucleotides differentiate three of four separate repeat units (FA, a repeat unit found in the PI 88788 genotype; P, a repeat unit found in the Peking genotype; FB, another repeat unit found in the PI 88788 genotype; and W, a single-copy version of the sequence found in the susceptible Williams 82 genotype). Positions are given relative to the first nucleotide (1 632 225 bp on chromosome 18) of the 31.2-kb repeat in the Williams 82 genome assembly. (b) Classification of repeat units using maximum parsimony analysis of sequences. Reconstructed individual Glyma18g02590 genes from the repeats are labelled according to copy number and relative abundance in the accession (e.g. ‘4-low’ means the less abundant sequence present in a four-copy genotype) or by position relative to the telomere if equally abundant (e.g. 2-first). Bootstrap support values are given above key nodes. (c) Interpretation of the Rhg1 repeat structure. Bold black labels represent sequences with position known from large insert cloning; grey labels are inferred from short-read shotgun sequence data classified by the parsimony analysis in ‘b’. Rhg1 copy number in the Glycine max accession genome is denoted on the left. Three different fusion sequences at the centromere-proximal end are marked by open squares, filled squares and filled circles. tel: telomere. (d) Amino acid variation in the predicted α-SNAP protein, Glyma18g02590. Amino acid positions are from the Williams 82 reference. Bold lines represent exons 6 and 9, respectively.
SNV detection
Single nucleotide variants were predicted from aligned read data using varscan version 2.3.5 (Koboldt et al. 2012). Command line options are as follows: mpileup2snp –min-coverage [8] –min-ave-qual [20] –min-var-freq [0.01] –p-value [0.01]. For all accessions of a given copy number, the frequency of reads carrying a SNV that differs from the Williams 82 reference divided by the total number of reads (hence, the proportion of SNVs in reads at any given nucleotide) was plotted against position in the 31.2-kb repeat unit (Fig. S1, Supporting information). The frequency of variants at each SNV, and thus the number of repeats present each carrying a given variant, was estimated using alignment of Illumina reads.
Repeat subunit assembly and type definitions
Phasing analysis using informative bases derived from paired-end reads from single molecules was used to reconstruct the individual repeated sequence units in the Rhg1 locus in a manual process of assembly similar to that used for haplotype reconstruction or phasing. For each germplasm accession, mapped, paired reads that possessed variants from the reference sequence (the Williams 82 single-copy sequence, W) were merged into a single data file using in-house Perl scripts. Only the reads with bases varying from the 31.2-kb single-copy region from Williams 82 with Phred q ≥ 20 were selected for assembly steps. Variants located at the start position of a read or within three bases at the end of read were ignored. Two SNVs that reside on the same read or the corresponding mate in paired-end reads were considered to have originated from the same molecule and can thus be used to define a given 31.2-kb repeat unit in an Rhg1 repeat genotype. Thus, only reads or mate pairs with two or more than two SNVs derived from the same molecule were used for phasing. To obtain accurate and complete phasing of each repeat unit, multiple possible phasing configurations were validated by fosmid clone sequences from our previous study (Cook et al. 2012). Firstly, we configured three different repeat unit subtypes within Rhg1 from PI 88788. Across all SNVs in PI 88788 (with the exception of the SNV at 1 657 025 bp), the proportion (expressed as an observed probability) of SNVs within Rhg1 are either 1 or ∼0.9, indicating either a single type of sequence present in 10 copies per haploid genome, or two distinguishable repeats, one with a single copy and one with nine copies. These reads could be assembled consistently across the variable regions of the repeat, including all of the Glyma18g02590 gene. To confirm the homogeneity of the repeat units, two fosmid clones, fosmids #2 and #3 in fig. 2a in Cook et al. (2012); carried all of the predicted single-copy SNVs including five positions identical to the reference sequence (Fig. 2a), confirming that the single-copy SNVs are all within a single repeat unit. In addition, the fosmid #2 (Cook et al. 2012) spans the last duplicated repeat copy at the centromeric end and the nonduplicated region. This indicates that the single-copy (W type) repeat subunit is located at the centromeric end of the repeat in the PI 88788 haplotype. Thirty-three per cent of PI 88788 reads are T (thymine) at the 1 657 025 bp SNV (Fig. S2b, Supporting information), suggesting three of the nine repeat units are distinguishable at this location. The three copies per haploid genome carrying T at 1 657 025 bp were used to define subtype FA (Fayette, 10 copies, is directly derived from PI 88788, which carries nine copies of the repeat; the two were previously assumed to carry the same Rhg1 allele). One copy is subtype W. The remaining five copies in PI 88788 were identified as subtype FB. Fosmids #4 and #5 in fig. 2a in Cook et al. (2012) confirmed complete sequences of repeat subtypes FB and FA, respectively. Thus, we reconstructed the repeat at Rhg1 from PI 88788 as three subtype FA, five subtype FB, and a single subtype W at the centromeric end. Three-copy type germplasm collections (Peking, PI 90763, PI 437654, PI 467327, PI 89772, & Jidong5) showed only a single genotype at each SNV. This sequence is distinctive from W, FA and FB, resulting in a single subtype P (Peking). Phasing steps for other germplasm accessions were performed in the same way described above. Phylogenetic analysis using the parsimony method was conducted on the phased SNV locations in and around Glyma18g02590 to validate the classification of four subtypes, once phasing was completed for all of the accessions for which whole-genome sequence data were available (Fig. 2b). Glyma18g02590 gene sequence, including coding DNA sequence (CDS), untranslated region (UTR) and introns from each sequenced accession, was used for the parsimony analysis. For each genotype, we were able to manually assemble a set of sequences derived from a single molecule into a contig by phasing of the paired-end reads. To identify the subtype located at the very 3′ end of the Rhg1 repeat in individual accessions, a 200-bp region spanning the junction between the end of the repeat and the neighbouring, nonrepeated sequence was amplified by PCR and sequenced by the Sanger method (Table S2, Supporting information; Fig. 2c).
Evolutionary analysis
Two separate nucleotide sequence data sets were prepared for phylogenetic analysis. Firstly, the SNVs in genes surrounding the Rhg1 locus were determined using alignments of the high-coverage and quality Illumina WGS reads from 18 germplasm accessions with experimentally validated Rhg1 copy number (hereafter, this data set is termed WGS data). The germplasm accessions in the WGS data are as follows: three single-copy germplasm accessions (Williams 82, PI 427136, & PI 518751), one 2 copy (PI 438489 B), three 3 copy (PI 467327, Peking, & PI 89772), one 4 copy (PI 89008), three 6 copy (PI 87631-1, PI 461509, & PI 467332), two 7 copy (PI 92720 & Cloud), one 9 copy (PI 88788) and four 10 copy (PI 209332, LD10-30036, LD09-15087a, & LD00-3309). SNPs within CDSs of 38 genes (a total of 54 771 bp) across a 400-kb region centred on Rhg1 were prepared using the methods from the step ‘SNV detection’ described above. Two genes (Glyma18g02730 & Glyma18g02750) that consistently gave read depth below the threshold applied in all analysis (sequence depth-of-coverage minimum 8×) were excluded in this study. Secondly, the complete data set for 19 652 Glycine max and G. soja germplasm accessions genotyped using the Illumina Infinium II BeadChip array, which carries 52 041 SNP probes targeting genic and intergenic regions, was obtained from SoyBase (http://www.soybase.org). Note the distinction we use here between SNV (any single nucleotide variant, such as those between repeat units in the same genome) and SNP (a single nucleotide shown to be polymorphic within a population and used for mapping). A set of 19 652 Infinium data sets was supplied by Perry Cregan of the USDA-ARS. Of these, a set of 19 548 germplasm accessions with high-quality, mono-allelic SNP analysis results was used as the main data set (hereafter, Infinium data). For population structure analysis, a subset of the Infinium data of 15 452 accessions with accompanying geographic origin data was used. For phylogenetic analysis, a subset of 15 996 accessions of the Infinium data with high-quality SNP calls at all of the 10 SNPs described was used. One hundred and thirty-five SNPs from the array, across the 1.5-Mbp region centred on Rhg1, but outside the repeat, were used for analysis of soybean populations. For WGS data, phylogenetic analysis was performed using the maximum parsimony (MP) method with 10 000 bootstrap replicates to assess reliability of clustering, using mega 6.0 (Tamura et al. 2013). Clustering of germplasm accessions from the much larger Infinium data set was performed using parsimonator version 1.0.2 (https://github.com/stamatak/Parsimonator-1.0.2). Ten SNPs for each taxon located between 1 620 585 and 1 712 832 bp on chromosome 18 were used for the phylogenetic analysis. For SNVs located in the repeat unit, apparent heterozygosity (i.e. diversity between repeat units) was validated using WGS data. SNVs with apparent heterozygosity were removed before analysis. All phylogenetic trees were visualized using geneious 5.6.5 (http://www.biomatters.com).
Selection analysis
For both data sets, WGS and Infinium, the nucleotide diversity was quantified as π (Nei & Li 1979) and θ (Watterson 1975). Tajima's test of neutrality (Tajima 1989) was additionally performed for the Infinium data. We used two software packages for these analysis (π, θ, Tajima's D): mega 6.0 (Tamura et al. 2013) and variscan 2.0.3 (Hutter et al. 2006) for the Linux platform; results were confirmed using both packages. Nucleotide diversity was calculated for each gene (WGS data) or a sliding window (Infinium data: 10 SNP window size and 5 SNP window increment). MS (Hudson 2002) was used to calculate 10 000 replicate simulations of a neutral model with the values of θ generated by variscan. As soybean is known to be subject to recent population bottlenecks as a result of domestication, we ran a range of simulations modelling instantaneous bottlenecks from 10 000 to 100 generations before present in order-of-magnitude increments. This and a similar range of recent population growth models consistent with expansion of a cultivated population over the same time frame produced lower threshold values than the neutral model. The more stringent neutral model values were used to create a P-value lookup table, P-values were calculated for each window, and a false discovery rate correction was applied to the P-values. tassel 3.0 (Bradbury et al. 2007) was used to evaluate linkage disequilibrium (LD) for both data sets. The full matrix was selected for comparisons. The fixation index (FST) was calculated for the Infinium data as follows: two separated populations, the first population with 46 single-copy germplasm accessions and the second with 48 multiple-copy accessions, were selected based on the genomic qPCR and whole-genome shotgun sequencing results. Significant values of FST were determined by calculating a P-value using a log-likelihood (G)-based significance test and then applying the Bonferroni correction (alpha level 0.01) on P-values obtained for each locus. For comparisons between soybean populations based on their origin, three major countries (China, Japan and Korea) were selected as potentially geographically distinct populations. FST was calculated for each SNP using genepop 4.3 (Rousset 2008).
Population structure analysis
Using the admixture model (structure version 2.3.4; Pritchard et al. 2000; Falush et al. 2003), we estimated the shared genetic structure of the G. max and G. soja population of 15 452 accessions with geographic origin data genotyped using the soybean Infinium array. These individuals were analysed using 42 509 Infinium SNPs. All structure runs used 10 000 iterations after a burn-in of length 10 000. The number of clusters considered was set from 2 to 8. The number of individuals in each subpopulation clustered by structure is as follows: 5.A (3776 accessions), 6.A (1230), 6.B (2906), 7.A (2900), 7.B (913), 8.A (2865) and 8.B (1115). Each subpopulation was used for the Tajima's test as described above.
Results
Copy number variation at Rhg1
We investigated copy number variation of the previously described Rhg1 repeat (Cook et al. 2012) within soybean germplasm accessions (Table S1, Supporting information). While the two genotypes PI 88788 and Peking represent the two main sources of Rhg1-mediated SCN resistance, several other germplasm accessions, not known to be related by pedigree, have been shown to have genetic SCN resistance, possibly mediated by Rhg1 (Concibido et al. 2004). We investigated lines that were previously described to show SCN resistance interactions of any type (Diers et al. 1997; Chen et al. 2006). Firstly, we ascertained the presence/absence of the previously described CNV event (Cook et al. 2012) in the SCN-resistant soybean germplasm, using a PCR assay specific to the fusion site common to the previously characterized repeats. As we only investigated accessions positive for the canonical repeat junction characteristic of the cloned Rhg1 locus, we cannot rule out the possibility that other copy number variations occur at this locus that do not have the same repeat fusion site. However, from a total of 89 accessions subjected to WGS, all 26 accessions that showed greater than one copy at the Rhg1 locus were also positive in the fusion site assay.
Of 106 accessions, 62 showed a product in a PCR that targeted the unique fusion site between tandemly duplicated copies at Rhg1. This indicates the presence of the repeat described by Cook et al. (2012); with the same junction sequence at the Rhg1 locus (Table S1, Supporting information). Secondly, copy number of the Rhg1 repeat in each accession was estimated using genomic qPCR. A wide distribution of copy number was found among the accessions, with known copy number variants possessing three copies (Peking, PI 89772, PI 437654, PI 90763), 7 (Cloud), 9 (PI 88788) and 10 (Fayette, PI 209332) showing the expected copy numbers (Fig.1; Cook et al. 2012, 2014). Estimated copy number was then independently confirmed in fourteen selected lines using WGS and calculating relative read depths acquired from alignments to the Williams 82 reference genome. A broad diversity of CNV among Rhg1 loci was detected; 2–4, 6, 7, 9 and 10 copies were detected in different Glycine max accessions and one three-copy variant in a Glycine soja accession (Fig.1; Table S1, Supporting information). Notably, despite its pedigree derived from PI 88788, we found that Fayette had 10 copies of the repeat as previously described, but PI 88788 had nine copies, consistent with Fiber-FISH data (Cook et al. 2014). This suggests that an event that increased copy number by one unit occurred during the process of selection for the Fayette cultivar.
Fig 1.
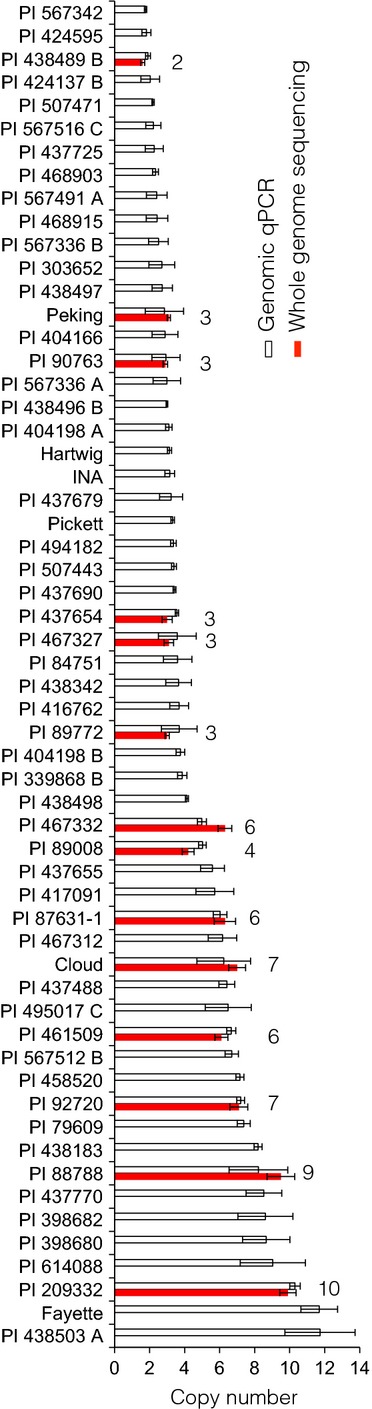
Distribution of copy number at the Rhg1 locus in soybean accessions with soybean cyst nematode resistance. Estimates of copy number of the tandem duplication at Rhg1 were obtained using genomic qPCR analysis targeting a gene (Glyma18g02590) in the repeat, and the mean ± 95% confidence interval plotted as white bars in order of estimated copy number. A second estimate of the Rhg1 copy number based on read depth of whole-genome sequencing was performed where data were available and plotted as mean ± standard deviation (red bars). The estimated copy number for lines with both types of data available (based on the whole-genome coverage data) is to the right of the bars.
Sequence variation in Rhg1
The WGS data were analysed for nucleotide sequence variations within Rhg1, and amino acid variants inferred from the nucleotide sequence. In total, 149 positions that harbour SNVs were identified within the sequence that comprises the Rhg1 repeat across accessions carrying eight separate copy number variants, including the susceptible single-copy versions of the sequence (Fig. S1, Supporting information). We detected several patterns of sequence variant within and between copy numbers. Firstly, the patterns of SNPs in each genotype were correlated with copy number of the genotype. For variants with three copies or more, the first 8 kb of the repeat was clearly differentiated from the Williams 82 sequence in all copies (region ‘i’ in Fig. S1, Supporting information). After the first 8 kb (region ‘ii’ in Fig. S1, Supporting information), in types with 4–10 copies, all copies but one of the repeat carry the same variants from the reference [the exception being the SNP at 1 657 025 bp (labelled ‘iii’ in Fig. S1, Supporting information), which likely arose after the origin of the repeat]. In the three-copy variant, the entire 31.2-kb unit is distinguished from the reference by multiple sequence variations. Variations throughout the Glyma18g02590 gene encoding an α-SNAP protein were observed in all multiple-copy lines (‘iv’ in Fig. S1, Supporting information). No sequence variants, however, were found in the next gene, a protein of unknown function (Glyma18g02600; region ‘v’ in Fig. S1, Supporting information). For one genotype with two copies, all of the nucleotide variants showed 0.5 probability of occurring in the shotgun genome sequence, suggesting two distinct repeat sequences, only one of which differs from the Williams 82 reference. In a comparison between the single-copy types, 24 SNPs differing from Williams 82 were found (‘vi’ in Fig. S1, Supporting information). These 24 variants were identified in the accession PI 518751 and, significantly, were also detected in resistant, multiple-copy loci (2 through 10 copies). These variants lie from the 5′ end of Glyma18g02610 (a wound-inducible protein) to the end of the repeat.
Reconstruction of individual repeat units
Figure S1 (Supporting information) reveals the physical distribution of variants in the tandemly duplicated blocks and thus provides intriguing evidence for specific crossover points during the evolution of the repeat. However, as the reads are far shorter than the length of the repeat, it was unclear whether these sequences represent distinct repeat subtypes, or whether individual repeat units show extensive variation within themselves. A previous study (Cook et al. 2014) also reported SNP heterogeneity between repeats in high-copy Rhg1 loci, but without determining the origin of these SNPs within the structure of the repeat. Therefore, we set out to determine variants that differentiate the multiple repeated loci. To investigate the diversity within the individual copies, we employed a haplotype phasing technique, using paired reads to connect variants that are present in the same copy among multiple copies (Fig. S2a, Supporting information). Four subtype configurations (referred to henceforth as subtypes W, P, FA, and FB) were obtained by phasing 149 SNPs in the repeats from Williams 82 (W), Peking (P) and PI 88788 (F). A SNP located in an intergenic region (1 657 025 bp on the chromosome 18) between two genes, Glyma18g02610 and Glyma18g02620, was useful in developing the hypothesis that there are four general types of repeat (W, P, FA and FB; Fig.2a) in the multiple-copy versions of the locus. We interpreted the data in Fig. S1 (Supporting information) as Williams 82 carrying only a single copy of the W type of the 31.2-kb sequence, Peking having three copies all of the P type, and PI 88788 having eight FA or FB copies and a copy of W. As individual copies could not be definitively linked to the rest of the repeat sequence as a result of zero sequence diversity in the vicinity of Glyma18g02600, we then performed a phylogenetic analysis of the sequences of the Glyma18g02590 gene. The four versions of the repeat present in the previously characterized PI 88788, Peking and Williams 82 genotypes (W, P, FA and FB) were found to be representative of all of the repeat units in other copy number alleles, all of the repeat units falling into one of these four categories (Fig.2b).
Repeat composition of different Rhg1 alleles
By combining data on the frequency of sequence variants in different germplasm accessions with different repeat composition (Fig. S2b, Supporting information) and using Sanger sequencing (Fig. S2c, Supporting information) to confirm the presence of SNP variants, the composition of repeat subtypes within each Rhg1 allele was estimated (Fig.2c). The Williams 82 and other single-copy genotypes investigated appear to only have the W type present. The three-copy accessions all had only one subtype (P) as in the Peking genotype. The one two-copy accession had one copy of P and one of W. The 6, 7, 9 and 10 copy alleles all have the same three subtypes present as PI 88788 (FA, FB & W), with W always present in one, partial copy. As indicated in Fig. S1 (Supporting information), the centromere-proximal repeat copy in these accessions has a 5′ (telomere proximal) sequence identical to FA or FB up to and including the variant at 8068 bp. The sequence then becomes highly similar to the W sequence beginning with base 8114 and continuing to the fusion site at the end of the repeat. Thus, the PI 88788 genome has 9 FA and FB type sequences of the Glyma18g02580 gene but eight FA and FB copies, and one copy of the W sequence for Glyma18g02590, −2600 and −2610. The germplasm accession PI 89008, with four copies, had three copies of subtype FB, again with just one copy of W. By re-analysis of the fosmids previously used to clone the repeat sequence (Cook et al. 2012), the partial subtype W sequence in the PI 88788 genotype was found to be located at the centromere-proximal end of the repeat. Sanger sequencing was also used on DNA amplified from the very 3′ end of the Rhg1 repeat (Table S2, Supporting information) to confirm the presence of the variant at this position. It is therefore likely that the subtype W sequence is also centromere-proximal in the genotypes containing FA and FB in multiple copies with one copy of the W subtype of Glyma18g02590 (Fig.2c).
Amino acid variation in Rhg1-coding regions
We then investigated the predicted amino acid sequences of the four genes in the repeat and how these varied between the duplicated copies. From all available Rhg1 repeat subtypes in all genotypes, the predicted amino acid sequences relative to the Williams 82 reference genome (subtype W) were investigated. No differences in encoded amino acid sequence were identified for Glyma18g02580 (where there were two synonymous substitutions), Glyma18g02600 (no variants at all) or Glyma18g02610 (four synonymous substitutions; Fig. S3, Supporting information). However, several variants exist in amino acid sequence of the α-SNAP protein (Glyma18g02590; Fig.2d; Fig. S3, Supporting information). Subtypes FA and FB are different from W at several locations. The amino acid sequence is identical between subtypes FA and FB. Subtype P has a distinctive amino acid sequence for Glyma18g02590, with some amino acids resembling FA and FB, some resembling W and some unique to P (Fig.2d). It is interesting to note that PI 88788-type Rhg1 alleles likely express two different forms of the α-SNAP protein and that Peking-derived germplasm has a third version of this protein which is distinct from either of the above.
Relationship between CNV and SCN resistance reactions
We selected nine germplasm accessions with validated CNVs at the Rhg1 locus where complete data for resistance to diverse SCN types are available (Fig. S4, Supporting information). So far, Rhg1-b is the only SCN resistance locus discovered in PI 88788 (9 copies; Concibido et al. 2004; Glover et al. 2004). PI 88788 shows resistance to both types 3 and 14. PI 209332 (10 copies), which harbours one more repeat unit than PI 88788, shows a similar resistance reaction to PI 88788 (Niblack et al. 2002; Colgrove & Niblack 2008), but this accession also shows resistance to an additional SCN type, 5, to which Peking (3 copies) and PI 438489 B (2 copies) are strongly resistant, likely because of one or more additional loci such as Rhg4 (Concibido et al. 2004). Surprisingly, as it only has two copies at the Rhg1 locus, PI 438489 B shows strong resistance to all investigated SCN types. It is interesting to note that two seven-copy accessions (Cloud & PI 92720) show nonidentical resistance reactions. This result is consistent with previous findings that while Rhg1 is usually necessary for effective nematode resistance, this resistance is modified by other resistance loci.
Diversity and disequilibrium at Rhg1
Previously, network analysis of shared variants within the genic regions of the repeat sequence was used to investigate relationships between high, low and single-copy Rhg1 accessions (Cook et al. 2014). However, the reconstruction of the repeat sequences using phasing allowed us to use phylogenetic approaches to infer ancestry of the entire 31.2-kb sequence of individual repeated units within each Rhg1 locus. We initially analysed sequence variant data from 18 soybean accessions where we had analysed WGS data and validated the presence of the repeat in the genome. As the repeat itself is variable in gene dosage and thus difficult to accurately genotype, we used surrounding sequences in LD with Rhg1 as a proxy for SNP genotyping. Protein-coding exons were analysed for variants in 38 genes flanking the Rhg1 repeat in the single-copy 400-kb region either side of the repeat. Nucleotide diversity (π) ranged between 0 and 0.00205 in coding sequence in this region across these accessions. As the location neared the Rhg1 locus, π rose sharply, most notably in the 70- to 80-kb region closest to the telomere-proximal end of the repeat (top graph in Fig.3a). The nucleotide diversity rose to almost six times the G. max average, 0.00053 (Zhu et al. 2003) in the Rhg1 flanking regions of accessions with 3, and 9 and 10 copies (middle and bottom, respectively, in Fig.3a). In contrast, low and even zero values of sequence diversity were seen at greater distances from the locus. We thus investigated LD surrounding the Rhg1 locus. The LD (measured by r2) within the ∼150 kb of the S2 region containing the Rhg1 locus is strong and statistically significant (Fig.3b; Fig. S5, Supporting information). Thus, we concluded that a block of strong LD extended for 70–80 kb either side of the repeat. We then used the apparent boundaries of the LD block, combined with the regions where nucleotide diversity quickly rose or fell (vertical lines in Fig.3a), to define three linkage blocks (S1, S2 and S3; Fig.3a, b) surrounding the repeat, with S2 being the block containing the Rhg1 repeat itself.
Fig 3.
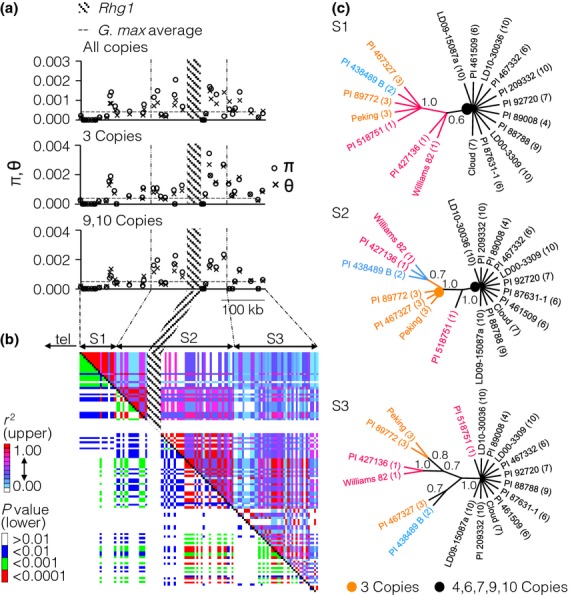
Diversity, linkage disequilibrium (LD) and sequence analysis of the region surrounding the Rhg1 locus. (a) Nucleotide diversity within 38 protein-coding genes surrounding Rhg1 in eighteen germplasm accessions (with 1–10 copies at Rhg1) is displayed in the uppermost graph. Those accessions with three copies (centre graph) and nine and ten copies (bottom graph) were also analysed separately. The average nucleotide diversity (π; 0.00053) of all coding regions in Glycine max is marked by a horizontal line. (b) LD plot using the r2 metric for the 400-kb region surrounding the Rhg1 locus. The same 18 accessions were used. Regions S1, S2 and S3 represent three linkage blocks used in further analysis. tel: telomere. (c) Phylogenetic trees derived from parsimony analysis of the three LD blocks. The tree for the region of linkage block S2 that contains Rhg1 is consistent with the analysis of the repeat sequence (Fig.2). The copy number of each accession is in parentheses. The consensus trees were created after collapsing branches with bootstrap values <60%, based on 10 000 replications. The orange and black circles indicate nodes that differentiate the three-copy and 4- to 10-copy versions of the Rhg1 locus. Bootstrap support values are shown above key nodes.
Evolutionary analysis of Rhg1 repeat units
While the Rhg1 repeat itself is polymorphically repetitive and thus not readily amenable to phylogenetic analysis, the surrounding regions, if in strong LD, can be used to determine the relatedness of the Rhg1 genomic regions in the accessions carrying the repeat. Within each of the three regions S1, S2 and S3, we performed phylogenetic analysis of the 18 accessions that underwent WGS using the MP method, in the case of the S2 region using only sequences outside the repeat. The resulting phylogenetic trees clearly showed that three groups were found in the S2 region (the Rhg1 locus and a ∼70-kb region extending either side; Fig.3c). The tree for the S2 region, while derived only using the genomic sequence outside the repeat, corresponds well to both the copy number data and the phylogenetic analysis of the repeat subtypes. Accessions with more than three copies (with FA + FB and W repeat types) form a distinct clade, as do all those with three copies (P repeat type; Figs2b, c and 3c). The single-copy types do not cluster into a monophyletic group. We do not see precisely the same clustering in the S1 and S3 regions, which are likely sufficiently distant that LD around the repeat has broken down (Fig.3b). Nonetheless, genotyping outside the repeat in the S2 region can be used to detect the Rhg1 accessions with either three or more than three copies, which correspond to all the Rhg1 alleles so far found to be useful in plant breeding.
Signatures of selection at Rhg1
We next investigated signatures of selection for Rhg1 in soybean populations by testing neutrality and population differentiation. Using high-density SNP genotyping data generated using the soybean 50K Illumina SNP array (Song et al. 2013) for the soybean germplasm collection (http://www.soybase.org), we initially analysed the Infinium data for 19 548 accessions. High nucleotide diversity (π) and positive Tajima's D (both statistically significant and well above the average for the soybean genome) were apparent near the Rhg1 locus in soybean (G. max & G. soja; top graph in Fig.4a). Wild soybean G. soja, also showed a very similar selection signature to the whole population of germplasm accessions (bottom graph in Fig.4a).
Fig 4.

Signatures of selection at the Rhg1 locus. (a) Nucleotide diversity (π and θ), Tajima's D and linkage disequilibrium were measured in the 1.5-Mbp region across the locus in 19 548 accessions (18 383 Glycine max & 1165 Glycine soja). The mean and 75th and 95th percentiles of whole-genome Tajima's D are marked by horizontal lines in corresponding graphs. (b) FST was calculated for lines with experimentally determined copy number (46 single copy vs. 48 multiple copy). Direction of telomere = ‘tel’. The mean and 95th percentile of whole-genome FST are marked by horizontal lines. Red marks indicates statistical significance. (c) FST between geographic subpopulations. One hundred and thirty-five single nucleotide polymorphisms were used to compare: Top graph, between 3311 germplasm accessions from Korea and 3855 from China; centre, between 3311 from Korea and 2466 from Japan; bottom, between 3855 from China and 2466 from Japan. The mean and 95th percentile values of whole-genome FST are marked by a horizontal line in the corresponding graphs.
A high FST, significantly (P ≤ 0.01) associated with population differentiation near the locus (Fig.4b), was also observed at Rhg1 when the Rhg1-carrying genotypes were considered as a separate population, to test whether FST is higher within the repeat-carrying genomes than expected if all the polymorphisms were randomly distributed among accessions. By treating the repeat-carrying accessions as a separate population, surrounding nucleotides fixed relative to the repeat could indicate either fixation at the locus due to selection, or a recent, common origin of Rhg1 with which surrounding nucleotides are still in LD. The shared fusion site sequence of all Rhg1 repeats (Cook et al. 2012) indicates that these repeats are likely of common origin, although this origin may pre-date soybean domestication. LD surrounding the Rhg1 gene was also detected using the r2 method (Fig.4b). Interestingly, the LD around Rhg1 was less marked on the centromere-proximal side of the repeat (Figs3b and 4b). Four indicators (Tajima's D, π, r2 LD and FST) suggest that differential selection may have occurred around the Rhg1 locus.
Geographic and genetic structure of nematode-resistant populations
To test whether the signatures of selection could be affected by the geographic area of origin for the accessions, the entire soybean germplasm high-density SNP data set was regrouped according to the origin of each germplasm accession; then, three major accession groups, originating in China (3855 accessions), Korea (3311) and Japan (2466), were selected. The repeat was present in all of these populations, in both the three-copy and more than three-copy versions. Overall, no significant population differentiation was detected between Korean and Chinese accessions (KR vs. CH), and Chinese and Japanese (CH vs. JP; top and bottom graph in Fig.4c, respectively). A lower degree of population differentiation compared to KR vs. CH and CH vs. JP was observed between Korean and Japanese accessions (KR vs. JP; middle graph in Fig.4c).
As false signals of selection can be caused by population structure, we evaluated whether the signatures of non-neutral selection at the Rhg1 locus could be related to population demography. We thus clustered the population according to Infinium SNP data (Fig.5a; Fig. S6, Supporting information). We observed a strongly positive value of Tajima's D (in all cases above the G. max genome average for the population) in all but one subpopulation (6.A in Fig.5b). The multiple-copy accessions are not confined to 6.A but are present in multiple subpopulations. In each case, the pattern resembles that obtained from the full population data described in Fig.4a. As most subpopulations are dominated by accessions from one country, we tested whether removal of those accessions altered the result; this did not appreciably change the pattern (Fig.5b). Therefore, population structure is not primarily responsible for the observed signature.
Fig 5.
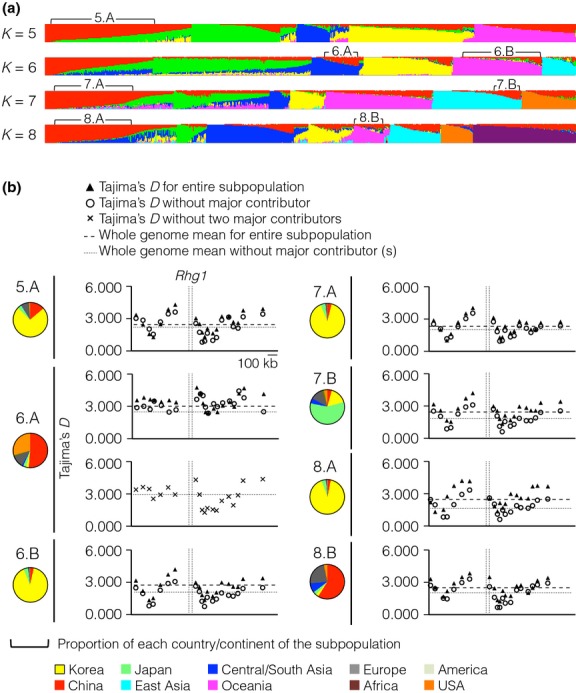
Signatures of selection at the Rhg1 locus are independent of population demography. (a) soybean population clustered by genetic structure. Each individual is represented by a thin vertical line, which is partitioned into K coloured segments that represent the individual's estimated membership fractions in K clusters. Selected clusters were labelled according to the K value and by ‘A’ or ‘B’ if two clusters were selected from a given value of K (e.g. ‘6.A’ means one of the two subpopulations selected from the K = 6 analysis). (b) Neutrality in subpopulations with shared genetic structure was tested by Tajima's D. The pie chart represents the geographic origin of the members of each subpopulation. Two separate D values are displayed for each subpopulation: one calculated from all accessions and one calculated excluding accessions from the largest single geographic origin in the cluster. An additional test without either Chinese or USA accessions was performed on the 6.A subpopulation. The East Asia group represents countries other than Korea, Japan and China. America represents South and North America except the USA. The whole-genome mean Tajima's D of each subpopulation is marked by a horizontal line.
The strong LD around the Rhg1 locus, and the shared ancestry of the SNPs within the range of LD, means that high-density genotyping data can potentially be used to classify accessions that likely carry alleles of Rhg1. To determine how common the Rhg1 genotype is within the germplasm collection, a maximum parsimony phylogenetic approach was applied to cluster the SNPs informative for copy-number alleles within the S2 LD block across the entire germplasm collection. A total of ten SNPs, nine located in sequence region S2 in Fig.3 and one within the Rhg1 repeat unit, formed 89 distinct combinations among 15 996 germplasm accessions (all accessions with high confidence data for all ten SNPs; Fig.6; Table S3, Supporting information). Four distinct SNP combinations corresponded to the clusters identified in Fig.3c. Validated multiple-copy accessions fell into one of two clusters, one containing 160 accessions and the other 707. A cluster with 160 accessions in Fig.6 corresponds to accessions with 4–10 copies, while a second cluster with 707 accessions corresponds to two- or three-copy accessions. Other clusters, including one that lies on the same branch as the two multicopy accessions, have either one or an unknown number of copies. Thus, at least 867 accessions have been identified as likely carriers of Rhg1 alleles, most of which are likely to represent new sources of the resistance gene, and several divergent but related groups have been identified that may be investigated for the presence of new alleles of Rhg1.
Fig 6.
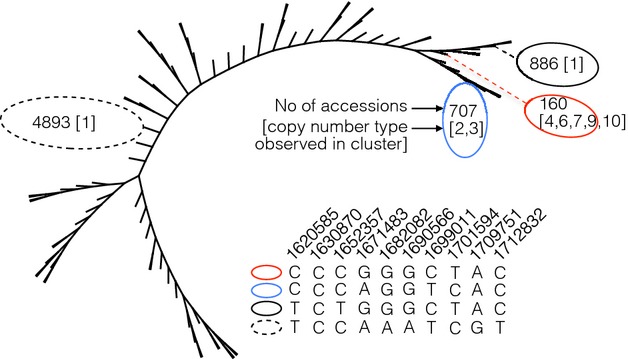
Parsimony tree of 15 996 soybean accessions with high confidence data at all single nucleotide polymorphisms (SNPs) in the S2 linkage disequilibrium block surrounding Rhg1. The four terminal branches containing all germplasm accessions described elsewhere in the manuscript are labelled, together with the number of accessions carrying the same combination of SNPs.
Discussion
In the current study, we report several major new findings: (i) Rhg1 is a highly variable repeat region that can be accurately genotyped by genomic qPCR. We add to previous knowledge of the diversity of this repeat, Rhg1 within the lines investigated having 2, 3, 4, 6, 7, 9 or 10 tandem repeats of just over 31 kb each; (ii) the individual repeated units of known Rhg1 alleles can be classified into four types based on sequence. Some Rhg1 loci carry up to three different types of repeat unit; (iii) the Rhg1 locus is in LD with the surrounding region of the genome; (iv) clustering of accessions by flanking sequence matches the phylogenetic analysis of the individual repeat units, and thus, existing high-density SNP data on flanking regions can be used to classify in silico thousand more accessions for Rhg1 presence and type; and (v) analysis of variants in the region around Rhg1 shows signatures of selection. The implications of these findings are discussed below.
Origin of Rhg1
The cloning of Rhg1 was the first observation that plant disease resistance loci can consist of a multigene cluster CNV of noncanonical resistance genes in tandem formation (Cook et al. 2012). The Rhg1 locus is common among nematode-resistant Glycine max accessions, because over half (58%; 62 of 106) of screened SCN-resistant germplasm is positive for the presence of the repeat junction. It is possible that other copy number variations exist at this locus and this number could be higher, but we have found no evidence for repeats at Rhg1 that do not contain the canonical fusion site, despite investigating a total of 89 whole-genome sequences for such repeats.
Soybean originated geographically in East Asia, where wild Glycine grows naturally. The PIs or germplasm accessions carrying Rhg1 (not including lines submitted as US cultivars) used experimentally in this study originate from distributed locations across East Asia (22 from China; 8 from Japan; 7 from Korea; 1 from Russia; Fig. S7, Supporting information). These 62 lines share a common repeat junction, strongly suggesting they share a common origin. Most likely the repeat originally arose as a duplication caused by unequal crossover, with subsequent illegitimate recombination events then giving rise to versions with more than two copies. Taken together, the evidence suggests that the hypothetical duplication event that created the copy number variation in Rhg1 happened sufficiently long ago in soybean evolution for it to be distributed across the area where soybean are endemic. In contrast to this, it has been reported that the CNV locus conferring the maize aluminium tolerance trait is detected only in maize lines sharing the same geographic origin (Maron et al. 2013). It has been estimated that the divergence of the progenitors of domesticated G. max and one modern wild Glycine soja line was 0.27 million years ago (Kim et al. 2010b); domestication itself is much more recent, occurring within the last 10 000 years. One of the G. soja accessions analysed by WGS carries three copies of the tandemly duplicated unit at Rhg1 and shows the same repeat structure as five of the three-copy G. max accessions, and the G. soja population shows the same signature of selection as the G. max population at Rhg1. This provides evidence that the origin of the tandem duplications of the 31.2-kb region at the Rhg1 locus occurred before the divergence of the common ancestors of cultivated soybean and one sequenced G. soja line, that is long before domestication. As the Rhg1 repeat is distributed in both G. max and G. soja lines throughout East Asia, we postulate that the origin of Rhg1 is likely to pre-date this divergence. As thousands of generations have likely passed since the repeat came into being, the LD and FST signatures in the population may indicate selection for the Rhg1 repeat.
Selection of Rhg1
We show strong LD surrounding the Rhg1 locus in both the SCN-resistant accessions and the population of all soybean germplasm accessions. However, complete LD extends for <100 kb, which implies that the locus has been under selection for a large number of meiotic cycles; many more than are conceivable as purposeful selection for SCN resistance by breeders began. On the other hand, other indicators of selection (such as Tajima's D and the FST signal) extend significantly further from the locus. This strong signature of selection is likely the result of pathogen pressure from SCN. This in turn provides evidence that SCN and resistance to SCN have been a major selective force for some time during evolution and artificial selection of G. max and G. soja.
Using phylogenetic analysis of the individual repeat sequences we assembled, and the flanking region in LD, we show that the individual repeat units in Rhg1 can be categorized into three lineages (three, more than three and two copies; P, FA/FB and W repeat types). Evidence for potentially divergent function comes from genes within the repeat, primarily the predicted α-SNAP protein, which also can be classified into three groups according to predicted amino acid sequence. Two of these variants are present together in the most widely used Rhg1 alleles from Fayette and PI 88788.
Population genetic analyses of the SNPs in a 1.5-Mbp region around Rhg1 revealed positive Tajima's D statistics, which along with LD around the locus, high nucleotide diversity and FST make positive selection likely at this locus. Population structure is unlikely to have resulted in a false signature of selection, as subpopulations derived from genotype-based clustering show the same signature. Just one subpopulation cluster (6.A in Fig.5b), which is composed of about 45% of US and European accessions, showed very minimal signs of selection on this locus. As positive selection at this locus is likely the result of pathogen pressure from SCN, the first report of SCN in the USA was 1954 (Winstead et al. 1955), and there has been no outbreak of SCN throughout Europe at the time of writing; this observation fits the conclusion that positive selection at the locus is a result of SCN pathogen pressure in areas with a longer history of soybean and SCN populations.
The FST statistic is significantly increased around the locus if the lines carrying Rhg1 are regarded as a separate population. We regard this as a signature of positive selection, as it is either a result of fixation around the locus or of LD around a relatively old event of common origin. However, when accessions are compared between countries of origin as separate populations, FST gives ambiguous results, with some comparisons showing reduced fixation around the locus. Segregation distortion at the Rhg1 locus has been reported in modern soybean breeding populations (Kopisch-Obuch & Diers 2006). Significantly fewer homozygous resistant plants were observed in analysed F4 populations, and seedling emergence was significantly lower for SCN-resistant plants. If the presence of Rhg1 reduces the survival of plants in the next generation, the frequency of Rhg1 in the population is expected to fall if SCN is absent and the locus has no other selective advantage. This could produce balancing selection, with selection for Rhg1 in the presence of SCN and selection against Rhg1 in its absence. Combining the evidence for wide variation in copy number at the locus, high LD and Tajima's D, the selection signature independent of the domestication bottleneck, relatively low or ambiguous FST between geographic populations, and reduced viability of Rhg1 homozygotes, we conclude that the Rhg1 locus may be subject to balancing selection within populations in East Asia. We also observed population differentiation in FST, which varied across geographic location comparisons. A likely explanation of this is unequal pathogen pressure of SCN in different geographic areas.
Mechanism of repeat origin and variation
Although recent CNV surveys in plants are increasing our knowledge of the extent and patterns of CNV in plant genomes such as soybean (McHale et al. 2012), the mechanisms of CNV generation remain unknown in most cases. All Rhg1 sequences examined so far possess the same junction point between the repeat and the genome, strongly implying a common origin, most likely a single duplication event by unequal crossover. A partial sequence (185 bp) having ∼75% identity to the 5′ and 3′ long terminal repeat (LTR) regions of Ty1/copia-like retrotransposons RTvr1 or RTvr2 is present within 400 bp of the duplication junction across all germplasm investigated. The Rhg1 locus is located close to the telomere (within 3% of the chromosome length) of chromosome 18. It is known that higher rates of recombination occur towards the telomere (Ott et al. 2011). It has been suggested that high levels of CNVs in crop genomes are located preferentially in regions of high recombination (Muñoz-Amatriaín et al. 2013). The source of the first duplication event to arise at Rhg1 could therefore be the result of Ty1/copia-like retrotransposon RTvr1 or RTvr2 activity in a sequence region with high recombination, which provided a similar sequence at the beginning and end of the repeated unit to allow illegitimate crossover. Once two copies of the unit were present, additional copies could readily be generated by slippage at the repeat during meiosis. The high rate of recombination at this locus, combined with strong positive selection pressure for high copy number, then led to the wide range of repeat copy number observed in the population. We found SNPs in a single-copy cultivar (PI 518751) that are shared with some multiple-copy types, which may represent evidence of recent crossover between repeat-carrying and single-copy lines during natural or artificial crossing and selection.
Implications for soybean breeding
Fayette (10 copies) is a cultivar developed from Williams (2) × PI 88788, with the objective of transferring the SCN resistance of PI 88788 (9 copies at Rhg1; Fig.1) to a US-adapted cultivar (Bernard et al. 1988). Given the wide range of observed copy number and this observed change during a soybean breeding programme, we speculate that alteration of the copy number at Rhg1 is rapid and continual. This suggests that manipulation of the repeat by artificial crossing and marker-assisted selection to obtain other repeat architectures is possible. For example, it may be possible to combine two different Rhg1 subtypes (e.g. subtype P and subtypes FA/FB) in a single line, if enough progeny are screened from an appropriate cross. Considering that variation in copy number has been observed within a population derived from a single Rhg1 allele (Fayette/PI 88788), it is possible that changes of copy number at Rhg1 may be a cause of variation in the effectiveness of nematode resistance observed in soybean lines.
Our data also give molecular evidence to support correlation between Rhg1 copy number and female indices (FI) observed from virulence assays. It has previously been shown that FIs from Cloud (7 copies), PI 88788 (9) and PI 209332 (10) were highly correlated, as were those of PI 438489 B (2), PI 90763 (3), PI 89772 (3) and Peking (3; Colgrove & Niblack 2008). It is now clear from the data presented here that the first three germplasm accessions have relatively high copy numbers compared to the second four and carry three repeat sequence subtypes (FA, FB, and W) corresponding to two distinct types of α-SNAP protein. On the contrary, the second four germplasm accessions have a different subtype (P), which has a third type of the α-SNAP protein. This finding strongly suggests that either the number of copies in the Rhg1 haplotype, the sequence of the α-SNAP protein or both have a strong effect on SCN type-specific resistance. The accession PI 438489 B possesses just two copies of the Rhg1 repeat. Its repeat is composed of subtype W, which is nearly identical in sequence to susceptible single-copy germplasm, and subtype P, which encodes an α-SNAP protein identical to that found in three-copy alleles of Rhg1 such as Peking. Thus, only two copies are present at Rhg1 in the line, with a single copy of repeat type P. Nonetheless, the resistance spectrum is similar to that observed in germplasm with three copies of subtype P (Colgrove & Niblack 2008). This suggests that the sequence of the individual repeat units, as well as copy number, plays a role in the type specificity of Rhg1-mediated nematode resistance.
Conclusions
Copy number variation at the Rhg1 locus was selected for and retained within the population of wild soybean prior to domestication. High levels of sequence and copy number diversity exist within the repeat, but surrounding SNPs are strongly linked to different repeat types. This strong LD around the locus allows classification of many soybean germplasm accessions as likely Rhg1 alleles according to the public high-density SNP genotyping data. The complex sequence and structural diversity at this locus likely has had a large impact on population-level nematode resistance, potentially allowing the rapid evolution of the repeat to compete with the evolution of virulence genes within the nematode. However, previously observed fitness penalties of the Rhg1 locus combined with our observations of limited fixation within individual populations imply that the susceptible alleles may be maintained in the population by balancing selection.
Acknowledgments
The authors would like to thank Andrew Bent, David Cook and Perry Cregan for sharing unpublished data and members of the MEH laboratory, especially Gopal Battu and Mohammad B. Belaffif, for helpful discussion.
Data accessibility
DNA sequence data are available at the NCBI Sequence Read Archive, study accession SRP053114. The complete data set for 19 652 Glycine max and Glycine soja accessions genotyped with SNPs is available at the SoyBase (http://www.soybase.org). The taxon and origin for the genus Glycine are available in the multicrop passport descriptor (MCPD) format at the Germplasm Resources Information Network (http://www.ars-grin.gov). The analyses-ready files are available at Dryad Digital Repository (doi:10.5061/dryad.g6311).
Supporting information
Additional supporting information may be found in the online version of this article.
Fig. S1 Sequence variation within the Rhg1 repeat.
Fig. S2 Reconstruction of the sequences of individual repeat units.
Fig. S3 Single nucleotide variations (SNVs) identified at three genes within the Rhg1 repeat in 22 soybean germplasm accessions.
Fig. S4 Soybean cyst nematode (SCN) resistant germplasm with copy number confirmed by two methods, and their resistance reaction to SCN types.
Fig. S5 Sequence variants in the 400 kb region across the Rhg1 locus, displayed according to copy number at Rhg1.
Fig. S6 Population structure estimation using K = 2 through 4, using the same population and parameters as for Fig. 5a.
Fig. S7 Map of East Asian collection localities for SCN resistant soybean germplasm, showing copy number variation in the Rhg1 locus.
Table S1 Presence/Absence of repeat junction(s) and copy number estimation using whole genome sequencing (WGS) or genomic qPCR amplification of the gene Glyma18g02590 in Rhg1.
Table S2 Sequence variants in the repeat junction: the sequence region that spans the centromere-proximal repeat and the adjoining non-duplicated region of the genome adjacent to Rhg1.
Table S3 A total of 15 996 soybean germplasm accessions clustered by maximum parsimony phylogenetic analysis of the sequence region near the Rhg1 allele, indicating germplasm accessions predicted to carry Rhg1.
Table S4 DNA sequences of oligonucleotide primers used for assays.
Table S5 Summary of whole-genome shotgun sequencing data generated in this study from multiple-copy Rhg1 germplasm accessions.
References
- Bernard RL, Noel GR, Anand SC, Shannon JG. Registration of Fayette soybean. Crop Science. 1988;28:1028–1029. [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nature Genetics. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang D, Arelli P, et al. Molecular marker diversity of SCN-resistant sources in soybean. Genome. 2006;49:938–949. doi: 10.1139/g06-057. [DOI] [PubMed] [Google Scholar]
- Colgrove AL, Niblack TL. Correlation of female indices from virulence assays on inbred lines and field populations of Heterodera glycines. Journal of Nematology. 2008;40:39–45. [PMC free article] [PubMed] [Google Scholar]
- Concibido VC, Diers BW, Arelli PR. A decade of QTL mapping for cyst nematode resistance in soybean. Crop Science. 2004;44:1121–1131. [Google Scholar]
- Conrad DF, Andrews TD, Carter NP, et al. A high-resolution survey of deletion polymorphism in the human genome. Nature Genetics. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- Cook DE, Lee TG, Guo X, et al. Copy number variation of multiple genes at Rhg1 mediates nematode resistance in soybean. Science. 2012;338:1206–1209. doi: 10.1126/science.1228746. [DOI] [PubMed] [Google Scholar]
- Cook DE, Bayless A, Wang K, et al. Distinct copy number, coding sequence and locus methylation patterns underlie Rhg1-mediated soybean resistance to soybean cyst nematode. Plant Physiology. 2014;165:630–647. doi: 10.1104/pp.114.235952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan PB, Mudge J, Fickus EW, et al. Two simple sequence repeat markers to select for soybean cyst nematode resistance conditioned by the rhg1 locus. Theoretical and Applied Genetics. 1999;99:811–818. [Google Scholar]
- Dı′az A, Zikhali M, Turner AS, et al. Copy number variation affecting the Photoperiod-B1 and Vernalization-A1 genes is associated with altered flowering time in wheat (Triticum aestivum. PLoS One. 2012;7:e33234. doi: 10.1371/journal.pone.0033234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers BW, Skorupska HT, Rao-Arelli AP, Cianzio SR. Genetic relationships among soybean plant introductions with resistance to soybean cyst nematodes. Crop Science. 1997;37:1966–1972. [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover KD, Wang D, Arelli PR, et al. Near isogenic lines confirm a soybean cyst nematode resistance gene from PI 88788 on linkage group J. Crop Science. 2004;44:936–941. [Google Scholar]
- Hanikenne M, Kroymann J, Trampczynska A, et al. Hard selective sweep and ectopic gene conversion in a gene cluster affording environmental adaptation. PLoS Genetics. 2013;9:e1003707. doi: 10.1371/journal.pgen.1003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Hutter S, Vilella AJ, Rozas J. Genome-wide DNA polymorphism analyses using VariScan. BMC Bioinformatics. 2006;7:409. doi: 10.1186/1471-2105-7-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovene M, Zhang T, Lou Q, et al. Copy number variation in potato—an asexually propagated autotetraploid species. The Plant Journal. 2013;75:80–89. doi: 10.1111/tpj.12200. [DOI] [PubMed] [Google Scholar]
- Kim M, Hyten DL, Bent AF, Diers BW. Fine mapping of the SCN resistance locus rhg1-b from PI 88788. The Plant Genome. 2010a;3:81–89. [Google Scholar]
- Kim MY, Lee S, Van K, et al. Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proceedings of the National Academy of Sciences of the United States of America. 2010b;107:22032–22037. doi: 10.1073/pnas.1009526107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Research. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopisch-Obuch FJ, Diers BW. Segregation at the SCN resistance locus rhg1 in soybean is distorted by an association between the resistance allele and reduced field emergence. Theoretical and Applied Genetics. 2006;112:199–207. doi: 10.1007/s00122-005-0104-2. [DOI] [PubMed] [Google Scholar]
- Lam HM, Xu X, Liu X, et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nature Genetics. 2010;42:1053–1059. doi: 10.1038/ng.715. [DOI] [PubMed] [Google Scholar]
- Li Z, Xing A, Moon BP, et al. Site-specific integration of transgenes in soybean via recombinase-mediated DNA cassette exchange. Plant Physiology. 2009;151:1087–1095. doi: 10.1104/pp.109.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maron LG, Guimarães CT, Kirst M, et al. Aluminum tolerance in maize is associated with higher MATE1 gene copy number. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5241–5246. doi: 10.1073/pnas.1220766110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH, et al. Common deletion polymorphisms in the human genome. Nature Genetics. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- McHale L, Tan X, Koehl P, et al. Plant NBS-LRR proteins: adaptable guards. Genome Biology. 2006;7:212. doi: 10.1186/gb-2006-7-4-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale LK, Haun WJ, Xu WW, et al. Structural variants in the soybean genome localize to clusters of biotic stress–response genes. Plant Physiology. 2012;159:1295–1308. doi: 10.1104/pp.112.194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Amatriaín M, Eichten SR, Wicker T, et al. Distribution, functional impact, and origin mechanisms of copy number variation in the barley genome. Genome Biology. 2013;14:R58. doi: 10.1186/gb-2013-14-6-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DQ, Webber C, Ponting CP. Bias of selection on human copy-number variants. PLoS Genetics. 2006;2:e20. doi: 10.1371/journal.pgen.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niblack TL, Arelli PR, Noel GR, et al. A revised classification scheme for genetically diverse populations of Heterodera glycines. Journal of Nematology. 2002;34:279–288. [PMC free article] [PubMed] [Google Scholar]
- Niblack TL, Lambert KN, Tylka GL. A model plant pathogen from the kingdom animalia: Heterodera glycines, the soybean cyst nematode. Annual Review of Phytopathology. 2006;44:283–303. doi: 10.1146/annurev.phyto.43.040204.140218. [DOI] [PubMed] [Google Scholar]
- Ott A, Trautschold B, Sandhu D. Using microsatellites to understand the physical distribution of recombination on soybean chromosomes. PLoS One. 2011;6:e22306. doi: 10.1371/journal.pone.0022306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repping S, van Daalen SK, Brown LG, et al. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nature Genetics. 2006;38:463–467. doi: 10.1038/ng1754. [DOI] [PubMed] [Google Scholar]
- Rousset F. GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Schmitz RJ, He Y, Valdés-López O, et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Research. 2013;23:1663–1674. doi: 10.1101/gr.152538.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Song Q, Hyten DL, Jia G, et al. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean. PLoS One. 2013;8:e54985. doi: 10.1371/journal.pone.0054985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton T, Baumann U, Hayes J, et al. Boron-toxicity tolerance in barley arising from efflux transporter amplification. Science. 2007;318:1446–1449. doi: 10.1126/science.1146853. [DOI] [PubMed] [Google Scholar]
- Swanson-Wagner RA, Eichten SR, Kumari S, et al. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Research. 2010;20:1689–1699. doi: 10.1101/gr.109165.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theoretical Population Biology. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Winstead NN, Skotland CB, Sasser JN. Soybean cyst nematode in North Carolina. Plant Disease Reporter. 1955;39:9–11. [Google Scholar]
- Zheng LY, Guo XS, He B, et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor. Genome Biology. 2011;12:R114. doi: 10.1186/gb-2011-12-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YL, Song QJ, Hyten DL, et al. Single-nucleotide polymorphisms in soybean. Genetics. 2003;163:1123–1134. doi: 10.1093/genetics/163.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Sequence variation within the Rhg1 repeat.
Fig. S2 Reconstruction of the sequences of individual repeat units.
Fig. S3 Single nucleotide variations (SNVs) identified at three genes within the Rhg1 repeat in 22 soybean germplasm accessions.
Fig. S4 Soybean cyst nematode (SCN) resistant germplasm with copy number confirmed by two methods, and their resistance reaction to SCN types.
Fig. S5 Sequence variants in the 400 kb region across the Rhg1 locus, displayed according to copy number at Rhg1.
Fig. S6 Population structure estimation using K = 2 through 4, using the same population and parameters as for Fig. 5a.
Fig. S7 Map of East Asian collection localities for SCN resistant soybean germplasm, showing copy number variation in the Rhg1 locus.
Table S1 Presence/Absence of repeat junction(s) and copy number estimation using whole genome sequencing (WGS) or genomic qPCR amplification of the gene Glyma18g02590 in Rhg1.
Table S2 Sequence variants in the repeat junction: the sequence region that spans the centromere-proximal repeat and the adjoining non-duplicated region of the genome adjacent to Rhg1.
Table S3 A total of 15 996 soybean germplasm accessions clustered by maximum parsimony phylogenetic analysis of the sequence region near the Rhg1 allele, indicating germplasm accessions predicted to carry Rhg1.
Table S4 DNA sequences of oligonucleotide primers used for assays.
Table S5 Summary of whole-genome shotgun sequencing data generated in this study from multiple-copy Rhg1 germplasm accessions.
Data Availability Statement
DNA sequence data are available at the NCBI Sequence Read Archive, study accession SRP053114. The complete data set for 19 652 Glycine max and Glycine soja accessions genotyped with SNPs is available at the SoyBase (http://www.soybase.org). The taxon and origin for the genus Glycine are available in the multicrop passport descriptor (MCPD) format at the Germplasm Resources Information Network (http://www.ars-grin.gov). The analyses-ready files are available at Dryad Digital Repository (doi:10.5061/dryad.g6311).