

Abstract
Aldose reductase (AR) is thought to play a role in the pathogenesis of diabetic eye diseases, including cataract and retinopathy. However, not all diabetics develop ocular complications. Paradoxically, some diabetics with poor metabolic control appear to be protected against retinopathy, while others with a history of excellent metabolic control develop severe complications. These observations indicate that one or more risk factors may influence the likelihood that an individual with diabetes will develop cataracts and/or retinopathy. We hypothesize that an elevated level of AR gene expression could confer higher risk for development of diabetic eye disease. To investigate this hypothesis, we examined the onset and severity of diabetes-induced cataract in transgenic mice, designated AR-TG, that were either heterozygous or homozygous for the human AR (AKR1B1) transgene construct. AR-TG mice homozygous for the transgene demonstrated a conditional cataract phenotype, whereby they developed lens vacuoles and cataract-associated structural changes only after induction of experimental diabetes; no such changes were observed in AR-TG heterozygotes or nontransgenic mice with or without experimental diabetes induction. We observed that nondiabetic AR-TG mice did not show lens structural changes even though they had lenticular sorbitol levels almost as high as the diabetic AR-TG lenses that showed early signs of cataract. Over-expression of AR led to increases in the ratio of activated to total levels of extracellular signal-regulated kinase (ERK1/2) and c-Jun N-terminal (JNK1/2), which are known to be involved in cell growth and apoptosis respectively. After diabetes induction, AR-TG but not WT controls had decreased levels of phosphorylated as well as total ERK1/2 and JNK1/2 compared to their nondiabetic counterparts. These results indicate that high AR expression in the context of hyperglycemia and insulin deficiency may constitute a risk factor that could predispose the lens to disturbances in signaling through the ERK and JNK pathways and thereby alter the balance of cell growth and apoptosis that is critical to lens transparency and homeostasis.
Keywords: Aldose reductase, AKR1B1, Cataract, Diabetes
1. Introduction
Over 29.1 million people in the United States and 285 million people worldwide are affected with diabetes mellitus (DM) [1, 2]. Nearly all of those with Type 1 diabetes and greater than 60% of those with Type 2 diabetes will have some form of retinopathy in the first decade of being diagnosed [3]. Furthermore, DM is the leading cause of preventable blindness in the United States [4, 5]. Each year, there are over 4 million new cases of blindness related to DM [3]. The cost alone in the United States is estimated at over 500 million dollars per year [6]. Increasingly worrisome, diabetes is rapidly growing around the world, making it a significant public health issue [7]. Thus, it is important to investigate the risk factors involved in diabetic eye disease and advance research in medical treatments that delay or inhibit diabetic eye complications.
There are three major eye conditions associated with diabetes: glaucoma, cataracts, and retinopathy. This study will focus on diabetic cataracts. A cataract is an opacification of the lens leading to visual impairment. It affects the ability of the eye to focus light and create clear images [8]. Duration of diabetes and glycemic control are important risk factors in the development of diabetic cataracts [9, 10]. Interestingly, diabetic cataracts are more likely to form in those with type 2 diabetes under the age of 18 and in those with type 1 diabetes from ages 18 to 44 years old [11]. There are several theories on the pathogenesis of diabetic cataracts, but the activation of the polyol pathway and its enzyme aldose reductase (AR) is of particular interest. In the polyol pathway, AR uses NADPH to reduce glucose into sorbitol. Next, sorbitol dehydrogenase reduces sorbitol into fructose with NAD+ acting as a cofactor [12]. High glucose conditions can activate the polyol pathway causing oxidative stress [13]. There are several ways the polyol pathway induces oxidative stress. First, sorbitol accumulation acts as an osmotic stressor, which leads to the production of reactive oxygen species (ROS) [10]. Second, depletion of NADPH, which is essential to the production of GSH an intracellular antioxidant, causes increased oxidative stress. Third, the production of NADH increases ROS production [10, 14]. Finally, fructose can be metabolized into fructose-3-phospate and 3-deoxyglucosone, which are potent non-enzymatic glycation agents. Fructose-3-phospate and 3-deoxyglucosone increase the amount of Advanced Glycation Endproducts (AGEs) leading to ROS generation [14, 15]. Furthermore, these oxidative stresses along with hyperglycemia are thought to activate mitogen-activated protein kinases (MAP kinases) which are involved in cell proliferation, survival, and differentiation [16, 17]. There are three groups of MAP kinases, which are regulated and activated by phosphorylation: extracellular signal regulated kinases (ERK), c-Jun N-terminal kinases (JNK) and p38 kinases [16]. ERK has two important forms p44 ERK1 and p42 ERK2 (ERK1/2). These kinases are primarily thought of as growth factor signaling kinases that regulate cell proliferation and survival [17, 18]. JNK has three forms JNK1, JNK2, and JNK3. JNK is thought to be a stress-activated protein kinase (SAPK) that can activate apoptosis [18]. Since AR is the rate-limiting step in the polyol pathway [19], inhibitors of AR have become an important topic of research.
Preventing diabetic eye disease is complicated. There are many risk factors that contribute to the process as demonstrated by a study finding those with optimal glucose control reduced their risk of developing retinopathy by 76 percent and their progression to retinopathy by 54 percent [20, 21]. Even though tight glucose control led to better outcomes in most patients, some patients still developed retinopathy. Thus understanding risk factors that can limit the progression of diabetic eye disease other than glucose control is of fundamental importance. In particular, determining whether specific metabolic pathways and enzymes can be targeted by medical interventions is essential.
Lee et al. [22] previously showed that overexpression of AKR1B1 is associated with cataract development in transgenic mice induced to develop hexosemia from galactose-overfeeding or from experimentally-induced diabetes. Our current study utilized a strain of AKR1B1 transgenic mice we produced using a different promoter construct designed for expression in lens epithelium and outer cortical fiber cells. Our results revealed that AR overexpression in the context of hyperglycemia and insulin dysregulation leads to changes to lens organization as well as significant alterations to ERK and JNK signaling pathways as potentially important elements in the pathogenesis of diabetic cataract.
2. Materials and Methods
2.1. Transgenic Mice
Transgenic mice designed for lens-specific expression of AKR1B1 were produced by standard methods on a C57BL/6 strain background essentially as described previously [23] with the long term goal of utilizing a human AR (AKR1B1)-expressing mouse model as a screening platform to validate new drug candidates against diabetic cataract formation. The current study was carried out using the strain designated PAR40. AR expression in this stain is controlled by a hybrid α/δ crystallin enhancer/promoter designed to drive transgene expression in lens epithelium and fiber cells in a manner similar to the expression pattern of AKR1B1 in the human lens [24–26]. Transgenic animals were identified by a PCR-based genotyping assay and were backcrossed to generate the transgene in a homozygous state as determined by a quantitative PCR assay using an ABI 7900 qPCR instrument (TaqMan Copy Number Assay and Copy Caller v2.0, Applied Biosystems, Inc. Foster City, CA). Homozygosity of such animals was verified by a backcross to wild type C57BL6 breeders (Jackson Laboratories, Bar Harbor, ME), which resulted in progeny that were verified to carry hemizygous levels of the AR transgene. In all cases, transmission of the transgene followed the expected Mendelian pattern. Unless noted otherwise, all studies with AR-TG mice in this report were carried out with animals homozygous for the AR transgene. PCR genotyping was also used to verify that our AR-TG strain does not carry the Rd8 mutation of the Crb1 gene that has been associated with ocular induced phenotypes (data not shown) [27].
Experimental diabetes was induced in AR-TG and nontransgenic mice using a high-dose streptozotocin induction protocol, essentially as described by Graham et al. [28]. Accordingly, AR-TG and C57BL/6 mice, 6 weeks old, were fasted for 4 hours, anesthetized by Isoflurane, and injected with streptozotocin (STZ, 160 mg/kg in Na-Citrate, Sigma-Aldrich, MO, USA). Mice were supplied with 10% sucrose water overnight to avoid post-injection hypoglycemia. Blood glucose levels were measured after three days by clipping the tail and using a OneTouch Ultra Mini glucometer (LifeScan, Milpitas, CA). Those with >500 mg/dL blood glucose levels were used for the study. After 16 days of diabetes (or euglycemia in nondiabetic controls), animals were euthanized by CO2 inhalation and lenses removed from each of four experimental groups: Wild type (WT, C57BL/6), WT with STZ, AR-TG, and AR-TG with STZ (total n=12). A pair of lenses from the same mouse was placed into 300 μL of radioimmunoprecipitation assay buffer (RIPA buffer, Boston BioProducts, MA, USA) and sonicated. Samples were centrifuged at 14000 rpm at 4° C for 10 minutes, and the supernatants (lens cell lysate) were then stored at −20° C until use.
Expression levels of AKR1B1 in transgenic strains were determined by measuring the rate of DL-glyceraldehyde reduction in the presence of NADPH using lens homogenates in a standard reaction mixture as described previously [29]. Transgene expression levels were also determined by semiquantitative Western blotting as described below.
This research was conducted in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All mice were handled in strict accordance with good animal practice, and all animal work was approved by Institutional Animal Care and Use Committee at The University of Colorado Anschutz Medical Campus (Aurora, CO, USA).
2.2. Sorbitol Concentration Assay
Lens cell lysates were deproteinized with Deproteinizing Sample Preparation Kit (BioVision, Milpitas, CA). Sorbitol in neutralized samples was measured using a D-Sorbitol Colorimetric Assay Kit as described by the manufacturer (BioVision).
2.3. Protein Concentration Assay
Protein concentration of lens cell lysate samples were determined using BCA (bicinchonic acid) Protein Assay Reagent (Thermo Scientific, IL, USA).
2.4. Western Blotting
Lens cell lysate samples were mixed with Laemmli buffer (Sigma-Aldrich) and were heated to 95° C for 10 minutes. After resolution with Mini Protean TGX Precast gel 4–15%, proteins from gels were transferred onto nitrocellulose membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The following primary antibodies were used for immunodetection: mouse anti-human AR (1:1000; Santa Cruz, TX, USA), mouse anti-actin (1:1000; Sigma-Aldrich). For detection of extracellular signal-related kinases 1 and 2 (ERK1/2) and c-Jun N-terminal kinases (JNK), we used rabbit anti-phospho ERK1/2 (1:1000; Cell Signaling, MA, USA), rabbit anti-ERK1/2 (1:1000; Cell Signaling), rabbit anti-phospho JNK (1:1000; Cell Signaling), and rabbit anti-JNK (1:1000; Cell Signaling), respectively. Secondary anti-mouse and anti-rabbit antibodies conjugated to horseradish peroxidase (1:2000; Millipore, Bedford, MA, USA), as well as the Immun-Star WesternC Kit (Bio-Rad, CA, USA) were used to detect chemilluminescence using a BioRad ChemiDoc XRS+ imaging system.
2.5. Histology
Mouse eyes were treated with buffered formalin and embedded in paraffin by standard procedures. Eyes were sectioned and stained using routine hematoxylin and eosin (H&E) staining. Morphology was characterized by light microscopy using a Nikon Eclipse 80i microscope fitted to a Nikon DS-Fi1 camera.
2.6. Statistical Analysis
Results are shown as the mean ± SEM of at least three experiments (n=3), except for Western blot data involving ERK and JNK, which are the mean ± SEM of at least two experiments (n=2). Data was analyzed by student’s t test with P value of < 0.05 considered significant.
3. Results
3.1. Increased AR expression leads to high levels of sorbitol with and without diabetes induction
We used several methods to evaluate the impact of AR gene over-expression in the lenses of transgenic mice. Western blotting confirmed that AR levels in animals heterozygous for the AR transgene are roughly half that observed in homozygotes (Fig. 1). Very low levels of AR were found in crude lens extracts from nontransgenic C57/BL6 control mice using either Western blotting (Fig. 1A) or enzyme activity (Fig. 1B) measurements. However, substantially increased levels were detected in transgenic mice that were heterozygous for the AR transgene, and roughly twice that amount in lenses from mice that were homozygous for the AR transgene (Fig. 1).
Figure 1.

AR expression in AR-TG mouse lenses. (A) Western blot of mouse lens homogenates. Heterozygous and homozygous AR-TG mice have increased AR expression compared to wild type mice. (B) Enzymatic activity measurement. Wild type mice have very little AR expression. AR-TG mice heterozygous for the AR transgene have increased AR expression, whereas AR expression is almost double in transgene homozygotes. Data are The mean ± SEM of three experiments (n=3).
To test the hypothesis that elevated levels of AR lead to activation of the polyol pathway, we measured sorbitol levels in lenses of AR-TG mice before and after diabetes-induced elevation of circulating glucose levels. In the absence of experimental diabetes, sorbitol levels were elevated approximately 4-fold in AR-TG lenses as compared to nontransgenic controls (Fig. 2). STZ-induced diabetes of 12-day duration led to somewhat higher levels of lens sorbitol, but the diabetes-influenced levels were not significantly different from those of AR-TG without diabetes.
Figure 2.

Sorbitol concentration of mice lenses. Sorbitol levels in AR-TG and AR-TG/STZ mice were significantly higher than wild type and wild type STZ mice p ≤ 0.01 (**) and p ≤ 0.005 (***). Data are The mean ± SEM of three experiments (n=3).
3.2. Increased AR expression causes diabetes-dependent formation of lens vacuoles
Sorbitol accumulation is thought to cause increased hydration and swelling of lens fiber cells, which is considered an early stage of cataract formation [30–32]. Hydration and swelling can result in the formation of vacuoles in lens tissue that is otherwise characterized by densely packed fiber cells. Thus we performed H&E staining on histological sections produced from eyes of our experimental mice. Normal, undisturbed ocular morphology was observed in wild type and AR-TG mice without diabetes induction (Fig. 3A–C). The lens fiber cells were observed in a regular packing array typical of the normal lens. In contrast, lenses of AR-TG mice after 12 days of streptozotocin-induced diabetes contained numerous vacuoles in the outer cortical fiber layer (Fig. 3D). Such vacuoles create disturbances in the refractive gradient index typical of the clear lens, causing disturbances in the transmission of light through the lens. This is the cause of light scattering and decreased visual acuity recognized clinically as the early stage of cataract formation. No additional morphological abnormalities were observed in other parts of the eye of the AR-TG strain, even after 12 days of diabetes. Tissue layers in the cornea, iris and ciliary body, and retina were uniformly intact and appeared completely normal in all respects (Fig. 3D). The morphological organization of cornea, iris, retina and lens in all other treatment groups appeared to be normal.
Figure 3.

H&E staining of mice lenses. (A) Wild type (C57/bl6) mouse with normal lens fibers. (B) Diabetic wild type (C57/bl6) mouse with normal lens fibers. (C) AR-TG mouse with normal lens fibers. (D) Diabetic AR-TG mouse with vacuolization of cortical lens fibers. Arrows demonstrate vacuolization. All images were taken at 40x magnification. Images are typical of multiple different animals from each group.
3.3. AR expression is not upregulated in diabetic mice lenses
Hodgkinson et al. [33] found that hyperglycemia upregulates expression of human AR (AKR1B1) compared to patients without diabetes. Interestingly, Lee et al [22] found that hyperglycemia in AR transgenic mice did not increase AR expression. We found similar result in our Western blot analysis of AR. WT STZ mice did not show any increase in AR expression compared to WT mice without diabetes induction (Fig 4). In our AR-TG mice, transgenic AR expression is controlled by promoter elements derived from lens crystallin genes. As expected we did not see diabetes-induced changes in expression of AR at the protein level.
Figure 4.
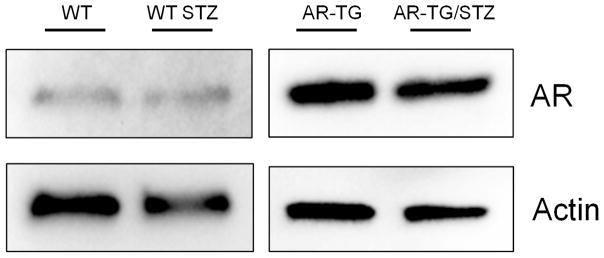
Western blot of lens homogenates. AR-TG and AR-TG/STZ treated mice show increased AR expression compared to wild type (WT) and wild type streptomycin (WT-STZ). WT STZ have similar levels of AR expression compared to WT.
3.4. Increased AR expression leads to an increase in pERK
It has been shown that the MAPK pathway can regulate glucose metabolism and can cause cataract formation when constitutively active [34]. Therefore, we investigated whether increased AR expression would activate ERK. We found that AR-TG mice had an increase in the ratio of activated or phosphorylated ERK (pERK1/2) relative to total ERK (tERK1/2) (Fig 5). Diabetes induction in AR-TG mice caused a 44 percent decrease in levels of total ERK compared to nondiabetic AR-TG mice. Even with this decrease in total ERK (the denominator), diabetes induction caused the levels of pERK1/2 to fall far enough to drop the pERK/tERK ratio to approximately 40% compared with lenses from AR-TG without diabetes. Thus, over-expression of AR causes a STZ diabetes-dependent disruption in the regulation of ERK expression and activation.
Figure 5.

Western blot and densitometry of mice lenses. (A) Western blot of pERK1/2, tERK1/2, and actin. (B) The ratio of pERK/tERK was significantly lower in wild type mice compared to AR-TG mice p ≤ 0.05. The ratio of pERK/tERK was significantly lower in AR-TG/STZ mice compared to AR-TG mice p ≤ 0.05 (*) and p ≤ 0.01 (**). Levels were normalized to actin. Data are the mean ± SEM of two experiments (n=2).
3.5. Increased AR expression leads to an increase in JNK
Studies have demonstrated that apoptosis of lens epithelial cell can lead to lens pathology [35, 36]. Therefore we investigated whether increased AR expression would lead to activation of JNK, a known apoptotic signaling pathway. We found that AR-TG mice without diabetes had an increase in the ratio of activated or phosphorylated JNK (pJNK1/2) to total JNK (tJNK1/2) (Fig 6). However, STZ diabetes induction in AR-TG/STZ mice caused about 50% drop in in the ratio of pJNK1/2 to tJNK1/2. Similar to our observations with total ERK expression, diabetes induction in AR-TG mice led to a 50 percent decrease in levels of total JNK compared to AR-TG mice. Thus, AR over-expression leads to significantly elevated JNK activation and also disrupts the regulation of JNK gene expression in the context of STZ-induced diabetes.
Figure 6.

Western blot and densitometry of mice lenses. (A) Western blot of pJNK1/2, tJNK1/2, and actin. (B) There was a significant difference between pJNK/tJNK in wild type (WT) and AR-TG mice with p ≤ 0.01 (**). The ratio of pJNK/tJNK was significantly lower in ARTG/STZ) mice compared to AR-TG mice with p ≤ 0.001 (***)[20]. Levels were normalized to actin. Data are the mean ± SEM of two experiments (n=2).
4. Discussion
Diabetes is a multifactorial disease and a single factor cannot predict the onset and progression of the major complications of diabetes, including diabetic cataract and retinopathy. AR is thought to be involved in the pathogenesis of diabetic eye disease, as evidenced by observations in animal models that AR inhibitors substantially prevent some complications of diabetes [37, 38], and AR null animals are largely protected from development of diabetic complications such as retinal capillary degeneration [39], visual function deficits [40] and onset of metabolic abnormalities associated with activation of the polyol pathway [41, 42]. Although the promise of newer generation AR inhibitors is still being examined [43, 44], understanding AR and its role in diabetic eye pathogenesis may lead to its use as a potential modifiable risk factor. Thus, we sought to test the hypothesis that AR expression is a risk factor in the pathogenesis of diabetic cataracts.
Several interesting observations were made in our experiments. First, under the conditions of short-term diabetes used in our studies, elevated sorbitol alone is not enough to cause diabetic cataracts. Sorbitol levels in AR-TG and AR-TG/STZ strains were not significantly different, but only AR-TG/STZ mice developed vacuoles that are characteristic of diabetic cataracts. Thus, it is possible that additional diabetes-induced disturbances such as elevated production of reactive oxygen species and oxidative stress may be secondary factors in diabetic cataract pathogenesis. It is also possible that under conditions of hyperglycemia and insulin deficiency, such as in STZ diabetes, AR-overexpression sensitizes the lens to disturbances in signaling mediated by ERK and JNK pathways. In addition to disrupted regulation of signaling cascades, early hyperglycemic damaged caused by decreases in cellular NAD+/NADH ratios are thought to contribute to mitochondrial damage especially in the early stages of diabetes [45]. Hyperglycemia itself can cause oxidative stress by reducing oxygen to create superoxide anions and hydroxyl radicals. In addition, hyperglycemia may decrease levels of antioxidant enzymatic activity placing cells at increased risk of damage from free radials [46, 47].
A second finding of our study was that elevation of AR alone is not sufficient to induce cataract formation in the absence of diabetes, as cataracts did not develop at an elevated frequency in AR-TG mice compared to nontransgenic controls even though sorbitol levels were significantly elevated in AR-TG lenses with or without STZ diabetes. Mice are thought to be inherently resistant to diabetic cataracts, because they have naturally low levels of AR in their lenses [22]. Our studies confirm previous observations that transgenic mice with lens-directed overexpression of AR develop cataracts only when given streptozotocin or high galactose diets to induce diabetes or galactosemia, respectively [22, 48]. Diabetes is needed to induce cataract formation since it has been shown that 3% of glucose enters the polyol pathway in euglycemia, while 30% of glucose enters the polyol pathway in hyperglycemia [49, 50]. Therefore, increased shuttling of glucose into the polyol pathway increases metabolic imbalances, but also likely leads to other secondary effects involving kinase signaling that can lead to cataract development [34, 51].
It is reasonable to conjecture that increased AR expression leads to increases in pERK most likely through increased production of ROS, which leads to cataract formation. Both high glucose and oxidants can activate ERK and JNK [52]. Mice with constitutively active MEK, and consequent high levels of activated ERK, developed lens opacities with increased hydration and vacuole formation. In addition, lenses from these mice contained elevated glucose levels and GLUT-1 expression [34]. ERK activation may be involved in the formation of diabetic cataracts by upregulating GLUT-1 in the lens, which increases glucose uptake for the polyol pathway [34, 51]. Interestingly, we found that when we induced diabetes in AR-TG mice, the pERK/tERK ratio was significantly decreased. This finding was puzzling since there are several published studies showing that diabetes increases ERK activation [16, 51, 53]. Future studies will explore the possibility that ERK degradation is increased in the setting of increased AR and high glucose. Loss of Insulin-Like Growth Factor (IGF-1) could also be an explanation for low ERK in our STZ model system. The liver produces most of the IGF-1 in the body, but it is also found in the pancreas and other tissues and may be involved in growth, differentiation, and other metabolic processes [52]. Since STZ is toxic to pancreatic beta cells, it is possible that our model results in decreased levels of IGF-1, which is known to activate ERK [52]. Clearly, elevated AR plays a role in the diabetes-induced drop in ERK, since no such decrease was observed in diabetic nontransgenic mice. A similar situation was observed with the effect of AR over-expression on activation of JNK. In the absence of diabetes, AR over-expression led to increases in the ratio of pJNK and total JNK. However, pJNK was significantly reduced when AR was over-expressed in the diabetic lens. Currently the role of JNK in diabetic complications is controversial. Diabetic rats that were treated with α-lipoic acid and γ-linolenic acid, which have antioxidant properties and protect against negative biochemical effects in diabetic rats, showed increased JNK levels in sciatic nerve samples. This observation led to the conclusion that activated JNK may have a role in response to oxidative stress [16]. More recent studies showed that hyperglycemia-induced oxidative stresses activates JNK, leading to apoptosis in human endothelial cells. JNK activation and hyperglycemic-induced apoptosis were diminished respectively by vitamin C [54]. Furthermore, inhibition of AR prevented activation of JNK, p38, and PKC suggesting that AR could have an important role in high glucose mediated cell death [55, 56]. Our finding of reduced pJNK and pERK in AR transgenic lenses point to an added layer of complexity in trying to understand how elevated AR expression under diabetes conditions may disrupt the normal upstream signaling components that feed into the ERK and JNK pathways. Future studies will be necessary to better understand the impact of insulin deficiency and elevated AR activity on MAPK signaling in the lens and other ocular tissues affected by diabetes.
We examined the effects of aldose reductase over-expression in transgenic mice.
Cataract-associated vacuoles were observed in diabetic transgenic mice.
Transgenic mice showed high levels of activated ERK and JNK.
Diabetes caused a reduction in activated ERK and JNK in transgenic mice.
Acknowledgments
The authors gratefully acknowledge support from the University of Colorado School of Medicine Research Track and Vision Scholars Program and NIH grants T35 EY021455 and EY005856.
Footnotes
Conflict of Interest
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.C.f.D. Control, Prevention. National diabetes statistics report: estimates of diabetes and its burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2014. [Google Scholar]
- 2.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes research and clinical practice. 2010;87:4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Tarr JM, Kaul K, Chopra M, Kohner EM, Chibber R. Pathophysiology of diabetic retinopathy. ISRN ophthalmology. 2013;2013:343560. doi: 10.1155/2013/343560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caspersen CJ, Thomas GD, Boseman LA, Beckles GLA, Albright AL. Aging, Diabetes, and the Public Health System in the United States. American Journal of Public Health. 2012;102:1482–1497. doi: 10.2105/AJPH.2011.300616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta A, Styles C. An overview of diabetic eye disease. The British Journal of Diabetes & Vascular Disease. 2010;10:224–230. [Google Scholar]
- 6.Zhang X, Saaddine JB, Chou CF, Cotch MF, Cheng YJ, Geiss LS, Gregg EW, Albright AL, Klein BE, Klein R. Prevalence of diabetic retinopathy in the United States, 2005–2008. JAMA: the journal of the American Medical Association. 2010;304:649–656. doi: 10.1001/jama.2010.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kempen JH, O’Colmain BJ, Leske MC, Haffner SM, Klein R, Moss SE, Taylor HR, Hamman RF. The prevalence of diabetic retinopathy among adults in the United States. Archives of ophthalmology. 2004;122:552–563. doi: 10.1001/archopht.122.4.552. [DOI] [PubMed] [Google Scholar]
- 8.Moemen LA, Mahmoud AM, Mostafa AM, Ghaleb F, Aziz MA, Abdelhamid MA, Farrag MY, Fahmy IA. The Relation Between Advanced Glycation End Products and Cataractogensis in Diabetics. World Journal of Medical Sciences. 2014;10:368–374. [Google Scholar]
- 9.Kim SI, Kim SJ. Prevalence and risk factors for cataracts in persons with type 2 diabetes mellitus. Korean journal of ophthalmology: KJO. 2006;20:201–204. doi: 10.3341/kjo.2006.20.4.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollreisz A, Schmidt-Erfurth U. Diabetic cataract-pathogenesis, epidemiology and treatment. J Ophthalmol. 2010;2010:608751. doi: 10.1155/2010/608751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obrosova IG, Chung SS, Kador PF. Diabetic cataracts: mechanisms and management. Diabetes/metabolism research and reviews. 2010;26:172–180. doi: 10.1002/dmrr.1075. [DOI] [PubMed] [Google Scholar]
- 12.Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacological reviews. 1998;50:21–33. [PubMed] [Google Scholar]
- 13.Lee AYW, Chung SSM. Contributions of polyol pathway to oxidative stress in diabetic cataract. The FASEB Journal. 1999;13:23–30. doi: 10.1096/fasebj.13.1.23. [DOI] [PubMed] [Google Scholar]
- 14.Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Frontiers in pharmacology. 2012;3:87. doi: 10.3389/fphar.2012.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stitt AW. The maillard reaction in eye diseases. Annals of the New York Academy of Sciences. 2005;1043:582–597. doi: 10.1196/annals.1338.066. [DOI] [PubMed] [Google Scholar]
- 16.Tomlinson DR. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia. 1999;42:1271–1281. doi: 10.1007/s001250051439. [DOI] [PubMed] [Google Scholar]
- 17.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 18.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 19.Reddy AB, Tammali R, Mishra R, Srivastava S, Srivastava SK, Ramana KV. Aldose reductase deficiency protects sugar-induced lens opacification in rats. Chemico-biological interactions. 2011;191:346–350. doi: 10.1016/j.cbi.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group, The New England journal of medicine. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 21.Heng LZ, Comyn O, Peto T, Tadros C, Ng E, Sivaprasad S, Hykin PG. Diabetic retinopathy: pathogenesis, clinical grading, management and future developments. Diabetic medicine: a journal of the British Diabetic Association. 2013;30:640–650. doi: 10.1111/dme.12089. [DOI] [PubMed] [Google Scholar]
- 22.Lee AY, Chung SK, Chung SS. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:2780–2784. doi: 10.1073/pnas.92.7.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zablocki GJ, Ruzycki PA, Overturf MA, Palla S, Reddy GB, Petrash JM. Aldose reductase-mediated induction of epithelium-to-mesenchymal transition (EMT) in lens. Chemico-biological interactions. 2011;191:351–356. doi: 10.1016/j.cbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reneker LW, Chen Q, Bloch A, Xie L, Schuster G, Overbeek PA. Chick delta1-crystallin enhancer influences mouse alphaA-crystallin promoter activity in transgenic mice. Invest Ophthalmol Vis Sci. 2004;45:4083–4090. doi: 10.1167/iovs.03-1270. [DOI] [PubMed] [Google Scholar]
- 25.Zablocki GJ, Ruzycki PA, Overturf MA, Palla S, Reddy GB, Petrash JM. Aldose reductase-mediated induction of epithelium-to-mesenchymal transition (EMT) in lens. Chemico-Biological Interactions. 2011;191:351–356. doi: 10.1016/j.cbi.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang SP, Palla S, Ruzycki P, Varma A, Harter T, Reddy GB, Petrash JM. Aldo-keto reductases in the eye. Journal of Ophthalmology. 2010 doi: 10.1155/2010/521204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham ML, Janecek JL, Kittredge JA, Hering BJ, Schuurman HJ. The streptozotocin-induced diabetic nude mouse model: differences between animals from different sources. Comparative medicine. 2011;61:356–360. [PMC free article] [PubMed] [Google Scholar]
- 29.Nakano T, Petrash JM. Kinetic and spectroscopic evidence for active site inhibition of human aldose reductase. Biochemistry. 1996;35:11196–11202. doi: 10.1021/bi9608121. [DOI] [PubMed] [Google Scholar]
- 30.Kawakubo K, Mori A, Sakamoto K, Nakahara T, Ishii K. GP-1447, an inhibitor of aldose reductase, prevents the progression of diabetic cataract in rats. Biological & pharmaceutical bulletin. 2012;35:866–872. doi: 10.1248/bpb.35.866. [DOI] [PubMed] [Google Scholar]
- 31.Kyselova Z, Stefek M, Bauer V. Pharmacological prevention of diabetic cataract. Journal of diabetes and its complications. 2004;18:129–140. doi: 10.1016/S1056-8727(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 32.Burg MB, Kador PF. Sorbitol, osmoregulation, and the complications of diabetes. The Journal of clinical investigation. 1988;81:635–640. doi: 10.1172/JCI113366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodgkinson AD, Sondergaard KL, Yang B, Cross DF, Millward BA, Demaine AG. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int. 2001;60:211–218. doi: 10.1046/j.1523-1755.2001.00788.x. [DOI] [PubMed] [Google Scholar]
- 34.Gong X, Wang X, Han J, Niesman I, Huang Q, Horwitz J. Development of cataractous macrophthalmia in mice expressing an active MEK1 in the lens. Investigative ophthalmology & visual science. 2001;42:539–548. [PubMed] [Google Scholar]
- 35.Long AC, Colitz CM, Bomser JA. Apoptotic and necrotic mechanisms of stress-induced human lens epithelial cell death. Experimental biology and medicine. 2004;229:1072–1080. doi: 10.1177/153537020422901012. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Kim CS, Sohn E, Kim H, Jeong IH, Kim JS. Lens epithelial cell apoptosis initiates diabetic cataractogenesis in the Zucker diabetic fatty rat. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2010;248:811–818. doi: 10.1007/s00417-010-1313-1. [DOI] [PubMed] [Google Scholar]
- 37.Ramana KV, Srivastava SK. Aldose reductase: a novel therapeutic target for inflammatory pathologies. The international journal of biochemistry & cell biology. 2010;42:17–20. doi: 10.1016/j.biocel.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghahary A, Luo JM, Gong YW, Chakrabarti S, Sima AA, Murphy LJ. Increased renal aldose reductase activity, immunoreactivity, and mRNA in streptozocin-induced diabetic rats. Diabetes. 1989;38:1067–1071. doi: 10.2337/diab.38.8.1067. [DOI] [PubMed] [Google Scholar]
- 39.Tang J, Du Y, Petrash JM, Sheibani N, Kern TS. Deletion of aldose reductase from mice inhibits diabetes-induced retinal capillary degeneration and superoxide generation. PloS one. 2013;8:e62081. doi: 10.1371/journal.pone.0062081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee CA, Li G, Patel MD, Petrash JM, Benetz BA, Veenstra A, Amengual J, von Lintig J, Burant CJ, Tang J, Kern TS. Diabetes-induced impairment in visual function in mice: contributions of p38 MAPK, rage, leukocytes, and aldose reductase. Investigative ophthalmology & visual science. 2014;55:2904–2910. doi: 10.1167/iovs.13-11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, Tanabe K, Andres-Hernando A, Orlicky DJ, Finol E, Inaba S, Li N, Rivard CJ, Kosugi T, Sanchez-Lozada LG, Petrash JM, Sautin YY, Ejaz AA, Kitagawa W, Garcia GE, Bonthron DT, Asipu A, Diggle CP, Rodriguez-Iturbe B, Nakagawa T, Johnson RJ. Endogenous Fructose Production and Fructokinase Activation Mediate Renal Injury in Diabetic Nephropathy. Journal of the American Society of Nephrology: JASN. 2014 doi: 10.1681/ASN.2013080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Rivard C, Inaba S, Roncal-Jimenez CA, Bales ES, Diggle CP, Asipu A, Petrash JM, Kosugi T, Maruyama S, Sanchez-Lozada LG, McManaman JL, Bonthron DT, Sautin YY, Johnson RJ. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nature communications. 2013;4:2434. doi: 10.1038/ncomms3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puppala M, Ponder J, Suryanarayana P, Reddy GB, Petrash JM, LaBarbera DV. The Isolation and Characterization of β-Glucogallin as a Novel Aldose Reductase Inhibitor from Emblica officinalis. PloS one. 2012;7:e31399. doi: 10.1371/journal.pone.0031399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Chang KC, Zhou Y, Shieh B, Ponder J, Abraham AD, Ali H, Snow A, Petrash JM, LaBarbera DV. Design of an amide N-glycoside derivative of beta-glucogallin: a stable, potent, and specific inhibitor of aldose reductase. Journal of medicinal chemistry. 2014;57:71–77. doi: 10.1021/jm401311d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teodoro JS, Gomes AP, Varela AT, Duarte FV, Rolo AP, Palmeira CM. Uncovering the beginning of diabetes: the cellular redox status and oxidative stress as starting players in hyperglycemic damage. Molecular and cellular biochemistry. 2013;376:103–110. doi: 10.1007/s11010-012-1555-9. [DOI] [PubMed] [Google Scholar]
- 46.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 47.Kumar P, Rao GN, Pal BB, Pal A. Hyperglycemia-induced oxidative stress induces apoptosis by inhibiting PI3-kinase/Akt and ERK1/2 MAPK mediated signaling pathway causing downregulation of 8-oxoG-DNA glycosylase levels in glial cells. Int J Biochem Cell Biol. 2014;53:302–319. doi: 10.1016/j.biocel.2014.05.038. [DOI] [PubMed] [Google Scholar]
- 48.Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes-induced oxidative stress. Journal of the American Society of Nephrology: JASN. 2003;14:S233–236. doi: 10.1097/01.asn.0000077408.15865.06. [DOI] [PubMed] [Google Scholar]
- 49.Anusha Chauhan GKH, Chidrawar Vijay R, Uma Maheswara Rao V. Polyol Pathway: A Review on the Potential Target for the Prevention of Diabetic Complications International Journal of Inventions in Pharmaceutical. Sciences. 2014;2:696–711. [Google Scholar]
- 50.Cheng HM, Gonzalez RG. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metabolism. 1986;35:10–14. doi: 10.1016/0026-0495(86)90180-0. [DOI] [PubMed] [Google Scholar]
- 51.Zatechka DS, Jr, Kador PF, Garcia-Castineiras S, Lou MF. Diabetes can alter the signal transduction pathways in the lens of rats. Diabetes. 2003;52:1014–1022. doi: 10.2337/diabetes.52.4.1014. [DOI] [PubMed] [Google Scholar]
- 52.George M, Ayuso E, Casellas A, Costa C, Devedjian JC, Bosch F. β cell expression of IGF-I leads to recovery from type 1 diabetes. The Journal of clinical investigation. 2002;109:1153–1163. doi: 10.1172/JCI12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernyhough P, Gallagher A, Averill SA, Priestley JV, Hounsom L, Patel J, Tomlinson DR. Aberrant neurofilament phosphorylation in sensory neurons of rats with diabetic neuropathy. Diabetes. 1999;48:881–889. doi: 10.2337/diabetes.48.4.881. [DOI] [PubMed] [Google Scholar]
- 54.Evans GI, Maddux JL, Grodsky BA, GM Oxidative Stress and Stress-Activated Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes. Endocrine Reviews. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 55.Ramana KV, Friedrich B, Bhatnagar A, Srivastava SK. Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2003;17:315–317. doi: 10.1096/fj.02-0568fje. [DOI] [PubMed] [Google Scholar]
- 56.Zhang P, Xing K, Randazzo J, Blessing K, Lou MF, Kador PF. Osmotic stress, not aldose reductase activity, directly induces growth factors and MAPK signaling changes during sugar cataract formation. Experimental eye research. 2012;101:36–43. doi: 10.1016/j.exer.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]