

Abstract
To examine the impact of common somatic mutations in prostate cancer (PCa) on androgen receptor (AR) signaling, mouse models were designed to perturb sequentially the AR, ETV1, and PTEN pathways. Mice with “humanized” AR (hAR) alleles that modified AR transcriptional strength by varying polyglutamine tract (Q-tract) length were crossed with mice expressing a prostate-specific, AR-responsive ETV1 transgene (ETV1 Tg). While hAR allele did not grossly affect ETV1-induced neoplasia, ETV1 strongly antagonized global AR regulation and repressed critical androgen-induced differentiation and tumor suppressor genes, such as Nkx3-1 and Hoxb13. When Pten was varied to determine its impact on disease progression, mice lacking one Pten allele (Pten +/−) developed more frequent prostatic intraepithelial neoplasia (PIN). Yet, only those with the ETV1 transgene progressed to invasive adenocarcinoma. Furthermore, progression was more frequent with the short Q-tract (stronger) AR, suggesting that the AR, ETV1, and PTEN pathways cooperate in aggressive disease. On the Pten +/− background, ETV1 had markedly less effect on AR target genes. However, a strong inflammatory gene expression signature, notably upregulation of Cxcl16, was induced by ETV1. Comparison of mouse and human patient data stratified by the presence of E26 transformation-specific ETS fusion genes highlighted additional factors, some not previously associated with prostate cancer but for which targeted therapies are in development for other diseases. In sum, concerted use of these mouse models illuminates the complex interplay of AR, ETV1, and PTEN pathways in pre-cancerous neoplasia and early tumorigenesis, disease stages difficult to analyze in man.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-014-0215-9) contains supplementary material, which is available to authorized users.
Keywords: Androgen Receptor, Prostatic Intraepithelial Neoplasia, PTEN Loss, Androgen Receptor Signaling, Tramp Mouse
Introduction
Signaling through the androgen receptor (AR), a hormone-activated transcription factor in the nuclear receptor superfamily, is critical for normal prostate development and prostate cancer (PCa) progression [2]. Androgen ablation therapy initially abrogates AR signaling and induces tumor regression, but tumors invariably recur despite ongoing therapy and with continued AR expression and signaling [63]. Even with recently developed therapies, disease progressing despite castrate levels of serum testosterone (termed castration-resistant prostate cancer [CRPC]) remains incurable [40]. A more thorough understanding of the interaction between AR and additional oncogenic pathways commonly dysregulated in PCa will be critical in identifying novel targets for therapeutic intervention.
Well-known genetic variation in the AR protein occurs in the length of a polyglutamine tract (Q-tract, CAG repeat) in the N-terminal domain. As shown by us and others, Q-tract length is inversely proportional to AR transcriptional strength in vitro and in vivo, as seen molecularly in differential target gene expression and physiologically in seminal vesicle weight [4, 17, 65]. Q-tract length alone has not proven to be associated with PCa risk in humans but in combination with alleles of the androgen synthesis genes CYP17 and SRD5A2, the stronger AR increases PCa risk, supporting the notion that the androgen axis overall influences disease [44, 45]. We have previously shown that altering AR strength via Q-tract length variation on a homogenous genetic background modifies PCa onset and progression in an aggressive transgenic mouse model [3]. Interestingly, AR genetic variation has recently been reported to be associated with the frequency of recurrent gene fusions in PCa (see below) [9, 82]. This led us to examine the in vivo effect of AR variation on PCa driven by alterations commonly seen in human tumors.
Recurrent genomic rearrangements that fuse 5′ regulatory elements of androgen-responsive genes to the coding region of E26 transformation-specific (ETS) transcription factor genes occur in approximately half of human prostate tumors [74, 76]. These fusions drive robust androgen-induced prostatic expression of ETS factors otherwise not expressed in the prostate. Fusions involving the ERG and ETV1 genes occur in approximately 50 and 5–10 % of PCa, respectively [74–76]. ERG fusion events themselves are AR-mediated [9], and prostate tumors are more likely to be ERG+ in patients harboring a short Q-tract (strong) AR allele [9, 82], further suggesting cooperation between ETS and AR pathways. These fusions occur early in PCa ontogeny, and individual cancer foci or metastases are generally clonal for a given rearrangement, suggesting selection [48, 70]. Prostatic overexpression of ETS factors induces PIN in some mouse models [8, 16, 39, 64, 74], and ETS factors promote invasion and alter gene expression in vitro [8, 29, 74, 85]. However, it is unknown how modulation of the androgen axis, such as by AR genetic variation, alters the effect of ETS overexpression on tumorigenesis.
Perturbation of AR and ETS pathways alone does not lead to PCa in mice but additional genetic events cooperate in tumorigenesis. The tumor suppressor PTEN is frequently deleted or inactivated in a variety of human cancers, including PCa [73]. PTEN is a phosphatase that inactivates the PI3K target PIP3, in turn preventing phosphorylation of AKT and activation of downstream proliferation and survival pathways [69]. Furthermore, the AKT and AR signaling pathways can repress one another in a reciprocal manner [15, 50]. In PCa, PTEN loss is associated with poor prognosis and progression to CRPC [12, 14]. In mice, Pten reduction causes PIN in a dose-dependent manner, while prostate-targeted Pten homozygous deletion leads to adenocarcinoma [5, 16, 43, 77]. Several additional genetic hits can cooperate with Pten reduction to promote PCa, such as further tumor suppressor loss (as of Nkx3-1) [1] or oncogene activation (as of ERG) [16, 39]. ETV1 overexpression cooperates with prostate-specific Pten deletion to drive aggressive adenocarcinoma [8]. However, the interaction of ETV1 overexpression and Pten reduction, which is more common in patients than homozygous loss [81], is not fully understood, with recently reported results [8] differing from the findings described here. Furthermore, the extent to which altered AR signaling influences disease progression in these contexts has not been addressed.
In the current study, we used gene expression profiling to determine how varying the strength of the androgen axis would alter prostate pathology initiated by ETV1 overexpression and/or PTEN reduction in mice. A marked antagonism of the normal AR-regulated transcriptome in ETV1-transgenic prostates was largely abrogated on a Pten-hemizygous background. With reduced PTEN, ETV1 overexpression induced disease progression, as well as a pro-inflammatory gene signature, both of which were impacted by AR strength. Integration of mouse and patient data revealed potential ETV1 regulation of known and novel PCa-associated genes, highlighting novel targets for therapeutic intervention.
Methods
Mice
All animal work was performed in accordance with protocols approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan. ETV1-transgenic FVB mice expressing the human ETV1 coding region driven by the ARR2-Pb promoter (ETV1 Tg) were a gift from Dr. Arul Chinnaiyan (University of Michigan) [74]. In the first experimental cohort, ETV1 Tg males were crossed with C57BL/6 females expressing a “humanized” androgen receptor (hAR) [4] with a short (12Q) or long (48Q) polyglutamine tract. Transgenic males (hAR;ETV1 Tg) were castrated at 12 weeks or left intact, and aged to 24 weeks. Intact, non-transgenic hAR mice were used as controls. For the second series, Pten flox/flox mice [43], on an FVB background, a gift from Dr. William Muller (McGill University), were crossed with FVB/N-Tg(EIIa-cre)C5379Lmgd/J mice (Jackson Labs # 003314) to excise the floxed Pten. Pten-deleted offspring were maintained as hemizygotes (Pten +/−). hAR;ETV1 Tg mice carrying 12Q, 21Q, or 48Q hAR alleles were backcrossed for at least five generations onto the FVB background and then female transgenics crossed with Pten +/− males. Resulting hAR;ETV1 Tg ;Pten +/− and hAR;Pten +/− males were either castrated at 12 weeks or left intact, and aged to a median of 41 weeks (range 20–83 weeks). Genotyping primers are listed in Online Resource 1.
Microdissection and Sample Preparation
Anterior prostate (AP), dorsolateral prostate (DLP), and ventral prostate (VP) lobes were individually microdissected from experimental mice. For the hAR; ETV1 Tg mice, lobes were frozen in optimal cutting temperature (OCT) compound (Sakura). Five-micrometer sections were cut and stained with hematoxylin and eosin (H&E) for histology, and RNA was extracted from the remaining tissue (see below). For hAR;ETV1 Tg ;Pten +/− mice, one lobe per pair was fixed in 10 % neutral-buffered formalin for 24 h, then transferred to 70 % ethanol until embedding in paraffin and cutting of 5-μm sections for H&E staining. The other lobe was frozen in RNA-later (Ambion). All prostate RNA was purified with the RNeasy kit with on-column DNase digestion (QIAGEN) following tissue homogenization in buffer RLT. Macroscopic prostate tumors were divided in half, with one half prepared for histology and the other half prepared for RNA analysis as described above.
qRT-PCR
Complementary DNA (cDNA) was reverse-transcribed from total RNA using random hexamers and either the SuperScript II 1st-Strand Synthesis kit (Invitrogen) for mouse prostate RNA or the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) for human cell line RNA. qRT-PCR samples were run on an AB7300 or AB7500 thermal cycler (Applied Biosystems) using Power SYBR Green Master Mix (Applied Biosystems) or ABSolute Blue SYBR Green Rox Master Mix (Thermo Scientific) according to the manufacturer’s instructions. Following normalization to Actb for mice, and ACTB or GAPDH for human cells, relative expression was calculated using the 2-ΔΔCt method. All primer sequences are listed in Online Resource 1.
RNA-seq Analysis
Mouse DLP RNA was used for Illumina mRNA-seq library construction. For each genotype/treatment group (n = 4–13 mice per group), individual RNA samples were divided among three unique pools (two unique pools for castrated groups due to low RNA yield), and a library was prepared from each pool. Individual libraries were prepared for each macroscopic DLP tumor as well (n = 2). For each library, poly-A+ RNA was first purified from 200 ng total RNA with Dynabeads Oligo(dT)25 (Invitrogen). Libraries were constructed with NEBNext mRNA-seq Master Mix reagents and NEBNext Multiplex Oligos for Illumina (New England Biolabs). Multiplexed libraries were sequenced on an Illumina Hi-Seq 2000 with the SE-50 module at the University of Michigan Sequencing Core. Four lanes were sequenced, each containing 11–12 indexed libraries. Reads were aligned to the mouse mm10 genome with TopHat2 [38], transcript counts were generated with the HT-Seq Python script (http://www-huber.embl.de/users/anders/HTSeq/doc/index.html), and differential expression was calculated in the Bioconductor package edgeR, which utilizes counts rather than FPKM [6]. Contrasts were performed between pairs of genotype/treatment groups, or between multiple genotype/treatment groups using the generalized linear model (GLM) tools in edgeR. Genes differentially expressed at a false-discovery rate (FDR) ≤ 0.05 were considered significant.
Bioinformatics Functional Annotation and Pathway Analysis
Functional annotation of significant gene sets was performed with Database for Annotation, Visualization, and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov) [20]. The following gene ontology (GO) terms and pathways were tested for enrichment: GOTERM_BP_ALL, GOTERM_CC_ALL, GOTERM_MF_ALL, PANTHER_BP, PANTHER_MF, BIOCARTA, KEGG_PATHWAY, and PANTHER_PATHWAY. GO terms and pathways with a Benjamini-adjusted FDR ≤ 0.05 were considered significantly enriched relative to the Mus musculus background gene list.
Enrichment with human cancer-related gene sets was performed with Oncomine (https://www.oncomine.org) [59]. Mouse gene IDs from significant gene sets were converted to unique human gene symbols using the HCOP tool at www.genenames.org, and uploaded to Oncomine as “custom concepts.” For gene sets with more than 1000 genes, the top 1000 were used for custom concepts. Each custom concept was queried against the “Biology Concepts,” “My Concepts,” “Literature-defined Concepts,” and “Oncomine Gene Expression Signatures” concept databases in Oncomine with “All Entrez Genes” as the background list. Significant enrichment was set as an odds ratio (OR) ≥ 4 and a p value ≤10−6. Oncomine output was used to generate molecular concept maps [58] in Cytoscape (www.cytoscape.org), with edges representing significant enrichment between concepts and node size proportional to overlap with the primary custom concept.
Pathological Analysis of Mouse Prostates
Mouse prostate lesions were classified and graded based on the recent consensus criteria established by the Mouse Models of Human Cancer prostate pathology committee [31, 55].
Immunohistochemistry
Slides were prepared with 5-μM sections from FFPE mouse prostates. The antibodies used in this study included AR N-20 (Santa Cruz # sc-816, 1:500 dilution), phospho-AKT (Ser473) (Cell Signaling #9271, 1:100 dilution), and PTEN (Cell Signaling #9188, 1:80 dilution). Immunohistochemistry (IHC) was performed essentially as described for AR and pAKT [3], and for PTEN [13, 61].
In situ Hybridization
A custom RNA probe against human ETV1 transcript (Advanced Cell Diagnostics) was hybridized to 5 μM FFPE mouse prostate sections essentially as described [41].
Cell Lines
RWPE-1 cells stably overexpressing ETV1 or LACZ [74, 75] were a gift from Dr. Arul Chinnaiyan (University of Michigan) and were maintained in KSFM (Gibco # 10724–011) supplemented with BPE and EGF, 1:100 penicillin/streptomycin (Gibco # 15140–122), and 3 μg/ml blasticidin (Life Technologies # R21001) to maintain stable expression of the transduced ETS construct or control. 293T cells were a gift from Dr. Margaret Gnegy (University of Michigan) and were maintained in DMEM (Gibco # 11995–065) with 10 % FBS (GeneMate # S-1200-500) and 1:100 penicillin/streptomycin.
Lentiviral Packaging and Transduction
The FG9 lentiviral expression plasmid was previously derived from FUGW (Addgene # 14883) [46, 57] and was a gift from Dr. David Baltimore (California Institute of Technology). FG9 was linearized with BamHI, and a BglII-3xFLAG-BamHI fragment was inserted, destroying the 5′ BamHI site but preserving the 3′ site. AR was excised as a BamHI fragment from the pCMV5-AR plasmid [68] and ligated into FG9-3xFLAG to produce the final FG9-3xFLAG-AR plasmid. For transduction, 5 × 106 293T cells were seeded in a poly-l-lysine-coated 10-cm dish. The following day, cells were transfected with 10 μg FG9-3xFLAG-AR plasmid or FG9-vector control, 3 μg pHCMVG, 2 μg pRSV-rev, 6 μg pRRE, and 80 μg polyethylenimine, and suspended in 150 mM NaCl. Virus-containing medium was harvested 48 and 72-h post-transfection, filtered through a 0.45 μM filter, and applied directly to RWPE-ETV1 or RWPE-LACZ cells. After transduction of the AR construct, 100 μg/ml Hygromycin B (Life Technologies #10687-010) was added to maintain expression of AR or the vector control.
Androgen Stimulation and RNA Purification
5 × 104 RWPE-ETV1-AR or RWPE-LACZ-AR cells were seeded in 12-well dishes in complete growth medium without antibiotic selection. Beginning the following day, cells were deprived of BPE for 48 h. Cells were then stimulated with 1 nM R1881 or methanol vehicle for 24 h before lysis and RNA purification with TRIzol reagent (Ambion # 15596018). The experiment was performed in biological triplicate.
Results
Effect of AR Allele Strength on ETV1-Induced PIN
To test the role of AR in PCa initiation, we examined the effect of variation in AR transcriptional strength on ETV1-induced neoplasia. “Humanized” AR (hAR) mice were previously engineered by germline knockin to express an AR protein essentially identical to human, including either a short (hAR 12Q), average (hAR 21Q), or long (hAR 48Q) polyglutamine tract (Q-tract) in the N-terminal domain, modeling the extremes of human variation [4]. As the mouse Ar lacks a contiguous Q-tract, which is known to affect AR activity, the hAR mice more accurately reflect human AR variation and biology. Q-tract length inversely correlates with AR transcriptional activity in vitro, seen as altered prostatic gene expression and endpoints of androgen action such as seminal vesicle weight [4, 17, 65]. Previously, we showed that AR Q-tract length modifies PCa onset, progression, and treatment response in the aggressive TRAMP mouse model [3]. Thus, variable AR transcriptional activity has the potential to modulate the effect of additional oncogenic events and alter the course of disease.
To test the interaction of AR with an oncogene activated in human PCa, hAR 12Q and hAR 48Q mice were crossed with ETV1 transgenic (ETV1 Tg) mice, in which a probasin-derived promoter drives androgen-responsive prostatic expression of human ETV1 (Fig. 1a) [74]. hAR;ETV1 Tg male offspring were castrated at 12 weeks (to ablate androgen signaling) or left intact, and prostate lobes were microdissected at 24 weeks and compared to those of intact non-transgenic hAR controls. Unlike the alobular human prostate, the mouse gland is comprised of three paired dorsolateral, ventral, and anterior prostate lobes, referred to as DLP, VP, and AP, respectively. Morphologically, there is not a particular mouse prostate lobe that is most similar to the human prostate [62].
Fig. 1.
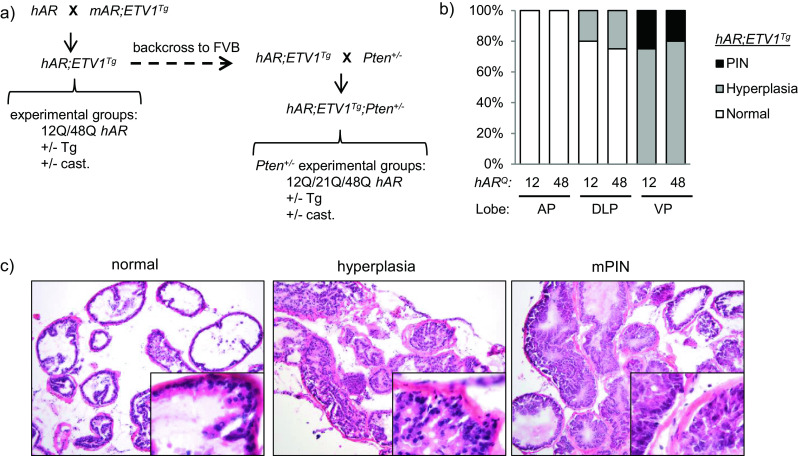
Effect of AR allele strength on ETV1-induced PIN. a C57BL/6 mice carrying a “humanized” AR gene with a short (hAR 12Q) or long (hAR 48Q) polyglutamine tract were crossed with ETV1-transgenic FVB mice (ETV1 Tg) to generate hAR;ETV1 Tg mice. Experimental mice were castrated or left intact at 12 weeks, aged to 24 weeks and compared to intact hAR controls. hAR;ETV1 Tg mice were then backcrossed to FVB and crossed with Pten +/− mice to generate hAR;ETV1 Tg ;Pten +/− experimental mice with short (12Q), median (21Q) or long (48Q) Q-tracts. Experimental hAR;ETV1 Tg ;Pten +/− mice were castrated or left intact at 12 weeks, aged to an average of 43 weeks and compared to hAR;Pten +/− controls. b Representative H&E stained prostates from hAR;ETV1 Tg mice. Normal tissue (DLP), hyperplasia (DLP), and mPIN (VP) are shown. Images are ×10 with ×40 insets. c The proportion of mice with hyperplasia and mPIN in AP, DLP, and VP among hAR 12Q ;ETV1 Tg and hAR 48Q ;ETV1 Tg mice. Analysis was of 5-8 lobes per group
Histopathological analysis (n = 5–8 lobes per group) revealed PIN and substantial hyperplasia in prostates of intact hAR;ETV1 Tg mice (Fig. 1b). Prostates of intact non-transgenic hAR controls were either normal or showed focal hyperplasia, and castrated hAR;ETV1 Tg mice had atrophic glands as expected (data not shown). In intact mice, 25 % of hAR 12Q;ETV1 Tg and 20 % of hAR 48Q ;ETV1 Tg animals developed PIN by 24 weeks in the VP (Fig. 1b). All of the remaining intact transgenic mice showed hyperplasia in either the DLP or VP (Fig. 1b). Representative examples of each stage are shown in Fig. 1c. These results are consistent with previous reports in which mice overexpressing ETV1 in the prostate via transgene or knockin developed PIN but did not progress to adenocarcinoma [8, 64, 74]. Here, we show that the incidence of PIN was not affected substantially by AR Q-tract length.
AR Regulation Is Antagonized by Overexpression of ETV1
Despite driving only early neoplasia, the ETV1 transgene had a pronounced effect on prostatic gene expression. The DLP was chosen for analysis because the expression pattern in mouse DLP is most similar to that of the human prostate peripheral zone [11], where most human PCa arises. Additionally, as shown below in Pten-hemizygous mice, the DLP harbored substantially more PIN and adenocarcinoma than other lobes (Online Resource 4). In the previously reported hAR TRAMP mice, PIN is most apparent in the DLP as well [3].
qRT-PCR analysis showed that the androgen-induced genes, namely Tmprss2, Pbsn, and Nkx3-1, were downregulated in prostates of intact hAR;ETV1 Tg mice, similar to the expected decrease in castrated controls (Fig. 2a). Conversely, Clu, which is normally repressed by AR and thus upregulated following androgen ablation, was upregulated in prostates of intact ETV1 Tg mice as well as in castrated mice (Fig. 2a). Nkx3-1 is a well-established tumor suppressor gene in the prostate and a marker of luminal epithelial cell differentiation [78]. Nkx3-1 null mice develop PIN, and Nkx3-1 is frequently downregulated in human PCa [37]. Its downregulation in hAR;ETV1 Tg mice here suggested a role in ETV1-mediated oncogenesis.
Fig. 2.
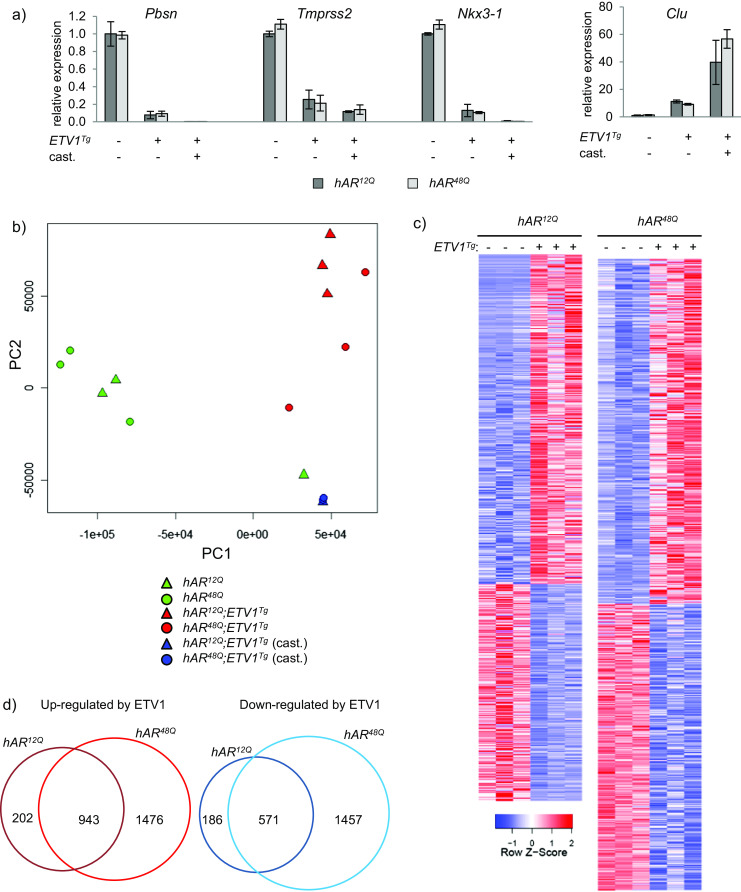
ETV1 overexpression alters prostatic gene expression. a Two unique pools of DLP RNA from each group were analyzed by qRT-PCR. Expression of the AR target genes Pbsn, Tmprss2, Nkx3-1, and Clu was normalized to Actb expression and plotted relative to the intact hAR 12Q group using the 2-ddCt method. Mean +/− SEM are plotted. b Principal component analysis was performed for all genes among the 16 individual RNA-seq libraries. hAR 12Q and hAR 48Q mice are represented as triangles and circles, respectively. Green, red, and blue represent intact non-transgenic, intact transgenic, and castrated transgenic mice, respectively. c RNA-seq analysis was performed on DLP RNA. Samples from intact groups were divided into three unique RNA pools for library construction (two unique pools for castrated groups). Heatmaps include differentially expressed genes between the groups shown (FDR ≤ 0.05). Individual libraries are shown for differentially expressed genes among intact hAR 12Q ;ETV1 Tg vs. hAR 12Q (1145 up, 757 down) or intact hAR 48Q ;ETV1 Tg vs. hAR 48Q (2419 up, 2028 down). Red and blue represent high or low expression, respectively, relative to the mean expression level in the samples shown. Columns and rows represent samples and genes, respectively. d Overlap between genes differentially expressed in hAR 12Q ;ETV1 Tg and hAR 48Q ;ETV1 Tg is illustrated by Venn diagram
Examination of global gene expression differences by RNA-seq revealed striking and widespread molecular changes in the prostates of hAR;ETV1 Tg relative to hAR mice. DLP RNA samples were divided into three unique pools per group (two pools for castrate groups), and a library was prepared for each pool. Along with samples from the Pten +/− mice (see below), indexed libraries were divided across four lanes on the Illumina Hi-Seq 2000 for an average of 11.94 million reads per library. Furthermore, 95.8 % of bases had quality scores ≥Q30. Principal component analysis of all genes in all libraries was performed to gauge overall expression differences between groups. Three distinct clusters were observed, with samples segregated by ETV1 transgene and castration, while AR genotype had a substantially smaller effect (hAR 12Q and hAR 48Q samples were intermixed), as shown in Fig. 2b.
Differential expression was performed between pairs of groups, or between multiple groups using a generalized linear model (GLM). Genes with a false-discovery rate (FDR) of 0.05 or less were considered significant. When the hAR 12Q and hAR 48Q mice were analyzed separately, 1145 and 2419 genes were upregulated and 757 and 2028 genes were downregulated, respectively, in transgenics vs. non-transgenics (Fig. 2a, c). The majority of genes, 943 upregulated (82 %) and 571 downregulated (75 %), were shared among the hAR 12Q and hAR 48Q mice (Fig. 2d), consistent with the principal component plot. However, a greater number of genes were significantly altered in the hAR 48Q mice. This may be because the weaker hAR48Q is more susceptible to perturbation by ETV1 overexpression, resulting in greater differential expression in transgenic mice.
Using the GLM tools to combine hAR 12Q and hAR 48Q mice for differential expression, 2368 genes were upregulated and 2196 downregulated in intact hAR;ETV1 Tg relative to hAR mice (Fig. 3a). A comparable number of genes were differentially expressed among the castrated mice relative to intact, serving to highlight androgen-regulated genes (whether direct or indirect targets) by the effects of androgen ablation (data not shown). Such extensive differential expression in castrated mice was expected, but the magnitude of the effect in intact ETV1 transgenics was striking. The differential expression patterns between transgenic and non-transgenic DLP revealed that the intact hAR;ETV1 Tg mice displayed a pattern similar to that of castrated mice, with many expression changes in the same direction (Fig. 3a). Along with the initial qRT-PCR data, this suggested that ETV1 may inhibit normal AR function in a manner akin to androgen ablation. However, some ETV1-induced changes were “reverted” by castration (middle portion of Fig. 3a), suggesting that ETV1 also controls a number of non-AR target genes.
Fig. 3.
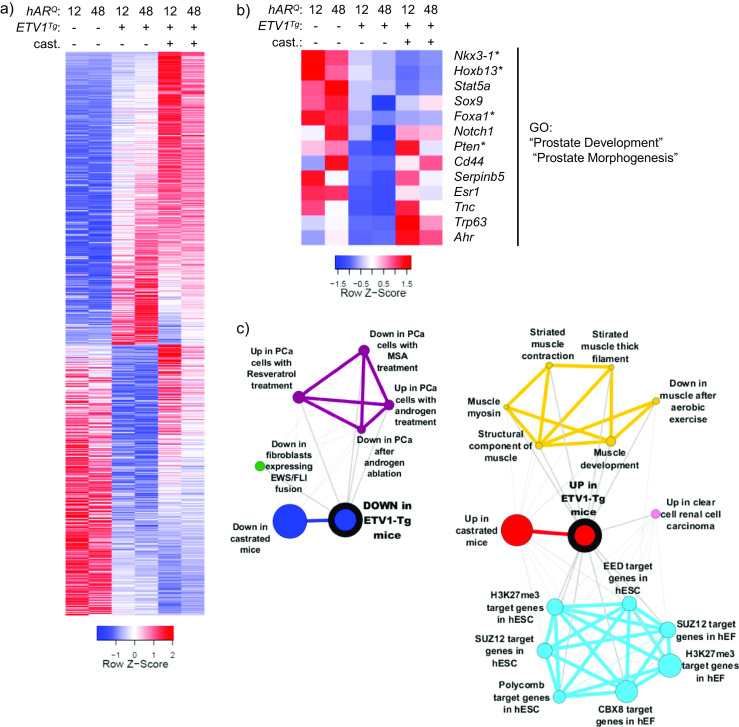
AR regulation is antagonized by ETV1 overexpression. a The generalized linear model (GLM) tools in edgeR were used to test differential expression between all hAR;ETV1 Tg and all hAR mice. In prostates of hAR;ETV1 Tg mice, 2368 genes were upregulated and 2196 downregulated (FDR<0.05) and are shown in the heat map Relative expression in prostates of castrated mice is included. The heatmap is plotted as in Fig. 2a, except that each column represents the average expression among biological replicate libraries. b Genes significantly upregulated or downregulated in prostates of hAR;ETV1 Tg relative to hAR mice were uploaded to DAVID for functional annotation with gene ontology (GO) terms. The heatmap shows genes annotated with select significantly enriched GO terms (Benjamini FDR ≤ 0.05) among the downregulated genes. See Online Resource 2 for the complete list of significant GO terms. c The top 1000 upregulated and downregulated genes in prostates of hAR;ETV1 Tg relative to hAR mice were converted to human gene symbols, uploaded to Oncomine as “custom concepts” and queried against the Oncomine concept database. Significantly enriched concepts, defined as having an odds ratio (OR) ≥ 4 and p ≤ 10−6, are shown as molecular concept maps. Node size is proportional to overlap with the primary concept. Similar concepts have the same color and are clustered together. See Online Resource 3 for a complete list of significant concepts
To determine which pathways were altered in hAR;ETV1 Tg DLP, functional annotation of differentially expressed genes was performed with DAVID [20] (http://david.abcc.ncifcrf.gov). “Prostate development” and “prostate morphogenesis” were among the gene ontology (GO) terms significantly enriched among ETV1-downregulated genes in intact mice. Relative expression of genes annotated with these GO terms is shown in Fig. 3b, and a complete list of enriched GO terms is in Online Resource 2. Importantly, this gene list includes the tumor suppressors Nkx3-1 and Pten. Furthermore, the AR pioneer factor Foxa1 [23, 47] was downregulated in hAR;ETV1 Tg mice. As downregulation of Pten or Foxa1 would be predicted to result in diminished AR transcriptional activity [15, 50], the expression pattern in hAR;ETV1 Tg mice suggests a possible upstream mechanism by which ETV1 overexpression could antagonize AR.
To examine the significance of these DLP expression differences in the context of human cancer, differentially expressed mouse gene sets were converted to human official gene symbols and uploaded to Oncomine [59]. The Oncomine database contains thousands of “molecular concepts,” which are gene sets annotated as being overexpressed in a certain cancer subtype, induced by a certain drug treatment, or some other biologically meaningful annotation. Mouse gene IDs from significant gene sets were converted to unique human gene symbols using the HCOP tool at www.genenames.org, and then uploaded to Oncomine as “custom concepts.” Each custom concept was queried against the public Oncomine concept database and our custom concepts. Notably, genes downregulated in hAR;ETV1 Tg mice relative to hAR mice were significantly associated with the following concepts: “upregulated genes in prostate cancer cells in response to synthetic androgen R1881” [21], “upregulated genes (time dependent) in prostate cancer cells in response to androgen” [33], “downregulated genes in prostate cancer after androgen ablation therapy” [30], and custom concepts containing genes downregulated in the castrated mice (Fig. 3b). Similarly, genes upregulated in hAR;ETV1 Tg mice relative to hAR mice were significantly associated with the concept: “upregulated genes in prostate cancer after androgen ablation therapy” [30] as well as custom concepts containing genes upregulated in the castrated mice (Fig. 3b). A complete list of significantly associated concepts can be found in Online Resource 3. Thus, a significant subset of androgen-responsive genes, including those altered in human PCa patients, are affected by the ETV1 transgene in these mice. Furthermore, a large number of muscle-associated genes were upregulated in the prostates of hAR;ETV1 Tg mice (Fig. 3b). This could reflect stromal smooth muscle cell gene expression that is affected in a paracrine manner by ETV1 activity in the epithelial compartment, disrupting normal pro-differentiation signals [66]. Finally, ETV1-upregulated genes were significantly enriched for targets of the polycomb repressive complex (PRC) (Fig. 3c). Co-regulation by the PRC, AR, and the ETS factor ERG has been reported previously; however, ERG repressed those shared targets in human PCa cells [85]. These results could indicate divergent functions of ETV1 and ERG, their differential action at early vs. late disease stages or discrepancies between in vitro and in vivo results.
In sum, ETV1 overexpression in mouse prostate antagonized AR transcriptional activity and disrupted the normal prostate expression program. This included the repression of important prostate tumor suppressors, differentiation genes, and AR coregulators as well as derepression of polycomb target genes.
Pten Reduction and ETV1 Overexpression Cooperate to Promote PCa Progression
ETV1 overexpression was previously shown to exacerbate PCa progression in mice with total knockout of Pten in the prostate [8]. Because many human prostate tumors show deletion of only one allele or a partial decrease in expression of PTEN, we asked whether reduction of this key tumor suppressor would cooperate with ETV1 overexpression in oncogenesis. Furthermore, we queried whether varying AR transcriptional strength would impact disease progression. ETV1 transgenic mice carrying short, median, or long Q-tract AR alleles were backcrossed at least five generations onto the FVB background, and then crossed with FVB mice bearing germline deletion of one Pten allele (Pten +/−) for optimal genetic homogeneity (Fig. 1a). Cohorts of each genotype were castrated at 12 weeks or left intact, and aged until moribund or until PTEN-dependent disease (generally lymphoma [77]) necessitated euthanization (median age of 41 weeks, range 20–83 weeks).
Pten +/− mice developed more frequent PIN than the Pten +/+ mice, and the overall rate was not notably affected by ETV1 transgene or AR allele (Fig. 4, compare to Fig. 1b). The majority of intact mice developed PIN, as did a subset of castrates. Of note, the hAR 21Q mice developed slightly less PIN overall, regardless of the ETV1 transgene, and showed no PIN in the absence of androgen. PIN was graded from PIN2 to PIN4, with PIN4 being the most severe [31, 55]. Among intact mice, progression to adenocarcinoma occurred exclusively in ETV1 transgenics (Fig. 4). Macroscopic DLP tumors were identified upon dissection in one hAR 12Q and one hAR 21Q mouse. Histopathological analysis detected adenocarcinoma in the VP of one additional transgenic hAR 12Q mouse (Online Resources 1 and 6). Intriguingly, adenocarcinoma was observed in two castrated hAR 12Q mice (one with and one without the ETV1 transgene) (Fig. 4). Castration-resistant disease has been observed in TRAMP mice [3] and in prostate-specific Pten knockout mice [50], but not in Pten heterozygous animals. These data suggest that even a partial reduction in PTEN may be sufficient to promote CRPC.
Fig. 4.

ETV1 overexpression with Pten reduction promotes progression to adenocarcinoma. H&E stained DLP and VP sections were scored by the pathologist as normal, mPIN2-4 or adenocarcinoma (ADC). The graph represents the proportion of mice per group (n = 5–14) at each disease stage, with the most severely diseased prostate lobe per mouse used to plot the data. See Online Resource 4 for stratification by age and individual prostate lobe
The DLP had markedly greater frequency and severity of neoplasia than the VP among intact mice, while among castrated mice, the rates of PIN were lower overall but similar between lobes (Online Resource 4). There was a trend toward more frequent and severe disease in intact mice older than the median age of 41 weeks, while among castrates, there was a greater trend for disease incidence in mice 41 weeks or younger (Online Resource 4). While it was previously reported that ETV1 promoted PCa progression in Pten-null mouse prostates [8], these results show that ETV1 overexpression cooperated with even partial PTEN loss (see below) to promote aggressive PCa. Additionally, these results indicated that even heterozygous PTEN loss promoted castration-resistant neoplasia and adenocarcinoma. Furthermore, the stronger AR carried by hAR 12Q mice promoted greater PCa progression despite similar overall rates of PIN.
Immunohistochemical (IHC) staining for AR protein revealed nuclear expression in prostate epithelial cells in normal regions as well as in PIN and adenocarcinoma foci, similar to human disease where AR is present throughout PCa progression (Fig. 5). Analysis of ETV1 expression by RNA in situ hybridization (RNA-ISH), performed by a pathologist blinded to genotype, confirmed ETV1 expression in prostate epithelial cells of hAR;ETV1 Tg ;Pten +/− mice and undetectable ETV1 expression in non-transgenics (Fig. 5, Online Resource 5). ETV1 expression varied somewhat between individual transgenic animals, with a trend for higher expression in mice with more advanced PIN or adenocarcinoma. This could be because high ETV1 expression promotes more aggressive disease, or alternatively because AR signaling is partially increased in neoplastic cells, resulting in increased expression of the AR-driven ETV1 transgene. Interestingly, there was substantial heterogeneity between the two DLP tumors, with the hAR 12Q tumor (Fig. 5, row 4) appearing less differentiated than the hAR 21Q tumor (Fig. 5, row 5). Thus, in this small sample, adenocarcinoma tended to be more frequent and aggressive in the hAR 12Q mice.
Fig. 5.
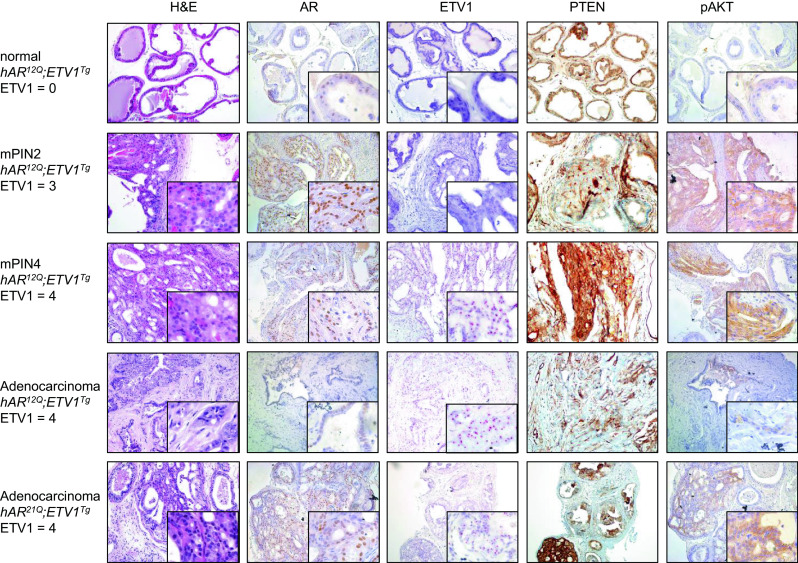
Histopathology in Pten-hemizygous mice. Representative prostate sections are shown from, in order, intact hAR 12Q ;ETV1 Tg ;Pten +/− mice with normal, PIN2 or PIN4 DLP, as well as macroscopic DLP tumors (adenocarcinoma) from hAR 12Q ;ETV1 Tg ;Pten +/− and hAR 21Q ;ETV1 Tg ;Pten +/− mice. Sections were stained with H&E. Immunohistochemical (IHC) staining was performed with antibodies to AR, PTEN, or pAKT protein (brown staining). In situ hybridization (ISH) was performed with a probe against the human ETV1 transcript (red staining). ETV1 expression level is scored as 0–4, with 4 being highest. Images are shown at ×10–20 magnification with ×60 inset. Consecutive sections were used when possible, and each row of images shows stained sections from a single mouse prostate
Despite the germline loss of one Pten allele and an approximately 50 % reduction in Pten mRNA (see below), robust PTEN protein expression was detected in all samples tested. However, locally reduced PTEN expression was seen in a subset of PIN and adenocarcinoma foci (Fig. 5). PTEN normally dephosphorylates and inactivates PI3K, in turn preventing phosphorylation and activation of AKT and thus inhibiting AKT-mediated cell growth and proliferation [67]. Despite maintenance of PTEN protein expression even in neoplastic foci, phospho-AKT (pAKT) protein was present exclusively in regions of PIN or adenocarcinoma (Fig. 5). Greater pAKT staining correlated with more advanced disease, as expected. Therefore, in this model, complete PTEN loss is not necessary for AKT activation.
Reduction of Pten Leads to Abrogated Effect of ETV1 on AR
We next examined the effect of ETV1 on gene expression in this model of PCa progression. qRT-PCR analysis of AR target genes Tmprss2, Pbsn, and Nkx3-1 revealed markedly attenuated ETV1 influence in prostates of Pten +/− relative to Pten +/+ mice (Fig. 6a, note that expression in Pten +/+ mice is shown in lanes 1–3 of each gene for reference). However, overall repression of AR target genes was observed when one Pten allele was deleted, and those targets were still strongly repressed in castrated Pten +/− mice (Fig. 6a). Prostatic Pten expression was indeed about half on the Pten +/− background, and was repressed by ETV1 (or castration) in prostates of Pten +/+ but not Pten +/− mice (Fig. 6b). This observation revealed that even a partial reduction in prostatic PTEN inhibited AR, extending a previous finding that total PTEN loss in the prostate leads to reduced AR activity [15, 50].
Fig. 6.
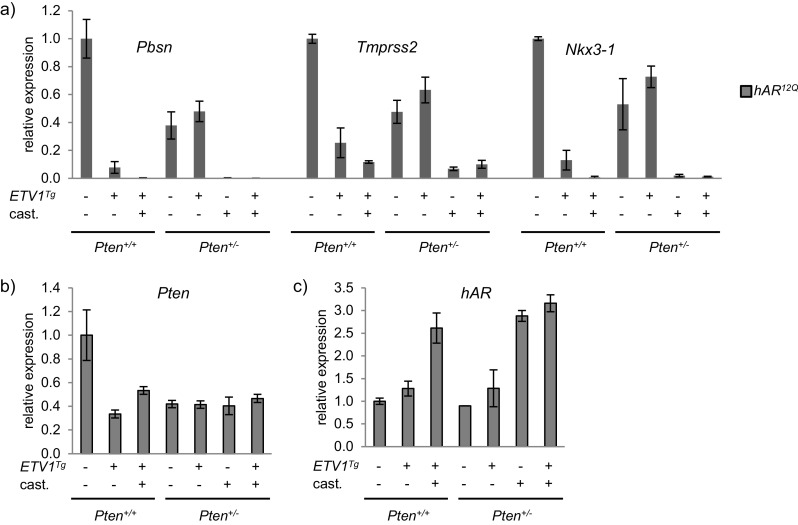
Effect of ETV1 on AR targets is muted with reduced Pten. a hAR 12Q mice are shown as representative data in all panels. Expression of AR target genes Pbsn, Tmprss2, and Nkx3-1 in DLP was measured by qRT-PCR and normalized to Actb. Expression levels in intact and castrate hAR 12Q ;ETV1 Tg, hAR 12Q ;Pten +/− and hAR 12Q ;ETV1 Tg ;Pten +/− are plotted relative to intact hAR 12Q. The first three lanes for each gene are repeated from Fig. 2a for comparison between backgrounds. b Pten expression in DLP was measured by qRT-PCR as above. c hAR expression was measured by qRT-PCR as above. For all qRT-PCR, DLP RNA samples were divided into two unique pools for analysis. Mean and SEM are plotted. The 2-ddCt method was used to calculate relative expression
To further compare the molecular effects of hAR allele and ETV1 overexpression in the Pten +/− background, global gene expression was analyzed by RNA-seq. DLP RNA samples from ETV1 transgenic vs. non-transgenic mice as well as from the individual macroscopic tumors that were assayed. Principal component analysis of the RNA-seq data showed that samples from all castrated mice and one macroscopic tumor clustered together (Fig. 7a). The remaining tumor from a hAR 12Q mouse mapped separately, suggesting a disparate expression pattern, as might be expected from the poorly differentiated phenotype (see Fig. 5). Samples from intact mice showed substantial heterogeneity within and between groups and did not form distinct clusters regardless of the transgene, suggesting that ETV1 had a more moderate and/or variable effect in this background (Fig. 7a).
Fig. 7.
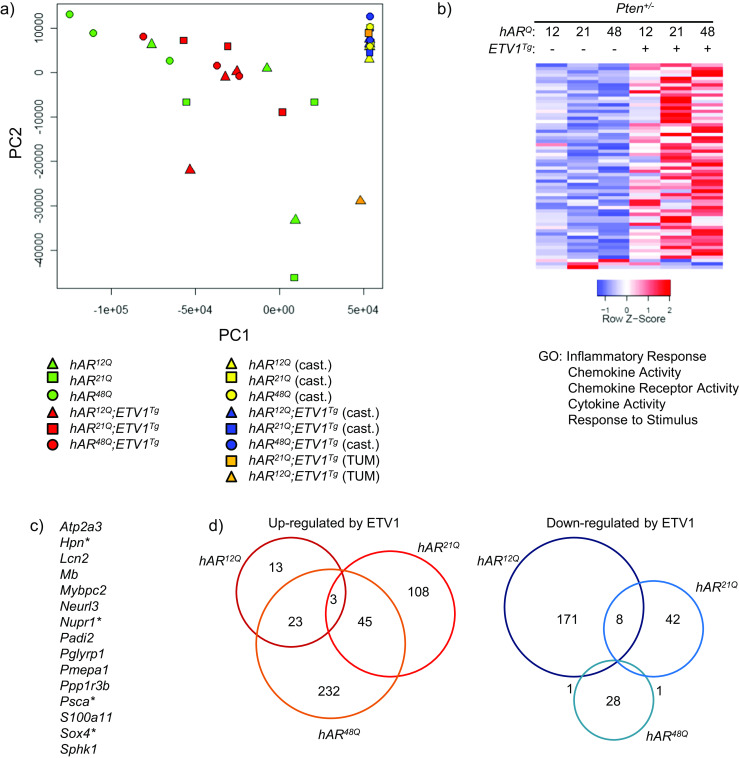
ETV1 overexpression alters genome-wide expression in Pten-hemizygous mice. a Principal component analysis was performed for all genes among the individual RNA-seq libraries. hAR 12Q, hAR 21Q, and hAR 48Q mice are represented as triangles, squares, and circles, respectively. Green and red represent intact non-transgenic and transgenic mice, yellow and blue represent castrated non-transgenic and transgenic mice, and orange represents macroscopic tumors, respectively. b RNA-seq analysis was performed on DLP RNA as before. Heatmaps include differentially expressed genes between the groups shown (FDR ≤ 0.05), with columns representing the average among biological replicate libraries. By GLM analysis of all hAR;ETV1 Tg ;Pten +/− libraries vs. all hAR;Pten +/− libraries, 59 genes are upregulated and 3 downregulated in prostates of hAR;ETV1 Tg ;Pten +/− mice. Select significantly enriched GO terms are listed. c ETV1-upregulated genes shared among Pten +/− and Pten +/+ mice are listed. d Differential expression was performed between transgenics and non-transgenics for each AR Q-tract group individually. Overlap between genes differentially expressed in hAR 12Q ;ETV1 Tg, hAR 21Q ;ETV1 Tg or hAR 48Q ;ETV1 Tg relative to non-transgenics is illustrated by Venn diagram
Overall, fewer genes were significantly differentially expressed between the prostates of transgenic and non-transgenic mice on the Pten +/− background. Fifty-nine genes were upregulated and three downregulated when all intact transgenics were contrasted with all intact non-transgenics by GLM analysis (Fig. 7b). Interestingly, this set was enriched with 15 genes also upregulated by ETV1 in the Pten +/+ mice, including orthologs of the PCa-associated genes, namely PSCA, SOX4, and HPN (Fig. 7c). In particular, HPN is highly overexpressed in human PCa, promotes metastasis in a mouse model, and can be effectively targeted with a small molecule inhibitor [71]. Additionally, these 59 genes were enriched for genes further upregulated in the macroscopic DLP tumors (see below), suggesting that ETV1 induces oncogenic expression changes prior to invasive disease, and that these genes may serve as biomarkers of impending tumorigenesis. Androgen ablation affected a similar number of genes as in the Pten +/+ mice (data not shown).
In contrast to little apparent effect of Q-tract length on the Pten +/+ background (see Fig. 2a), separating the groups by hAR allele revealed notable differences in expression due to androgen axis variation in Pten +/− mice. Analysis of individual hAR genotypes revealed 39, 156, and 303 genes upregulated and 180, 51, and 30 genes downregulated between transgenic and non-transgenic hAR 12Q, hAR 21Q, and hAR 48Q prostates, respectively (Fig. 7d and Online Resource 6). Unlike in the Pten +/+ background, the majority of differentially expressed genes showed little overlap, indicating they were unique to a single hAR genotype. There was a general trend toward greater repression by ETV1 in the context of the stronger hAR 12Q allele, and greater activation by ETV1 with the weaker hAR 48Q allele. Fewer differences in gene expression between ETV1-transgenic and non-transgenic Pten +/− mice could result in part from larger variance in age and disease stage among mice, or more heterogeneous ETV1 effects overall, in turn resulting in the variation seen in Fig. 7b and thus reduced statistical sensitivity. Additionally, ETV1 could have a directly reduced influence on gene expression on the Pten +/− background. This latter explanation is supported by the qPCR data (Fig. 6a), suggesting an abrogation of the effect of ETV1 on AR targets. Concept mapping confirmed that the broad ETV1-AR antagonism observed in the Pten +/+ mice was diminished by loss of one Pten allele (Online Resource 3).
ETV1-Induced Inflammatory Gene Expression
Genes upregulated in prostates of ETV1-transgenic vs. non-transgenic hAR;Pten +/− mice were significantly enriched with inflammation-associated GO terms (Fig. 7a). Relative expression of these genes is illustrated in the heatmap of Fig. 8a, including expression levels in the macroscopic DLP tumors. Many of the inflammatory genes were upregulated further in one or both tumors. The chemokines CXCL5 and CXCL16 are known to be associated with human PCa progression [10, 34], and NUPR1 is associated with a variety of cancers including breast, pancreatic, and lung cancer [27, 28, 35]. Because a significant inflammation signature was seen in prostates of ETV1 transgenic mice prior to overt tumorigenesis, it is possible that ETV1-dependent inflammation may contribute to oncogenesis. Regardless, these factors may be biomarkers of impending tumorigenesis in ETV1+ prostates. In support of the functional significance of this gene signature, histologic evidence of inflammation has been previously noted in ETV1 knockin mice [8].
Fig. 8.
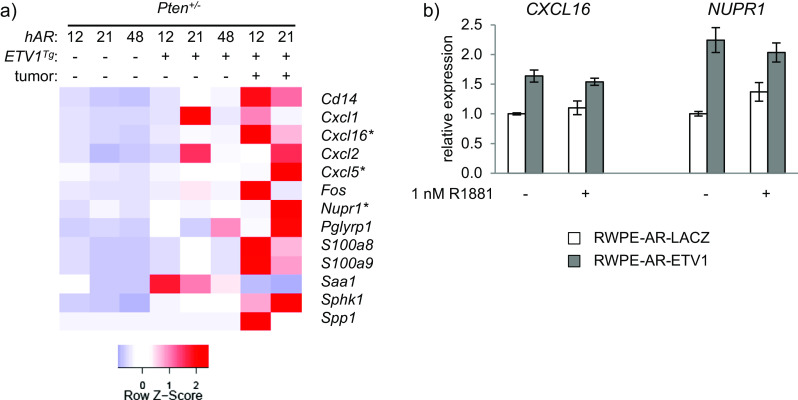
ETV1-induced inflammatory expression signature precedes tumorigenesis. a Genes that are differentially expressed between hAR;Pten +/− and hAR;ETV1 Tg ;Pten +/− mice and annotated with inflammation-related GO terms (see Fig. 7a) are shown in the heatmap. Relative expression in prostates of hAR;Pten +/− and hAR;ETV1 Tg;Pten +/− mice is shown, as well as expression in the macroscopic prostate tumors. b RWPE-1 cells stably expressing AR and either ETV1 or LACZ were stimulated with 1 nM R1881 or methanol vehicle, and RNA was prepared for qRT-PCR analysis. Expression of the inflammatory genes CXCL16 and NUPR1 were normalized to GAPDH or ACTB, respectively, and plotted relative to RWPE-AR-LACZ cells. Mean and SEM are plotted for biological triplicate samples
Direct ETV1 regulation of CXCL16 and NUPR1 was tested in human prostate cells. The benign human prostate epithelial cell line RWPE-1 was transduced with lentiviral vectors that stably express AR, as well as either ETV1 or LACZ [74, 75]. qRT-PCR revealed that the ETV1-transduced cells expressed the endogenous CXCL16 and NUPR1 genes at significantly higher levels than the LACZ controls (Fig. 8b), providing evidence of direct gene regulation by ETV1. This upregulation was observed with or without stimulation with the synthetic androgen R1881, suggesting that ETV1 induction of CXCL16 in these cells is not AR-dependent. In contrast, NUPR1 was sensitive to androgen in the absence but not the presence of exogenous ETV1.
ETV1 Effects in Mouse DLP Parallel Those in ETS+ and Aggressive Human PCa
We explored the significance of these data by comparison to human PCa expression patterns and sought novel ETV1-regulated genes. Molecular concept analysis in Oncomine revealed significant overlap between androgen-induced genes (see Fig. 3), genes downregulated in DLP tumors, and genes downregulated by ETV1 in Pten +/+ mice. Relative expression of these shared genes is shown in the heatmap in Fig. 9a. This pattern suggests that expression patterns in late ETV1/PTEN-induced tumorigenesis recapitulate early ETV1-induced signatures. Thus, selective AR repression may be important at multiple disease stages. Notably, the tumor suppressor and luminal epithelial differentiation marker Nkx3-1 [78] is among androgen-induced transcripts downregulated in the tumors as well as prostates of early-stage ETV1 Tg mice. Genes differentially expressed in ETV1 Tg ;Pten +/− prostates were not significantly enriched for this set of androgen-induced genes, yet overlapped with early-stage mouse disease, DLP tumors, and human PCa (see Figs. 7 and 8). When genes differentially expressed in the macroscopic DLP tumors were further queried in Oncomine, significant enrichment with aggressive human PCa was observed. Genes downregulated in the hAR 21Q;ETV1 Tg ;Pten +/− DLP tumor were significantly enriched for downregulated genes in human PCa metastases and advanced Gleason grade tumors (Fig. 9b) [25, 30, 42, 51, 84].
Fig. 9.
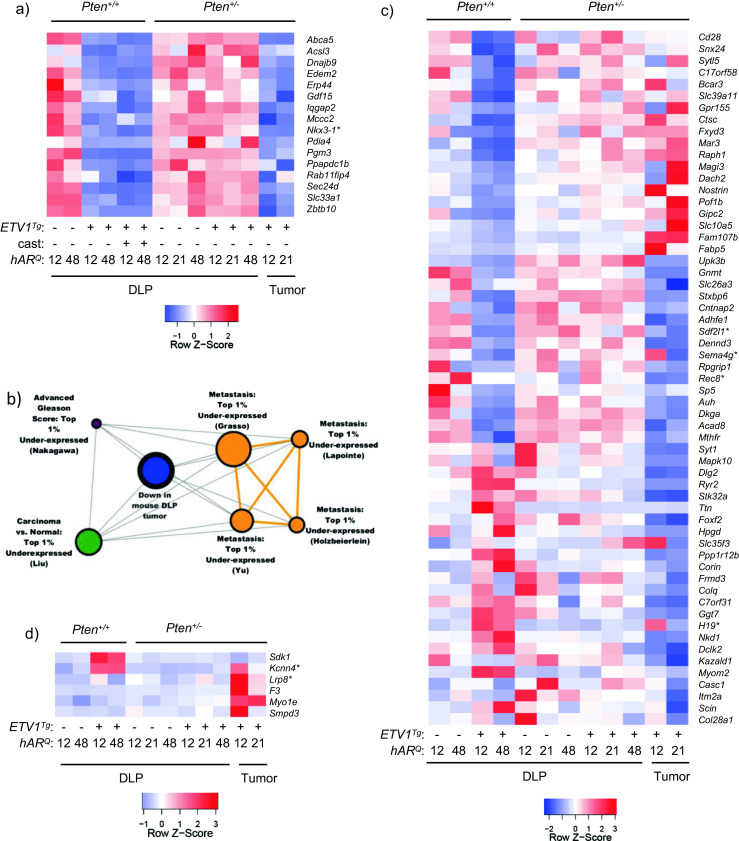
ETV1 effects in mouse DLP parallel those in ETS+ and aggressive human PCa. a A set of androgen-induced genes is significantly repressed in hAR;ETV1 Tg prostates and in Pten +/− tumors. Relative expression across groups is represented in the heatmap. b Genes downregulated in hAR 21Q ;ETV1 Tg ;Pten +/− tumor tissue were uploaded to Oncomine and queried against “Oncomine gene expression signatures.” Significant enrichment of genes downregulated in human PCa metastases and advanced Gleason grade PCa was observed, and select concepts are shown in the molecular concept map. Node size is proportional to the number of genes shared with the primary concept. OR ≥ 4.0, p ≤ 10−6. c The heatmap shows the relative expression pattern of genes that are significantly downregulated in ETS+ vs. non-ETS localized tumors from the Taylor Prostate data set as well as in hAR;ETV1 Tg mouse prostates or DLP tumors. d The heatmap shows relative expression of genes that are significantly overexpressed in ETV1+ vs. non-ETS localized tumors from the Grasso Prostate data set as well as in hAR;ETV1 Tg mouse prostates or DLP tumors
Two recent large-scale studies profiled gene expression in localized and metastatic PCa from over 200 total patients [25, 73]. These microarray data were stratified by ETS status, PTEN expression level, and localized vs. metastatic tumors, and compared with the mouse RNA-seq data. Molecular concept analysis of the dataset from Taylor et al., with ERG+ and ETV1+ patients combined as “ETS+,” revealed that genes downregulated in ETS+, PTEN-normal localized tumors vs. non-ETS tumors, were significantly enriched among genes downregulated in prostates of hAR;ETV1 Tg mice or in DLP tumors (Fig. 9c). While many transcripts were present in patterns unique to DLP tumors or hAR;ETV1 Tg prostates, a number were repressed in both groups (Fig. 9c, middle horizontal rows). As with important androgen-regulated genes (see Fig. 9a), this overlap once again suggests common ETV1 action in early neoplasia and in tumors, in contrast to intermediate stages of tumorigenesis.
Genes downregulated in ETS+ patients that are also downregulated in hAR;ETV1 Tg mice as well as DLP tumors include a number of genes associated with various cancers, but not yet with PCa (Fig. 9c). For example, REC8 is a meiotic recombination gene whose hypermethylation is associated with poor prognosis in gastrointestinal stromal tumors [54]. Downregulation of the stromal cell-derived factor 2-like 1 (SDF2L1) gene is associated with poor prognosis in breast cancer [36]. SEMA4G was one of the few candidate tumor suppressor genes on the colorectal cancer (CRC) associated chromosome 10 loss of heterozygosity (LOH) region to be significantly downregulated in CRC tumors [80]. This suggests potential roles for these genes in PCa, and potential regulation by ETS factors.
Of note, several genes were dysregulated in opposite directions in the two DLP tumors, including H19, SEMA4G, and KCNN4 (Fig. 9c, d). This highlights the heterogeneity between the tumors and among these mice in general, similar to heterogeneity among human PCa patients. Furthermore, many genes were repressed in hAR;ETV1 Tg mice but upregulated in DLP tumors, or vice-versa (Fig. 9c). This pattern emphasizes that while there is substantial overlap between early and late stages, as described above, there is a great deal of divergence as well, and different ETV1 targets may be more important or play different roles at one stage of disease progression than another.
PCa patient expression data from the study by Grasso et al. [25] was stratified as above by ETS status, PTEN expression level, and localized vs. metastatic tumor site, and differential expression was re-calculated. Overlap with the mouse RNA-seq data is shown in Fig. 9d. KCNN4, a potassium channel gene, was upregulated in localized ETV1+ vs. ETS− tumors in human PCa patients with normal PTEN levels [25], as well as in hAR;ETV1 Tg vs. prostates and in DLP tumors (Fig. 9d). KCNN4 expression is increased in benign prostatic hyperplasia (BPH) and intermediate Gleason grade PCa, but not high Gleason grade PCa [52, 53]. Additionally, LRP8, a low-density lipoprotein receptor, is upregulated in localized ETV1+ human tumors with normal PTEN [25] and in mouse DLP tumors (Fig. 9d). High LRP8 expression is associated with several human cancers, including lung and gastric cancer, but has not yet been linked to PCa [22, 24, 56]. As before, these genes showed variable upregulation between the DLP tumors, highlighting heterogeneity in mouse as well as in human patients.
Expression in the intermediate-stage ETV1 Tg /Pten +/− mice showed little overlap with the human data in these particular comparisons. In part, this could be due to fewer differentially expressed genes at that intermediate stage (see above). However, as seen in Figs. 7 and 8, ETV1 induced a notable inflammatory signature in Pten-hemizygous mice, along with a number of individual PCa-associated genes and genes shared with both the early ETV1 Tg mice and the later DLP tumors. In sum, these studies shed light on distinct ETV1 gene regulatory networks at different disease stages, and highlight novel ETV1 targets when filtered against human data.
Discussion
This study examined in mice the degree to which AR genetic variation and ETV1 overexpression interact early in neoplasia, and their subsequent interaction with PTEN reduction in PCa progression. In both patients and transgenic mice, ETS overexpression is AR-driven, and AR activity is affected by factors downstream of PTEN. The molecular consequences of altering AR and ETV1 transcriptional regulation was queried with high-throughput gene expression analysis. Marked antagonism of the normal AR-regulated transcriptome occurred in prostates of ETV1 transgenic mice despite mild pathology. This antagonism was largely abrogated on a Pten-hemizygous background. In the Pten-hemizygous mice, concurrent ETV1 overexpression induced progression to adenocarcinoma in a subset of mice as well as a striking pro-inflammatory gene signature. AR variation had little impact on early neoplasia driven by ETV1, but a stronger AR allele impacted gene expression and PCa progression in the Pten-hemizygous model. These results suggest that ETV1 cooperates with even reduced PTEN signaling to drive cancer progression in mice, and that partially antagonizing AR and promoting inflammation may be key components of the ETV1-driven oncogenic program. Integration of mouse and patient data revealed potential ETV1 regulation of known and novel PCa-associated genes, suggesting novel targets for therapeutic intervention.
Despite substantial AR antagonism similar to the effects of castration, prostates of ETV1 Tg mice do not show atrophy, suggesting selective action favoring survival and proliferation. While ETV1 overexpression is insufficient to cause invasive PCa in mice, disruption of the normal AR-regulated, pro-differentiation gene expression program drives PIN and hyperplasia. This may “prime” the prostate for further dedifferentiation and tumor progression following additional oncogenic insults, such as PTEN reduction or loss. In the case of ERG, it has been reported that PTEN loss is correlated with ERG fusions in patients [39] and that ERG fusion precedes PTEN loss [26].
We have previously shown an influence of the androgen axis on PCa onset and progression in TRAMP mice, where oncogenesis is driven by prostatic expression of an SV40 Large-T antigen transgene [3]. While AR strength does not impact the early stages of ETV1-induced neoplasia in the current model, the rate of progression to adenocarcinoma is impacted when combined with Pten reduction. Confirmation here of the influence of AR in a model reflecting recurrent events in human PCa adds to the recent discovery that ETS+ PCa is more common in men with a shorter Q-tract (stronger) AR [82].
While fewer genes are affected by ETV1 overexpression in the Pten +/− background overall, a greater number of differentially expressed genes are identified when Q-tract variants are analyzed individually, revealing divergent effects of the androgen axis on gene expression and disease progression. The hAR 12Q mice show significant repression of polycomb target genes with ETV1 overexpression, as do the DLP tumors, where most other groups show the opposite pattern (Online Resource 3). Since ETS factors can interact with the polycomb repressive complex [85], perhaps the greater repression seen with hAR 12Q indicates a greater cooperation in the context of the stronger AR and low PTEN, and could signify impending tumorigenesis. Induction of inflammatory genes is more pronounced in the hAR 21Q and hAR 48Q mice (Online Resources 2 and 3). A number of those inflammatory genes are further upregulated in the tumors, indicating that they may be potential early biomarkers of aggressive disease. While a number of mouse models of PCa show inflammation during tumorigenesis, the expression signature specifically in ETV1 Tg mice relative to non-transgenics suggests a role of ETV1. Confirmation here of regulation by ETV1 in RWPE cells supports inflammation as a factor in ETV1+ neoplasia.
A “core” set of targets is upregulated by ETV1 in both Pten +/+ and Pten +/− mice, including a number of genes known to be upregulated in human PCa. Several of these genes including PSCA and HPN encode proteins that are druggable in vitro and in preclinical models [71, 83]. ETS+ patients with particularly high expression of these genes may respond more favorably in future targeted therapy. ERG promotes SOX4 expression to stimulate epithelial to mesenchymal transition in PCa [79], and these mouse data suggest that ETV1 may promote SOX4 expression as a common component of ETS-driven oncogenesis.
Combined inhibition of the AR and PI3K/AKT pathways in PCa has been recently shown to be a potentially effective treatment strategy, as these pathways reciprocally repress one another, and either pathway can compensate when the other is inhibited [15, 50]. Since the AR, PTEN, and ETV1 pathways all cooperate in PCa progression, stratification by ETS fusion status may enhance the efficacy of dual inhibition of AR and PI3K/AKT for some patients.
The CRPC seen in castrated Pten-hemizygous mice highlights the fact that hormone ablation generally slows disease progression in mice and patients, but in some cases can accelerate it [3, 32]. In the previous TRAMP study, CRPC varied with AR strength, with half of castrated hAR 12Q mice failing to evince a palpable tumor prior to dying of metastatic castrate-resistant disease [3]. Not only did a number of castrated Pten-hemizygous mice develop PIN or adenocarcinoma, but disease tended to arise earlier than in the intact mice, and greater progression was seen with the stronger hAR allele. While CRPC has been previously reported in mice with a prostate-specific Pten deletion, these results suggest that even Pten reduction can promote CRPC. These findings support clinical data that hemizygous PTEN deletion predicts more rapid disease recurrence following radical prostatectomy and hormone therapy [19]. Furthermore, this raises the question of whether AR genotyping might have predictive value in these situations.
Whether ETS fusion status in patients is associated with poor prognosis remains controversial and depends heavily on cohorts studied and definitions of disease aggressiveness [60, 72]. Results from the current study suggest that a stronger AR promotes ETV1-driven PCa progression, in accord with the androgen regulation of the transgene in mice and of relevance to fusion genes in patients. Castration prevents ETV1-driven disease in Pten +/+ mice, and significantly reduces overall disease in Pten +/− mice. In a clinical study, a subset of ERG+ patients responded very favorably to abiraterone acetate, which targets androgen synthesis via CYP17 inhibition [7]. This underscores the androgen regulation of ETS fusion genes, and suggests that those tumors may be more dependent on AR activity than fusion-negative tumors.
Genes downregulated in the DLP tumors relative to PIN show the same expression pattern in high Gleason grade and metastatic human PCa (see Fig. 9b). This finding suggests that the genes and pathways altered in the mouse models described here are not only generally relevant to human PCa, but may be informative for aggressive disease. Genes downregulated in DLP tumors are enriched for androgen-induced targets, and in particular those that are also repressed by ETV1 in prostates of Pten +/+ mice. These data suggest that the selective antagonism of AR in early neoplasia recurs as a redirection of AR activity in later stage tumorigenesis.
Direct comparison of the mouse data to several human PCa patient data sets stratified by ETS status and PTEN expression level [25, 73] reveal a number of known and novel PCa-associated genes potentially regulated by ETV1. One of these, KCNN4, is targetable with a potassium channel inhibitor in a rat model of BPH [53], suggesting that it may have utility in ETV1+ PCa patients. Additional genes including LRP8, SEMA4G, REC8, and SDF2L1 are associated with other cancers but not yet PCa, and not yet with ETS regulation [22, 24, 36, 54, 56, 80]. H19 has a known role in PCa [86] but no known ETS regulation. These data suggest potentially novel roles for these genes in PCa, as well as regulation by ETV1, warranting their further exploration as biomarkers or in therapeutic strategies.
Recently, ERG overexpression was shown to have little effect on AR signaling in the mouse prostate, but enhances AR activity and stimulates tumorigenesis following deletion of Pten [18]. The same effect is observed in human PCa expression sets stratified by these factors, i.e., ERG and AR cooperate in PTEN-low tumors [18]. In vitro, ERG is a strong repressor of AR in several PCa cell lines with or without functional PTEN [85]. Further data show that ERG blocks AR-induced differentiation in luminal epithelial cells [49]. These studies suggest that the ERG-AR interaction is context-dependent, with different outcomes observed in vitro and in vivo, and dependent on PTEN status and cancer stage. Based on findings here, ETV1 also appears to behave in a highly context-dependent manner. In part, our in vivo AR-ETV1 results follow a similar “spectrum” as the AR-ERG interaction described by Chen et al. [18]. However, we observe ETV1 antagonism of AR that is lost with reduced Pten, yet in the DLP tumors, strong repression of AR targets recurs. The different interactions could result from PTEN dose (hemizygous vs. prostate-specific knockout) or reflect distinct properties of ETV1 vs. ERG. ETV1 has been reported to enhance AR activity in vitro [8, 18, 64]. However, such studies have been limited to LNCaP cells, which lack functional PTEN. Further investigation of the AR-ETV1 interaction in additional models is needed to clarify the effect in vivo vs. in vitro, as well as the influence of AR variation and PTEN status.
In conclusion, the mouse models described here provide novel insight into the interaction of key pathways driving early-intermediate stage PCa, and also reflect aggressive human disease. We confirm in these models a role of the androgen axis in disease progression as recently shown to be significant in patients bearing ETS gene fusions. A number of known and druggable PCa-associated factors show potential ETV1 regulation and warrant further investigation in patients stratified by AR, ETS, and PTEN status as novel therapeutic targets or prognostic biomarkers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Primer sequences for genotyping and real-time PCR (PDF 81 kb)
GO terms significantly enriched (Benjamini FDR ≤ 0.05) among genes differentially expressed in the mouse RNA-seq data. A separate tab is included with significant GO terms for each contrast (XLS 850 kb)
Oncomine molecular concepts significantly enriched (p ≤ 10−6, OR ≥ 4) among genes differentially expressed in the mouse RNA-seq data. A separate tab is included with significant molecular concepts for each contrast (XLS 343 kb)
Prostate pathology in Pten-hemizygous mice varies with age and lobe: a DLP and VP sections were scored as normal or atrophy (N + A), mPIN2-4 (P2-4) or adenocarcinoma (ADC). The median age at sacrifice was 41 weeks, and samples were divided into subsets ≤41 weeks or >41 weeks of age. The proportion of samples at each stage was plotted per experimental group. Samples were plotted according to the most severely diseased lobe. Prostates from 5–14 mice per group were analyzed. b As in a, but DLP and VP sections from each mouse were plotted separately for mice of any age (PDF 304 kb)
Histopathology in Pten-hemizygous mice. Additional prostate sections are shown from, in order, an intact hAR 12Q ;Pten +/− DLP, a castrated hAR 12Q ;ETV1 Tg ;Pten +/− DLP, an intact hAR 12Q ;Pten +/− VP, and a normal region of a macroscopic hAR 12Q ;ETV1 Tg ;Pten +/− DLP tumor. Sections were stained with H&E. Immunohistochemical (IHC) staining was performed with antibodies to AR, PTEN, or pAKT protein (brown staining). In situ hybridization (ISH) was performed with a probe against the human ETV1 transcript (red staining). ETV1 expression level is scored as 0–4, with 4 being highest. Images are shown at ×10–20 magnification with ×60 inset. Consecutive sections were used when possible, and each row of images shows sections from a single mouse prostate (PDF 306 kb)
The heatmaps show relative expression of genes differentially expressed (FDR ≤ 0.05) in intact ETV1-transgenic vs. non-transgenic mice on the Pten-hemizygous background. Each hAR Q-tract allele was analyzed separately. For each group, relative expression in castrated mice is included. Each lane represents the average expression from of 2–3 pools of RNA per group. See Fig. 7d for the number of shared and unique genes in each contrast (PDF 61 kb)
Acknowledgements
This work was supported by grants from the Department of Defense (W81XWH-08-1-0034) and the National Institutes of Health (NIH) (R01-CA144032) to D.M.R. Additional funding was provided by a pilot grant from the Michigan Institute for Clinical and Health Research. Core support for RNA-seq (UM DNA Sequencing Core), and histology (UM Cancer Center Tissue Core and the UM Microscopy and Imaging Laboratory) was provided by the Cancer Center Support Grant (5P30-CA46592). J.H. was supported in part by the NIH Predoctoral Genetics Training Grant (T32-GM07544) and by a fellowship from the UM Endowment for the Development of Graduation Education. S.A.T was supported by the A. Alfred Taubman Medical Research Institute and the Prostate Cancer Foundation. We thank Sravanthi Gutta for technical assistance.
Conflict of Interest
The University of Michigan has been issued a patent on the detection of ETS gene fusions in prostate cancer, on which R.M. and S.A.T are listed as co-inventors. The University of Michigan licensed the diagnostic field of use to Gen-Probe, Inc., who has sublicensed some rights to Ventana/Roche. S.A.T. has served as a consultant to, and has received honoraria from, Ventana/Roche. The remaining authors declare that they have no conflicts of interest.
References
- 1.Abate-Shen C, Banach-Petrosky WA, Sun X, Economides KD, Desai N, Gregg JP, Borowsky AD, Cardiff RD, Shen MM. Nkx3.1; Pten mutant mice develop invasive prostate adenocarcinoma and lymph node metastases. Cancer Res. 2003;63(14):3886–3890. [PubMed] [Google Scholar]
- 2.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14(19):2410–2434. doi: 10.1101/gad.819500. [DOI] [PubMed] [Google Scholar]
- 3.Albertelli MA, O’Mahony OA, Brogley M, Tosoian J, Steinkamp M, Daignault S, Wojno K, Robins DM. Glutamine tract length of human androgen receptors affects hormone-dependent and -independent prostate cancer in mice. Hum Mol Genet. 2008;17(1):98–110. doi: 10.1093/hmg/ddm287. [DOI] [PubMed] [Google Scholar]
- 4.Albertelli MA, Scheller A, Brogley M, Robins DM. Replacing the mouse androgen receptor with human alleles demonstrates glutamine tract length-dependent effects on physiology and tumorigenesis in mice. Mol Endocrinol. 2006;20(6):1248–1260. doi: 10.1210/me.2006-0021. [DOI] [PubMed] [Google Scholar]
- 5.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42(5):454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8(9):1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 7.Attard G, Reid AH, Olmos D, de Bono JS. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009;69(12):4937–4940. doi: 10.1158/0008-5472.CAN-08-4531. [DOI] [PubMed] [Google Scholar]
- 8.Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, et al. ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev. 2013;27(6):683–698. doi: 10.1101/gad.211011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bastus NC, Boyd LK, Mao X, Stankiewicz E, Kudahetti SC, Oliver RT, Berney DM, Lu YJ. Androgen-induced TMPRSS2:ERG fusion in nonmalignant prostate epithelial cells. Cancer Res. 2010;70(23):9544–9548. doi: 10.1158/0008-5472.CAN-10-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begley LA, Kasina S, Mehra R, Adsule S, Admon AJ, Lonigro RJ, Chinnaiyan AM, Macoska JA. CXCL5 promotes prostate cancer progression. Neoplasia. 2008;10(3):244–254. doi: 10.1593/neo.07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berquin IM, Min Y, Wu R, Wu H, Chen YQ. Expression signature of the mouse prostate. J Biol Chem. 2005;280(43):36442–36451. doi: 10.1074/jbc.M504945200. [DOI] [PubMed] [Google Scholar]
- 12.Bertram J, Peacock JW, Fazli L, Mui AL, Chung SW, Cox ME, Monia B, Gleave ME, Ong CJ. Loss of PTEN is associated with progression to androgen independence. Prostate. 2006;66(9):895–902. doi: 10.1002/pros.20411. [DOI] [PubMed] [Google Scholar]
- 13.Bhalla R, Kunju LP, Tomlins SA, Christopherson K, Cortez C, Carskadon S, Siddiqui J, et al. Novel dual-color immunohistochemical methods for detecting ERG-PTEN and ERG-SPINK1 status in prostate carcinoma. Mod Pathol. 2013 doi: 10.1038/modpathol.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bismar TA, Yoshimoto M, Vollmer RT, Duan Q, Firszt M, Corcos J, Squire JA. PTEN genomic deletion is an early event associated with ERG gene rearrangements in prostate cancer. BJU Int. 2011;107(3):477–485. doi: 10.1111/j.1464-410X.2010.09470.x. [DOI] [PubMed] [Google Scholar]
- 15.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, Alimonti A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41(5):619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain NL, Driver ED, Miesfeld RL. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994;22(15):3181–3186. doi: 10.1093/nar/22.15.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, et al. ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med. 2013 doi: 10.1038/nm.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choucair K, Ejdelman J, Brimo F, Aprikian A, Chevalier S, Lapointe J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer. 2012;12(1):543. doi: 10.1186/1471-2407-12-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Huangda W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 21.DePrimo, S. E., M. Diehn, J. B. Nelson, R. E. Reiter, J. Matese, M. Fero, R. Tibshirani, P. O. Brown, and J. D. Brooks (2002) Transcriptional programs activated by exposure of human prostate cancer cells to androgen. Genome Biol 3 (7):RESEARCH0032 [DOI] [PMC free article] [PubMed]
- 22.Dun B, Sharma A, Teng Y, Liu H, Purohit S, Xu H, Zeng L, She JX. Mycophenolic acid inhibits migration and invasion of gastric cancer cells via multiple molecular pathways. PLoS One. 2013;8(11):e81702. doi: 10.1371/journal.pone.0081702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y, Jin R, Gupta A, Rennie PS, Matusik RJ. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17(8):1484–1507. doi: 10.1210/me.2003-0020. [DOI] [PubMed] [Google Scholar]
- 24.Garnis C, Campbell J, Davies JJ, Macaulay C, Lam S, Lam WL. Involvement of multiple developmental genes on chromosome 1p in lung tumorigenesis. Hum Mol Genet. 2005;14(4):475–482. doi: 10.1093/hmg/ddi043. [DOI] [PubMed] [Google Scholar]
- 25.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012 doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gumuskaya B, Gurel B, Fedor H, Tan HL, Weier CA, Hicks JL, Haffner MC, Lotan TL, De Marzo AM. Assessing the order of critical alterations in prostate cancer development and progression by IHC: further evidence that PTEN loss occurs subsequent to ERG gene fusion. Prostate Cancer Prostatic Dis. 2013 doi: 10.1038/pcan.2013.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo X, Wang W, Hu J, Feng K, Pan Y, Zhang L, Feng Y. Lentivirus-mediated RNAi knockdown of NUPR1 inhibits human nonsmall cell lung cancer growth in vitro and in vivo. Anat Rec (Hoboken) 2012;295(12):2114–2121. doi: 10.1002/ar.22571. [DOI] [PubMed] [Google Scholar]
- 28.Hamidi T, Algul H, Cano CE, Sandi MJ, Molejon MI, Riemann M, Calvo EL, et al. Nuclear protein 1 promotes pancreatic cancer development and protects cells from stress by inhibiting apoptosis. J Clin Invest. 2012;122(6):2092–2103. doi: 10.1172/JCI60144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25(20):2147–2157. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164(1):217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, Simons BW. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73(9):2718–2736. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson MA, Iversen P, Schwier P, Corn AL, Sandusky G, Graff J, Neubauer BL. Castration triggers growth of previously static androgen-independent lesions in the transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Prostate. 2005;62(4):322–338. doi: 10.1002/pros.20148. [DOI] [PubMed] [Google Scholar]
- 33.Jones SB, DePrimo SE, Whitfield ML, Brooks JD. Resveratrol-induced gene expression profiles in human prostate cancer cells. Cancer Epidemiol Biomarkers Prev. 2005;14(3):596–604. doi: 10.1158/1055-9965.EPI-04-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, Berry JE, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung SH, Lee A, Yim SH, Hu HJ, Choe C, Chung YJ. Simultaneous copy number gains of NUPR1 and ERBB2 predicting poor prognosis in early-stage breast cancer. BMC Cancer. 2012;12:382. doi: 10.1186/1471-2407-12-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang H, Escudero-Esparza A, Douglas-Jones A, Mansel RE, Jiang WG. Transcript analyses of stromal cell derived factors (SDFs): SDF-2, SDF-4 and SDF-5 reveal a different pattern of expression and prognostic association in human breast cancer. Int J Oncol. 2009;35(1):205–211. doi: 10.3892/ijo_00000330. [DOI] [PubMed] [Google Scholar]
- 37.Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, Desai N, Wang Y, Hayward SW, Cunha GR, Cardiff RD, Shen MM, Abate-Shen C. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res. 2002;62(11):2999–3004. [PubMed] [Google Scholar]
- 38.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, Taylor BS, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet. 2009;41(5):524–526. doi: 10.1038/ng.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knudsen, K. E., and W.K. Kelly (2011) Outsmarting androgen receptor: creative approaches for targeting aberrant androgen signaling in advances prostate cancer [DOI] [PMC free article] [PubMed]
- 41.Kunju LP, Carskadon S, Siddiqui J, Tomlins SA, Chinnaiyan AM, Palanisamy N. Novel RNA hybridization method for the in situ detection of ETV1, ETV4, and ETV5 gene fusions in prostate cancer. Appl Immunohistochem Mol Morphol. 2014;22(8):e32–e40. doi: 10.1097/PAI.0000000000000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci U S A. 2004;101(3):811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32(2):148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 44.Lindstrom S, Wiklund F, Adami HO, Balter KA, Adolfsson J, Gronberg H. Germ-line genetic variation in the key androgen-regulating genes androgen receptor, cytochrome P450, and steroid-5-alpha-reductase type 2 is important for prostate cancer development. Cancer Res. 2006;66(22):11077–11083. doi: 10.1158/0008-5472.CAN-06-3024. [DOI] [PubMed] [Google Scholar]
- 45.Lindstrom S, Zheng SL, Wiklund F, Jonsson BA, Adami HO, Balter KA, Brookes AJ, et al. Systematic replication study of reported genetic associations in prostate cancer: strong support for genetic variation in the androgen pathway. Prostate. 2006;66(16):1729–1743. doi: 10.1002/pros.20489. [DOI] [PubMed] [Google Scholar]
- 46.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295(5556):868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 47.Lupien M, Brown M. Cistromics of hormone-dependent cancer. Endocr Relat Cancer. 2009;16(2):381–389. doi: 10.1677/ERC-09-0038. [DOI] [PubMed] [Google Scholar]
- 48.Mehra R, Han B, Tomlins SA, Wang L, Menon A, Wasco MJ, Shen R, Montie JE, Chinnaiyan AM, Shah RB. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: molecular evidence for an independent group of diseases. Cancer Res. 2007;67(17):7991–7995. doi: 10.1158/0008-5472.CAN-07-2043. [DOI] [PubMed] [Google Scholar]
- 49.Mounir Z, Lin F, Lin VG, Korn JM, Yu Y, Valdez R, Aina OH, et al. TMPRSS2:ERG blocks neuroendocrine and luminal cell differentiation to maintain prostate cancer proliferation. Oncogene. 2014 doi: 10.1038/onc.2014.308. [DOI] [PubMed] [Google Scholar]
- 50.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011 doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakagawa T, Kollmeyer TM, Morlan BW, Anderson SK, Bergstralh EJ, Davis BJ, Asmann YW, Klee GG, Ballman KV, Jenkins RB. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS One. 2008;3(5):e2318. doi: 10.1371/journal.pone.0002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohya S, Kimura K, Niwa S, Ohno A, Kojima Y, Sasaki S, Kohri K, Imaizumi Y. Malignancy grade-dependent expression of K+−channel subtypes in human prostate cancer. J Pharmacol Sci. 2009;109(1):148–151. doi: 10.1254/jphs.08208SC. [DOI] [PubMed] [Google Scholar]
- 53.Ohya S, Niwa S, Kojima Y, Sasaki S, Sakuragi M, Kohri K, Imaizumi Y. Intermediate-conductance Ca2+−activated K+ channel, KCa3.1, as a novel therapeutic target for benign prostatic hyperplasia. J Pharmacol Exp Ther. 2011;338(2):528–536. doi: 10.1124/jpet.111.182782. [DOI] [PubMed] [Google Scholar]
- 54.Okamoto Y, Sawaki A, Ito S, Nishida T, Takahashi T, Toyota M, Suzuki H, et al. Aberrant DNA methylation associated with aggressiveness of gastrointestinal stromal tumour. Gut. 2012;61(3):392–401. doi: 10.1136/gut.2011.241034. [DOI] [PubMed] [Google Scholar]
- 55.Park JH, Walls JE, Galvez JJ, Kim M, Abate-Shen C, Shen MM, Cardiff RD. Prostatic intraepithelial neoplasia in genetically engineered mice. Am J Pathol. 2002;161(2):727–735. doi: 10.1016/S0002-9440(10)64228-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151(5):1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci U S A. 2003;100(1):183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhodes DR, Kalyana-Sundaram S, Tomlins SA, Mahavisno V, Kasper N, Varambally R, Barrette TR, Ghosh D, Varambally S, Chinnaiyan AM. Molecular concepts analysis links tumors, pathways, mechanisms, and drugs. Neoplasia. 2007;9(5):443–454. doi: 10.1593/neo.07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi: 10.1016/S1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol. 2011;29(27):3659–3668. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sathyanarayana UG, Birch C, Nagle RB, Tomlins SA, Palanisamy N, Zhang W, Hubbard A, Brunhoeber P, Wang Y, Tang L. Determination of optimum formalin fixation duration for prostate needle biopsies for immunohistochemistry and quantum dot FISH analysis. Appl Immunohistochem Mol Morphol. 2014 doi: 10.1097/PAI.0000000000000085. [DOI] [PubMed] [Google Scholar]
- 62.Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, Humphrey PA, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64(6):2270–2305. doi: 10.1158/0008-5472.CAN-03-0946. [DOI] [PubMed] [Google Scholar]
- 63.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24(18):1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shin S, Kim TD, Jin F, van Deursen JM, Dehm SM, Tindall DJ, Grande JP, Munz JM, Vasmatzis G, Janknecht R. Induction of prostatic intraepithelial neoplasia and modulation of androgen receptor by ETS variant 1/ETS-related protein 81. Cancer Res. 2009;69(20):8102–8110. doi: 10.1158/0008-5472.CAN-09-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simanainen U, Brogley M, Gao YR, Jimenez M, Harwood DT, Handelsman DJ, Robins DM. Length of the human androgen receptor glutamine tract determines androgen sensitivity in vivo. Mol Cell Endocrinol. 2011;342(1–2):81–86. doi: 10.1016/j.mce.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh M, Jha R, Melamed J, Shapiro E, Hayward SW, Lee P. Stromal androgen receptor in prostate development and cancer. Am J Pathol. 2014 doi: 10.1016/j.ajpath.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 68.Steinkamp MP, O’Mahony OA, Brogley M, Rehman H, Lapensee EW, Dhanasekaran S, Hofer MD, et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. 2009;69(10):4434–4442. doi: 10.1158/0008-5472.CAN-08-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273(2):175–184. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Svensson MA, LaFargue CJ, MacDonald TY, Pflueger D, Kitabayashi N, Santa-Cruz AM, Garsha KE, et al. Testing mutual exclusivity of ETS rearranged prostate cancer. Lab Invest. 2011;91(3):404–412. doi: 10.1038/labinvest.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tang X, Mahajan SS, Nguyen LT, Beliveau F, Leduc R, Simon JA, Vasioukhin V. Targeted inhibition of cell-surface serine protease Hepsin blocks prostate cancer bone metastasis. Oncotarget. 2014;5(5):1352–1362. doi: 10.18632/oncotarget.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taris M, Irani J, Blanchet P, Multigner L, Cathelineau X, Fromont G. ERG expression in prostate cancer: the prognostic paradox. Prostate. 2014 doi: 10.1002/pros.22863. [DOI] [PubMed] [Google Scholar]
- 73.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448(7153):595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 75.Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE, Cao Q, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10(2):177–188. doi: 10.1593/neo.07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 77.Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1(3):E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461(7263):495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang L, Li Y, Yang X, Yuan H, Li X, Qi M, Chang YW, et al. ERG-SOX4 interaction promotes epithelial-mesenchymal transition in prostate cancer cells. Prostate. 2014 doi: 10.1002/pros.22783. [DOI] [PubMed] [Google Scholar]
- 80.Wang X, Zbou C, Qiu G, Fan J, Tang H, Peng Z. Screening of new tumor suppressor genes in sporadic colorectal cancer patients. Hepatogastroenterology. 2008;55(88):2039–2044. [PubMed] [Google Scholar]
- 81.Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 1998;95(9):5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoo S, Pettersson A, Jordahl KM, Lis RT, Lindstrom S, Meisner A, Nuttall EJ, et al. Androgen receptor CAG repeat polymorphism and risk of TMPRSS2:ERG positive prostate cancer. Cancer Epidemiol Biomarkers Prev. 2014 doi: 10.1158/1055-9965.EPI-14-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu S, Feng F, Wang K, Men C, Lin C, Liu Q, Yang D, Gao Z. The therapeutic efficacy of I131-PSCA-mAb in orthotopic mouse models of prostate cancer. Eur J Med Res. 2013;18:56. doi: 10.1186/2047-783X-18-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, McDonald C, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22(14):2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 85.Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17(5):443–454. doi: 10.1016/j.ccr.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu M, Chen Q, Liu X, Sun Q, Zhao X, Deng R, Wang Y, et al. lncRNA H19/miR-675 axis represses prostate cancer metastasis by targeting TGFBI. FEBS J. 2014;281(16):3766–3775. doi: 10.1111/febs.12902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences for genotyping and real-time PCR (PDF 81 kb)
GO terms significantly enriched (Benjamini FDR ≤ 0.05) among genes differentially expressed in the mouse RNA-seq data. A separate tab is included with significant GO terms for each contrast (XLS 850 kb)
Oncomine molecular concepts significantly enriched (p ≤ 10−6, OR ≥ 4) among genes differentially expressed in the mouse RNA-seq data. A separate tab is included with significant molecular concepts for each contrast (XLS 343 kb)
Prostate pathology in Pten-hemizygous mice varies with age and lobe: a DLP and VP sections were scored as normal or atrophy (N + A), mPIN2-4 (P2-4) or adenocarcinoma (ADC). The median age at sacrifice was 41 weeks, and samples were divided into subsets ≤41 weeks or >41 weeks of age. The proportion of samples at each stage was plotted per experimental group. Samples were plotted according to the most severely diseased lobe. Prostates from 5–14 mice per group were analyzed. b As in a, but DLP and VP sections from each mouse were plotted separately for mice of any age (PDF 304 kb)
Histopathology in Pten-hemizygous mice. Additional prostate sections are shown from, in order, an intact hAR 12Q ;Pten +/− DLP, a castrated hAR 12Q ;ETV1 Tg ;Pten +/− DLP, an intact hAR 12Q ;Pten +/− VP, and a normal region of a macroscopic hAR 12Q ;ETV1 Tg ;Pten +/− DLP tumor. Sections were stained with H&E. Immunohistochemical (IHC) staining was performed with antibodies to AR, PTEN, or pAKT protein (brown staining). In situ hybridization (ISH) was performed with a probe against the human ETV1 transcript (red staining). ETV1 expression level is scored as 0–4, with 4 being highest. Images are shown at ×10–20 magnification with ×60 inset. Consecutive sections were used when possible, and each row of images shows sections from a single mouse prostate (PDF 306 kb)
The heatmaps show relative expression of genes differentially expressed (FDR ≤ 0.05) in intact ETV1-transgenic vs. non-transgenic mice on the Pten-hemizygous background. Each hAR Q-tract allele was analyzed separately. For each group, relative expression in castrated mice is included. Each lane represents the average expression from of 2–3 pools of RNA per group. See Fig. 7d for the number of shared and unique genes in each contrast (PDF 61 kb)