

Abstract
Glioblastoma (GBM), the most common primary adult malignant brain tumor, is associated with a poor prognosis due, in part, to tumor recurrence mediated by chemotherapy and radiation resistant glioma stem-like cells (GSCs). The metabolic and epigenetic state of GSCs differs from their non-GSC counterparts, with GSCs exhibiting greater glycolytic metabolism and global hypoacetylation. However, little attention has been focused on the potential use of acetate supplementation as a therapeutic approach. N-acetyl-L-aspartate (NAA), the primary storage form of brain acetate, and aspartoacylase (ASPA), the enzyme responsible for NAA catalysis, are significantly reduced in GBM tumors. We recently demonstrated that NAA supplementation is not an appropriate therapeutic approach since it increases GSC proliferation and pursued an alternative acetate source. The FDA approved food additive Triacetin (glyceryl triacetate, GTA) has been safely used for acetate supplementation therapy in Canavan disease, a leukodystrophy due to ASPA mutation. This study characterized the effects of GTA on the proliferation and differentiation of six primary GBM-derived GSCs relative to established U87 and U251 GBM cell lines, normal human cerebral cortical astrocytes, and murine neural stem cells. GTA reduced proliferation of GSCs greater than established GBM lines. Moreover, GTA reduced growth of the more aggressive mesenchymal GSCs greater than proneural GSCs. Although sodium acetate induced a dose-dependent reduction of GSC growth, it also reduced cell viability. GTA-mediated growth inhibition was not associated with differentiation, but increased protein acetylation. These data suggest that GTA-mediated acetate supplementation is a novel therapeutic strategy to inhibit GSC growth.
Keywords: acetyl coenzyme A synthetase, aspartoacylase, differentiation, epigenetics, glioblastoma, glioma, glyceryl triacetate, metabolism, N-acetyl-L-aspartate, proliferation, triacetin
Introduction
Glioblastoma (GBM, WHO grade IV astrocytoma), the most common primary brain tumor of the adult central nervous system, is associated with a poor prognosis. The standard of care of maximal surgical resection followed by concurrent radiotherapy and temozolomide (TMZ, Temodar®) chemotherapy is associated with a median survival of ~14 months (Stupp et al., 2005). Despite advances in adjuvant therapy, there has been little improvement in outcomes primarily due to the post-surgical persistence of chemotherapy and radiation resistant glioma stem-like cells (GSCs) that contribute to recurrence (Singh et al., 2004). Induction of GSC differentiation appeared to be a logical therapeutic approach. However, it is now evident that GSCs are highly plastic and their differentiation into “terminally” differentiated cells is reversible (Chaffer et al., 2011; Gupta et al., 2011; Jiang et al., 2011). Thus, therapeutic approaches that target both GSCs and their progeny, while sparing resident neural stem cells (NSCs) and oligodendrocyte progenitor cells (OPCs), the most abundant cycling population in the adult brain (Dawson et al., 2003), are of considerable interest.
The extent of histone hypoacetylation is linked to promoter CpG hypermethylation, silencing of tumor-suppressor genes, and poorer clinical outcomes (Seligson et al., 2009). Several histone deacetylase inhibitors (HDACi) (e.g., sodium butyrate, trichostatin A, Vorinostat) have shown promise in glioma treatment (Roesler et al., 2010). However, HDACi have serious limitations, including ineffective penetrance into solid tumors, cardiotoxicity, and thrombocytopenia (Duvic et al., 2007; Gryder et al., 2012). Safe alternative approaches to promote and/or maintain histone acetylation are required; yet, to date researchers have not investigated acetate supplementation as a therapeutic approach. Normally, most of the nuclear acetate required for histone acetylation is derived from glucose via ATP-citrate lyase (ACL)-mediated conversion of mitochondrial-derived citrate to acetyl-Coenzyme A (acetyl-CoA) (Fig. 1) (Wellen et al., 2009). However, in highly proliferative, glycolytic tumor cells, citrate is exported from the mitochondria to the cytosol to support lipid synthesis and biomass accumulation (i.e., Warburg effect) (Hatzivassiliou et al., 2005; Koppenol et al., 2011). This suggests that an alternative nucleocytosolic acetyl-CoA synthetic pathway, distinct from ACL, may provide a therapeutic target.
Figure 1. Acetyl-CoA Synthesis and Metabolism.
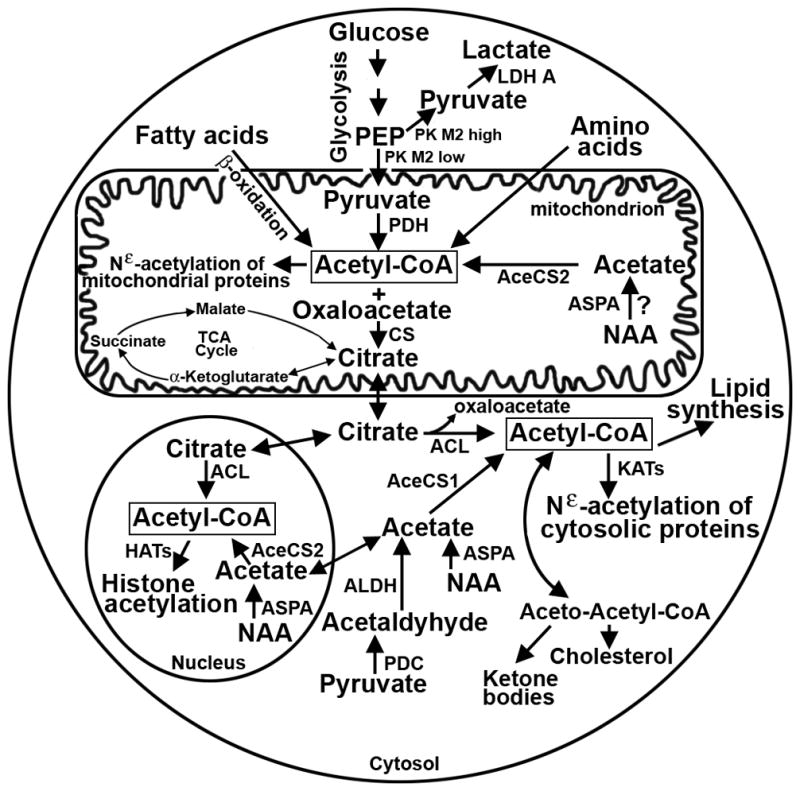
Acetyl-CoA is synthesized within mitochondrial, cytosolic and nuclear subcellular compartments. Under glycolytic conditions, typified by rapidly dividing transformed cells (i.e., high pyruvate kinase M2 activity), glucose is converted to lactate. In contrast, under oxidative conditions (i.e., low pyruvate kinase M2 activity) glucose generates mitochondrial acetyl-CoA to drive energy production via the tricarboxylic acid (TCA) cycle and for the Nε-acetylation of mitochondrial proteins. Mitochondrial acetyl-CoA may also be generated via amino acid metabolism and the β-oxidation of fatty acids. To support biomass accumulation of rapidly dividing cells (e.g., cholesterol and lipid synthesis), cytosolic and nuclear acetyl-CoA is derived from citrate that is exported from the mitochondria, thereby creating a more glycolytic metabolic state. Cytosolic and nuclear acetyl-CoA may also be generated from pyruvate- or NAA-derived acetate. Although it is well accepted that NAA is synthesized in neuronal mitochondria, its mitochondrial synthesis or ASPA-mediated catalysis in other cells has not been investigated. However, NAA provides an important alternative source of acetyl-CoA within the cytosol and nucleus. Abbreviations: AceCS1/2 - acetyl-CoA synthetase 1/2, ACL - ATP-citrate lyase, ADLH - aldehyde dehydrogenase, ASPA - aspartoacylase, CS - citrate synthase, HATs - histone acetyltransferases, KATs - protein acetyltransferases, LDH - lactate dehydrogenase, NAA - N-acetyl-L-aspartate, PDH - pyruvate dehydrogenase, PEP - phosphoenolpyruvate, PK - pyruvate kinase,
N-acetyl-L-aspartate (NAA) is the most concentrated source of acetate in the human brain (12.1 ± 1.5 mM) (Rigotti et al., 2011). Aspartoacylase (ASPA) catalyzes the breakdown of NAA, its only known substrate (Kaul et al., 1991), to L-aspartate, for use in protein synthesis and the Krebs cycle, and acetate (Fig. 1). Free acetate is then converted to acetyl-CoA via cytosolic/nuclear acetyl-CoA synthetase-1 (AceCS1) for lipid biosynthesis and histone/protein acetylation (Goldberg and Brunengraber, 1980) and via mitochondrial AceCS2 for ATP production (Fig. 1) (Fujino et al., 2001). Both NAA and ASPA protein levels are decreased in glioma (Moffett et al., 2007; Tsen et al., 2014). Thus, decreased NAA-derived acetate bioavailability could contribute to histone/protein hypoacetylation as well as an increased glycolytic state by further exhausting the mitochondrial citrate supply. NAA-mediated acetate supplementation is not a candidate therapeutic approach since NAA catalysis requires ASPA, which is also decreased in glioma. Furthermore, we recently demonstrated that NAA promotes GSC proliferation in vitro via mechanisms that remain to be determined (Long et al., 2013a). Hence, an alternative acetate source is required.
Triacetin (glyceryl triacetate, GTA) is ideal for therapeutic acetate supplementation. Unlike free acetate, GTA is readily absorbed by the gastrointestinal tract, freely crosses the blood-brain barrier and plasma membranes, and is hydrolyzed by non-specific lipases and esterases in all cell types to liberate glycerol and acetate. Glycerol can either participate in de novo triglyceride synthesis or be used for glycolysis after conversion to glyceraldehyde-3-phosphate, while the acetate generates acetyl-CoA (Fig. 1) (Reisenauer et al., 2011). Thus, GTA serves as an ideal acetate delivery vehicle.
We demonstrate that GTA induces cytostatic growth arrest of primary tumor-derived GSCs more than established human GBM cells (U87 and U251), but has less effect on normal cells (i.e., normal human astrocytes and murine neural stem cells [NSCs]). Interestingly, GTA is more effective at growth arrest than sodium acetate and glycerol comparable to that generated by complete GTA catalysis. GTA-mediated acetate supplementation does not induce anti-proliferative effects via the promotion of differentiation or apoptosis, but increased acetylation of several proteins, suggesting an epigenetic mechanism of action.
Materials and Methods
Cell Culture
Established glioblastoma cell lines, U87 and U251, were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Mediatech; Herndon, VA) supplemented with 10% fetal bovine serum (FBS; Hyclone; Logan, UT) on untreated cell culture dishes. Human cerebral cortical astrocytes (HA#1800 ScienCell; Carlsbad, CA) were cultured in basal medium with 2% FBS and astrocyte growth supplement (AM#1801 ScienCell). Mouse neural stem cells (NSCs), kindly provided by Dr. Jeffrey Spees (University of Vermont Department of Medicine), were prepared from postnatal day 4 cerebral cortex as described (Shimada et al., 2012). GSCs, kindly provided by Dr. Antonio Chiocca (Brigham and Women’s Hospital Department of Neurosurgery), were isolated from surgical specimens using previously described methodology (Godlewski et al., 2008). The GSCs were derived from frontal lobe GBM tumors: GBM2 from a 47-year-old male, GBM8 from a 70-year-old female, GBM34 from a 78-year-old female, and GBM44 from a 44-year-old male. GBM12 was derived from a recurrent tumor in a 64-year-old female from which GBM9 GSCs were originally established. GSCs were maintained as free-floating spheres in stem cell medium (SCM) consisting of DMEM/F12 (Mediatech) supplemented with 1X B27 supplement (Life Technologies; Carlsbad, CA), 20 ng/ml EGF and 20 ng/ml bFGF (PeproTech; Rocky Hill, NJ) on non-adhesive plastic (Falcon petri dish). GSC differentiation was induced by culturing in DMEM with 10% FBS (differentiation medium, DM). For pharmacological induction of differentiation, GBM12 and GBM34 GSCs were plated in DM in the absence or presence of dibutyryl cAMP (1 mM in water, Sigma; St. Louis, MO), forskolin (10 μM in ethanol, Sigma), all-trans retinoic acid (10 μM in ethanol, Sigma) or the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 (10 μM in dimethylsulfoxide [DMSO], Tocris Bioscience/R & D Systems; Minneapolis, MN) plus the mammalian target of rapamycin (mTOR) inhibitor rapamycin (20 nM in ethanol, Sigma) and the mitogen-activated protein kinase (MEK1/2) inhibitor PD035901 (1 μM in DMSO, Tocris) for 6 days. Since the final DMSO concentration for PD035901 and LY294002 was 0.1% each, 0.2% DMSO was used as a control for PI3K/mTOR/MEK inhibition. All media contained 50 U/ml penicillin and 50 μg/ml streptomycin (Life Technologies) and was replenished every 48 hours.
mRNA and DNA analysis
Reverse transcription polymerase chain reaction (rtPCR) for proneural and mesenchymal antigenic profiling and whole genome cytogenetic analysis using GeneChip® Human Mapping 250K Nsp Arrays (Affymetrix; Santa Clara, CA) were performed as previously described (Tsen et al., 2014).
Growth Assessment
For cell cycle kinetics, cells (0.5 × 106 per 3.5-cm dish) were analyzed 24 hours after treatment with 0.25% GTA (12 mM), 0.25% glycerol, 0.25% triglyceride (canola oil), 12 mM sodium acetate (equivalent to 0.25% GTA), 36 mM sodium acetate (since 3 moles of acetate are derived per mole of GTA) or 36 mM sodium acetate plus 0.25% glycerol. The GTA concentration was selected based on a dose response and represents the lowest concentration that induced growth arrest in both SCM and DM (Tsen et al., 2014). Flow cytometric analysis of propidium iodide labeled cells was performed as described (Long et al., 2013a).
Growth dynamics were assessed using unbiased trypan blue exclusion based cytometry. Cells were plated at 10,000 cells per well of a 24-well plate and treated as described above with medium replenished every 48 hours. After 1, 3, and 5 days of treatment, cells were trypsinized, collected via centrifugation, and counted according to the manufacturer’s instructions (Countess Automated Cell Counter; Life Technologies). Medium was collected, centrifuged briefly to remove cellular debris, and pH measured using a standard pH meter.
Protein Analysis
For western blot analysis, cells were plated at a density of 10,000 cells per cm2 per 6-cm dish and the cells were harvested with Triton Lysis Buffer (Lluri et al., 2008) after 1, 3 and 5 days of treatment. SDS-PAGE (25 μg protein from whole cell lysates) and western blotting were performed as previously described (Lluri et al., 2008). To detect proteins acetylated consequent to GTA treatment, cells were incubated with 0.25% GTA, in the absence of a HDACi, and the cells were harvested in Triton Lysis Buffer 6, 12, or 24 hours later. Because protein acetylation is transient, cells were also treated with 0.25% GTA in the absence or presence of the HDACi Vorinostat (suberoylanilide hydroxamic acid, SAHA; 1 μM, Sigma). Twenty-four hours later, the cells were harvested in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) to increase the extraction of nuclear proteins, particularly histones. Immunocomplexes were visualized by enhanced chemiluminescence (PerkinElmer Life Sciences; Boston, MA). Semi-quantitative densitometry was performed using Quantity One software (Bio-Rad; Hercules, CA) as described (Long et al., 2013b).
For immunocytochemistry (ICC), cells were plated at a density of 20,000 cells per well of 24-well plate, fixed with 2% paraformaldehyde after 1, 3, 5 days of treatment and processed as previously described (Jaworski and Pérez-Martínez, 2006). Immunoreactivity was visualized with a Nikon epifluorescence microscope (MicroVideo Instruments; Avon, MA) and all images were acquired with identical exposure settings using a SPOT RT digital camera (Diagnostic Instruments; Sterling Heights, MI).
The following antibodies were used: rabbit anti-human ASPA (GTX13389 GeneTex, Irvine, CA; 7,500X for western blot, 500X for ICC), rabbit anti-human AceCS1 (2,500X for western blot, 150X for ICC; #6516-1 Epitomics; Burlingame, CA), rabbit anti-human AceCS2 (500X for western blot, 100X for ICC; C14234 Assay Biotech; Sunnyvale, CA), and rabbit anti-E. coli Glutathione S-Transferase pi (GSTπ, 800X; AB8902 Millipore; Billerica, MA). Mouse anti-porcine glial fibrillary acidic protein (GFAP, 5,000X for western blot and ICC; G3893), mouse anti-porcine vimentin (200X; V6630), and rabbit anti-mouse laminin (200X; L9393) were from Sigma. Rabbit anti-acetylated lysine (1,000X; #9441) and mouse anti-CD44 (2,000X, #5640) were from Cell Signaling Technology (Danvers, MA). Goat anti-human actin (1,000X, sc-1616) and rabbit anti-mouse 2′,3′-Cyclic-nucleotide 3′-phosphodiesterase (CNPase; 250X, sc-30158) were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-human Sox2 (1,000X, ab97959), mouse anti-human nestin (1,000X, ab22035), rabbit anti-human Ki67 (50X, ab833), and mouse anti-human neuron-specific βIII tubulin (Tuj1, 3,000X; ab775) were from Abcam (Cambridge, MA). Species-specific HRP- (3,000X) and Cy3- (500X) conjugated secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
Statistical Analysis
Statistical analyses were performed using a minimum of three independently prepared cultures. Data are expressed as mean ± standard error of the mean. Significant differences were determined by either one-way or two-way ANOVA and Bonferroni multiple comparison tests using Prism software (GraphPad; San Diego, CA). p < 0.05 was considered statistically significant.
Results
Mesenchymal GSCs share a common cytogenetic signature identified by rtPCR and Nsp array mapping
Inasmuch as mesenchymal and proneural glioma subtypes exhibit distinct metabolic states (Chinnaiyan et al., 2012; Mao et al., 2013), the cell lines used in the study were subjected to in-depth DNA analysis to determine whether GTA responsiveness was correlated with glioma subtype. First, primary tumor-derived GSCs from 5 distinct patient’s tumors (GBM12 was derived from a recurrent tumor from which GBM9 was derived) were characterized by rtPCR with well-accepted markers of proneural (i.e., CD133, Notch 1, SOX2, platelet-derived growth factor receptor-alpha [PDGFRα], nestin, and Olig2) and mesenchymal (i.e., BCL2A1, Wilms Tumor 1, CD44, and CD44v6 (Jijiwa et al., 2011)) GBM tumors (Phillips et al., 2006; Verhaak et al., 2010) (Supplementary Fig. 1A). GBM8, GBM44, and GBM2 GSCs exhibited a proneural gene profile whereas GBM12, GBM34, and GBM9 GSCs exhibited a mesenchymal gene profile. Immunocytochemistry confirmed the expression of SOX2 and nestin in proneural GSCs and CD44 in mesenchymal GSCs (Supplementary Fig. 1B). Next, whole genome cytogenetic analysis was performed using GeneChip® Human Mapping 250K Nsp arrays to characterize chromosomal amplifications and deletions. The principal component analysis plot of SNP raw intensity data indicated that the established U87 and U251 GBM lines more closely resembled proneural GSCs than mesenchymal GSCs and that mesenchymal GSCs share common chromosomal alterations (Supplementary Fig. 1C). However, examination of individual chromosomal maps (Supplementary Figure 2, (Tsen et al., 2014)) revealed unique copy number variations in each GBM GSC, confirming that each line was derived from a distinct individual.
GTA induces growth arrest of GBM GSCs
Given the decreased NAA levels in glioma tumors (McBride et al., 1995) and that NAA treatment increased GSC proliferation (Long et al., 2013a), the effect of GTA-mediated acetate supplementation on the growth of GBM-derived GSCs was examined. Flow cytometric analysis of cells treated with 0.25% GTA for 24 hours revealed that GTA did not alter proliferation of U87 or U251 cells, NSCs or primary astrocyte (i.e., no change in the percent of cells in S phase) (Fig. 2A), whereas GTA consistently induced G0 growth arrest of all GSCs in SCM (Fig. 2B). Growth arrest was more pronounced for the mesenchymal GSCs than the proneural GSCs. In DM, GTA reduced proliferation of the proneural GSCs and increased proliferation of GBM12 and GBM34 GSCs (Fig. 2C). GTA is cleaved by non-specific lipases and esterases in all cells into acetate and glycerol, yet the active molecule of GTA is widely believed to be acetate (Arun et al., 2010a; Arun et al., 2010b; Bhatt et al., 2013; Brissette et al., 2012; Madhavarao et al., 2009; Mathew et al., 2005; Reisenauer et al., 2011; Soliman and Rosenberger, 2011; Soliman et al., 2012). Hence, the growth inhibitory effect of 0.25% GTA (~12 mM) was compared to 12 mM sodium acetate and 36 mM sodium acetate (since 3 moles of acetate are derived per mole of GTA) in GBM8 and GBM12 GSCs as representative proneural and mesenchymal GBM subtypes, respectively (Figs. 2D, E). The growth effects of 0.25% glycerol alone and 0.25% glycerol plus 36 mM sodium acetate, equivalent to the complete catalysis of 0.25% GTA, were also tested. As an intact molecule, GTA is a short chain triglyceride. All triglycerides shorter than palmitate (C16) are synthetic, with GTA being the shortest. Rather than testing another synthetic triglyceride as a control, we tested the growth effects of canola oil, a naturally occurring, frequently consumed triglyceride that is touted for its high levels of cholesterol-lowering fats (i.e., low in saturated fats and contains omega-6 and omega-3 fatty acids). In SCM, 36 mM sodium acetate alone, 36 mM sodium acetate plus 0.25% glycerol, and GTA reduced proliferation of GBM8 GSCs (Fig. 2D). Interestingly, the inclusion of glycerol decreased the growth inhibitory effects of 36 mM sodium acetate. In GBM12 GSCs, GTA was the only treatment to decrease cell proliferation, suggesting that GTA may exert functions distinct from serving as an acetate delivery vehicle. In DM, GTA-mediated growth inhibition in GBM8 GSCs was greater than 36 mM sodium acetate or 36 mM sodium acetate plus 0.25% glycerol, while in GBM12 GSCs, only 36 mM sodium acetate plus 0.25% glycerol induce G0 growth arrest (Fig. 2E).
Figure 2. GTA induces G0 growth arrest of glioblastoma-derived GSCs in vitro.

Established GBM cell lines in growth medium (A), free-floating GSCs in SCM (B, D), and adherent GSCs in DM (C, E) were treated with 0.25% GTA and, after 24 hours, the cell cycle profile of propidium iodide-labeled cells was analyzed by flow cytometry. A) GTA did not alter the percent of proliferating U87 and U251 GBM cells, mouse neural stem cells (NSCs) or human cerebral cortical astrocytes. B) GTA significantly reduced proliferation via G0 growth arrest of all GSCs except GBM8, which displayed G2/M arrest. The anti-proliferative effect of GTA was more pronounced in aggressive GSCs with a mesenchymal genetic signature (GBM12, GBM34, GBM9). C) In DM, GTA reduced proliferation only in proneural GSCs (GBM8, GBM44, GBM2) and actually increased proliferation of GBM12 and GBM34 GSCs. D, E) Cell cycle profile of a representative proneural GSC (GBM8) and mesenchymal GSC (GBM12) in SCM (D) or DM (E) after 24 hours of treatment with 0.25% triglycerides (canola oil), 0.25% glycerol, 12 mM sodium acetate (NaAc), 36 mM NaAc (equivalent acetate to 0.25% GTA), 36 mM NaAc plus 0.25% glycerol (equivalent to complete catalysis of 0.25% GTA) or 0.25% GTA. Only GTA reduced proliferation of both GSCs in SCM (D). E) In DM, acetate supplementation was more effective at inhibiting proliferation of proneural GBM8 cells than mesenchymal GBM12 cells. n ≥ 3 independent experiments. *p < 0.05, **p ≤ 0.01, #p ≤ 0.001, ##p ≤ 0.0001.
The long-term growth inhibitory effect of 0.25% GTA was tested by unbiased cytometry over 5 days (Fig. 3). GTA reduced growth of U251 cells (mutant p53) more than U87 cells (wild-type p53), modestly reduced growth of human cerebral cortical astrocytes, and actually promoted expansion of NSCs (Fig. 3A). Similar to the growth effects observed after 24 hours, protracted GTA treatment in SCM significantly reduced growth of mesenchymal GSCs and had less effect on proneural GSCs. Interestingly, the mesenchymal GSCs increased their growth rate ~2-fold in DM (Fig. 3C) and were more responsiveness to GTA-mediated growth inhibition than in SCM (Fig. 3B). In contrast, the growth rate of the proneural GSCs decreased in DM. Unlike in SCM, GTA significantly decreased the growth of proneural GSCs in DM. The differences in growth effects between sodium acetate and GTA became more apparent following continuous treatment (Fig. 4). Sodium acetate induced a dose-dependent growth reduction of GBM8 and GBM12 GSCs in both SCM (Fig. 4A) and DM (Fig. 4B). With the exception of GBM12 cells in DM, 36 mM sodium acetate plus 0.25% glycerol exerted greater growth inhibitory effects than GTA. However, it also was associated with a significant decrease in cell viability. While triglycerides promoted the growth of oligodendroglioma-derived GSCs in SCM (Long et al., 2013b), it had no effect on the growth of GBM-derived GSCs in SCM or DM. Similar to oligodendroglioma-derived GSCs (Long et al., 2013b), glycerol largely had no effect on the growth of GBM GSCs (except of GBM8 cells in DM), supporting the contention that acetate is the active component of GTA. Because GTA appeared to promote medium acidification (Supplementary Fig. 3), medium pH was assessed after 1 and 5 days of treatment (Fig. 5). In SCM (Fig. 5A), medium pH did not differ between treatment groups after 1 day, but sodium acetate and GTA treated medium was more basic than control medium after 5 days of treatment (i.e., showed a time-dependent increase in pH). Since medium is changed every 48 hours, the pH of medium with GTA was tested after 2 days incubation. Medium pH after 2 days incubation did not differ from medium incubated for 1 day with GTA. As empirically observed (Supplementary Fig. 3), by 1 day of GTA treatment in DM, the medium was more acidic than other treatments (Fig. 5B). Almost all treatments displayed a time-dependent medium acidification that was more pronounced in rapidly dividing, mesenchymal GBM12 cells than proneural GBM8 cells. Interestingly, the pH of SCM medium increased (Fig. 5C) while the pH of DM medium decreased (Fig. 5D) when incubated with 0.25% GTA in the absence of cells for 5 days, suggesting that GTA may be interacting with medium components to alter pH. In sum, GTA exerts growth inhibitory effects on GSCs and their differentiated progeny comparable to 36 mM sodium acetate plus 0.25% glycerol without decreasing cell viability and has little to no effect on normal NSCs or astrocytes, suggesting GTA would have less off target effects in vivo.
Figure 3. GTA induces growth inhibition of GSCs and their differentiated progeny.

Established GBM cell lines in growth medium (A), free-floating GSCs in SCM (B), and adherent GSCs in DM (C) were treated with 0.25% GTA for 5 days, with medium replenished every 48 hours. GTA-mediated growth dynamics were assessed by unbiased trypan blue exclusion based cytometry after 1, 3, and 5 days of treatment. A) GTA reduced growth of U251 cells greater than U87 cells and only had a modest effect on normal human cerebral cortical astrocytes. In contrast, GTA slightly increased NSC expansion. B) GTA significantly reduced growth of mesenchymal GSCs (GBM12, GBM34, GBM9). However, similar to flow cytometric results at 24 hours (Fig. 2B), GTA was less effective at reducing proneural GSC growth (GBM8, GBM44, modest GBM2 effect). C) Similar to SCM, GTA reduced growth of mesenchymal GSCs in DM. In contrast to growth in SCM, GTA indeed reduced proneural GSC growth in DM. n ≥ 3 independent experiments. *p < 0.05, **p ≤ 0.01, #p ≤ 0.001, ##p ≤ 0.0001.
Figure 4. Sodium acetate is less effective at growth inhibition than GTA.

Free-floating GSCs in SCM (A), and adherent GSCs in DM (B) were treated with 0.25% triglycerides (canola oil), 0.25% glycerol, 12 mM sodium acetate (NaAc), 36 mM NaAc (equivalent acetate to 0.25% GTA), 36 mM NaAc plus 0.25% glycerol (equivalent to complete catalysis of 025% GTA) or 0.25% GTA for 5 days, with medium replenished every 48 hours. Growth dynamics were assessed by unbiased trypan blue exclusion based cytometry after 1, 3, and 5 days of treatment. A) In SCM, 36 mM NaAc plus 0.25% glycerol was more effective at growth reduction than GTA, but was also associated with decreased cell viability. B) In DM, GTA reduced growth of mesenchymal GBM12 GSCs greater than 36 mM NaAc plus 0.25% glycerol and did not reduce cell viability. n ≥ 3 independent experiments. *p < 0.05, **p ≤ 0.01, #p ≤ 0.001, ##p ≤ 0.0001, n.s.- not significant.
Figure 5. Acetate supplementation alters medium pH.

Free-floating GSCs in SCM (A), and adherent GSCs in DM (B) were treated with 0.25% triglycerides (canola oil), 0.25% glycerol, 12 mM sodium acetate (NaAc), 36 mM NaAc (equivalent acetate to 0.25% GTA), 36 mM NaAc plus 0.25% glycerol (equivalent to complete catalysis of 025% GTA) or 0.25% GTA for 5 days, with medium replenished every 48 hours. Medium pH was measured after 1 and 5 days of treatment. A) In SCM, the media pH was comparable after 1 day of treatment. However, after 5 days, acetate supplemented medium was more basic than control, likely due to growth reduction (Fig. 4A). B) In DM, after 1 day GTA medium was already more acidic than other treatments. Medium pH of mesenchymal GBM12 GSCs became more acidified under most treatment conditions. C, D) The effect of GTA on medium pH in the absence cells was assessed in SCM (C) and DM (D). GTA-induced changes in medium pH was independent of cell metabolism. n ≥ 3 independent experiments. *p < 0.05, **p ≤ 0.01, #p ≤ 0.001, ##p ≤ 0.0001, n.s.- not significant.
GTA does not significantly alter ASPA, AceCS1, or AceCS2 protein levels
To begin to address the mechanism by which GTA reduced cell growth, the expression and spatial localization of ASPA and acetyl-CoA synthesizing enzymes, AceCS1 and AceCS2, were examined. Mesenchymal GSCs expressed more ASPA protein than proneural GSCs in SCM (p=0.01), but not DM (p=0.12) (Fig. 6A). GTA decreased ASPA protein levels in GBM44 and GBM2 cells in DM, but otherwise had no effect on ASPA protein levels. Because ASPA undergoes cytosolic-nuclear shuttling (Hershfield et al., 2006), it is critical to examine its spatial localization (Figs. 6B, C). Immunocytochemistry of GBM9 and GBM8 cells, as representative mesenchymal and proneural GSCs, respectively, revealed that ASPA was expressed in the cytosol and nucleus of cells in SCM (Fig. 6B). Other than inducing a more adherent and flattened cell morphology, GTA had no discernable effect on ASPA spatial localization. In DM, ASPA appeared primarily nuclear in GBM9 cells and more cytosolic in GBM8 cells. GTA appeared to increase cytosolic ASPA in both cells. This apparent difference is spatial localization in DM was further examined in all cell lines at higher magnification (Fig. 6C). Indeed, in the proneural lines (GBM2, GBM8, GBM44), ASPA was enriched in the cytosol, and this was enhanced by GTA. In contrast, in the rapidly proliferating mesenchymal lines (GBM9, GBM12, GBM34), ASPA was enriched within the nucleus. GTA-mediated growth arrest was associated with a loss of nuclear ASPA, except in the small proportion of cells that continue to divide in the presence of GTA (Fig. 6C insets). Thus, the spatial localization of ASPA, and not the total level of ASPA protein, appears to be regulated by GTA.
Figure 6. GTA treatment alters ASPA cytosolic-nuclear spatial localization.

A) Western blots (25 μg whole cell lysate) and densitometric analysis of ASPA protein levels normalized to actin. Treatment with 0.25% GTA had no effect on ASPA protein levels in SCM and reduced ASPA protein levels in DM in GBM44 and GBM2 cells. B) ASPA immunocytochemistry in representative mesenchymal (GBM9) and proneural (GBM8) cells after 3 days of GTA treatment. In SCM (GSCs grown adherently on PLL), ASPA was abundantly expressed in the cytosol and nucleus of GSCs, even in GTA treated cells. In DM, ASPA was similarly detected in the cytosol and nucleus in untreated cells, but appeared more cytosolic in GTA treated cells. C) ASPA immunoreactivity in cells treated with 0.25% GTA in DM for 3 days. Examination at higher magnification revealed that ASPA was primarily cytosolic in the proneural cells, regardless of GTA treatment. In contrast, in mesenchymal cells, ASPA was abundantly detected in the nucleus in the absence of GTA and was almost exclusively cytosolic in GTA treated, growth arrested cells. The exception was the presence of nuclear ASPA in dividing cells (insets). n ≥ 3 independent experiments. Unless otherwise indicated, symbols represent differences relative to day 1 cells of the same treatment condition. *p < 0.05. Scale bar = 100 μm (B), 50 μm (C).
Since acetate generated via GTA cleavage must be converted to acetyl-CoA via cytosolic/nuclear AceCS1 and/or mitochondrial AceCS2, the expression and spatial localization of these acetyl-CoA synthesizing enzymes were examined. Mesenchymal and proneural GSCs had comparable AceCS1 levels in SCM (p=0.41) and DM (p=0.76) (Fig. 7A). Growth in DM decreased AceCS1 protein levels in most of the cell lines. GTA did not alter AceCS1 protein levels either in SCM or DM. Although AceCS1 is abundantly expressed in the nucleus of oligodendroglial cells (Ariyannur et al., 2010; Moffett et al., 2011) and oligodendroglioma-derived GSCs (Long et al., 2013b), it was primarily cytosolic in both mesenchymal and proneural GBM GSCs under all culture conditions (Fig. 7B). AceCS2 levels were greater in proneural GSCs both in SCM (p=0.0006) and DM (p<0.0001) (Fig. 8A). GTA modestly reduced AceCS2 levels in GBM2 cells in DM; otherwise, GTA did not appreciably alter AceCS2 protein levels or spatial localization (Fig. 8B). Hence, GTA-mediated growth arrest had no effect on the levels of acetyl-CoA synthesizing enzymes.
Figure 7. GTA treatment does not alter acetyl-CoA synthetase 1 expression.

A) Western blots (25 μg whole cell lysate) and densitometric analysis of AceCS1 protein levels normalized to actin. Most GSC lines expressed more AceCS1 in SCM than DM. Treatment with 0.25% GTA had no effect on AceCS1 protein levels in SCM or DM. B) AceCS1 immunocytochemistry in representative mesenchymal (GBM9) and proneural (GBM8) cells after 3 days of GTA treatment. In all cases, AceCS1 was primarily cytosolic. n ≥ 3 independent experiments. Unless otherwise indicated, symbols represent differences relative to day 1 cells of the same treatment condition. *p < 0.05, **p ≤ 0.01, #p ≤ 0.001. Scale bar = 100 μm.
Figure 8. GTA treatment does not alter acetyl-CoA synthetase 2 expression.

A) Western blots (25 μg whole cell lysate) and densitometric analysis of AceCS2 protein levels normalized to actin. AceCS2 was comparably expressed in SCM and DM, but expression was greater in proneural than mesenchymal cells. Treatment with 0.25% GTA had no effect on AceCS2 protein levels in SCM and reduced AceCS2 protein levels in DM in GBM34 and GBM2 cells. B) AceCS2 immunocytochemistry in representative mesenchymal (GBM9) and proneural (GBM8) cells after 3 days of GTA treatment. AceCS2 displayed punctate cytosolic expression indicative of mitochondrial localization, which was unaffected by GTA treatment. n ≥ 3 independent experiments. Unless otherwise indicated, symbols represent differences relative to day 1 cells of the same treatment condition. *p < 0.05, **p ≤ 0.01. Scale bar = 100 μm.
Mesenchymal GSCs are recalcitrant to serum-induced differentiation
To assess the differentiation capacity of the GSCs, the expression of the astrocytic marker GFAP was examined (Fig. 9). Growth factor depletion (i.e., growth in medium with 10% FBS without addition of bFGF or EGF) failed to promote GFAP protein levels in the mesenchymal GSCs (GBM12, GBM34, GBM9), but significantly increased GFAP protein levels in proneural GSCs (GBM8, GBM44, GBM2) (Fig. 9A). GTA decreased GFAP protein levels in GBM44 GSCs in SCM and attenuated DM-induced GFAP in GBM2 GSCs. Although GTA induced morphological alterations in GBM9 cells after 3 days in DM, no GFAP-positive cells were detectable (Fig. 9B). Immunocytochemistry of cells cultured in DM for 5 days further confirmed the differentiation capacity of the GSCs (Fig. 10). While the proneural GSCs differentiated into GFAP-positive astrocytes, Tuj1-positive neurons, and CNPase-positive oligodendrocytes, the mesenchymal lines failed to express these markers of differentiation (Fig. 10A). Rather, the mesenchymal GSCs retained the expression of CD44 and abundantly expressed the proliferation marker Ki67 despite growth in differentiation permissive conditions (Fig. 10B); thus, supporting the increased proliferation observed when cells were grown in DM relative to SCM (Figs. 3B, C). Treatment with 0.25% GTA appeared to reduce immunoreactivity for CD44, the drug resistance protein GST π (Calatozzolo et al., 2005), laminin, and Ki67 (Fig. 10B), but failed to promote immunoreactivity for GFAP, Tuj1, or CNPase (Supplemental Figs. 4, 5). Finally, we attempted to induce differentiation using agonists and antagonists of known signaling pathways dysregulated in glioma (Supplemental Figs. 4, 5). Activation of adenylyl cyclase or inhibition of PI3K, mTOR, and MAPK (Ras-Raf-MEK-ERK pathway) signaling failed to promote differentiation of GBM12 (Supplementary Fig. 4) or GBM34 (Supplementary Fig. 5) cells. Although some treatments were associated with morphological alterations, these had little or no effect on the expression of GFAP, TuJ1, or CNPase. Therefore, GTA-mediated growth arrest is not due to the promotion of differentiation.
Figure 9. GTA-mediated growth inhibition is not due to the promotion of differentiation.

A) Western blots (25 μg whole cell lysate) and densitometric analysis of GFAP protein levels normalized to actin. No GFAP expression, either in SCM or DM, was detected in the GSCs with a mesenchymal signature (GBM12, GBM34, GBM9). In contrast, growth factor depletion of proneural GSCs (GBM8, GBM44, GBM2) was associated with significantly increased GFAP expression. Treatment with 0.25% GTA had no effect on differentiation, except in GBM44 GSCs in SCM where GTA decreased GFAP levels and in GBM2 GSCs where GTA attenuated DM-induced GFAP expression. B) GFAP immunocytochemistry in representative mesenchymal (GBM9) and proneural (GBM8) cells after 3 days of GTA treatment in DM. GBM9 cells failed to express GFAP under conditions permissive to differentiation (i.e., 10% FBS). n ≥ 3 independent experiments. Unless otherwise indicated, symbols represent differences relative to day 1 cells of the same treatment condition. *p < 0.05, **p ≤ 0.01. Scale bar = 100 μm.
Figure 10. Mesenchymal GSCs display reduced differentiation capacity.

Immunocytochemistry of cells cultured in DM for 5 days in the absence (A) or presence (B) of GTA. Human cerebral cortical astrocytes were used as a control. A) In the absence of GTA, the proneural GSCs differentiated into GFAP-positive astrocytes, Tuj1-positive neurons, and CNPase-positive oligodendrocytes; yet, a subset of cells retained expression of the progenitor marker nestin. In contrast, the mesenchymal GSCs failed to exhibit markers of differentiation. Moreover, the cells abundantly expressed the proliferation marker Ki67, even under growth conditions that promote differentiation. B) Immunocytochemistry of cells cultured with 0.25% GTA in DM for 5 days. Both mesenchymal GBM12 and proneural GBM8 cells exhibited decreased immunoreactivity for the hyaluronan receptor CD44, drug resistance protein GSTπ, extracellular matrix protein laminin, and proliferation marker Ki67. Scale bars = 100 μm.
GTA promotes protein acetylation
Previous studies have demonstrated a role for GTA in histone acetylation in vivo (Soliman and Rosenberger, 2011; Soliman et al., 2012). To identify other proteins that may be acetylated by GTA in vitro, proneural (GBM8) and mesenchymal (GBM12) GSCs were treated with 0.25% GTA, in the absence of an HDACi, for 6, 12 or 24 hours and the extent of lysine acetylation was assessed by western blot analysis using an antibody specific for acetylated lysine residues (Figs. 11). Even in the absence of an HDACi to prevent in vitro deacetylation, GTA increased acetylation of several proteins. Acetylation was more pronounced in GBM8 cells (Fig. 11A) than GBM12 cells (Fig. 11B) and more pronounced in SCM than DM. The acetylation was specific since actin acetylation was not increased. Inasmuch as acetylation is a dynamic process, it is possible that proteins acetylated by GTA could have become deacetylated either in culture or subsequent to extraction in Triton Lysis Buffer. Therefore, cells were treated with 0.25% GTA in the absence or presence of the HDACi SAHA (1 μM) for 24 hours, extracted with RIPA buffer to increase histone protein extraction, and analyzed by western blot analysis (Fig. 11C). Not unexpectedly, there was a significant increase in the number of acetylated proteins in SAHA treated cells. Preliminary mass spectrometry analysis of GBM12 treated cells has characterized the identity of some of the proteins whose acetylation was increased by GTA, including increased acetylation of histone H4K8, H4K12, and H4K16 as well as several other proteins involved in cell cycle regulation (Lam & Jaworski, unpublished observation). In sum, GTA induced growth arrest, which was more pronounced in rapidly proliferating mesenchymal GSCs, without altering the levels of acetyl-CoA synthesizing enzymes. Furthermore, GTA increased acetylation of several proteins without promoting differentiation, suggesting GTA exerts it growth inhibitory effect via an epigenetic mechanism involving the promotion of lysine acetylation of proteins involved in cell cycle regulation.
Figure 11. GTA increases protein acetylation in GSCs in vitro.
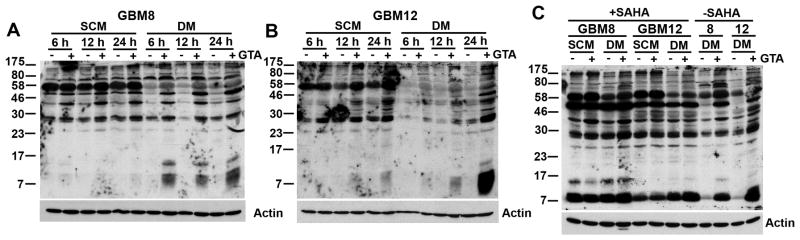
GBM8 (A) and GBM12 (B) GSCs were treated with 0.25% GTA in the absence of an HDACi, the cells were harvested at the indicated time points, and analyzed by western blot analysis (25 μg protein, whole cell Triton Lysis Buffer lysate) with an antibody specific to acetylated lysine residues. Both GSCs displayed a time-dependent increased acetylation of several proteins. C) Cells were treated with 0.25% GTA in the absence or presence of the HDACi SAHA (1 μM) for 24 hours and analyzed by western blot analysis (25 μg protein, whole cell RIPA Buffer lysate). There was a significant increase in the number of acetylated proteins in SAHA treated cells. Even in the absence of SAHA, GTA enhanced acetylation of several proteins, including within the histone molecular weight range (H1-21.3kDa, H2a-14.3kDa, H2b-13.8kDa, H3-15.0kDa, and H4-11.3kDa).
DISCUSSION
GTA displays the greatest efficacy on cancer cells with the most aggressive phenotype, including greatest growth rate, glycolytic metabolic state, and genetic alteration (e.g., mutant p53). GTA induced growth arrest of GSCs to a greater extent than established glioma cell lines (U87, U251) and had little to no effect on normal cells (human cerebral cortical astrocytes, murine NSCs). GTA also displayed greater efficacy on more aggressive mesenchymal GSCs than proneural GSCs (Colman et al., 2010) and on U251 cells, which possess mutant p53, more than U87 cells, which possess wild-type p53. We showed that proneural GSCs expressed more mitochondrial AceCS2 suggesting they had greater capacity to oxidize acetate than mesenchymal GSCs, supporting previous reports that mesenchymal glioma cells are more glycolytic (Chinnaiyan et al., 2012; Mao et al., 2013). Rapidly dividing cells are more glycolytic because they divert mitochondrial citrate to the cytosol to support biomass accumulation (Hatzivassiliou et al., 2005; Koppenol et al., 2011). We believe that GTA had little effect on NSC and normal astrocyte proliferation because these slowly cycling cells are more oxidative; thus, GTA should not negatively affect normal brain cells in vivo when administered therapeutically. By serving as an exogenous source of free acetate, GTA may permit citrate to remain within the mitochondria and promote oxidative phosphorylation and thereby antagonize the Warburg effect. However, a recent study demonstrated that GTA does not alter mitochondrial biogenesis under physiological conditions in vivo (Bhatt et al., 2013). Therefore, we propose that in cells that preferentially express AceCS1, as opposed to AceCS2, GTA-derived acetate primarily supports cytoplasmic and nuclear acetyl-CoA synthesis to promote protein acetylation.
GTA likely contributes to the acetylation of a plethora of proteins since it is primarily viewed as a delivery vehicle for acetate (Arun et al., 2010a; Arun et al., 2010b; Bhatt et al., 2013; Brissette et al., 2012; Madhavarao et al., 2009; Mathew et al., 2005; Reisenauer et al., 2011; Soliman and Rosenberger, 2011; Soliman et al., 2012). The identification of 3600 acetylation sites on 1750 proteins indicates that the prevalence of acetylation rivals that of phosphorylation as a post-translational modification (Choudhary et al., 2009). In particular, N-terminal acetylation at the ε-amino group of lysine residues is one of the most common protein modifications, occurring on 80–90% of cytosolic mammalian proteins (Persson et al., 1985; Starheim et al., 2012). Lysine acetylation regulates diverse protein properties, including DNA-protein interactions (Yang and Seto, 2008), DNA repair response (Li et al., 2010; Robert et al., 2011; Shubassi et al., 2012), autophagy (Bánréti et al., 2013), subcellular localization (Forte et al., 2011), protein stability (Abe et al., 2000; Hwang et al., 2010) and protein-protein interaction (Scott et al., 2011).
Thus far, the only identified substrates for GTA-mediated acetylation are histones (i.e., H3K9, H4K8, and H4K16) (Soliman and Rosenberger, 2011; Soliman et al., 2012). Acetylation of H4K16 is particularly noteworthy since loss of H4K16 acetylation is a common hallmark of cancer (Fraga et al., 2005), while acetylated H4K16 is an important epigenetic regulator that marks actively transcribed euchromatin. The nuclear co-localization of ASPA and AceCS1 is thought to support AceCS1-dependent acetyl-CoA synthesis and histone acetylation subsequent to ASPA-mediated NAA catalysis (Ariyannur et al., 2010; Hershfield et al., 2006; Moffett et al., 2011; Takahashi et al., 2006). Nuclear AceCS1 may also exert ASPA/NAA-independent functions (e.g., recycling HDAC-derived free acetate). In contrast to the abundant nuclear AceCS1 immunoreactivity observed in oligodendroglioma-derived GSCs (Long et al., 2013b), nuclear AceCS1 was not as prominent in GBM-derived GSCs, even in mesenchymal GSCs in DM that abundantly expressed nuclear ASPA. This glial-specific difference could be due to the fact that extensive histone acetylation maintains OPCs in a proliferative state whereas astrocytic acetylation promotes differentiation (Marin-Husstege et al., 2002). We recently performed an acetylome mass spectrometry analysis of GTA treated cells and have identified several candidate cell cycle regulatory proteins that may contribute to GTA-mediated growth arrest.
Acetylation of non-histone proteins may also underlie the growth inhibitory effects of GTA (Singh et al., 2010). For example, acetylation enhances p53-dependent transcription and facilitates the cellular response to genotoxic stress (Marouco et al., 2013; Pisano et al., 2010; Tang et al., 2008). Recently, mitochondrial protein acetylation has garnered considerable interest as a potential therapeutic approach to restore dysregulated metabolism in cancer (Xu et al., 2013). Under basal conditions, over one-third of all proteins in mitochondria are acetylated (Anderson and Hirschey 2012; Choudhary et al., 2009; Kim et al., 2006; Newman et al., 2012). However, in response to nutrient fluctuations, almost every metabolic enzyme involved in the Krebs cycle, fatty acid metabolism, urea cycle and glycogen metabolism becomes acetylated (Wang et al., 2010; Xu et al., 2013). Hence, we do not rule out the possibility that GTA-derived acetate promotes oxidative phosphorylation similar to dichloroacetate, which promotes mitochondrial pyruvate oxidation by stimulating activity of the enzyme pyruvate dehydrogenase (Michelakis et al., 2010; Michelakis et al., 2008; Xie et al., 2011). GTA-dependent media acidification raised concerns since extracellular acidification rate in vitro is primarily driven by lactic acid release, suggesting that GTA treated cells exhibited accelerated glycolysis. However, GTA incubated with DM in the absence of cells also showed media acidification, suggesting that GTA was reacting with a media component. Treatment with media acidified with hydrochloric acid to a comparable pH failed to induce growth arrest, indicating that media pH alone was not the basis for GTA-induced growth arrest.
Acetyl-CoA levels increase over 2-fold in brain, heart and liver following GTA administration (Reisenauer et al., 2011); yet, we did not observe increased AceCS protein levels in GBM GSCs. We acknowledge that AceCS enzymatic activity may be increased without increasing total protein levels. Alternatively, the lack of increased AceCS protein levels could be due to a feedback mechanism since acetylation inactivates AceCS1 and AceCS2 (Hallows et al., 2006; Starai et al., 2002). However, this would decrease the cell’s ability to convert GTA-derived acetate to biologically active acetyl-CoA and would beg the question of what the cell does with the additional acetate.
An alternative hypothesis to GTA serving as an acetate source is that, as a short-chain fatty acid, GTA may function as an HDACi, similar to sodium butyrate. In fact, GTA inhibits HDAC activity and expression (Soliman and Rosenberger, 2011). Studies have never examined whether GTA remains intact within cells because the abundance of intracellular lipases and esterase would likely result in complete catalysis. Three features distinguish GTA and sodium acetate treated cells, suggesting that GTA may subserve functions other than as an acetate source. First, GTA (0.25%, 12 mM) was as, or more, effective at growth arrest than 12 mM glycerol plus 36 mM sodium acetate, concentrations that would be generated by complete catalysis of 0.25% GTA. GTA has also been shown to be a more effective than calcium acetate as a method of increasing brain acetate levels (Mathew et al., 2005). Second, GTA, but not 12 mM glycerol plus 36 mM sodium acetate, resulted in media acidification. Finally, whereas GTA induced growth arrest without inducing apoptosis, as demonstrated by trypan blue staining and lack of activated caspase-3 expression, 12 mM glycerol plus 36 mM sodium acetate significantly increased cell death. However, HDACi induce terminal cell differentiation and apoptosis (Marks et al., 2000), neither of which was observed in GTA treated cells. Thus, further studies are needed to determine the exact mechanism of GTA-induced cytostasis.
GTA is not the only dietary agent that displays chemotherapeutic effects. Numerous natural and synthetic products, including a broad range of dietary agents (e.g., short-chain fatty acids sodium butyrate and sodium proprionate, genistein, quercetin, trichostatin A, sulforaphane, resveratrol, caffeine) exhibit HDAC inhibitory activity in vitro and/or in vivo (Myzak et al., 2006; Seidel et al., 2012). HDAC inhibition has also been considered a therapeutic approach for a number of neuropathologies, including neurodegenerative disorders (e.g., Alzheimer’s disease, Huntington’s disease, Parkinson’s disease) (Konsoula and Barile, 2012), cerebral ischemia (Schweizer et al., 2013), acute central nervous system injury (Moffett et al., 2013; Shein and Shohami, 2011), neuropsychiatric disorders (Ariyannur et al., 2013), muscular dystrophies (Consalvi et al., 2011) and multiple sclerosis (Faraco et al., 2011). We acknowledge that GTA provides acetate globally; thus, we cannot target specific cells or proteins. Since GTA-mediated acetylation could transcriptionally activate oncogenes (Lin et al., 2013; Ohshiro et al., 2010) as well as tumor suppressors and increased histone acetylation of myelin basic protein may exacerbate multiple sclerosis (Koch et al., 2013), we are cautious not to view GTA as a panacea. Inasmuch as GTA is an FDA approved food additive with “generally regarded as safe” status that has been tested for parenteral nutrition in a wide variety of species with no adverse effects (Fiume and Panel., 2003) and has been chronically administered to infants with Canavan disease without cardio-, hemo- or hepatotoxicity (Segel et al., 2011), we assert that identification of GTA’s targets and further investigations of GTA as a chemotherapeutic adjuvant are warranted.
Supplementary Material
Acknowledgments
This work was supported by R01NS045225 co-funded by NINDS and NCRR, and Pilot Project grants from the Vermont Cancer Center/Lake Champlain Cancer Research Organization, Neuroscience COBRE (NIH NCRR P20 RR016435), and UVM College of Medicine (DMJ). Facilities and equipment supported by the Neuroscience COBRE Molecular Core Facility (NIH NCRR P20 RR016435), Vermont Cancer Center DNA Analysis Facility (NIH P30 CA22435), Vermont Genetics Network Bioinformatics Core and Microarray Facility (NIH NIGMS 8P20GM103449), and the Harry Hood Bassett Flow Cytometry and Cell Sorting Facility were instrumental to the completion of the study.
The corresponding author wishes to thank Professor Dylan R. Edwards and Dr. Caroline J. Pennington (University of East Anglia School of Biological Sciences, Norwich UK) for the fabulous sabbatical experience performing the TLDA-based degradome profiling that identified ASPA dysregulation in glioma, Drs. William C. Broaddus and Helen L. Fillmore (Virginia Commonwealth University Division of Neurosurgery) for providing the necessary surgical samples, and Dr. John R. Moffett (Uniformed Services University of the Health Sciences Department of Anatomy, Physiology & Genetics) for insightful discussions regarding acetate metabolism and therapeutic uses of GTA. We acknowledge Dr. Antonio Chiocca (Brigham and Women’s Department of Neurosurgery) for kindly providing the GSCs and Dr. Jeffrey Spees (UVM Department of Medicine) for providing mouse neural stem cells and Sox2 antibody.
Abbreviations
- acetyl-CoA
acetyl coenzyme A
- AceCS1
acetyl coenzyme A synthetase 1
- AceCS2
acetyl coenzyme A synthetase 2
- ASPA
aspartoacylase
- ACL
ATP-citrate lyase
- CNPase
2′, 3′-cyclic nucleotide 3′-phosphodiesterase
- DM
differentiation medium
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
fetal bovine serum
- GBM
glioblastoma
- GFAP
glial fibrillary acidic protein
- GSC
glioma stem-like cell
- GTA
glyceryl triacetate
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- ICC
immunocytochemistry
- NAA
N-acetyl-L-aspartate
- NSC
neural stem cell
- PCR
polymerase chain reaction
- PLL
poly-L-lysine
- SAHA
suberoylanilide hydroxamic acid
- SCM
stem cell medium
- SNP
single nucleotide polymorphism
- STR
short tandem repeat
References
- Abe A, Saeki K, Yasunaga T, Wakabayashi T. Acetylation at the N-terminus of actin strengthens weak interaction between actin and myosin. Biochem Biophys Res Commun. 2000;268:14–19. doi: 10.1006/bbrc.1999.2069. [DOI] [PubMed] [Google Scholar]
- Anderson KA, Hirschey MD. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012;52:23–35. doi: 10.1042/bse0520023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyannur PS, Arun P, Barry ES, Andrews-Shigaki B, Bosomtwi A, Tang H, Selwyn R, Grunberg NE, Moffett JR, Namboodiri AM. Do reductions in brain N-acetylaspartate levels contribute to the etiology of some neuropsychiatric disorders? J Neurosci Res. 2013;91:934–942. doi: 10.1002/jnr.23234. [DOI] [PubMed] [Google Scholar]
- Ariyannur PS, Moffett JR, Madhavarao CN, Arun P, Vishnu N, Jacobowitz DM, Hallows WC, Denu JM, Namboodiri AM. Nuclear-cytoplasmic localization of acetyl coenzyme a synthetase-1 in the rat brain. J Comp Neurol. 2010;518:2952–2977. doi: 10.1002/cne.22373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Ariyannur PS, Moffett JR, Xing G, Hamilton K, Grunberg NE, Ives JA, Namboodiri AM. Metabolic acetate therapy for the treatment of traumatic brain injury. J Neurotrauma. 2010a;27:293–298. doi: 10.1089/neu.2009.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Madhavarao CN, Moffett JR, Hamilton K, Grunberg NE, Ariyannur P, Gahl WA, Anikster Y, Mog S, Hallows WC, Denu JM, Namboodiri AM. Metabolic acetate therapy improves phenotype in the tremor rat model of Canavan disease. J Inherit Metab Dis. 2010b;33:195–210. doi: 10.1007/s10545-010-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bánréti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy. 2013;9:819–829. doi: 10.4161/auto.23908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DP, Houdek HM, Watt JA, Rosenberger TA. Acetate supplementation increases brain phosphocreatine and reduces AMP levels with no effect on mitochondrial biogenesis. Neurochem Int. 2013;62:296–305. doi: 10.1016/j.neuint.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissette CA, Houdek HM, Floden AM, Rosenberger TA. Acetate supplementation reduces microglia activation and brain interleukin-1β levels in a rat model of Lyme neuroborreliosis. J Neuroinflammation. 2012;9:249. doi: 10.1186/1742-2094-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calatozzolo C, Gelati M, Ciusani E, Sciacca FL, Pollo B, Cajola L, Marras C, Silvani A, Vitellaro-Zuccarello L, Croci D, Boiardi A, Salmaggi A. Expression of drug resistance proteins Pgp, MRP1, MRP3, MRP5 and GST-pi in human glioma. J Neurooncol. 2005;74:113–121. doi: 10.1007/s11060-004-6152-7. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan P, Kensicki E, Bloom G, Prabhu A, Sarcar B, Kahali S, Eschrich S, Qu X, Forsyth P, Gillies R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012;72:5878–5888. doi: 10.1158/0008-5472.CAN-12-1572-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A, Popoff S, Nutt CL, Louis DN, Cairncross J, Gilbert MR, Phillips HS, Mehta MP, Chakravarti A, Pelloski CE, Bhat K, Feuerstein BG, Jenkins RB, Aldape K. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12:49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consalvi S, Saccone V, Giordani L, Minetti G, Mozzetta C, Puri PL. Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol Med. 2011;17:457–465. doi: 10.2119/molmed.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Cavone L, Chiarugi A. The therapeutic potential of HDAC inhibitors in the treatment of multiple sclerosis. Mol Med. 2011;17:442–447. doi: 10.2119/molmed.2011.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiume MZ CIRE Panel. Final report on the safety assessment of triacetin. Int J Toxicol. 2003;22(Suppl 2):1–10. [PubMed] [Google Scholar]
- Forte GM, Pool MR, Stirling CJ. N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLoS Biol. 2011;9:e1001073. doi: 10.1371/journal.pbio.1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer N, Pérez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem. 2001;276:11420–11426. doi: 10.1074/jbc.M008782200. [DOI] [PubMed] [Google Scholar]
- Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, Raychaudhury A, Newton HB, Chiocca EA, Lawler S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- Goldberg RP, Brunengraber H. Contributions of cytosolic and mitochondrial acetyl-CoA syntheses to the activation of lipogenic acetate in rat liver. Adv Exp Med Biol. 1980;132:413–418. doi: 10.1007/978-1-4757-1419-7_41. [DOI] [PubMed] [Google Scholar]
- Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Fillmore CM, Jiang G, Shapira S, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Hershfield JR, Madhavarao CN, Moffett JR, Benjamins JA, Garbern JY, Namboodiri A. Aspartoacylase is a regulated nuclear-cytoplasmic enzyme. FASEB J. 2006;20:2139–2141. doi: 10.1096/fj.05-5358fje. [DOI] [PubMed] [Google Scholar]
- Hwang CS, Shemorry A, Varshavsky A. N-terminal acetylation of cellular proteins creates specific degradation signals. Science. 2010;327:973–977. doi: 10.1126/science.1183147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski DM, Pérez-Martínez L. Tissue inhibitor of metalloproteinase-2 (TIMP-2) expression is regulated by multiple neural differentiation signals. J Neurochem. 2006;98:234–247. doi: 10.1111/j.1471-4159.2006.03855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B Can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia. 2011;13:492–503. doi: 10.1593/neo.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jijiwa M, Demir H, Gupta S, Leung C, Joshi K, Orozco N, Huang T, Yildiz VO, Shibahara I, de Jesus JA, Yong WH, Mischel PS, Fernandez S, Kornblum HI, Nakano I. CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS One. 2011;6:e24217. doi: 10.1371/journal.pone.0024217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul R, Casanova J, Johnson AB, Tang P, Matalon R. Purification, characterization, and localization of aspartoacylase from bovine brain. J Neurochem. 1991;56:129–135. doi: 10.1111/j.1471-4159.1991.tb02571.x. [DOI] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Koch MW, Metz LM, Kovalchuk O. Epigenetic changes in patients with multiple sclerosis. Nat Rev Neurol. 2013;9:35–43. doi: 10.1038/nrneurol.2012.226. [DOI] [PubMed] [Google Scholar]
- Konsoula Z, Barile FA. Epigenetic histone acetylation and deacetylation mechanisms in experimental models of neurodegenerative disorders. J Pharmacol Toxicol Methods. 2012;66:215–220. doi: 10.1016/j.vascn.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, Min J, Dou Y. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010;30:5335–5347. doi: 10.1128/MCB.00350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, Lei QY. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell. 2013;51:506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluri G, Langlois GD, Soloway PD, Jaworski DM. Tissue inhibitor of metalloproteinase-2 (TIMP-2) regulates myogenesis and b1 integrin expression in vitro. Exp Cell Res. 2008;314:11–24. doi: 10.1016/j.yexcr.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long PM, Moffett JR, Namboodiri AMA, Viapiano MS, Lawler SE, Jaworski DM. N-acetylaspartate (NAA) and N-acetylaspartylglutamate (NAAG) promote growth and inhibit differentiation of glioma stem-like cells. J Biol Chem. 2013a;288:26188–26200. doi: 10.1074/jbc.M113.487553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long PM, Tighe SW, Driscoll HE, Moffett JR, Namboodiri AMA, Viapiano MS, Lawler SE, Jaworski DM. Acetate supplementation induces growth arrest of NG2/PDGFRα-positive oligodendroglioma-derived tumor-initiating cells. PLoS One. 2013b;8:e80714. doi: 10.1371/journal.pone.0080714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Anikster Y, Mog SR, Staretz-Chacham O, Moffett JR, Grunberg NE, Gahl WA, Namboodiri AM. Glyceryl triacetate for Canavan disease: a low-dose trial in infants and evaluation of a higher dose for toxicity in the tremor rat model. J Inherit Metab Dis. 2009;32:640–650. doi: 10.1007/s10545-009-1155-3. [DOI] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, Wang M, Hu B, Cheng SY, Sobol RW, Nakano I. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci USA. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Husstege M, Muggironi M, Liu A, Casaccia-Bonnefil P. Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J Neurosci. 2002;22:10333–10345. doi: 10.1523/JNEUROSCI.22-23-10333.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- Marouco D, Garabadgiu AV, Melino G, Barlev NA. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 2013;4:1556–1571. doi: 10.18632/oncotarget.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Arun P, Madhavarao CN, Moffett JR, Namboodiri MA. Progress toward acetate supplementation therapy for Canavan disease: glyceryl triacetate administration increases acetate, but not N-acetylaspartate, levels in brain. J Pharmacol Exp Ther. 2005;315:297–303. doi: 10.1124/jpet.105.087536. [DOI] [PubMed] [Google Scholar]
- McBride DQ, Miller BL, Nikas DL, Buchthal S, Chang L, Chiang F, Booth RA. Analysis of brain tumors using 1H magnetic resonance spectroscopy. Surg Neurol. 1995;44:137–144. doi: 10.1016/0090-3019(95)00139-5. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Arun P, Ariyannur PS, Garbern J, Jacobowitz DM, Namboodiri AM. Extensive aspartoacylase expression in the rat central nervous system. Glia. 2011;59:1414–1434. doi: 10.1002/glia.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Arun P, Ariyannur PS, Namboodiri AM. N-Acetylaspartate reductions in brain injury: impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation. Front Neuroenergetics. 2013;5:11. doi: 10.3389/fnene.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol. 2007;81:89–131. doi: 10.1016/j.pneurobio.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myzak MC, Ho E, Dashwood RH. Dietary agents as histone deacetylase inhibitors. Mol Carcinog. 2006;45:443–446. doi: 10.1002/mc.20224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, He W, Verdin E. Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J Biol Chem. 2012;287:42436–42443. doi: 10.1074/jbc.R112.404863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshiro K, Rayala SK, Wigerup C, Pakala SB, Natha RS, Gururaj AE, Molli PR, Månsson SS, Ramezani A, Hawley RG, Landberg G, Lee NH, Kumar R. Acetylation-dependent oncogenic activity of metastasis-associated protein 1 co-regulator. EMBO Rep. 2010;11:691–697. doi: 10.1038/embor.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson B, Flinta C, von Heijne G, Jörnvall H. Structures of N-terminally acetylated proteins. Eur J Biochem. 1985;152:523–527. doi: 10.1111/j.1432-1033.1985.tb09227.x. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Pisano C, Vesci L, Milazzo FM, Guglielmi MB, Foderà R, Barbarino M, D’Incalci M, Zucchetti M, Petrangolini G, Tortoreto M, Perego P, Zuco V, Orlandi A, Passeri D, Carminati P, Cavazza C, Zunino F. Metabolic approach to the enhancement of antitumor effect of chemotherapy: a key role of acetyl-L-carnitine. Clin Cancer Res. 2010;16:3944–3953. doi: 10.1158/1078-0432.CCR-10-0964. [DOI] [PubMed] [Google Scholar]
- Reisenauer CJ, Bhatt DP, Mitteness DJ, Slanczka ER, Gienger HM, Watt JA, Rosenberger TA. Acetate supplementation attenuates lipopolysaccharide-induced neuroinflammation. J Neurochem. 2011;117:264–274. doi: 10.1111/j.1471-4159.2011.07198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigotti DJ, Kirov II, Djavadi B, Perry N, Babb JS, Gonen O. Longitudinal whole-brain N-acetylaspartate concentration in healthy adults. AJNR Am J Neuroradiol. 2011;32:1011–1015. doi: 10.3174/ajnr.A2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesler R, Brunetto AT, Abujamra AL, de Farias CB, Brunetto AL, Schwartsmann G. Current and emerging molecular targets in glioma. Expert Rev Anticancer Ther. 2010;10:1735–1751. doi: 10.1586/era.10.167. [DOI] [PubMed] [Google Scholar]
- Schweizer S, Meisel A, Märschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab. 2013;33:1335–1346. doi: 10.1038/jcbfm.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel R, Anikster Y, Zevin S, Steinberg A, Gahl WA, Fisher D, Staretz-Chacham O, Zimran A, Altarescu G. A safety trial of high dose glyceryl triacetate for Canavan disease. Mol Genet Metab. 2011;103:203–206. doi: 10.1016/j.ymgme.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012;7:357–367. doi: 10.1007/s12263-012-0283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, McBrian MA, Mah V, Yu H, Tze S, Wang Q, Chia D, Goodglick L, Kurdistani SK. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol. 2009;174:1619–1628. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shein NA, Shohami E. Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol Med. 2011;17:448–456. doi: 10.2119/molmed.2011.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, LeComte MD, Granger J, Quinlan NJ, Spees JL. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from cortical peri-infarct tissues after stroke. J Neurosci. 2012;32:7926–7940. doi: 10.1523/JNEUROSCI.4303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubassi G, Robert T, Vanoli F, Minucci S, Foiani M. Acetylation: a novel link between double-strand break repair and autophagy. Cancer Res. 2012;72:1332–1335. doi: 10.1158/0008-5472.CAN-11-3172. [DOI] [PubMed] [Google Scholar]
- Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert Rev Anticancer Ther. 2010;10:935–954. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Soliman ML, Rosenberger TA. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol Cell Biochem. 2011;352:173–180. doi: 10.1007/s11010-011-0751-3. [DOI] [PubMed] [Google Scholar]
- Soliman ML, Smith MD, Houdek HM, Rosenberger TA. Acetate supplementation modulates brain histone acetylation and decreases interleukin-1β expression in a rat model of neuroinflammation. J Neuroinflammation. 2012;9:51. doi: 10.1186/1742-2094-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci. 2012;37:152–161. doi: 10.1016/j.tibs.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO Groups. EOfRaToCBTaR, Group. NCIoCCT. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23:207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsen AR, Long PM, Driscoll HE, Davies MT, Teasdale BA, Penar PL, Pendlebury WW, Spees JL, Lawler SE, Viapiano MS, Jaworski DM. Triacetin-based acetate supplementation as a chemotherapeutic adjuvant therapy in glioma. Int J Cancer. 2014;134:1300–1310. doi: 10.1002/ijc.28465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Network CGAR. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C, Chen HZ. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int J Oncol. 2011;38:409–417. doi: 10.3892/ijo.2010.851. [DOI] [PubMed] [Google Scholar]
- Xu W, Li Y, Liu C, Zhao S. Protein lysine acetylation guards metabolic homeostasis to fight against cancer. Oncogene. 2013 doi: 10.1038/onc.2013.163. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.