

Abstract
Rem, Rad, Kir/Gem (RGK) proteins, including Rem2, mediate profound inhibition of high-voltage activated Ca2+ channels containing intracellular regulatory β subunits. All RGK proteins bind to voltage-gated Ca2+ channel β subunit (Cavβ) subunits in vitro, but the necessity of the interaction for current inhibition remains controversial. This study applies NMR and calorimetric techniques to map the binding site for Rem2 on human Cavβ4a and measure its binding affinity. Our experiments revealed 2 binding surfaces on the β4 guanylate kinase domain contributing to a 156 ± 18 µM Kd interaction: a hydrophobic pocket lined by 4 critical residues (L173, N261, H262, and V303), mutation of any of which completely disrupted binding, and a nearby surface containing 3 residues (D206, L209, and D258) that when individually mutated decreased affinity. Voltage-gated Ca2+ channel α1A subunit (Cav2.1) Ca2+ currents were completely inhibited by Rem2 when co-expressed with wild-type Cavβ4a, but were unaffected by Rem2 when coexpressed with a Cavβ4a site 1 (L173A/V303A) or site 2 (D258A) mutant. These results provide direct evidence for a low-affinity Rem2/Cavβ4 interaction and show definitively that the interaction is required for Cav2.1 inhibition.—Xu, X., Zhang, F., Zamponi, G. W., Horne, W. A. Solution NMR and calorimetric analysis of Rem2 binding to the Ca2+ channel β4 subunit: a low affinity interaction is required for inhibition of Cav2.1 Ca2+ currents.
Keywords: RGK proteins, brain, electrophysiology, ion channel, synaptic transmission
Ca2+ channel β subunits (Cavβ1–Cavβ4) are vital intracellular regulatory proteins that bind with nanomolar affinity to a diverse family of high-voltage activated (HVA) Ca2+ channels (1). These channels are found in the plasma membrane of excitable cells and are composed principally of a pore-forming α1 subunit (Cav1.1–Cav1.4; Cav2.1–Cav2.3) and an α2δ subunit (α2δ1−α2δ4) that is largely extracellular (2). Among their many regulatory functions, Ca2+ channel β subunits mediate potent RGK protein (Rem, Rem2, Rad, Gem/Kir) inhibition of HVA Ca2+ channels in muscle, neuronal, and endocrine cells (3–5). The molecular details of the Cavβ subunit-mediated RGK effects are incompletely understood, in large part because RGK inhibitory mechanisms are complex and appear to vary with RGK and Ca2+ channel α1 subunit subtypes (6–8). It has been shown that all RGK proteins can bind to all Cavβ subunits in vitro, but in some cases, direct binding to a Cavβ subunit is not required for RGK Ca2+ current inhibition. For instance, 1 study has shown that Gem inhibition of Cav2.1 requires a Cavβ3 subunit but that mutations that disrupt Cavβ3/Gem binding do not prevent Ca2+ channel inhibition (9). A model is proposed to suggest that Cavβ subunit binding to Cav2.1 induces an inhibitory site on Cav2.1 that serves as a target for the C terminus of Gem. This was supported by a further study showing that in the presence of a Cavβ subunit, a Gem C-terminal 12-amino-acid peptide could, on its own, inhibit Cav2.1 currents (10). By contrast, Cav1.2 channels, combined with Cavβ2a subunits rendered incapable of binding RGK proteins, were still inhibited by Rem and Rad, but not by Gem and Rem2 (6). Adding to this mechanistic diversity, there appears to be multiple ways in which a single RGK protein can inhibit a Ca2+ channel. Three non–mutually exclusive modes of channel inhibition have been identified to date: down-regulation of the number (N) of channels expressed in the plasma membrane by reduced trafficking (11) and accelerated dynamin-dependent endocytosis (6); partial immobilization of α1 subunit voltage sensors resulting in reduced gating current and Qmax (12); and direct inhibition of Ca2+ channel gating (stabilization of low PO gating mode) with no effect on the number of channels trafficked to the membrane (6, 7, 13, 14). Not all RGK inhibitory modes are Cavβ subunit dependent. For example, Rem coexpressed with Cav1.2 has been shown to require Cavβ2a for down-regulation of N and a decrease in Po gating, but not for a reduction in Qmax (6). Also, not all α1 subunits respond to RGKs in the same way. Rem, for instance, does not affect Cav1.1 charge movement as it does with Cav1.2, whereas Rad does so in a manner that requires the Rad N terminus (7). By contrast, Rem2 inhibits Cav1.2, Cav1.3, and Cav2.2 by reducing PO gating (7, 13). Moreover, Rem2 appears to be unique among the RGKs in that it also has direct effects on Ca2+ channel gating kinetics (8). Clearly, much more experimental work is required to fully understand the molecular details of the functional diversity of RGK inhibition of voltage-gated Ca2+ channel currents.
Our study uses techniques in solution NMR spectroscopy and isothermal titration calorimetry (ITC) to examine the structural details of Rem2 binding to the Ca2+ channel Cavβ4 subunit. All Cavβ subunits are members of the membrane-associated guanylate kinase (MAGUK) protein family that contain a core structure composed of Src homology (SH3) and guanylate kinase (GK) domains (1). Previous studies using site-directed mutagenesis and in vitro binding assays localized the Rem binding site to an ∼130 region of the Cavβ subunit GK domain that was distinct from the α1 subunit α1 subunit interaction domain (AID) peptide binding site (15). This was replicated in a modeling/mutagenesis study that more precisely localized the Gem binding region to a pocket in the GK domain that is composed of remnant structural motifs that formed the GMP binding site in ancestral guanylate kinase (16). Both studies provided structural information to support the notion that RGK binding does not interfere with Cavβ subunit binding to Cav1.2 and vice versa. The modeling/mutagenesis study identified 3 aspartate residues (D194, D270, and D272) and a hydrophobic pocket on the β3 subunit GK domain that support RGK binding (16). Our results confirm the general location of the GK binding site identified in previous studies and provide added detail regarding the amino acids that are affected by the interaction. Mutating β4 D258 (β3 D272 equivalent) to alanine lowers RGK binding affinity and also affects the ability of Rem2 to inhibit Cav2.1 Ca2+ channel currents. Mutating residues in the hydrophobic pocket, β4 L173 and V303 (β3 L187 and V317 equivalents), to alanines completely eliminates Rem2 binding, as well as the ability of Rem2 to inhibit CaV2.1 Ca2+ channel currents. Further mutagenesis reveals a surface for Rem2 binding to Cavβ4 that is somewhat different from that previously reported for Rem binding to Cavβ3 (16). This may partially explain why Rem2 has unique Ca2+ channel inhibitory properties relative to its RGK relatives.
MATERIALS AND METHODS
DNA subcloning and mutagenesis
The cDNAs encoding the SH3 domain of the Ca2+ channel β4a subunit (β4a38–133, residue numbers 38–133 of the full-length sequence) (17) and the G-domain of the human Rem2 protein (residue numbers 110–286 of the full-length sequence) (18) were subcloned into pET15b (Novagen, Madison, WI, USA) using NdeI and XhoI restriction sites. A cDNA encoding the GK domain of the human Ca2+ channel β4a subunit (residue numbers 167–374 of the full-length sequence) (17) was subcloned into pET-15b using NcoI and XhoI restriction sites to bypass the N-terminal 6His residues. Six histidine residues were incorporated into the C terminus of the GK construct to enable purification by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography. Site-directed mutagenesis was conducted with the Quickchange (Stratagene, La Jolla, CA, USA) mutagenesis kit according to the manufacturer’s instructions. All constructs were verified by DNA sequencing.
Protein expression and purification
Isotopic labeling and purification of a conjoined β4 SH3-GK core protein (329 amino acids; contains SH3 residues 18–134 and GK residues 166–374 of the full-length β4a sequence bridged by 2 serines) was performed according to protocols previously described (19), which contains resonance assignments for SH3-GK fusion protein residues 12–350. [All residue numbers in the manuscript refer to the fusion construct sequence (19). Add 22 or 14 to obtain full-length β4a (17) or β3 (16) GK domain residue numbers, respectively.] To create a SH3-GK core protein in which the GK domain was specifically labeled, the Cavβ438-133 SH3 (fusion protein amino acids 44–139) and Cavβ4167-374 GK (fusion protein residues 145–352) proteins were first expressed separately in the Escherichia coli strain BL21 Rosetta (DE3) pLysS (EMD, Millipore, Billerica, MA, USA). Subsequently, the SH3 protein was grown in isotope-free Luria broth (LB) medium, and the GK protein was grown in M9 minimal medium containing 15NH4Cl (1 g/L) and U-2H-13C-glucose (2 g/L) (Cambridge Isotope Laboratories, Andover, MA, USA). After sonication and centrifugation, the SH3 protein was purified from the supernatant by Ni-NTA affinity chromatography. Next, the protein was subjected to thrombin cleavage to remove the 6His tag and further purified by FPLC size exclusion chromatography using a Sephacryl 200 column (AKTA; GE Healthcare, Pittsburgh, PA, USA). For purification of the GK domain, a single colony of freshly transformed E. coli harboring pet15b-Cavβ4167-374 was inoculated into 20 ml LB medium in 80% (v/v) D2O containing 50 µg/ml ampicillin and grown overnight. One milliliter of the overnight culture was further inoculated into 100 ml M9 media supplemented with 15NH4Cl, 13C glucose, and 80% (v/v) D2O. After 12-hour growth at 37°C, the cells were pelleted by centrifugation at 4000 rpm and transferred into 1 L 99.9% D2O containing M9 media ingredients and 15NH4Cl (1 g/L) and U-2H-13C-glucose (2 g/L). The cells were further grown at 37°C until the OD600 reached 0.5 AU. Isopropyl-β-D-thiogalactopyranoside was added to a final concentration of 0.5 mM, and protein expression was induced at 37°C for 12 h before cell harvest. The perdeuterated 1H,15N,13C-labeled GK protein was purified from inclusion bodies after sonication and centrifugation. The inclusion bodies were dissolved in 6 M guanidium HCl, and the denatured GK protein was purified by Ni-NTA affinity chromatography. The denatured protein was then refolded at 4°C by rapid dilution (20-fold) of a 10 ml aliquot of 100 μM purified GK into a 200 ml solution containing 5 μM purified SH3, 500 mM NaCl, and 50 mM Tris-HCl, pH 8.0. After extensive dialysis against 500 mM NaCl and 50 mM Tris-HCl, pH 8.0, to remove the denaturant, the refolded labeled GK protein in complex with SH3 was further purified by Sephacryl 200 size exclusion chromatography.
The Rem2110-286 G-domain protein was expressed in the E. coli strain, Rosetta (DE3) pLysS as an N-terminal 6His fusion protein. The soluble lysate was purified by Ni-NTA affinity chromatography followed by thrombin cleavage and Sephacryal S-200 column size exclusion chromatography. The reagents 50 μM GDP and 5 mM MgCl2 were included in buffers in all purification steps to prevent Rem2 precipitation.
A 16-mer peptide (IERELNGYMEWISKAE) corresponding to the I–II loop AID of the α1A voltage-gated Ca2+ channel was custom synthesized to a purity level >95% (Genescript, Piscataway, NJ, USA). The peptide was used for NMR titration experiments without further purification.
Protein purity was analyzed on SDS-PAGE gels, and the protein samples were concentrated via centrifugal filtration using 10 kDa exclusion filters for Rem2, SH3, and GK and a 30 kDa exclusion filter for the SH3-GK complex (Amicon Ultra 15; EMD, Millipore). Protein concentrations were determined by the UV absorbance at 280 nm using theoretical extinction coefficients at 280 nm of (M-1 cm-1) 21,645 (Rem2), 27,055 (SH3-GK), 13,980 (SH3), 11,585 (GK), and 6990 (AID peptide).
NMR spectroscopy
NMR experiments were recorded at 298 K with a 900 MHz Bruker Avance spectrometer (Billerica, MA, USA) equipped with a triple resonance TCI CryoProbe. The data were processed with NMRPipe (20) and analyzed using Sparky assignment software (21). The amide backbone assignments of the TROSY-HSQC derived from the GK-specific labeled protein SH3-GK complex were largely transferred from the assignment of the 1H-15N TROSY-HSQC of the deuterated SH3-GK core (19). The assignments were confirmed by 1H-15N-13C-TROSY type 3-D experiments: HNCA, HNCOCA, and CBCACONH. The NMR samples contained 0.5 mM protein (unlabeled SH3 protein in complex with the 2H, 15N, and [13C]-labeled GK domain) in 100 mM NaCl, 2 mM DTT, 0.1% NaN3, 10% (v/v) D2O, and 50 mM sodium phosphate buffer, pH 7.0.
For AID peptide titrations, stock solutions of 10 mM peptide were prepared in 100 mM NaCl and 50 mM sodium phosphate buffer, pH 7.0. A baseline 1H-15N TROSY-HSQC spectrum was recorded with an initial sample containing 0.3 mM 2H,15N, and 13C-labeled SH3-GK or 2H,15N, and 13C-specific labeled GK in complex with unlabeled SH3 in 100 mM NaCl, 2 mM DTT, 10% (v/v) D2O, and 50 mM sodium phosphate buffer, pH 7.0. A series of 1H-15N TROSY-HSQC spectra was then recorded when 1 μl aliquots of unlabeled peptide were added to the solution to a final ratio of peptide to protein of 1.5. To analyze the amide backbone assignment of SH3-GK core in complex with the AID peptide, a 3D 1H-15N NOESY-HSQC was recorded. The averaged chemical shift change of 15N and 1H was calculated with the equation
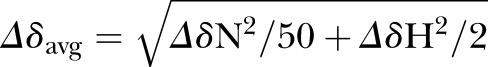 |
in which ΔδN and ΔδH represent the chemical shift change of the amide nitrogen and proton, respectively.
For Rem2 G-domain titrations, all proteins were exchanged into a buffer containing 50 mM NaCl, 5 mM MgCl2, 1 mM GDP, 2 mM DTT, 10% (v/v) D2O, and 25 mM sodium phosphate buffer, pH 7.4. The first HSQC was recorded with a sample containing 0.3 mM 2H, 15N, and 13C-labeled SH3-GK or 2H,15N, and 13C-labeled GK coupled to unlabeled SH3. A series of 1H-15N TROSY-HSQC spectra was recorded for samples when the unlabeled Rem2 protein was added to reach the ratios of 0.2, 0.5, 1.0, and 2.0.
ITC
ITC measurements were carried out at 293K using a MicroCal VP-ITC microcalorimeter (GE, Nothampton, MA, USA). All proteins were dialyzed against the same buffer, and all buffers were degassed before experiments. Repeats of each experiment were performed with different batches of protein. For the AID peptide titration, the AID peptide was buffer exchanged into ITC buffer containing 100 mM NaCl and 50 mM sodium phosphate, pH 7.0, using a PD10 column. Multiple injections of 5 μl of 350 μM peptide were titrated into 1.4 ml of 14 μM β4 SH3-GK core protein (or buffer only, for baseline correction) to a stoichiometric ratio of 3.8:1 (n = 3). For the Cavβ4 SH3-GK core protein (n = 3) and its mutants (n = 2 for each mutant) interacting with Rem2, the buffer contained 100 mM NaCl, 5 mM MgCl2, 50 μM GDP, and 25 mM sodium phosphate, pH 7.4. (Protein yields for N261A and H262A mutants were quite low compared with other Cavβ4 mutants. This mandated extensive concentration of the proteins and a number of additional repeat experiments to generate reliable ITC curves.) Multiple injections of Cavβ4 protein in the concentration range of 400–700 μM were titrated into 1.4 ml of 15–30 μM Rem2 G-domain protein. Data were processed and analyzed with MicroCal Origin software, v.7.0 (GE).
Cell culture and transient transfection
HEK tsA-201 cells were grown to 80–90% confluence at 37°C (5% CO2) in DMEM medium (Life Technologies, Grand Island, NY, USA), supplemented with 10% (v/v) fetal bovine serum (HyClone; Thermo Scientific, Pittsburgh, PA, USA), 200 U/ml penicillin, and 0.2 mg/ml streptomycin (Life Technologies). Cells were suspended with 0.25% trypsin/EDTA and plated onto glass coverslips in 35 mm culture dishes (Corning, Corning, NY, USA) at 10% confluence, 24 hours before transfection. The cells were transiently transfected with cDNAs encoding rat Cacna1A (GenBank accession number: NP_037050), rat Cacna2d1 (GenBank accession number: NP_037051), human Cacnb4 (GenBank accession number: NP_001005747) (wild-type, L173A/V303A double mutant, D256A mutant, or D258 mutant), mouse Rem2 (GenBank accession number: NP_542764) or empty vector [pcDNA3.1(+), Life Technologies], and pEGFP-N1 (Clontech Laboratories, Mountain View, CA, USA) as a marker at 1 + 1 + 1 + 0.1 + 0.1 μg with 6 μl Turbofect transfection reagent (Thermo Scientific). Cells were transferred to 30°C 6 hours after transfection and stored for 42 hours before recording.
Electrophysiology
Cells on a glass coverslip were transferred into an external bath solution of 20 mM BaCl2, 1 mM MgCl2, 40 mM tetraethylammonium chloride, 65 mM CsCl, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 10 mM glucose, pH 7.4. Borosilicate glass pipettes (Sutter Instrument Co., Novato, CA, USA) (2–4 MΩ) were filled with internal solution containing 140 mM CsCl, 2.5 mM CaCl2, 1 mM MgCl2, 5 mM EGTA, 10 mM HEPES, 2 mM Na-ATP, and 0.3 mM Na-GTP, pH 7.3. Whole-cell patch clamp recordings were performed by using an EPC 10 amplifier (HEKA Elektronik, Bellmore, NY, USA) linked to a personal computer equipped with Pulse (V8.65) software (HEKA Elektronik). After seal formation, the membrane beneath the pipette was ruptured, and the pipette solution was allowed to dialyze into the cell for 1–2 minutes before recording. Currents were elicited by depolarization from a holding potential of −90 mV to various test potentials for 100 ms with an interpulse interval of 1 s. Voltage-dependent currents were corrected for leak by using an online P/4 subtraction paradigm. Data were recorded at 10 kHz and filtered at 2.9 kHz. Data analysis was performed by using online analysis built in Pulse software, and graphs were prepared by using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Error bars plotted represent the mean values ± se.
RESULTS
GK domain specific labeling by in vitro corefolding
A Cavβ4 core domain construct (329 amino acids) composed of the SH3 domain (residues 18–134 of the full-length sequence) and GK domain (residues 166–374 of the full-length sequence) bridged by 2 serines was expressed, 15N and 13C labeled, and perdeuterated in E. coli. The removal of a large serine-rich loop (33 amino acids) in the hook region between the SH3 and GK domains resulted in expression of a stable, monomeric protein construct suitable for multidimensional NMR experiments. This is confirmed by the well-dispersed 1H, 15N-TROSY-HSQC spectrum of the fully labeled Cavβ4 SH3-GK construct (Fig. 1A). The backbone assignments of this construct were completed with a suite of 3-dimensional experiments and reported previously (19). As can be seen in the figure, there is considerable resonance overlap in the spectrum, especially in the central region. This has hindered the ability to perform chemical shift analysis using this labeled construct to study β4 protein-protein interactions.
Figure 1.

2H,15N -specific labeling of the Ca2+ channel β4 subunit GK domain. A) 1H-15N TROSY-HSQC spectra of the conjoined 2H,15N- labeled Cavβ4 SH3-GK core domains. B) 1H-15N TROSY-HSQC spectra of the specifically labeled (SH3)-2H,15N-GK domain bound to unlabeled Cavβ4 SH3. Note the substantially reduced resonance overlap in the HSQC of (SH3)-2H,15N GK.
To address the problem of resonance overlap, a domain-specific labeling protocol was developed for the Cavβ4 GK domain. After multiple failed attempts to express a singular GK domain that folded properly, we developed a refolding strategy in which the purified, folded, and unlabeled SH3 domain was used as a chaperone to assist in the folding of labeled GK. When the fifth β-strand (residues 166–171) of the SH3 domain was included on the N terminus of GK, a folded SH3 lacking the fifth strand was capable of assisting with in vitro refolding of GK. This enabled us to 2H,15N,13C label the GK domain specifically while leaving the SH3 unlabeled in the final Cavβ4 core construct: (SH3)-2H15N GK. As shown in Fig. 1B, this dramatically improved the spectral resolution of the GK domain 1H-15N TROSY-HSQC spectrum.
AID peptide chemical shift perturbations
To determine whether the purified conjoined SH3-GK and SH3 bound GK constructs were properly folded and able to bind protein we performed a chemical shift perturbation analysis with a Cavα peptide known to interact with the GK domain. A 16-mer peptide sequence derived from the Cav2.1 α1 subunit I–II loop was synthesized for these experiments. The peptide lacks 2 N-terminal Gln residues compared with an 18-mer AID peptide used by other groups (22, 23); however, as shown in a previous ITC analysis, these 2 residues were not absolutely required for AID binding to Cavβ subunits (24). Our experiments reveal large chemical shift changes in 1H,15N-TROSY-HSQC spectra following titration of the 16-mer peptide into NMR samples containing either the 2H,15N-labeled β4 conjoined SH3-GK core (Fig. 2A) or the core complex composed of the refolded and specifically labeled 2H,15N GK domain bound to SH3 (Fig. 2B). Residues M182, L186, L328, E329, and D330, shown previously to be 5 key residues in the Cavβ4 α1 subunit binding pocket (1), are highlighted in both figures as examples of the large chemical shifts that are typical of high-affinity binding. For each titration, only 2 sets of cross-peaks representing free and peptide bound states of Cavβ4 were detected, indicating that the interaction is in a slow exchange regime. Nearly all the chemical shift change patterns were maintained in the titrations using the labeled GK domain (Fig. 2B) compared with the titrations using the labeled Cavβ4 SH3-GK core (Fig. 2A). This demonstrates the equivalent folding and binding ability of the 2 Cavβ4 core constructs. Importantly, the ability to perform chemical shift perturbation analysis was dramatically enhanced when the SH3 resonances were removed by specifically labeling the GK domain. The complete AID peptide perturbation analysis is shown in Fig. 2C. Importantly, residues M182 and L328 are 2 of 5 residues in the β4 binding pocket shown in a previous study (24) to have the greatest impact on binding affinity (>10-fold reduction) when mutated to alanine.
Figure 2.
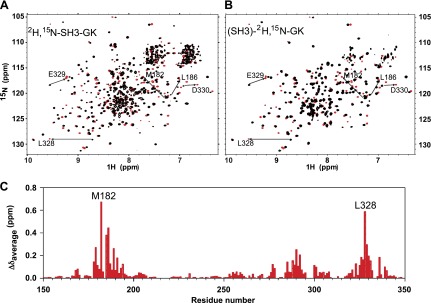
NMR chemical shift perturbations invoked by Ca2+ channel α1A AID peptide binding to the β4 subunit SH3-GK core structure. A) Overlay of the 1H-15N TROSY-HSQC spectra of the 2H,15N-labeled conjoined Cavβ4 SH3-GK core domains before (black) and after (red) the titration of the α1A AID peptide to the ratio of 1.5:1. Residues L186, M182, L328, E329, and D330, shown previously to be located in the Cavβ4 α1 subunit binding pocket, are labeled as examples of the largest chemical shifts (arrows). B) Overlay of the 1H-15N TROSY HSQC spectra of the 2H,15N labeled GK domain of the bound SH3-GK core before (black) and after (red) the titration of the peptide to the ratio of 1.5:1. Residues M182, L186, L328, E329, and D330 are again shown as examples of large chemical shifts (arrows). Note that they are identical to the shifts in A. C) Plot of all α1A AID-induced chemical shift changes (red bars) against the residues numbers of the Cavβ4 subunit GK domain. Residue numbers correspond to those of the conjoined SH3-GK fusion protein construct described previously (19). M182 and L328 are labeled as the largest chemical shifts. [In this and all subsequent figures, residue numbers refer to the fusion construct sequence (19). Add 22 or 14 to obtain full-length β4a (17) or β3 (16) GK domain residue numbers, respectively.]
Rem2 G-domain titration into Cavβ4 SH3-GK core structures
To determine whether Rem2 binds to Cavβ4, we used the GDP-bound form of the Rem2 G-domain to perform a chemical shift perturbation analysis. Both GDP and Mg2+ were included in the buffers throughout the purification process and in all NMR experiments to stabilize the Rem2 G-domain. When the unlabeled Rem2 G-domain was titrated into the perdeuterated, conjoined 2H,15N-labeled Cavβ4 SH3-GK core structure, complex formation was evident from general line broadening and disappearance of cross-peaks (Fig. 3A). Some cross-peaks showed very rapid chemical shift changes, as line broadening was evident as early as the third titration point. In contrast to results with the AID peptide, no changes representing population averaged chemical shifts of the bound state were detected for residues in the presence of Rem2. This indicates that the interaction is in an intermediate exchange regime. Fig. 3A clearly shows the difficulty in performing chemical shift perturbation analysis of the central region of the completely labeled SH3-GK core structure that results from severe resonance overlap. Interestingly, however, most of the cross-peaks in the well-dispersed region of the spectrum that undergo rapid disappearance can be assigned to the residues of GK domain. This suggests that the Rem2 binding site is located principally on the GK domain. This was confirmed by performing Rem2 G-domain titration against a core complex that contained the 2H,15N-labeled labeled GK domain bound to an unlabeled SH3 (Fig. 3B). GK domain residues experiencing significant line broadening at the third titration point have been identified as possible points of contact for Rem2 (see below).
Figure 3.
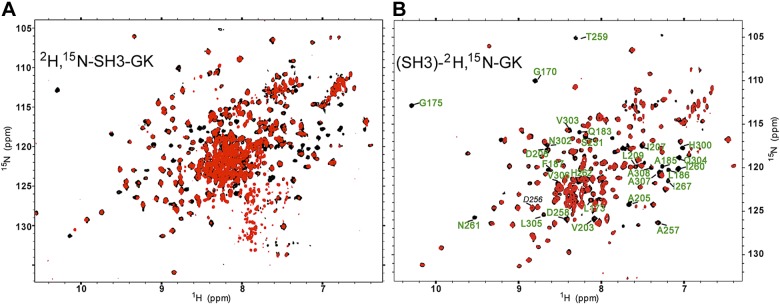
NMR evidence of Rem2 G domain binding to the Ca2+ channel β4 subunit GK domain. A) Overlay of 1H-15N TROSY-HSQC spectra of the conjoined 2H,15N-labeled Cavβ4 SH3-GK core structure before (black) and after (red) adding unlabeled Rem2 G-domain to a ratio 2:1. B) Overlay of 1H-15N TROSY-HSQC spectra of the 2H,15N-labeled GK domain bound to unlabeled SH3 before (black) and after (red) adding unlabeled Rem2 G-domain to 2:1 ratio. The cross-peaks experiencing substantial line broadening on Rem2 addition are labeled with residue assignments (green labels). Residue D256, which is not perturbed by Rem2 but is used in a later electrophysiology experiment, is also labeled (black label).
Comparison of the interface for AID and Rem2 binding to the Cavβ4 GK domain
To confirm that there is little overlap between the AID peptide and Rem2 binding sites on Cavβ4, we created a surface map for each site based on our perturbation analysis data. Residues on the Cavβ4 subunit that showed significant NMR chemical shift changes in response to binding of the α1 AID peptide are mapped onto the SH3-GK crystal structure in Fig. 4A. A continuous surface was found to surround an AID binding pocket previously identified by X-ray crystallography (1). Some residues such as F189, R193, F194, and L339 showing chemical shifts are located relatively far from the binding groove. These chemical shifts are likely caused by small side chain conformational changes of aromatic residues. The Rem2 G domain interaction site was mapped to a large concaved region on the flip side of the GK domain (180° rotation; Fig. 4B) that includes remnants of the nucleotide biding pocket of canonical GK proteins. The binding site also contains residues just proximal to 310 helix sequences 2 and 3 and residues in α helix 6. As seen in Fig. 5B, the residues are located in 5 small contiguous subdomains: 1 (G170, L173, and G175); 2 (Q183, A185, L186, and F187); 3 (A205, D206, I207, and L209); 4 (A257, D258, I260, N261, H262, Q265, and I267); and 5 (H300, N302, V303, Q304, L305, V306, A307, and A308). These surface representations clearly indicate that the Cavβ subunit binding regions for AID and Rem2 are distinct and that a direct competition for binding between the AID and Rem2 is unlikely.
Figure 4.

Solution NMR-derived surface representations of binding regions for the α1A AID peptide and Rem2 G-domain on the GK domain of the Ca2+ channel β4 subunit. A) Ca2+ channel α1A AID binding site derived from chemical shift perturbations and mapped onto the GK domain of the Cavβ4 subunit SH3-GK crystal structure (Protein Data Bank ID code 1VYV). Residues of the GK domain experiencing chemical shift changes >0.12 ppm on adding the AID peptide are labeled in red; those >0.08 ppm but <0.12 ppm are labeled in orange. The AID peptide in complex with β4 is shown in ribbon diagram form (blue) with side chains shown as sticks (modeled from Protein Data Bank ID code 1VYT using Modeler 9.10; University of California, San Francisco, CA, USA). B) Rem2 G-domain binding site on the GK domain of the Cavβ4 subunit SH3-GK core structure as derived from chemical shift perturbations. Residues showing Rem2 G-domain induced line broadening because of rapid intermediate chemical exchange are labeled in green.
Figure 5.

Isothermal titration calorimetry analysis of α1A AID peptide and Rem2 G-domain binding to the Cavβ4 SH3-GK core protein. A–L) (Upper) Exemplar raw thermograms after baseline correction. (Lower) Data processed with Microcal Origin software (7.0). Solid lines represent best fits to a 1-site binding model. A) α1A AID peptide (350 μM) titrated into Cavβ4 SH3-GK core (16 μM). B) Cavβ4 SH3-GK core (800 μM) titrated into Rem2 G-domain (45 μM). C–L) The core alanine mutant was titrated into the wild-type Rem2 G-domain. Protein amounts [expressed as SH3-GK (μM) and Rem2 (μM)] were as follows: L173A (550, 20); D206A (600, 30); L209A (600, 22); D256A (480, 18); D258A (700, 35); N261A (500, 15); H262A (400, 20); Q265A (600, 33); N302A (600, 21), and V303A (700, 30).
Identification of critical GK residues required for weak interaction of Rem2 with the Cavβ4 GK domain
Rapid signal broadening caused by intermediate exchange prevents an accurate NMR measurement of the binding affinity for the Rem2 G-domain/Cavβ4 subunit GK domain interaction. We therefore used ITC for this purpose (Fig. 5). For comparison purposes, we first performed an ITC experiment using the same 16-mer AID peptide used for NMR titrations against the β4 SH3-GHK construct. We observed a thermogram characteristic of a high-affinity, saturable interaction (Fig. 5A). The reaction is exothermic and has a calculated equilibration dissociation constant (Kd) of 341 ± 13 nM. This is weaker than a previously published ITC-determined affinity of an 18-mer Cav2.1 peptide binding to Cavβ2a (24) and is likely attributed to a charge difference (use of nonacetylated peptide) and the lack of 2 N-terminal Asn residues. For Rem2 binding, experiments were performed by including the Rem2 G-domain in the sample cell and titrating in wild-type or mutant Cavβ4 SH3-GK proteins (Fig. 5B–L). The wild-type SH3-GK thermogram reveals a typical weak interaction curve that does not reach saturation even at the final titration point (Fig. 5B). The interaction is endothermic with a calculated Kd of 156 ± 18 μM. This affinity falls into the low end of measuring limits of isothermal titration calorimetry (25).
Of the 26 Cavβ4 GK domain surface residues identified by NMR to be perturbed by the interaction with Rem2 (Fig. 4), we found 9 that had exposed side chains by examining the Cavβ4 core crystal structure. We posited that these surface residues were most likely to be involved in direct binding with Rem2. To test this hypothesis, we individually mutated each of the 9 residues to alanine and repeated the ITC experiments. These surface mutations would not be predicted to cause global structural changes given that their side chains are not involved in internal chemical bonds. Thus, changes in binding properties would serve as a good estimate of the contribution of each residue to binding affinity. The results of these experiments were as follows: thermograms of Q265A and N302A were not different than wild type, indicating that these residues are not directly involved in Rem2 binding; thermograms for L173A, N261A, H262A, and V303A (Figs. 5C, H, I, and L, respectively) were essentially flat (after correcting for the heat of dilution), indicating that these residues are essential for binding of Rem2; and thermograms for D206A, L209A, and D258A (Fig. 5D, E, and G, respectively) showed some evidence of very low-affinity binding, although too low to calculate an accurate Kd. This indicates that these residues are essential but may not be absolutely required for binding. Thus, the results of our alanine-scanning mutagenesis experiments led us to identify a Rem2 binding pocket formed by L173, V303, V306, N261, and H262 and a contiguous binding surface formed by D206, L209, and D258 (Figs. 6 and 7). As an additional experiment, we mutated D256 to alanine because, although not perturbed in our chemical shift analysis (Fig. 3B), it has previously been identified as important for the Kir/Gem interaction with Cavβ3 (16). Consistent with our NMR data, the D256A mutation had no effect on Rem2 binding to Cavβ4 (Fig. 5F).
Figure 6.
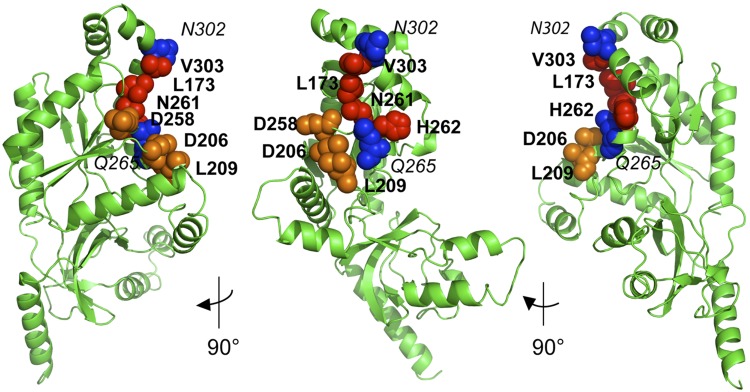
Ca2+ channel β4 subunit GK domain binding site for Rem2 G-domain identified by NMR-directed alanine scanning mutagenesis and isothermal titration calorimetry. Residues for which alanine substitutions eliminated Rem2 G domain binding, L173, V303, N261, and H262, are shown as red spheres. Residues for which individual substitutions resulted in decreased affinities, D206, L209, and D258, are shown as orange spheres. Residues for which substitutions had no effect on binding, N302 and Q265, are shown as blue spheres.
Figure 7.
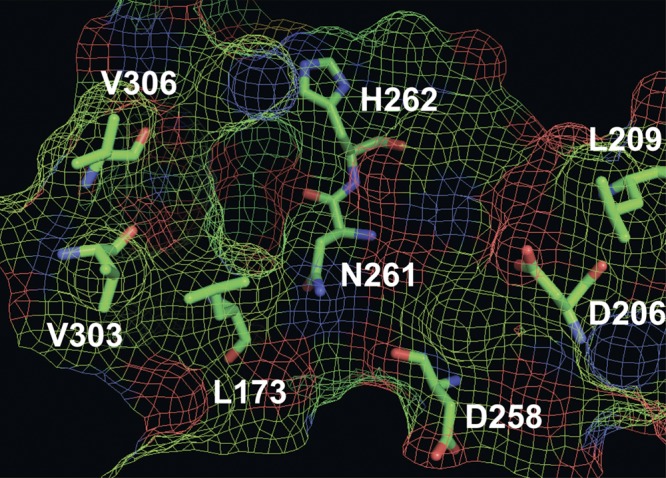
Mesh surface diagram of the Ca2+ channel β4 subunit-Rem2 G domain binding site. NMR perturbation analysis reveals that the site is composed of 1) a primary pocket formed by 1 residue on the loop between β-strand 6 and α-helix 3 (L173), 2 residues on the very distal portion of the loop between β-strand 8 and 310-helix 3 (N261 and H262), and 2 residues on the distal portion of the α-helix 6 (V303 and V306); and 2) a supporting surface formed by 2 residues from the loop between β-strand 5 and α-helix 4 (D206 and D209) and 1 residue from β-strand 6 (D258). Residue numbers shown are based on the CavSH3-GK core construct in ref. 19 and correspond to L195, D228, L231, D280, N283, H284, V325, and V328 of the full-length Cavβ4a sequence. The mesh diagram was created in PyMOL (v. 1.5) using the complete structure of the Cavβ4 GK domain. The α-helices, β-strands, and loops are hidden to highlight the binding pocket. Amino acid residues are shown as sticks.
To further validate our binding results, a double mutant, Cavβ4 SH3-GK-L173A/V303A, was expressed and 2H-15N-labeled, and used in Rem2 chemical shift perturbation analysis experiments. As seen in the 1H,15N-TROSY-HSQC overlay (Supplemental Fig. S1), small chemical shift changes were observed that were likely attributable to the 2 mutated residues. The fact that the mutations had no detectable effect on the overall dispersion of the remaining resonances indicates that they had minimal effects on protein folding. To determine whether the double mutation completely eliminated Rem2 G domain binding, the unlabeled G-domain was titrated into 2H,15N-labeled Cavβ4a SH3-GKL173A/V303A. As shown in Supplemental Fig. S2, neither chemical shift nor line broadening of NMR cross-peaks was observed, indicating that these 2 mutations completely abolished the interaction of the Rem2 G-domain with the Cavβ4 SH3-GK core domain.
Functional effects of mutating Cavβ4 residues required for Rem2 binding
To assess the functional importance of residues L173 and V303, the mutations were introduced into an expression construct for Cavβ4a. This construct (or wild-type Cavβ4a) was then coexpressed with Cav2.1 and Cavα2δ in tsA-201 cells in the presence and absence of a cotransfected Rem2 construct. In accordance with what we had observed previously with Cav2.2 + Cavβ3 channels (26), coexpression of Rem2 in cells expressing Cav2.1 channels + wild-type Cavβ4 shows that whole cell currents were virtually abolished in the presence of Rem2 (Fig. 8). By contrast, in cells expressing the L173/V303 double mutant, current densities were unaltered by Rem2, in agreement with the notion that this mutant was unable to interact with Rem2. The double mutant did not significantly alter current densities or voltage-dependent gating compared with the wild-type Cavβ4a subunit, indicating that the physical association of the Cβ4a subunit with Cav2.1 is not affected by the mutation. We also tested 2 additional Cavβ4a mutants (D256A and D258A) on Cav2.1 channel function. In the absence of Rem2, both of these mutants behaved like wild-type Cavβ4a with regard to current density. In the presence of the D256A mutant, Cav2.1 current densities were significantly reduced, whereas in the case of the D258A mutant, Rem2 had little effect. These data fit with the observation that strength of Rem2 binding affinity for Cavβ4 is important for Rem2 inhibition of Cav2.1 Ca2+ currents.
Figure 8.

Rem2-induced Cav2.1 current inhibition is reversed by certain Cavβ4a mutants. Average current-voltage relationships for currents recorded in cells expressing Cav2.1 with or without coexpressing Rem2. For wild-type Cavβ4a, Cav2.1 current densities were significantly smaller in Rem2 cotransfected cells than those in cells not expressing Rem2 (P < 0.0001 at −5 mV). For double mutant Cavβ4a (L173A/V303A), however, Cav2.1 current densities were similar between Rem2 coexpressing cells and cells not expressing Rem2 (P = 0.7061 at −5 mV). The double mutations in Cavβ4a per se did not affect the overall Cav2.1 current densities relative to those seen with wild-type Cavβ4a (P = 0.6198 at −5 mV). The D256A mutation did not affect the ability of Rem2 to reduce current densities (P = 0.003 at 15 mV), whereas D258A shows similar current densities in the presence and absence of Rem2 (P = 0.5151 at −5 mV). Ten experiments per condition are included in the current-voltage data.
DISCUSSION
NMR and functional assays to study weak protein-protein interactions
Weak (Kd ≥ 10−4) protein-protein interactions are poorly understood and yet extremely important for many cellular events, including rapid and transient assembly of regulatory protein complexes and signal transduction turnover (27). Such events play key roles in fine-tuning synaptic vesicle release at nerve terminals (2). The reason for our incomplete understanding of weak protein-protein interactions lies in the technical difficulty in characterizing them. They do not typically lend themselves to study by conventional biochemical methods, including X-ray crystallography. This barrier can be overcome by using techniques in NMR spectroscopy, especially when combined with highly sensitive functional assays (28); however, this approach is not without its limitations. In particular, the complexity of NMR spectra increases with protein size, often resulting in difficult to discern resonance overlaps in all dimensions of a 3D NMR experiment.
We studied the low-affinity interaction between the 37 kDa SH3-GK MAGUK core of the Ca2+ channel β4 subunit and the 22 kDa G-domain of the RGK protein, Rem2, by combining NMR chemical shift perturbation (CSP) analysis and ITC with site-directed mutagenesis to map and characterize the Rem2/Cavβ4 subunit interaction. At 37 kDa, resonance overlap was significant for the labeled SH3-GK protein and thus pushed the CSP analysis technique beyond its size limitation. We overcame this by purifying the SH3 domain separately and using it as an in vitro chaperone to fold the specifically labeled GK domain (which could not be folded on its own). This enabled nearly complete assignment of GK domain resonances and opened the way for clear CSP analysis. (It will be interesting to see whether this technique has wide applications in the field of MAGUK protein structure and function.) Thus, coupled with the patch-clamp electrophysiology technique, NMR and ITC have permitted us to show directly, and for the first time, that a weak protein-protein interaction is absolutely essential for potent Rem2 inhibition of Cav2.1 Ca2+ channel currents.
Comparison to other RGK/β subunit interaction studies
This study has provided the first NMR evidence for a direct low-affinity interaction of an RGK protein with a binding site on the voltage-gated Ca2+ channel β subunit. Interestingly, the surface map of the Rem2 binding site on Cavβ4 generated from our experiments (Fig. 7) differs somewhat from that determined by a mutagenesis/molecular modeling study of the interaction of Kir/Gem with Cavβ3 (16). Although both studies identify the hydrophobic pocket that is bordered by L173 and N261 (L187 and N275 in Cavβ3) as being critical for binding of Kir/Gem and Rem2, NMR analysis of Rem2 binding to Cavβ4 indicates that the negatively charged surface formed by D180, D256, and D258 (D194, D270, and D272 in Cavβ3) and important for Kir/Gem binding is not required for binding of Rem2 (D180 and D256 charges in particular). By contrast, the surface formed by L209, D206, and D258 in Cavβ4 contributes to binding of Rem2. Individual alanine substitutions of these residues reduce affinity but do not eliminate binding. Interestingly however, the reduced affinity caused by D258A was enough to eliminate Rem2’s ability to inhibit Cav2.1 Ca2+ currents in HEK cells (Fig. 8). This result supports the finding of previous laboratories showing that Cavβ constructs containing the D258A mutation in a triple mutant construct were unable to support Ca2+ current inhibition by Rem, Rem2, or Gem (6, 9, 16). Our results may explain why Béguin et al. concluded from their studies that there may be subtle differences in the way Rem2 associates with Cavβ3 compared with Rad and Rem (16); however, whether this explains the result that a Gem/β3 subunit interaction is not required for Gem inhibition of Cav2.1 (9) remains to be determined. Our contrasting results raise the interesting possibility that there is a yet to be fully characterized structural specificity underlying RGK inhibition of Ca2+ channels. The results also suggest that future studies examining the role of β4-Rem2 interactions in physiologic processes would benefit from using the β4 L173/V303 double mutant to ensure that Rem2 binding has been eliminated.
Diversity of RGK protein/β subunit interactions
Sequence comparisons of RGK proteins from a number of genetically diverse organisms suggest that their ability to modulate Ca2+ channels arose from an ancestor that predates the protostome and deuterostome split that occurred >500 million years ago (29). Key RGK G-domain loop residues important for binding to Ca2+ channel β subunits are highly conserved throughout evolution (R196, V/L223, and H225), arguing that the RGK-β subunit interaction is very important to the function of the RGK protein class. Ca2+ channel β subunit GK residues, including residues of the RGK binding site, are also highly conserved (30); however, this is a general feature of the entire GK domain, and sequence comparisons do not point to any one region as a potential binding site.
Our results add to the evidence that the Rem2 site has evolved as a distinct site from that of the ancestral GMP binding site of guanylate kinase. There is some structural overlap between this site and the location of Walker A and B sequences in P-loop kinases (β-strand 6 and β-strand 8 and proximal loop sequence between β-strands 8 and 9, respectively); however, the Cavβ4 sequence lacks key residues necessary for binding the α-phosphate of GMP. Moreover, although β-strands 2 and 4 (equivalents of β-strands 7 and 9 in the β4 subunit, respectively) are key structures involved in GMP binding, they do not play a role in the binding of Rem2. Identifying this as a distinct site is consistent with the notion that MAGUKs have evolved from GKs by accelerated divergence (31).
The physiologic importance of the RGK-Cavβ subunit interactions remains to be fully elucidated; however, preliminary studies have shown that cellular responses are both cell type and subcompartment specific. In the case of Rem2, direct inhibition of nerve terminal Cav2 channels would be expected to cause immediate inhibition of synaptic vesicle release (13), whereas negative feedback inhibition of cell soma Cav1 channels would have longer-term effects on activity-dependent Rem2 signaling pathways (18, 32, 33). As Rem2, Cavβ4, and Cav2.1 are all localized to neurons, and most excitatory CNS synapses contain Cav2.1, our results should contribute in a substantive way to the understanding of the role that neuronal voltage-gated Ca2+ channels play in health and disease.
Supplementary Material
Acknowledgments
The authors thank Dr. Li Li and Ms. Heather De Feijter-Rupp for excellent technical support in all aspects of the project. This work was supported in part by U.S. National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant NS42600 (to W.A.H.), and National Science Foundation Grant IOS 0719242 (to W.A.H.). F.Z. is supported by a postdoctoral fellowship from Alberta Innovates–Health Solutions. G.W.Z. is supported by grants from the Canadian Institutes for Health Research and from the Natural Sciences and Engineering Research Council and holds a Canada Research Chair. The authors declare no conflicts of interest.
Glossary
- AID
α1 subunit interaction domain
- Cav2.1
voltage-gated Ca2+ channel α1A subunit
- Cavβ
voltage-gated Ca2+ channel β subunit
- CSP
chemical shift perturbation
- GK
guanylate kinase
- HSQC
heteronuclear single quantum coherence spectroscopy
- HVA
high-voltage activated
- ITC
isothermal titration calorimetry
- MAGUK
membrane-associated guanylate kinase
- N
number of channels expressed at the cell surface
- Ni-NTA
nickel-nitroacetic acid
- Po
channel open probability
- RGK
Rem, Rad, Kir/Gem proteins
- TROSY
transverse relaxation-optimized spectroscopy
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Buraei Z., Yang J. (2010) The β subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simms B. A., Zamponi G. W. (2014) Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45 [DOI] [PubMed] [Google Scholar]
- 3.Correll R. N., Pang C., Niedowicz D. M., Finlin B. S., Andres D. A. (2008) The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell. Signal. 20, 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flynn R., Zamponi G. W. (2010) Regulation of calcium channels by RGK proteins. Channels (Austin) 4, 434–439 [DOI] [PubMed] [Google Scholar]
- 5.Yang T., Colecraft H. M. (2013) Regulation of voltage-dependent calcium channels by RGK proteins. Biochim. Biophys. Acta 1828, 1644–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang T., Puckerin A., Colecraft H. M. (2012) Distinct RGK GTPases differentially use α1- and auxiliary β-binding-dependent mechanisms to inhibit Cav1.2/Cav2.2 channels. PLoS ONE 7, e37079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beqollari D., Romberg C. F., Meza U., Papadopoulos S., Bannister R. A. (2014) Differential effects of RGK proteins on L-type channel function in adult mouse skeletal muscle. Biophys. J. 106, 1950–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seu L., Pitt G. S. (2006) Dose-dependent and isoform-specific modulation of Ca2+ channels by RGK GTPases. J. Gen. Physiol. 128, 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan M., Buraei Z., Luo H. R., Levenson-Palmer R., Yang J. (2010) Direct inhibition of P/Q-type voltage-gated Ca2+ channels by Gem does not require a direct Gem/Cavbeta interaction. Proc. Natl. Acad. Sci. USA 107, 14887–14892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan M., Zhang W. K., Buraei Z., Yang J. (2012) Molecular determinants of Gem protein inhibition of P/Q-type Ca2+ channels. J. Biol. Chem. 287, 22749–22758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Béguin P., Nagashima K., Gonoi T., Shibasaki T., Takahashi K., Kashima Y., Ozaki N., Geering K., Iwanaga T., Seino S. (2001) Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411, 701–706 [DOI] [PubMed] [Google Scholar]
- 12.Yang T., Xu X., Kernan T., Wu V., Colecraft H. M. (2010) Rem, a member of the RGK GTPases, inhibits recombinant Cav1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J. Physiol. 588, 1665–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H., Puhl H. L. III, Niu S. L., Mitchell D. C., Ikeda S. R. (2005) Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J. Neurosci. 25, 9762–9772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlin B. S., Mosley A. L., Crump S. M., Correll R. N., Ozcan S., Satin J., Andres D. A. (2005) Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J. Biol. Chem. 280, 41864–41871 [DOI] [PubMed] [Google Scholar]
- 15.Finlin B. S., Correll R. N., Pang C., Crump S. M., Satin J., Andres D. A. (2006) Analysis of the complex between Ca2+ channel β-subunit and the Rem GTPase. J. Biol. Chem. 281, 23557–23566 [DOI] [PubMed] [Google Scholar]
- 16.Béguin P., Ng Y. J., Krause C., Mahalakshmi R. N., Ng M. Y., Hunziker W. (2007) RGK small GTP-binding proteins interact with the nucleotide kinase domain of Ca2+-channel β-subunits via an uncommon effector binding domain. J. Biol. Chem. 282, 11509–11520 [DOI] [PubMed] [Google Scholar]
- 17.Helton T. D., Horne W. A. (2002) Alternative splicing of the β4 subunit has α1 subunit subtype-specific effects on Ca2+ channel gating. J. Neurosci. 22, 1573–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finlin B. S., Shao H., Kadono-Okuda K., Guo N., Andres D. A. (2000) Rem2, a new member of the Rem/Rad/Gem/Kir family of Ras-related GTPases. Biochem. J. 347, 223–231 [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X., Horne W. A. (2014) ¹H, ¹³C, and ¹⁵N backbone resonance assignments of the 37 kDa voltage-gated Ca²⁺ channel β4 subunit core SH3-GK domains. Biomol. NMR Assign. 8, 217–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 21.Goddard, T.D., Kneller, D.G., SPARKY 3, University of California, San Francisco.
- 22.Opatowsky Y., Chen C. C., Campbell K. P., Hirsch J. A. (2004) Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42, 387–399 [DOI] [PubMed] [Google Scholar]
- 23.Van Petegem F., Clark K. A., Chatelain F. C., Minor D. L. Jr (2004) Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Petegem F., Duderstadt K. E., Clark K. A., Wang M., Minor D. L. Jr (2008) Alanine-scanning mutagenesis defines a conserved energetic hotspot in the Cavalpha1 AID-Cavbeta interaction site that is critical for channel modulation. Structure 16, 280–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajarathnam K., Rösgen J. (2014) Isothermal titration calorimetry of membrane proteins - progress and challenges. Biochim. Biophys. Acta 1838(1 Pt A), 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flynn R., Chen L., Hameed S., Spafford J. D., Zamponi G. W. (2008) Molecular determinants of Rem2 regulation of N-type calcium channels. Biochem. Biophys. Res. Commun. 368, 827–831 [DOI] [PubMed] [Google Scholar]
- 27.Nooren I. M., Thornton J. M. (2003) Diversity of protein-protein interactions. EMBO J. 22, 3486–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaynberg J., Qin J. (2006) Weak protein-protein interactions as probed by NMR spectroscopy. Trends Biotechnol. 24, 22–27 [DOI] [PubMed] [Google Scholar]
- 29.Puhl H. L. III, Lu V. B., Won Y. J., Sasson Y., Hirsch J. A., Ono F., Ikeda S. R. (2014) Ancient origins of RGK protein function: modulation of voltage-gated calcium channels preceded the protostome and deuterostome split. PLoS ONE 9, e100694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebert A. M., McAnelly C. A., Handschy A. V., Mueller R. L., Horne W. A., Garrity D. M. (2008) Genomic organization, expression, and phylogenetic analysis of Ca2+ channel β4 genes in 13 vertebrate species. Physiol. Genomics 35, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leipe D. D., Koonin E. V., Aravind L. (2003) Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol. 333, 781–815 [DOI] [PubMed] [Google Scholar]
- 32.Flynn R., Labrie-Dion E., Bernier N., Colicos M. A., De Koninck P., Zamponi G. W. (2012) Activity-dependent subcellular cotrafficking of the small GTPase Rem2 and Ca2+/CaM-dependent protein kinase IIα. PLoS ONE 7, e41185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghiretti A. E., Moore A. R., Brenner R. G., Chen L.-F., West A. E., Lau N. C., Van Hooser S. D., Paradis S. (2014) Rem2 is an activity-dependent negative regulator of dendritic complexity in vivo. J. Neurosci. 34, 392–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.