

Abstract
During DNA replication, nucleosomes ahead of replication forks are disassembled to accommodate replication machinery. Following DNA replication, nucleosomes are then reassembled onto replicated DNA using both parental and newly synthesized histones. This process, termed DNA replication-coupled nucleosome assembly (RCNA), is critical for maintaining genome integrity and for the propagation of epigenetic information, dysfunctions of which have been implicated in cancers and aging. In recent years, it has been shown that RCNA is carefully orchestrated by a series of histone modifications, histone chaperones and histone-modifying enzymes. Interestingly, many features of RCNA are also found in processes involving DNA replication-independent nucleosome assembly like histone exchange and gene transcription. In yeast, histone H3 lysine K56 acetylation (H3K56ac) is found in newly synthesized histone H3 and is critical for proper nucleosome assembly and for maintaining genomic stability. The histone acetyltransferase (HAT) regulator of Ty1 transposition 109 (Rtt109) is the sole enzyme responsible for H3K56ac in yeast. Much research has centered on this particular histone modification and histone-modifying enzyme. This Critical Review summarizes much of our current understanding of nucleosome assembly and highlights many important insights learned from studying Rtt109 HATs in fungi. We highlight some seminal features in nucleosome assembly conserved in mammalian systems and describe some of the lingering questions in the field. Further studying fungal and mammalian chromatin assembly may have important public health implications, including deeper understandings of human cancers and aging as well as the pursuit of novel anti-fungal therapies.
Keywords: Chromatin, epigenetics, H3K56ac, histone acetyltransferases, nucleosome assembly, replication-coupled nucleosome assembly, Rtt109
Introduction
In eukaryotic cells, DNA is assembled into chromatin, which is composed of repeating structures called nucleosomes consisting of 147 bp of helical DNA wound around a histone octamer, the protein component of the nucleosome containing a (H3–H4)2 tetramer and two H2A–H2B dimers. Chromatin encodes epigenetic information and is the physiological substrate for gene transcription and DNA repair and replication. Therefore, chromatin is dynamically and tightly regulated with respect to DNA-related processes. This regulation is often through the presence or absence of histone post-translational modifications (PTMs) such as methylation, acetylation, phosphorylation, SUMOylation and ubiquitylation. Histone PTMs can be added or removed by histone-modifying enzymes. For histone acetylation, this is done by histone acetyltransferases (HAT) and histone deacetylases, respectively. Furthermore, certain proteins, called histone chaperones, can also regulate the trafficking and assembly of histones into nucleosomes by non-covalent interactions with histones and histone-modifying enzymes, thereby adding yet another layer of complexity and regulation to chromatin.
During the S phase of the cell cycle, nucleosomes located ahead of replication forks are disassembled so that DNA replication machinery can replicate DNA. Once DNA is replicated, it must then be reassembled into nucleosomes using the parental histones that were disassembled ahead of replication forks, as well as newly synthesized histones, in a process called DNA replication-coupled nucleosome assembly (RCNA; Figure 1, top). This process is important for epigenetic inheritance and genomic integrity (Groth et al., 2007; Ransom et al., 2010). During certain DNA damage responses, parental histones must also be removed to pave the way for the DNA repair machinery. This process is thought to have many parallels with RCNA (Morrison & Shen, 2009; Smerdon, 1991). Additionally, nucleosome assembly during gene transcription and histone exchange can occur throughout the cell cycle in a process called DNA replication-independent nucleosome assembly (RINA; Figure 1, bottom). An important question in the epigenetic and chromatin fields is how cells regulate and ultimately carry out the complex processes of nucleosome disassembly and assembly.
Figure 1.

General schematic of RCNA and RINA. (Top) Replication-coupled nucleosome assembly (RCNA). Nucleosomes must be disassembled to make way for DNA replication machinery. Nucleosomes are then (re)assembled in close concert with DNA replication on the leading and lagging strands. (Bottom) Replication-independent nucleosome assembly (RINA). Many of the same core principles of nucleosome disassembly, DNA access and nucleosome assembly are likely applicable to replication-independent processes like gene transcription.
In this Critical Review, we first summarize the current body of knowledge surrounding nucleosome assembly, describing the importance of several histone chaperones, histone modifications and histone-modifying enzymes to this crucial epigenetic process. While describing the current state of understanding with respect to RCNA and RINA in yeast, we will also describe conserved features in these processes that have been found in mammalian cells. A special emphasis will be placed on critically analyzing the insights learned from studying regulator of Ty1 transposition 109 (Rtt109) HATs and H3K56ac in fungi, as this histone-modifying enzyme and histone PTM are crucial for proper RCNA and for genome stability in yeast. We go on to synthesize the molecular, enzymatic and structural insights that are helping to explain the function and regulation of Rtt109 HATs and H3K56ac in nucleosome assembly and other cellular processes. We conclude by the describing recent efforts to exploit Rtt109-catalyzed histone acetylation for anti-fungal purposes, an important but challenging bench-to-bedside endeavor.
DNA replication-coupled nucleosome assembly
The incorporation of replicated DNA into nucleosomes has been linked to DNA replication for some time (McKnight & Miller, 1977; Stillman, 1986). By one estimate, RCNA needs to generate ~30 million nucleosomes per replication per human cell (Kadyrova et al., 2013). More recently, lagging-strand DNA (Okazaki fragments) synthesis has been more firmly linked to nucleosome assembly. Not coincidentally, these fragments in yeast have approximately the same length as the repeating unit of nucleosomal DNA (Smith & Whitehouse, 2012). When DNA synthesis and nucleosome assembly are uncoupled by removing nucleosome assembly factors, cells show phenotypes consistent with genomic instability (Tyler et al., 1999). During S phase, nucleosomes already present on genomic DNA (i.e. parental nucleosomes, which are ahead of the replication forks) must be disassembled to make way for replication machinery (Figure 1, top). After DNA replication, parental histones can be recycled and reloaded onto the nascent DNA strands, though many of the mechanistic details of this process are still actively being characterized (Henikoff, 2008). The doubling of genomic content in DNA replication necessitates the doubling of histones, meaning new histone proteins must be synthesized and assembled into nucleosomes (i.e. newly synthesized, or nascent nucleosomes).
Despite its complexity, it is thought the overall process of nucleosome assembly in mammalian cells is conceptually similar to that in yeast, and therefore many features of nucleosome assembly are likely conserved from yeast to human cells. For example, in both yeast and mammalian cell RCNA, nucleosomes must be disassembled by first removing H2A–H2B dimers, followed by removing (H3–H4)2 tetramers. Nucleosome assembly then occurs by the loading of (H3–H4)2 tetramers onto nascent DNA strands – believed to be a random process with respect to strands – which is then followed by the rapid deposition of two H2A–H2B dimers to form a complete nucleosome (Verreault, 2000). A complex system of histone chaperones, histone modifications and histone-modifying enzymes have been shown to coordinate this process (Figure 2) (Burgess & Zhang, 2013). The exact molecular details linking nucleosome assembly to DNA replication are still poorly understood due to complex interactions between the two sets of machinery (Burgess & Zhang, 2013).
Figure 2.
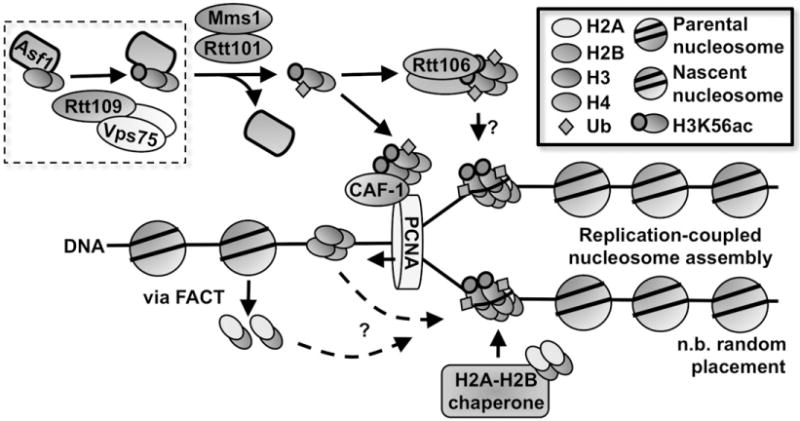
Fungal RCNA pathways. Shown is a schematic of RCNA in yeast. Note that the placement of parental and nascent nucleosomes is random with respect to strands.
In yeast, newly synthesized H3–H4 is bound to antisilencing function protein 1 (Asf1) and is imported from the cytoplasm to the nucleus in the heterotrimeric Asf1-H3–H4 form (English et al., 2006). Newly synthesized histone H3 is marked by H3K56ac prior to nucleosome assembly (Masumoto et al., 2005). H3K56ac is an atypical histone modification because it is located in the globular domain of the histone tetramer (Kenseth & Coldiron, 2004). H3K56ac levels in yeast peak during S phase and decrease in subsequent phases. Our group and others have shown that the acetylation of H3K56 (and other H3 residues as well) is catalyzed by the HAT Rtt109 in yeast, and that this HAT can form a complex with the histone chaperone vacuolar protein sorting 75 (Vps75) in vivo (Driscoll et al., 2007; Han et al., 2007). Rtt109 is unique among the known HATs because it is the sole enzyme responsible for H3K56ac in fungi and utilizes the Asf1-H3–H4 complex as the substrate for H3K56ac (Figure 2). Yeast with defects in Rtt109 or with certain H3K56 mutants are more sensitive to genotoxic stress (Han et al., 2007).
Histone chaperones such as Vps75 and Asf1 are crucial for RCNA, especially as they relate to Rtt109 HATs. Along with its role in stabilizing Rtt109, Vps75 helps traffic Rtt109 to the nucleus (Keck & Pemberton, 2011) and facilitates Rtt109-catalyzed acetylation of N-terminal lysines on H3. While Rtt109 catalyzes H3K56ac via the Asf1-H3-H4 substrate in vivo, the role of Vps75 appears to relate more to Rtt109-catalyzed acetylation of the H3 tail. Recently, a study involving the C-terminal carboxy domain of Rtt109 has provided evidence that Vps75 may play some role in promoting H3K56ac in vivo (Radovani et al., 2013), observations that demonstrate the complex interactions between Rtt109, histone chaperones and histone substrates.
As it is currently understood, the seminal role of Rtt109 in yeast RCNA is to catalyze the acetylation of H3K56 on newly synthesized H3–H4 during S phase. Defects in H3K56ac lead to reduced cell growth, genotoxic sensitivity and increased Rad52 foci (chromosomal breaks) – phenotypes linked to disruptions in RCNA (Han et al., 2007; Li et al., 2008). Given the critical importance of this modification, what exactly are the functional purposes of H3K56ac? Our group has recently shown H3K56ac promotes the interaction of H3–H4 with the Rtt101Mms1 E3 ubiquitin ligase in vivo and in vitro (Han et al., 2013). This interaction in turn promotes ubiquitination at H3K121, H3K122, and H3K125 by Rtt101Mms1. Yeast with disruptions in this process show phenotypes consistent with errors in RCNA. Importantly, H3K122ub weakens the Asf1-H3 interaction, which then facilitates the handoff of H3K56ac-H4 from Asf1 to the downstream chaperones regulator of Ty1 transposition 106 (Rtt106) and chromatin assembly factor-1 (CAF-1) (Figure 2) (Su et al., 2012). CAF-1 has well-known roles in targeting newly synthesized histone H3–H4 onto DNA (Krude, 1999; Smith & Stillman, 1991; Verreault et al., 1996). CAF-1 interacts with proliferation cell nuclear antigen (PCNA) via its largest subunit p150, effectively linking DNA replication and nucleosome assembly (Zhang et al., 2000). Based on several lines of evidence, including structural biology, Rtt106 likely promotes nucleosome assembly by aiding in the assembly of (H3–H4)2 tetramers (Fazly et al., 2012; Su et al., 2012; Zunder et al., 2012). Importantly, deposition of newly synthesized (H3–H4)2 tetramers by CAF-1 and Rtt106 should inhibit the formation of mixed nucleosomes containing a parental dimer and another dimer consisting of newly synthesized H3–H4 (Burgess & Zhang, 2013; Xu et al., 2010). Together these studies reveal that deposition of new H3–H4 is a highly regulated process during the S phase of the cell cycle.
After (H3–H4)2 deposition, two H2A–H2B dimers are deposited to complete the nucleosome octomer. In vitro experiments show the chaperone FACT can deposit H2A–H2B onto DNA (Belotserkovskaya et al., 2003). However, which histone chaperone(s) are involved in the deposition of histone H2A–H2B dimers (and also linker histone H1) onto newly replicated DNA in vivo is still not clear. Sometime during this complicated assembly process, H3K56ac is deacetylated by Hst3/Hst4, histone deacetylases (HDACs) whose expression peaks in G2/M and are regulated by Mec1 kinase (Celic et al., 2006; Haldar & Kamakaka, 2008; Maas et al., 2006; Thaminy et al., 2007; Yang et al., 2008). For completeness, we note that another HDAC, Sir2, has also been implicated in H3K56ac removal in telomeric heterochromatin (Xu et al., 2007). In addition to Rtt109 and H3K56ac, other HATs like Gcn5 and many other histone PTMs (e.g. H3K27ac, H4K5, 12ac) have been implicated in yeast RCNA, and for brevity, we refer the reader to recent reviews for more thorough discussions (Burgess et al., 2010; Burgess & Zhang, 2013).
RCNA in mammalian cells appears to be more complicated than in yeast. Compared to yeast cells where there is only one histone H3, mammalian cells express both canonical histone H3 (H3.1/H3.2) and the histone H3 variant H3.3. While “only” differing by five amino acids, canonical H3.1 is assembled into nucleosomes during S phase in the context of RCNA. In addition, the histone H3.1–H4 dimer is recruited by Asf1a and Asf1b and is transferred onto CAF-1 for deposition in mammalian cells (Alvarez et al., 2011). Furthermore, Cul4A-DDB1, a paralogue of yeast Rtt101Mms1, appears to also negatively regulate the interaction between Asf1 and histone H3–H4 (Han et al., 2013). Therefore, many features of RCNA are conserved from yeast to human cells.
Although they share many features, one of the differences between yeast and mammalian RCNA involves the associated histone modifications. Compared to yeast, the histone markers essential for mammalian nucleosome assembly are not as well characterized, along with the molecular mechanisms surrounding their role(s) in nucleosome assembly. While detectable, H3K56ac levels appear less abundant in human cells compared to yeast (Jasencakova et al., 2010). A more recent report show global levels of H3K56ac in mammalian cells represent less than 1% of total H3 content (Drogaris et al., 2012), suggesting that this modification may not be globally important, it is highly dynamic, or it has important functions in specialized contexts. Notably, there are conflicting reports as to whether its levels are coupled to the cell cycle (Xie et al., 2009; Yuan et al., 2009). But like in yeast, H3K56ac levels in mammalian cells appear to increase on chromatin following genotoxic treatment and also co-localize with sites of DNA repair (Das et al., 2009), though again there have been conflicting reports (Miller et al., 2010; Tjeertes et al., 2009). These observations suggest the role of H3K56ac in nucleosome assembly is either less critical – or perhaps more complicated, highly localized and/or a transient phenomenon – than that found in simpler model organisms. CAF-1 is necessary to incorporate H3K56ac into chromatin, and as in fungi, Asf1 (Asf1a) is crucial for H3K56ac in vivo (Das et al., 2009). The histone-modifying enzymes responsible for H3K56ac and its removal in mammalian cells have also been described. It is reported that the HATs p300 and Gcn5 can acetylate H3K56 in mammalian cells (Das et al., 2009; Tjeertes et al., 2009). There are reports showing H3K56ac is removed by different deacetylases (HDAC1, HDAC2, SIRT1, SIRT2 and SIRT6) in mammalian cells (Michishita et al., 2009; Miller et al., 2010; Toiber et al., 2013; Yang et al., 2009; Yuan et al., 2009).
In human cells, H3K56ac appears to have some relevance to human disease and development. H3K56ac levels are higher in certain human cancers and undifferentiated cells (Das et al., 2009), and they have been linked to unique global epigenetic signatures (Li et al., 2013) and pluripotency factors like NANOG, SOX2, and OCT4 in embryonic stem cells (Tan et al., 2013; Xie et al., 2009). Recently, several studies have been examining the role of H3K56ac in the context of obesity (Lo et al., 2011). For example, deletion of neural Sirt6 has been linked to hyperacetylation of H3K56, eventually leading to obesity in mice (Schwer et al., 2010).
A recent study raised concerns about many antibody-based studies (e.g. ChIP, immunofluorescence) focusing on mammalian H3K56ac (Drogaris et al., 2012). The chief concern is that several commercial anti-H3K56ac antibodies, while having specificity towards H3K56ac, also have the potential to recognize similar histone modifications such as H3K9ac, which are in much higher abundance than H3K56ac levels in mammalian cells. We believe these concerns should not be taken lightly, and it may be prudent for the field to reexamine (and hopefully reconfirm) some of the basic findings on H3K56ac in mammalian systems in light of these concerns. In addition, human cells contain both H3.1 and H3.3. It could be possible that these two histones are modified differently at H3K56, which may contribute to the discrepancies in various studies with H3K56ac. Therefore, we believe one of the major tasks in the future will be reconciling several conflicting reports with respect to the roles and regulation of H3K56ac in mammalian nucleosome assembly as well as other cellular processes. This will likely include better confirmation of detection method specificity (and sensitivity), as well as better characterization of H3K56ac localization in the genome, its dependence on the cell cycle and deacetylases and timing, and as discussed later, its role(s) in the DNA damage response.
In addition to H3K56ac, newly synthesized histone H3–H4 in mammalian cells appears to be labeled by acetylation on histone H4 lysines 5 and 12 (catalyzed by HAT1) and by methylation at H3K9 (Loyola et al., 2009; Nagarajan et al., 2013; Verreault et al., 1998). H4K12ac is conserved from yeast to humans, and the loss of HAT1 leads to defects in proliferation and the repair of DNA damage. However, this acetylation also appears dispensable for RCNA (Barman et al., 2006). It is likely that H4K5, K12ac regulate nucleosome assembly in part through modulating nuclear import of newly-synthesized H3.1–H4, as defects in these histone residues are linked to reduced levels of nuclear H3.1–H4 levels (Ejlassi-Lassallette et al., 2011). Additional studies are needed to determine the function of H3K9me1 in nucleosome assembly in human cells.
DNA replication-independent nucleosome assembly (RINA)
As in DNA replication, nucleosomes must be reassembled following gene transcription in a process linked to RINA (Figure 1, bottom). This category of nucleosome assembly also includes a phenomenon known as histone exchange whereby free histones are swapped for intact nucleosomes outside the context of DNA replication. Histone exchange may provide non-dividing cells with opportunities to dynamically regulate chromatin without resorting to replicating DNA, as well as a possible mechanism for replacing damaged histones. In mammalian cells, replication-coupled and RINA utilize distinct histone H3 molecules: canonical H3.1 and histone variant H3.3, respectively.
Many histone proteins are exchanged, including CenH3 (related to centromeres), H3.3 and some mammalian H2A variants. H2A–H2B is thought to exchange more readily than (H3–H4)2 (i.e. last on, first off), though there is evidence of (H3–H4)2 tetramer splitting and H3–H4 dimer histone exchange at actively-transcribed genes (Katan-Khaykovich & Struhl, 2011). While histone exchange is thought to occur frequently on highly transcribed genes, it has been observed in inactive promoters as well. For a thorough discussion of this important process, we point the reader to a well-written, comprehensive review (Das & Tyler, 2013).
It appears the deposition and exchange of H2A–H2B occurs similarly in both RCNA and in RINA processes such as histone exchange and gene transcription. In contrast to nucleosomal (H3–H4)2 tetramers which are relatively stable, nucleosomal H2A–H2B dimers can undergo rapid exchange with free (unbound) H2A–H2B. This occurs even in S phase, suggesting the deposition of H2A–H2B occurs similarly in the context of DNA replication and gene transcription. One of the most extensively studied H2A–H2B chaperones is Nap1, which preferentially binds H2A–H2B over H3-H4 in vivo (Mosammaparast et al., 2002). Nap1 contributes to H2A–H2B deposition and exchange on severallevels, including 1) the shuttling of H2A–H2B into the nucleus from the cytoplasm, 2) the disruption of non-productive histone-DNA interactions and 3) the direct deposition of both H2A–H2B and H3–H4 onto DNA for nucleosome assembly in vitro (Andrews et al., 2010; Ito et al., 1997; Mosammaparast et al., 2002). Another histone chaperone implicated in H2A–H2B nucleosome assembly is FACT, which favors binding H2A–H2B over H3–H4 in vitro. Based on early in vitro experiments, FACT has been proposed to facilitate the removal of H2A–H2B for RNA polymerase II. But more recently, FACT has been implicated in H3–H4 nucleosome assembly following gene transcription (Jamai et al., 2009).
In mammalian cells, histone H3.3 is deposited in RINA (along with H4) by chaperones such as HIRA, Daxx and DEK. HIRA is implicated in the assembly and exchange of histones H3.3–H4 in genic regions, since cells lacking this chaperone have reduced H3.3 levels at repressed and active genes. Daxx is thought to be involved in H3.3 deposition at telomeric regions, while DEK is thought to be involved in maintaining heterochromatin integrity. Therefore, with respect to RINA, H3.3 incorporation is likely regulated by different histone chaperones at different chromatin locations.
Compared to RCNA, there are some notable differences in how H3–H4 is incorporated in RINA. For example, in RCNA, (H3.1–H4)2 tetramers rarely split. However, during S phase, a small percentage of parental (H3.3–H4)2 tetramers split into H3.3–H4 dimers, which then form mixed nucleosomes containing nascent and parental H3.3–H4 dimers (Xu et al., 2010). In yeast, these mixed nucleosomes are concentrated in highly transcribed regions as well as regulatory regions (Katan-Khaykovich & Struhl, 2011). Therefore, unlike H3.1–H4, it may be possible that H3.3–H4 may be deposited in either dimeric or tetrameric form, and future studies will hopefully answer this important question.
Specific histone PTMs have been implicated in RINA. The roles of H3K56ac related to RINA are also intimately related to Asf1, as this chaperone is also known to be important for chromatin disassembly. Disruptions related to Asf1 are linked to reductions in nucleosome remodeling, histone eviction and the incorporation of new H3 at transcription sites (Gkikopoulos et al., 2009; Rufiange et al., 2007). Consistent with the interpretation that Asf1 can facilitate chromatin disassembly via promoting H3K56ac, H3K56Q mutants can correct defects in chromatin disassembly in asf1 mutants (Williams et al., 2008). Furthermore, when yeast are unable to acetylate H3K56, the global histone exchange rate decreases (Kaplan et al., 2008). This being said, Asf1 may have roles in chromatin assembly during RINA, independent of its role in chromatin disassembly. It is also hypothesized that the “looser” structure of chromatin enabled by H3K56ac may be important for encouraging multiple transcription rounds at transcribed genes (Dennehey & Tyler, 2014). At the same time, Rtt109 and Asf1 (but interestingly, not H3K56ac) are important for transcription repression in certain instances (Lin & Schultz, 2011), attesting to the complex interplay of histone exchange, gene transcription, histone chaperones and histone PTMs. Future studies may help to detail the contributions of Rtt109 and H3K56 in influencing gene transcription, – whether this is through histone exchange, altering DNA accessibility and/or some other mechanism(s) – and how dependent these mechanisms are to specific genes. There are also other histone PTMs that may affect RINA, specifically the deposition of newly synthesized H3.3–H4. The phosphorylation of histone H4K47 (H4K47ph) by Pak2 promotes the assembly of H3.3–H4 while inhibiting the assembly of H3.1–H4. Future studies will likely implicate more histone PTMs in RINA and gene transcription levels, and it may be important (as well as challenging) to differentiate between the modulation of histone–histone chaperone interactions versus the modulation of histone-DNA or histone-transcription machinery interactions.
Nucleosome assembly in the context of DNA repair
DNA integrity is continuously challenged on multiple fronts, from intrinsic errors in the cellular machinery that handles DNA to electromagnetic radiation to chemicals (e.g. mutagens). Deficiencies in DNA repair are one hallmark of human cancers (Jackson & Bartek, 2009). Several DNA repair pathways exist, notably homologous recombination (HR), non-homologous end joining, nucleotide excision repair (NER) and base excision repair. With respect to chromatin, the current model for DNA repair is “access, repair, restore” (Smerdon, 1991). That is, first chromatin must be disassembled for DNA repair machinery to access the damaged site(s) (access), then this machinery must perform the necessary repairs on the DNA (repair), and then the repair machinery must detach and the chromatin reassembled (restore). Errors in nucleosome assembly associated with the DNA repair process can have far-reaching consequences, as improper assembly could damage epigenetic inheritance (e.g. PTMs) or lead to genomic instability. For instance, DNA damage has the potential to propagate quickly, as nucleo-somes flanking a double-stranded break (DSB) can be disrupted during DNA repair (Tsukuda et al., 2005). While we will discuss nucleosome assembly in the context of DNA repair briefly, we point the reader to recent reviews for a comprehensive summary of this important topic (Adam et al., 2014; Dennehey & Tyler, 2014).
Many of the details regarding the histone chaperones involved in DNA repair are still being characterized (Dennehey & Tyler, 2014). As in RCNA, the details of how nucleosomes in the vicinity of DNA damage sites are dismantled are still relatively uncharacterized (Dennehey & Tyler, 2014). DNA-end resection may drive nucleosome disassembly via chromatin remodelers like INO80 (Chambers & Downs, 2012; Morrison et al., 2004), or this process may in fact be more complicated. To add to the complexity, there is also dynamic exchange of histone variants at sites of DNA damage and repair (Volle & Dalal, 2014). Histone H2A.Z is incorporated near DSBs in human cells (Xu et al., 2012), and defects in its incorporation lead to genotoxic sensitivity and defects in DNA repair. It has been hypothesized that nucleosomes with this histone variant are more easily disassembled, which can allow for better access of repair machinery (Dennehey & Tyler, 2014). Histone H2A.X, which has a random distribution across chromatin, becomes phosphorylated at serine 129/139 in yeast and mammals, respectively, by the DNA damage checkpoint system. This histone variant is therefore involved in signaling the presence of DNA damage, and is removed (via H2A.X–H2B dimers) by FACT and replaced with canonical H2A–H2B dimers (Heo et al., 2008).
The prevailing thought is that nucleosome assembly during DNA repair occurs similarly as compared to RCNA, perhaps because DNA repair involves DNA synthesis. Consistent with this idea, CAF-1 is recruited to sites of UV damage, where it functions to help deposit new H3.1–H4 at DNA repair sites with the aid of PCAF (Adam et al., 2014; Moggs et al., 2000). Recently, the histone H3.3 chaperone HIRA was shown to be enriched at DNA damage sites in human cells, prior to the arrival of CAF-1 (Adam et al., 2013). In yeast, an important component of nucleosome assembly after DNA repair is Asf1-dependent H3K56ac. Supporting this notion is that the H3K56Q mutation, which chemically resembles H3K56ac, removes the requirement for Asf1 in proper nucleosome assembly after DSB in yeast (Chen et al., 2008).
We and others acknowledge there is still much more to learn about nucleosome assembly in the context of DNA damage and repair (Adam et al., 2014). Advances in knowledge related to RCNA will almost certainly help our understanding of nucleosome assembly in the context of DNA repair. Just as the fate of parental histones is elusive in RCNA, their fate in the DNA damage responses will be an important question to address, as it may have important implications for epigenetic memory. We and others are interested to see the differences, if any, between nucleosome assembly in the context of specific types of DNA damage and repair pathways (Adam & Polo, 2012), most notably in mammalian systems.
Relevance of nucleosome assembly to human health
Given the integral roles of nucleosome assembly in gene transcription, DNA replication and DNA repair, it is not surprising that factors in nucleosome assembly have been linked to cancers and other diseases (Aqeilan et al., 2008; Ask et al., 2012; Burgess & Zhang, 2013; May et al., 2013; Polo et al., 2010; Wise-Draper et al., 2009; Yang et al., 2013), aging (Feser et al., 2010), and recently, cell differentiation and neuronal asymmetry (Nakan et al., 2011). For example, mutations in the histone chaperone ATRX, along with DAXX, have been identified in tumors exhibiting alternative lengthening of telomeres (ALT), including pancreatic neuroendocrine tumors and certain sets of pediatric brain tumors (Killela et al., 2013). The loss of wild-type ATRX is thought to promote cancer development by facilitating alternative lengthening of telomeres (ALT) pathway activation (Bower et al., 2012). As another example, the expression levels of the histone chaperone Asf1b has been correlated with the proliferative status of breast cancer cells, a finding which could aid breast cancer diagnosis and prognosis (Corpet et al., 2011). Mutations in Asf1 have also been predicted to drive cancers by altering DNA replication and by modifying gene expression during cell cycling to increase proliferation (Reimand & Bader, 2013). Other studies have linked factors involved in nucleosome assembly with Beckwith–Wiedemann syndrome, DiGeorge and Velocardiofacial syndromes and congenital dyserythropoietic anemia type I (Catchpool et al., 2000; DAntoni et al., 2004; Renella et al., 2011; Soekarman et al., 1992). Finally, other potential regulators of nucleosome assembly like p300/CBP and HAT-1, enzymes that are responsible for histone modifications linked to nucleosome assembly, have been implicated in developmental disorders and cancers (Nagarajan et al., 2013; Valor et al., 2013; Van Beekum & Kalkhoven, 2007). In the future, additional studies will almost surely identify more links between disruptions in nucleosome assembly and human pathologies. Mechanistic understanding of this critical cellular process (“basic science”) and identifying relevant patient subsets (patient stratification) may then provide opportunities for therapeutic interventions (Box 1).
Box 1. Other recent findings related to Rtt109 and H3K56.
Several recent studies are linking Rtt109 and H3K56 to other important cellular processes. For example, in Neurospora, H3K56ac is required homologous recombination and interestingly, this modification has been implicated in quelling-induced small RNA production (Zhang et al., 2014). Other reports have expanded the links between Rtt109 and/or H3K56ac to diverse biological processes such as Artemia development (Zhou et al., 2013), rRNA hyper-amplification (Ide et al., 2013), retrotransposon regulation (Tanaka et al., 2012), the Ras-PI3K pathway (Liu et al., 2012), angiogenesis (Dutta et al., 2010), circadian rhythms (Malapeira et al., 2012) and mTOR signaling and rRNA biogenesis (Chen et al., 2012). Some of these biological functions may be inherently linked to the role of H3K56ac in nucleosome assembly. Future studies are needed to address these possibilities.
A closer look at Rtt109 HATs: molecular, biochemical and structural insights
Our group and others have discovered and extensively studied Rtt109. These studies have shown Rtt109 HATs are subject to complex regulation, including some rather unique features. This section will synthesize the current knowledge of Rtt109 HATs, including key discoveries in molecular and cellular biology, enzymatic mechanisms and biomolecular structures. It is anticipated that deeper understandings of Rtt109 HATs at the molecular level may enhance our understanding of similar processes in mammalian systems, and may provide additional insights for therapeutically targeting HATs.
Complex chaperone and substrate dynamics
The substrates involved in Rtt109-catalyzed histone acetylation are complex, even in relatively simplified in vitro settings. Rtt109 is capable of acetylating H3K56 (Driscoll et al., 2007; Han et al., 2007; Tsubota et al., 2007), located in the globular core of the nucleosome. Rtt109 can also acetylate the N-terminal tail residues H3K9, H3K14, H3K23 and H3K27 in vitro (Figure 3A) (Berndsen & Denu, 2008; Berndsen et al., 2008; Burgess et al., 2010; Fillingham et al., 2008). The majority of Rtt109 substrates were determined by Western blotting or mass spectrometry using in vitro assays, and have been either qualitative or semi-quantitative in nature, which can make it more difficult to decipher and unambiguously quantify the contributions of various chaperones on specific histone modifications (Fulzele et al., 2012). A welcome advance has been the application of quantitative proteomics to characterize the acetylation patterns of Rtt109-Asf1 and Rtt109-Vps75 in vitro (Abshiru et al., 2012). Interestingly, it was found that Rtt109-Vps75 is capable of acetylating H4K12 (and possibly H4K5 and H4K8) in vitro (Abshiru et al., 2012), though the contribution of Rtt109 to these modifications in vivo is unknown. Future studies with quantitative proteomics may want to compare the effects of each chaperone-Rtt109 combination with Rtt109 itself (i.e. no chaperone present) to better gauge the effect of each chaperone. Rtt109 and its complexes do not significantly acetylate histone H2A, H2B, or H4 in vitro (Tsubota et al., 2007). In terms of other in vitro substrates, the Rtt109-Vps75 complex is capable of acetylating histone H1 linker under some conditions in vitro, though the functional significance of this ability is unknown (Radovani et al., 2013).
Figure 3.

Molecular biology of Rtt109 HATs. (A) Simplified substrate profile for Rtt109. (B) Model of Rtt109 regulation in vivo. Vps75 is linked to trafficking Rtt109 to the nucleus. We caution that the exact cellular location of Rtt109 autoacetylation and its removal has not been confirmed. The cellular levels of acetyl-CoA has been hypothesized as one regulator of Rtt109 autoacetylation, and thus Rtt109 activity, in vivo. Inhibition by product accumulation (CoA, acetylated histones) may also regulate Rtt109 activity.
The substrate patterns of Rtt109 are subject to complex modulation by chaperones (Abshiru et al., 2012). In vitro, Rtt109-Vps75 activity towards histone H3 is dramatically enhanced by the presence of Asf1 if H4 is also present (Han et al., 2007). Rtt109-Vps75 complexes acetylate non-nucleosomal histone H3, such as (H3–H4)2 tetramers or core histones (H3–H4 – H2A–H2B). However, Rtt109-Vps75 does not acetylate histone H3 efficiently in the context of mono-or dinucleosomes (Han et al., 2007). This observation suggests Rtt109 only acetylates histone H3 before it is incorporated into nucleosomes, which is consistent with H3K56ac being observed in newly-synthesized H3 in S phase (Celic et al., 2006; Masumoto et al., 2005; Recht et al., 2006). Therefore, substrate regulation in the form of available nucleosomal or non-nucleosomal H3 is likely another layer of regulating Rtt109 activity. Compared to Asf1, Vps75 seems to confer a broader substrate profile. While Vps75 seems to enhance the substrate profile of Rtt109, this same study showed Asf1 directs Rtt109 towards H3K56 in vitro (Abshiru et al., 2012). Together, the collective observations show Rtt109-substrate dynamics are complicated.
The substrate specificity patterns of Rtt109 and its chaperones in vivo are difficult to characterize (Figure 3A). Teasing apart the contributions of each chaperone on specific histone modifications is difficult, as the results can be confounded by HATs will overlapping substrates such as Gcn5 in yeast. It is well established that Rtt109 and Asf1 are both required for H3K56ac in vivo while Vps75 does not have as appreciable an effect on H3K56ac levels (Berndsen et al., 2008; Celic et al., 2006; Driscoll et al., 2007; Fillingham et al., 2008; Han et al., 2007; Recht et al., 2006; Tsubota et al., 2007). Many studies have also examined the effects of Rtt109, Vps75, Asf1 and Gcn5 deletion on the levels of specific N-terminal histone H3 acetylation modifications (Berndsen et al., 2008; Burgess et al., 2010; Fillingham et al., 2008; Keck & Pemberton, 2011; Tang et al., 2011). Our observation that gcn5∆ yeast also lacking vps75 are completely deficient in H3K27ac levels suggests Vps75 is required for acetylation of the H3 N-terminus (Burgess et al., 2010).
These studies demonstrate the contributions of these chaperones and HATs to individual H3 tail modifications in vivo is quite complex, and more studies are needed for clarification. A logical extension of the in vitro quantitative proteomics findings could be to apply quantitative proteomics to in vivo systems. It may be interesting to get a quantitative characterization of specific histone modifications in response to various chaperone perturbations or environmental stresses in vivo. Based on the breadth of data to date, the working model for Rtt109 substrate specificity is that the Rtt109-Vps75 complex is responsible for acetylation of the H3 N-terminus, while Rtt109 and Asf1 are responsible for H3K56ac. As of yet, there have not been any studies testing whether Asf1 is required for the acetylation of the H3 N-terminus in vivo.
Characterizing the Rtt109 chaperone interactions
The effects of the histone chaperones Vps75 and Asf1 on Rtt109 activity have been studied extensively. As mentioned previously, Rtt109 forms reversible complexes with the histone chaperone Vps75 (Table 1). Vps75 is a member of the NAP1 histone chaperone family, and it can also bind either (H3–H4)2 or H2A–H2B dimers (Park et al., 2008; Selth & Svejstrup, 2007). Vps75 forms a self-dimer in vitro, and this dimerization enhances Rtt109 HAT activity in vitro (Tang et al., 2011). Rtt109 and Vps75 can form a complex in vitro and in vivo with low nanomolar affinity (Albaugh et al., 2010; Krogan et al., 2006; Tsubota et al., 2007). Additionally, Vps75 protects Rtt109 from proteolysis in vitro, most likely by shielding the Vps75-binding loop (residues 130–179) from proteases (Tang et al., 2011). The binding of Vps75 to Rtt109 enhances enzymatic activity in vitro (Berndsen et al., 2008; Han et al., 2007; Tsubota et al., 2007), as reflected by a nearly 100-fold increase in kcat, but does not appear to dramatically alter the KM of either protein or acetyl coenzyme A (acetyl-CoA) substrate, suggesting Vps75 stabilizes a more active conformation of Rtt109 rather than enhancing substrate binding (Tables 1 and 2).
Table 1.
Summary of select Rtt109-related protein–protein interactions.
| Protein 1 | Protein 2 | appKd (nM) | Technical notes | Reference |
|---|---|---|---|---|
| Rtt109 | Vps75 | 10 ± 2 | FP; acetyl-CoA present | (Albaugh et al., 2010) |
| Rtt109 | Vps75 | 13 ± 3 | Steady-state kinetic method; acetyl-CoA and H3 present | (Kolonko et al., 2010) |
| Rtt109 | Vps75 | 10; 2700 | Kd1 and Kd2; modeled as Vps75 and Rtt109 dimers | (Tang et al., 2008) |
| Rtt109 | H3–H4 | 150 ± 10 | FT | (Park et al., 2008) |
| Rtt109 | H3 | 17 ± 8 | FP | (Berndsen et al., 2008) |
| Vps75 | H3–H4 | 25 ± 3 | FT | (Park et al., 2008) |
| Vps751–223 | H3–H4 | 13 ± 1 | FT; truncated C-terminal acidic domain | (Park et al., 2008) |
| Vps75 | H3 | 120 ± 10 | FT | (Park et al., 2008) |
| Vps75 | H3 | 56 ± 8 | FP | (Berndsen et al., 2008) |
| Vps75 | H2A–H2B | 4.5 ± 0.3 | FT | (Park et al., 2008) |
| Asf1 | H3–H4 | 10 ± 1 | FT | (Park et al., 2008) |
| H3–H4 | Asf1 | 2.5 ± 0.7 | FQ | (Rishton, 1997) |
FP, fluorescence polarization; FT, fluorescence titration method; FQ, fluorescence quenching method.
Table 2.
Summary of Rtt109 kinetic parameters.
| Enzyme form | Protein substrate | Protein KM (μM) | kcat (s−1) | acetyl-CoA KM (μM) | Technical notes | Reference |
|---|---|---|---|---|---|---|
| Rtt109 | H31–20 | 83 ± 29 | 0.0017 ± 0.0001 | 0.3 ± 0.1 | acetyl-CoA + protein substrate pairing unclear | (Berndsen et al., 2008) |
| H3 | 8.1 ± 0.1 | 0.0033 ± 0.0003 | 0.3 ± 0.1 | acetyl-CoA + protein substrate pairing unclear | (Berndsen et al., 2008) | |
| H3–H4 | 2.9 ± 0.6 | 0.0044 ± 0.0009 | 0.3 ± 0.1 | acetyl-CoA + protein substrate pairing unclear | (Berndsen et al., 2008) | |
| Rtt109-Vps75 | H31–20 | 75 ± 15 | 0.13 ± 0.04 | 1.0 ± 0.2 | acetyl-CoA + protein substrate pairing unclear | (Berndsen et al., 2008) |
| H31–20 | 112 ± 11 | 0.11 ± 0.01 | 0.3 ± 0.1 | – | (Albaugh et al., 2010) | |
| H3 | 5.8 ± 0.8 | 0.21 ± 0.04 | 1.0 ± 0.2 | acetyl-CoA + protein substrate pairing unclear | (Berndsen et al., 2008) | |
| H3 | – | 0.19 ± 0.01 | 1.0 ± 0.2 | – | (Tsubota et al., 2007) | |
| H3 | 3.9 ± 2 | 0.069 ± 0.001 | NR | 0.2 eq. Vps75 | (Kolonko et al., 2010) | |
| H3 | 6.5 ± 2 | 0.62 ± 0.02 | NR | 8 eq. Vps75 | (Kolonko et al., 2010) | |
| H3 | 7 ± 1 | 0.63 ± 0.02 | NR | – | (Albaugh et al., 2010) | |
| H3 | 8.5 ± 0.7 | 0.375 ± 0.027 | 8.0 ± 2.0 | – | (Tang et al., 2008) | |
| H3–H4 | 1.4 ± 0.4 | 0.11 ± 0.05 | 1.0 ± 0.2 | acetyl-CoA-protein substrate pairing unclear | (Berndsen et al., 2008) | |
| H3–H4 | 2.12 ± 0.63 | 0.068 ± 0.004 | NR | 6 eq. Vps75 | (Tsubota et al., 2007) | |
| H3–H4 | 0.84 ± 0.28 | 0.34 ± 0.04 | NR | – | (Tang et al., 2011) | |
| H3–H4 | 0.5 ± 0.3 | 0.041 ± 0.004 | NR | 0.2 eq. Vps75 | (Kolonko et al., 2010) | |
| H3–H4 | 0.1 ± 0.1 | 0.13 ± 0.007 | NR | 8 eq. Vps75 | (Kolonko et al., 2010) | |
| yH3–H4 | 1.4 ± 0.4 | 0.41 ± 0.05 | NR | Tetramer | (Kolonko et al., 2010) | |
| yH3(A110E)–H4 | 2.4 ± 0.7 | 0.39 ± 0.06 | NR | Dimeric form | (Kolonko et al., 2010) | |
| Rtt109-Asf1 | H3–H4 | 1.19 ± 0.34 | 0.021 ± 0.002 | Low micromolar | – | (Tsubota et al., 2007) |
| H3–H4 | 1.84 ± 0.81 | 0.015 ± 0.002 | NR | – | (Tang et al., 2011) |
In general, the KM for protein substrate decreases from the peptide to the full-length H3 to the H3–H4 tetramer form, with or without Vps75 present. In general, the addition of Vps75 leads to an increase in kcat regardless of protein substrate, while the KM for acetyl-CoA is relatively constant. NR, no report.
The other chaperone, Asf1, is highly conserved throughout eukaryotes and binds to the H3–H4 heterodimer, as opposed to (H3-H4)2 (Table 1). Rtt109 also interacts with Asf1, but the Rtt109-Asf1 interaction is hard to detect, which contrasts the Rtt109-Vps75 interaction (Han et al., 2007; Tsubota et al., 2007). In vitro, Rtt109 and Rtt109-Vps75 cannot pull-down Asf1 without H3–H4 bound to Asf1 (Han et al., 2007). However, different Rtt109 constructs have been reported to bind Asf1 in vitro in the absence of histones (Tsubota et al., 2007). Rtt109 and Asf1 do not co-purify in vivo without the aid of crosslinking, providing further evidence that this interaction is transient and is mediated in part through their interactions with H3–H4 (Han et al., 2007). These results strongly support a model whereby Rtt109-Vps75 utilizes the Asf1-H3–H4 complex, instead of H3–H4, as the substrate for H3K56ac. This model explains why Asf1 is essential for H3K56 acetylation in vivo. Compared to studies with Rtt109-Vps75, there is much less data about the effects of Asf1 on the kinetics of Rtt109 HAT activity (Table 2).
Proposed enzymatic mechanism of action
Understanding of the chemical mechanism of Rtt109-catalyzed histone acetylation can be important for several reasons. In structure-and mechanism-based drug design, transition-state analogs can be potent inhibitors of enzyme activity (Schramm, 2011). This could be important in the design of specific chemical probes to further study nucleosome assembly (Cole, 2008), or for developing Rtt109-specific inhibitors, which as described later on, have been hypothesized as anti-fungal agents. HATs catalyze the transfer of the acetyl moiety from acetyl-CoA to a lysine substrate, though the exact order of substrate binding and the nature of chemical catalysis can vary among HATs (Berndsen & Denu, 2008; Hodawadekar & Marmorstein, 2007; Marmorstein & Trievel, 2009; Roth & Denu, 2001; Vetting et al., 2005; Wang et al., 2008). For instance, in the Theorell-Chance mechanism, acetyltransferases bind both the acetyl-CoA and histone substrates prior to a direct nucleophilic attack by lysine residues to the acetyl moiety on acetyl-CoA (Tanner et al., 1999; Trievel et al., 1999). By contrast, other HATs like yeast Esa1 form an acyl-enzyme intermediate before the acyl moiety is transferred to lysine (i.e. Ping-Pong mechanism) (Yan et al., 2002). A common component of both mechanisms is that lysine, which is normally protonated at physiologic pH, must be deprotonated to sufficiently increase its nucleophilicity towards either the acyl-enzyme intermediate or acetyl-CoA.
Early on, the catalytic mechanism of yeast Rtt109 has been the subject of much investigation. Speculation has surrounded the role of residues D287 and D288 as potential general bases, as double mutations at these two residues can cripple Rtt109 activity in vitro, but this possibility has been ruled out. The decrease in activity with D287/D288 mutants has instead been attributed to reduced acetyl-CoA binding and/or suboptimal positioning of acetyl-CoA for enzymatic catalysis, presumably through an interaction with K290ac (Albaugh et al., 2010, 2011). Further examination of the putative Rtt109 active site does not reveal any obvious catalytic residues such as a strong nucleophiles or acids/bases. The search for a general base has even stretched to Vps75, but Rtt109 and Rtt109-Vps75 had similar pH-pKa profiles (Kolonko et al., 2010). There had also been speculation that the acetylation observed at K290, which happens to be near the acetyl-CoA binding domain, may represent an acyl-enzyme intermediate in a Ping-Pong mechanism. But again, this possibility was excluded by a thorough analysis of K290ac (Albaugh et al., 2011).
In a well-designed series of experiments, Albaugh et al. proposed a mechanism of action for the Rtt109-Vps75 complex (Albaugh et al., 2010). They proposed a sequential order of substrate binding where both substrates bind Rtt109-Vps75 to form a ternary complex, with the actual order of substrate binding being either random or not strictly ordered (Figure 4A). This is unlike some other known HATs, where binding has an obligate order (Liu et al., 2008; Tanner et al., 2000). Rapid-quench kinetic analysis was consistent with chemical catalysis being the rate-limiting step, rather than substrate binding or product release. In this proposed mechanism, the lysine substrate, whose primary amine side chain (pKa ~10) is typically protonated at physiologic pH, must have its positive charge neutralized to increase its nucleophilicity, quaternary amines are insufficiently nucleophilic (Figure 4B). As Rtt109 lacks a discernable general base in its active site, Albaugh et al. invoke a Born/desolvation effect to explain how the lysine substrate can become deprotonated and thereby increase its nucleophilicity enough for an acetylation reaction to occur at catalytic rates (Harris & Turner, 2002). In this model, the Rtt109-Vps75 hydrophobic active site perturbs the substrate lysine pKa from its typical value of 10 downwards to somewhere near 8.5, as it would be energetically unfavorable for a charged species to migrate from a high- to low-dielectric environment. Such a general mechanism of decreasing the lysine pKa fits with the ability of Rtt109 to acetylate multiple lysine residues, but a more detailed understanding of this proposed mechanism, such as which residues are involved in lysine and transition-state stabilization, will require more studies (Zhang et al., 2014). Several lingering questions remain with respect to the Rtt109 enzymatic mechanism. Due to some well-recognized technical difficulties, these studies were performed with H3 peptides or histones without Asf1, conditions that most likely do not match the key physiological substrate for Rtt109-Vps75 complex. Therefore, it remains to be seen what effects, if any, Asf1 may have on the catalytic mechanism and kinetics of Rtt109 and Rtt109-Vps75 HAT activity (Table 2). Answering these more detailed questions will require careful optimization of experimental conditions, as histones and histone-chaperone complexes like Asf1-H3–H4 are prone to precipitation under many in vitro testing conditions (Dahlin JL, Zhang Z, unpublished observations).
Figure 4.
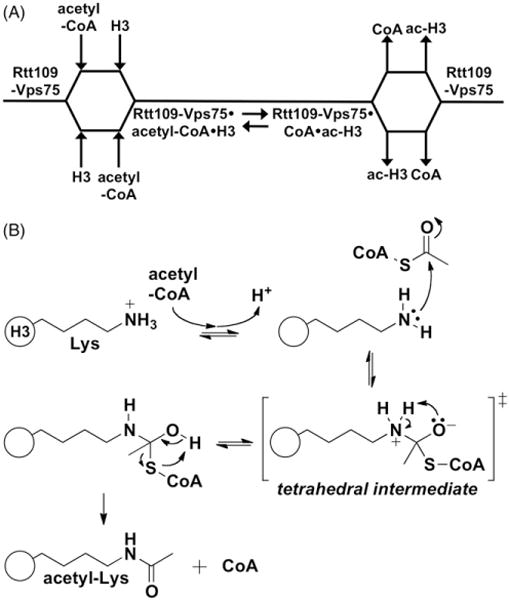
Current model for enzymatic mechanism of yeast Rtt109. (A) Cleland diagram for Rtt109-Vps75-catalyzed histone acetylation depicting the proposed order of substrate binding and product release. H3 is depicted as the substrate, but it can most likely include other substrates. Protein substrate and acetyl-CoA appear to bind and depart Rtt109-Vps75 in a non-obligate order. (B) Proposed catalytic model of Rtt109-catalyzed histone acetylation. The lysine substrate (pKa ~10) becomes deprotonated during the course of binding. In the absence of a general base catalyst, the lysine side chain is likely positioned at an optimal distance and orientation with respect to bound acetyl-CoA to undergo a nucleophilic attack at the acetyl carbonyl carbon. A tetrahedral intermediate would then be formed, followed by product release. The protein and/or solvent component(s) that likely stabilize the transition states are unknown.
Autoacetylation as a regulatory mechanism
An intriguing regulatory feature of yeast Rtt109 activity is lysine 290 acetylation (K290ac), a phenomenon first observed via autoradiography (Driscoll et al., 2007; Han et al., 2007). Several mass spectrometry studies have unambiguously identified K290ac (Lin & Yuan, 2008; Stavropoulos et al., 2008; Tang et al., 2008), and crystal structures show electron densities consistent with acetylation at K290 (Lin & Yuan, 2008; Stavropoulos et al., 2008; Tang et al., 2008). Functionally, K290 mutants show decreased HAT activity in vitro, and some are deficient in cellular functions such as genotoxin resistance (Lin & Yuan, 2008; Stavropoulos et al., 2008). Substitution with a thiocarbamate mimic of acetylated lysine can partially restore enzymatic activity in vitro (Huang et al., 2010). K290ac is surrounded by a cage of conserved hydrophobic residues, and the formation of this structure likely requires the neutralization of the positive lysine charge by acetylation. K290ac is located approximately 10 Å away from the acetyl-CoA aperture, and it unlikely to be directly involved in enzymatic catalysis. It has currently thought that one role of D288, whose carboxylate forms a hydrogen bond with the K290 amine, is to help properly position K290ac and thereby promote enzymatic activity. Indeed, the body of mutagenic studies for D288 and K290 suggests the precise positioning of the activation domain is crucial for Rtt109 activity and more efficient acetyl-CoA binding (Lin & Yuan, 2008; Stavropoulos et al., 2008; Tang et al., 2008).
Additional details concerning K290ac have recently been reported. While the majority of TAP-purified or recombinant Rtt109 is acetylated at K290 (Tang et al., 2008), this modification can be reversed by the histone deacetylase Hst2 in vitro (Albaugh et al., 2011). Subsequent experiments have shown K290ac is catalyzed by an intramolecular process (Albaugh et al., 2011). A series of well-designed experiments showed that K290ac is reversible and is critical for Rtt109 HAT activity in vitro by increasing the rate of catalysis and by enhancing acetyl-CoA binding, and this modification does not contribute to Rtt109-Vps75 stability or likely represent a catalytic intermediate (Albaugh et al., 2011). Interestingly, both enzymes known to acetylate H3K56 – Rtt109 and p300 – feature autoacetylation, though in p300 this process in intermolecular and contains several autoacetylation sites (Balasubramanyam et al., 2006; Thompson et al., 2004). An early study reported K290ac is not essential for H3K56ac in vivo or genotoxic resistance in yeast (Tang et al., 2008), which contrasts with subsequent data (Stavropoulos et al., 2008). It is unclear why this apparent discrepancy exists between the in vitro and in vivo data. Possible explanations include experimental considerations, or that the role of K290ac with respect to H3K56ac and other cellular functions related to nucleosome assembly are more nuanced than the in vitro data suggests. Therefore, additional experiments are likely needed to clarify the role of K290ac in an in vivo setting.
Clearly, there are still unanswered questions about the regulation and significance of Rtt109 autoacetylation. It could be that autoacetylation is one of the many layers of Rtt109 regulation in vivo (Figure 3B). Analogous to the autophosphorylation seen in protein kinases, autoacetylation may activate the Rtt109 enzyme. It has been hypothesized that Rtt109 may exist between two states, an inactive state without K290ac, and an active state with K290ac (Stavropoulos et al., 2008). The enzymes responsible for its deacetylation in vivo are unknown, though Hst3 and/or Hst4 may be responsible (Albaugh et al., 2011). This could explain how cells completing S phase nucleosome assembly decrease in both H3K56ac levels and Rtt109 activity. Another intriguing connection brought up by Albaugh et al. is that K290ac lowers the KM for acetyl-CoA, which may prime Rtt109 for the low levels of acetyl-CoA found during its peak expression in G1/S phase yeast. Autoacetylation appears to be a conserved feature among Rtt109 HATs, as K290 is highly conserved among fungal Rtt109s. Signs of autoacetylation has been observed in vitro by autoradiography in several other fungal Rtt109 HATs, including Schizosaccharomyces pombe, Candida albicans, Pneumocystis carinii, Pneumocystis jirovecii and Aspergillus fumigatus (Dahlin JL, Zhang Z, unpublished results). The continued understanding of Rtt109 autoacetylation may also have implications for HATs in general, as acetylation has been found in other HATs (Kadlec et al., 2011; Karanam et al., 2006; Santos-Rosa et al., 2003; Thompson et al., 2004; Wang & Chen, 2010).
Structural overview of yeast Rtt109
Several crystal structures have been solved for Rtt109 (Stavropoulos et al., 2008; Su et al., 2011; Tang et al., 2008, 2011) and many structural features have now been linked to specific protein functions. The overall structure revolves around a central plate-like eight-strand β-sheet (the “acetyltransferase domain”), which is surrounded by nine a-helices and some additional loops (Figure 5A). Borrowing the nomenclature from Tang et al., the helices α1–α4 link the bottom strands of Rtt109. Helices α5–α7 (which form part of the ~45-residue “activation domain”) and helices α8–α9 bundle along opposite ends of the protein. Two linkers connect the acetyltransferase domain to the activation domain, which sits atop the acetyltransferase domain. The acetyl, β-mercaptoethylamine and pantothenic acid moieties of acetyl-CoA are surrounded by the β4–α2 and β5–α4 segments on the interior side of the binding domain, and are encapsulated by the 21-residue L1 loop that connects β5 to α4. The adenosine diphosphate moiety of acetyl-CoA is solvent-exposed, while the acetyl moiety extends towards a narrow aperture approximately 6 Å wide. In terms of electrostatics, the histone-binding region proximal to the proposed lysine-binding site is relatively apolar with a nearby basic patch (Figure 5A). Many of the structures contain a truncated Vps75-binding loop (Stavropoulos et al., 2008; Tang et al., 2008), which normally consists of residues 130–179 but is intrinsically disordered and non-amenable to crystallization without Vps75 present (Su et al., 2011). Many of the active site residues that define the acetyl-CoA active site are conserved among putative Rtt109 orthologues (Tang et al., 2008).
Figure 5.
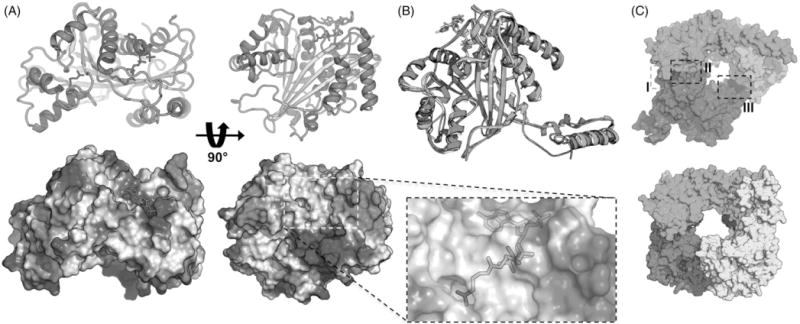
Structural overview of yeast Rtt109. (A) Secondary structure features (top) and electrostatic potentials (bottom) of yeast Rtt109 (PDB ID 3QMO). The inset highlights the acetyl-CoA binding mode. α-helices, red; β-sheets, yellow; loops, green; acetyl-CoA, magenta sticks; K2090ac, blue sticks. (B) Presence of Vps75 does not significantly alter secondary structure of Rtt109 in crystal structures. PDB IDs: green, 3Q33; yellow, 3QMO; magenta, 3Q66. (C) Comparison of yeast Rtt109-Vps75 crystal structures. (top) Su et al. structure, PDB ID 3Q66. Green and cyan, Vps75; magenta, Rtt109. I–III denotes the locations of the three protein-protein interfaces. (bottom) Tang et al. structure, PDB ID 3Q33. Yellow and green, Vps75; magenta and blue, Rtt109. All molecular representations were constructed using PyMOL.
Differences in Rtt109-Vps75 structures
Two series of crystal structures have been published of the Rtt109-Vps75 complex (Su et al., 2011; Tang et al., 2011), along with an excellent review on these structures (D’Arcy & Luger, 2011). Given that Vps75 increases the efficiency of Rtt109-catalyzed histone acetylation but not its binding affinity towards histones or acetyl-CoA, it has been thought that Vps75 stabilizes or induces an active Rtt109 conformation. In both series, the addition of Vps75 did not cause a significant shift in the conformation of the Rtt109 active sites, which suggests either Vps75 does not induce such a conformation or that this active conformation is not represented in the solved crystal structures (Figure 5B). The two series of structures have some notable differences. In one series of structures by Tang et al., Rtt109-Vps75 was crystallized with acetyl-CoA, and showed a 2:2 Rtt109: Vps75 ratio whereas the Su et al. structures show a 1:2 Rtt109:Vps75 ratio and do not model acetyl-CoA. The two series revealed two and three Rtt109-Vps75 interactions, respectively (Figure 5C). In both structures, Vps75 adopts a homodimeric headphone structure, with the earmuff domains binding to Rtt109, forming a central pore that presumably accommodates a histone substrate (Figure 5C).
The most significant differences between the two sets of structures is the Rtt109:Vps75 stoichiometry and the resulting remaining interfaces. Interface I involves the Rtt109 residues 130–175, which are susceptible to proteolysis and important for Rtt109 stabilization (Fillingham et al., 2008; Keck & Pemberton, 2011). This disordered loop was truncated in earlier Rtt109 structures, but becomes ordered when bound to Vps75 (Tang et al., 2011). In the Su et al. structures, there are two additional interfaces, II and III, while the Tang et al. structures contain only interfaces I and II. Interface II is between helices α8 and α9 of Rtt 109 and chain A of the Vps75 earmuff domain and involves a mix of hydrophobic contacts and electrostatic interactions (Figure 5C) (Su et al., 2011). Interface III is between the K290ac helix of Rtt109 and chain B in the Vps75 earmuff domain, as well as the N-terminal tail of chain A. With the exception of one salt bridge, Interface III is composed almost entirely of hydrophobic interactions (Figure 5C) (Su et al., 2011).
Comparison of the two structures shows this stoichiometry has a potentially significant effect on the modeled size and nature of the putative histone-binding region located in the pore formed by the proteins (Figure 5C). The differences between these two sets of structures may arise from different purification and crystallization procedures, or different protein constructs, as the Tang et al. structures used a truncated form of Vps75. Another possibility is that each set of structures may represent a different enzymatic state of the Rtt109-Vps75 complex (i.e. acetylation of H3K56 versus acetylation of H3 N-termini residues). Clearly, more experiments are needed to further address these questions.
The next structural questions that could be addressed involve the interactions among Rtt109, its chaperones, and histones. Detailed understanding of how chaperones and histones interact with Rtt109 and how this confers acetylation specificity is still a key unanswered question. Another key question is how Vps75 and Asf1 interact with respect to Rtt109? Fortunately, there is already structural information for chaperones and histones, including crystal structures for yeast Asf1 (Daganzo et al., 2003; Padmanabhan et al., 2005), yeast Asf1 bound to histone substrates (Antczak et al., 2006; Chavez et al., 2012; English et al., 2006) and yeast Vps75 (Tang et al., 2008). However, there remains no crystal or solution structures modeling Rtt109-Asf1 interactions, which may be due to the transient nature of this interaction, or perhaps experimental considerations.
The nature of the Rtt109-histone interaction is now beginning to be probed. In the Su et al. structures, the three proteins form a cylindrical enclosure ~30 × 35 Å, a spatial arrangement which is unlikely to accommodate a full (H3–H4)2 tetramer. The fact that the histone-binding pore domain is probably too small to accommodate a full (H3–H4)2 tetramer (and therefore H3K56) has led us to also hypothesize that Asf1 may function to dissociate (H3–H4)2 tetramers, which would presumably allow for H3K56 to be properly positioned for Rtt109-catalyzed acetylation. Given some of the difficulties in obtaining crystal structures with Rtt109-protein complexes, NMR-based methods may be useful for further probing Rtt109-related protein-protein interactions and protein dynamics (Box 2).
Box 2. Comparison of Rtt109 with p300.
An intriguing component of Rtt109 HATs lies in its comparison to other HATs. There are three major families of HATs: MYST, GNAT and the p300/CBP family, of which Rtt109 is a member. For a more thorough comparison of HATs, we point the reader to several well-written reviews (Berndsen & Denu, 2008; Dyda et al., 2000; Roth & Denu, 2001). In fungi, H3K56ac is catalyzed solely by Rtt109, while in mammalian cells this process may be carried out by p300 and/or Gcn5. Interestingly, organisms with p300 do not contain Rtt109 and vice-versa. It appears Rtt109 is an evolutionary precursor to p300. While both enzymes catalyze the formation of H3K56ac, there are some significant differences between Rtt109 and p300 (Table 3). Rtt109 does not have significant primary structure homology to p300, but its core (β2–β4 and α2) does share a general shape similarity with p300 (Tang et al., 2008). Both enzymes have a similar secondary structure arrangement featuring an ab core and helical bundles at opposing ends. Another common structural feature is the L1 and L2 loops that help bury the acetyl-CoA substrate. The electrostatic surface potentials of Rtt109 and p300 are also different in that the histone-binding region of p300 is predominately electronegative, while the same region is relatively apolar with an adjacent basic patch in Rtt109, making it more alike to Gcn5 (Tang et al., 2008). Rtt109 can bind to and co-crystallize with Vps75, whereas p300 does not feature such a chaperone. Like Rtt109, p300 has a hydrophobic active site, is autoacetylated, and requires an ionizible group at pH 8.4 (Karanam et al., 2006; Liu et al., 2008). Further understanding of the structural divergences between these two classes of enzymes – as well as other HATs – should enhance the overall understanding of histone-acetylation pathways, and as will be discussed below, has the potential to be exploited for anti-fungal purposes.
Therapeutic applications targeting Rtt109 and H3K56ac levels
The crucial roles of Rtt109 HATs and H3K56ac in yeast nucleosome assembly and genotoxic resistance may provide exploitable opportunities for therapeutics in humans. One of the recognized traits of good drug targets is that chemical and/or genetic modulation leads to a clear phenotype with a proven link to the pathophysiology of a disease (Gashaw et al., 2012). Indeed, the growth defects and increased sensitivities of yeast to genotoxins caused by mutant H3K56 or deletion of rtt109 demonstrate functional consequences of this pathway modulation. Another key observation is that Rtt109 HATs are not found in mammals, which several groups – including ours – have tried to exploit for anti-fungal purposes.
Anti-fungal hypothesis
Opportunistic fungal infections represent a significant source of morbidity and mortality in organ transplant patients, cancer patients and other immunocompromised populations (Fei et al., 2009; Marr et al., 2002; Steinbach, 2010). Mortality from these infections can exceed 50% in many human studies (Maschmeyer et al., 2007; Salman et al., 2011). The difficulty in successfully treating opportunistic fungal infections is due to several factors. First, fungi are eukaryotes, and many of their biologically crucial genes are conserved in humans. As a result, it has proven difficult to find fungi-specific therapeutic targets to help minimize toxicity to humans. Second, fungal populations can become resistant to therapeutics (Linden et al., 2011). Third, detection and diagnosis of opportunistic fungal infections can be difficult in the clinic (Carmona & Limper, 2011; Thornton, 2010). Finally, fungal pathogenesis is characterized by complicated host-pathogen interactions (Cramer et al., 2011; Thomas Jr & Limper, 2007). Therefore, there is still an unmet clinical need for novel, efficacious and minimally toxic anti-fungal treatments.
Several key observations have led to the hypothesis that modulating Rtt109-catalyzed histone acetylation may constitute an effective anti-fungal strategy. First, Rtt109 does not show any obvious primary sequence homology to mammalian HATs (Tang et al., 2008). Second, Rtt109 appears to utilize a different catalytic mechanism than p300/CBP, the closest known homologue found in mammals (Table 3). Third, potent inhibitors of p300 activity in vitro such as Lys-CoA are relatively ineffective inhibitors of Rtt109 activity in vitro (Tang et al., 2008). Based on the growing availability of sequenced fungal genomes, Rtt109 HATs appear to be conserved across the fungal kingdom (Figure 6). This conservation is also observed for the key histone chaperones Vps75 and Asf1 (Dahlin & Zhang, unpublished observations). This suggests that inhibitors targeting Rtt109 may be adaptable for use against different fungal species. For instance, small-molecule Rtt109 inhibitors developed against one fungal species may inhibit Rtt109 from other fungal species, depending on their mechanism(s) of inhibition, the degree of sequence and structural homology with other fungal species, as well as other species-specific factors. Based on available Rtt109 primary sequences, several features appear to be highly conserved in fungal Rtt109 HATs such as the autoacetylation site (K290 in Saccharomyces cerevisiae) and key catalytic residues (D89, W221, W222, D287 and D288 in S. cerevisiae; Figure 6). Interestingly, some fungal species do not appear to contain the known Vps75-binding motif found in yeast (Figure 6).
Table 3.
Generalized comparisons of Rtt109 and p300.
| Rtt109 | p300 | Key references | |
|---|---|---|---|
| Catalyzes H3K56ac | Y | Y | (Das et al., 2009; Han et al., 2007) |
| Found in mammalian cells | N | Y | – |
| Found in fungi | Y | N | – |
| Proposed enzymatic mechanism | Sequential (random) | Theorell-Chance | (Albaugh et al., 2010; Liu et al., 2008) |
| Substrate binding order | Non-obligate | Obligate | (Albaugh et al., 2010; Liu et al., 2008) |
| Multiple histone residue substrates | Y | Y | (McManus & Hendzel, 2003) |
| Known non-histone substrates | N | Y | (Barlev et al., 2001; Sakaguchi et al., 1998) |
| Inhibition by Lys-CoA | N | Y | (Cebrat et al., 2003; Sagar et al., 2004; Tang et al., 2008) |
| Acetylates nucleosomes | N | Y | (Han et al., 2007b; Ogryzko et al., 1996) |
| Contains bromodomain | N | Y | (Manning et al., 2001) |
| Autoacetylation | Y | Y | (Albaugh et al., 2010, 2011; Arif et al., 2007; Karanam et al., 2006) |
| Intra/intermolecular autoacetylation | Intra- | Inter- | (Albaugh et al., 2010, 2011; Karanam et al., 2006) |
| Other post-translational modifications | N | Y | (Girdwood et al., 2003) |
| Size (aa) | 436 | 2414 | (Chan & Thangue, 2001; Johnston et al., 1997) |
Figure 6.

Multiple-sequence alignment of Rttl09 from several fungal species. Sequence numbering and secondary structure annotation is based on S. cerevisiae Rttl09. Sequences were accessed from GenBank (NCBI). Alignment was performed with the T-Coffee algorithm (Di Tommaso et al., 2011; Notredame et al., 2000) and rendered with ENDscript (http://endscript.ibcp.fr) (Gouet et al., 2003). Secondary structure annotations were assigned based on the structure of PDB ID 3Q66. Organism abbreviations: SCER, S. cerevisiae; SPOM, S. pombe; CALB, C. albicans; CGLA, C. glabrata; PCAR, P. carinii; PJIR, P. jirovecii; PMUR, P. murina; AFUM, A. fumigatus; ANID, Aspergillus nidulans; NCRA, Neurospora crassa; RDEL, Rhizopus delemar.
Rtt109 has already been linked to virulence in the opportunistic fungi C. albicans, which is responsible for significant morbidity and mortality in certain immunocompromised populations (Baell et al., 2013; Tomašić & Mašič, 2012). Genetic deletion of rtt109 sensitizes C. albicans to genotoxic stress and select echinocandin anti-fungals, and leads to altered cellular phenotypes (Lopes da Rosa et al., 2010; Wurtele et al., 2010). In one of these studies, an rtt109Δ C. albicans strain was more susceptible to ROS-mediated macrophage clearance. Most significantly, mice infected with rtt109-deficient C. albicans showed decreased fungal burden, attenuated immune responses and increased survival compared to wild-type virulent strains (Lopes da Rosa et al., 2010; Wurtele et al., 2010), strongly suggesting Rtt109 contributes to fungal pathogenesis and is therefore a promising therapeutic target in C. albicans and perhaps other fungi. Interestingly, hyperacetylation of H3K56 via Hst3 histone deacetylase modulation also appears to be toxic for C. albicans (Wurtele et al., 2010), suggesting that cellular H3K56ac levels are subject to important regulations in order to maintain cell viability in this organism. Modulation of Hst3 activity with nicotinamide was cytotoxic to several fungal species, suggesting H3K56 hyperacetylation toxicity may be applicable to fungi in general. However, the concentrations used for these particular in vitro and in vivo experiments may not be therapeutically useful and may also contribute to off-target effects. It will be interesting to see whether this “opposing” approach targeting H3K56ac removal can also be exploited for anti-fungal purposes, as H3K56 hyperacetylation can also be detrimental to cell health (Celic et al., 2008). For this strategy, minimizing potential toxicity in humans would likely necessitate fungal H3K56ac deacetylase(s) be sufficiently structurally divergent from mammalian HDACs. Additionally, modulators of Rtt109-cataylzed histone acetylation could be used synergistically with other therapeutics (Pfaller et al., 2009; Singh & Pursell, 2008), as rtt109 deletion can increase yeast sensitivity to genotoxins.
Rtt109 HATs possess many other properties associated with good therapeutic targets. Multiple robust methods have been developed to assay HAT activity, as “assayability” is an important consideration for high-throughput screen (HTS) campaigns (Aherne et al., 2002). Additionally, multiple protein structures have been solved for yeast Rtt109, Rtt109-Vps75 and their protein substrates, which enable structure-based “druggability” assessments and pave the way for structure-based lead-finding and lead-optimization strategies. Lessons learned from studying nucleosome assembly and targeting Rtt109 HATs may also provide important insights relevant to human cancers and aging.
Rtt109 in clinically relevant fungal pathogens
Most studies of Rtt109 HATs have been performed in yeast, which are excellent model organisms but are generally not a significant source of deadly opportunistic fungal infections in humans compared to other pathogens like Pneumocystis and Aspergillus. To move towards more clinically relevant targets, our group has been steadily characterizing the Rtt109 system in Pneumocystis and other fungi. Pneumocystis pneumonia (PCP) is caused by the opportunistic pathogen P. jirovecii and is a significant source of mortality, especially in patients infected with HIV (Thomas Jr & Limper, 2007). Pneumocystis research is challenging because each host species is susceptible to a specific Pneumocystis species, and Pneumocystis cannot be continuously propagated by cell culture. This means most basic research is performed in vivo using surrogate organisms like P. carinii or Pneumocystis murina, which are responsible for PCP in rats and mice, respectively. A natural consequence is that it is difficult to perform many standard molecular biology techniques like genetic manipulation. However, using heterologous expression in budding yeast and in vitro HAT assays, we have shown that P. carinii contains a functional Rtt109 orthologue, PcRtt109, whose activity can be enhanced by the histone chaperones Vps75 and Asf1 derived from either yeast or P. carinii (Kottom et al., 2011; Pupaibool et al., 2013). With the recent availability of a P. jirovecii genome (Cissé et al., 2012), we have shown that P. jirovecii also contains a functional Rtt109 orthologue, PjRtt109, with similar in vitro properties as PcRtt109 (Dahlin et al., 2013). This supports the use of P. carinii as a surrogate model for P. jirovecii with regards to the Rtt109 system. Despite these studies, detailed knowledge of non-yeast Rtt109 HATs is relatively sparse. Towards the goals of developing species-specific or broad-spectrum Rtt109-based anti-fungal therapies, we believe additional studies are needed to clarify the inter-species variation in fungal Rtt109 HAT structures, function and regulation. Additionally, further examination of fungal H3K56ac HDACs and the nature of H3K56ac hyperacetylation toxicity may also be worthwhile pursuits as an alternative therapeutic strategy.
Medicinal chemistry considerations of Rtt109 HATs
In principle, there are multiple ways Rtt109-catalyzed histone acetylation could be modulated by a small-molecule or peptide. Compounds could bind in the acetyl-CoA binding domain (Figures 7 and 8), the histone-lysine binding region (Figure 8A, middle), or occupy both, as is the case for the p300 bi-substrate inhibitor Lys-CoA (see Table S1 for a listing these interaction surfaces). In yeast Rtt109, the acetyl-CoA active site is relatively hydrophobic and narrow at its interior region, which corresponds to the binding domain for the acetyl-pantetheine portion of acetyl-CoA. Anti-fungal agents that selectively target fungal Rtt109 over important human HATs such as p300 requires substrate selectivity. Figure 7 (middle) shows that in both human p300 and yeast Rtt109, the pantetheine portion of bound ligands are almost structurally identical, suggesting that targeting this portion of the enzyme would present selectivity challenges. Further removed from the active site, the 3′-phosphoadenosine-5′-diphosphate moiety does not appear to be rigidly bound in either of these enzymes. Its binding region appears to be relatively flat, hinting that binding here is fairly conserved between these two acetyl-CoA-utilizing enzymes and simply serves to direct the acetyl-CoA into the acetyltranferase active site (Figure 7, left). Probably the greatest challenge with targeting the CoA binding region is that CoA is used as a co-factor in up to 4% of all human enzymes. Therefore, developing selective inhibitors solely targeting this portion of the enzyme might be challenging. Avoiding potentially ubiquitous co-factor binding domains has also been applied to the somewhat related histone methyltransferases (Kubicek et al., 2007).
Figure 7.
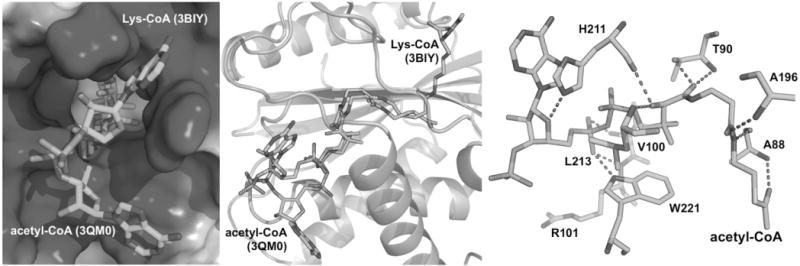
Binding mode analysis of acetyl-CoA to yeast Rtt109. (Left) Comparison of the site and modes of binding of the 3′-phosphoadenosine-5′-phosphate moiety in the related HATs Rtt109 and p300. Shown are the bisubstrate inhibitor Lys-CoA (PDB ID 3BIY, light blue sticks) and acetyl-CoA (PDB ID 3QM0, green sticks). The electrostatic surface (blue, positive potential) is p300 (3BIY). (Middle) Another view of the bound structures of acetyl-CoA and Lys-CoA. Tertiary structure shown is p300 (3BIY, light green). (Right) Polar contacts between acetyl-CoA (red dashed lines) and Rtt109 (acetyl-CoA, green sticks; contact residues in blue sticks; PDB ID 2ZFN). All biomolecular representations were constructed using PyMOL.
Figure 8.

Medicinal chemistry considerations of Rtt109 HATs. (A) Crystallographic binding sites of acetyl-CoA (left) and the presumed binding and contact sites (purple surfaces) of histone-lysine (middle) and histone substrates (right) using PDB ID 2ZFN (cartoon, light blue). (B) Structure of Rtt109-Vps75 complex (left, PDB ID 3Q66). Red circle demarks the putative H3-H4-binding pocket. Tertiary structure of Rtt109 (far right) with druggable binding site predicted by DoGSiteScorer. Volkamer A, Kuhn D, Grombacher T, et al. (2012). Combining global and local measures for structure-based druggability predictions. J Chem Inf Model 52:360–72. Shown as solid surface colored by atom type: N, dark blue; O, red; C, light blue. (C) Chemical structure of a reported inhibitor of Rtt109 HAT activity in vitro, identified by a CPM-based HTS (Lopes da Rosa et al., 2013). All biomolecular representations were constructed using PyMOL.
The fact that Rtt109 can acetylate multiple lysine residues suggests the histone-binding region(s) can accommodate a range of substrates and potential inhibitors. A detailed description of the histone-binding region is difficult, as there are currently no structures of histones or peptides crystallized in a physiologically relevant manner. However, we can infer the histone-binding region is located on the interior surface of the Rtt109-Vps75 complex (Figure 8A, right, and 8B) through which the lysine residue penetrates to access the poised acetyl group of the acetyl-CoA. This surface is highly conserved (Tang et al., 2008, 2011), and is lined by amino acids on non-contiguous chains of the Rtt109 enzyme. One prominent feature of this surface is a binding region that is predicted to be “druggable” by DoGSiteScorer (Volkamer et al., 2012), though the utility of targeting this pocket is still highly speculative (Figure 8B). Nevertheless, the histone-binding region of Rtt109, with its attendant “druggable pocket”, may be the best target for the preparation of selective fungal Rtt109 inhibitors.
Compounds could also bind to an allosteric site on Rtt109, though the prediction of such sites (if any) may be difficult a priori given the complexity of Rtt109 biochemistry (Fuller et al., 2009). Compounds could also modulate a key protein-protein interaction in the Rtt109-Vps75-Asf1-H3–H4 system. However, the utility of disrupting these interactions is difficult to gauge, given that Rtt109 can still be active even in the absence of chaperones in vitro and in vivo. Useful compounds inhibiting Rtt109 would most likely be reversible inhibitors, as none of the cysteine residues in Rtt109 has been implicated directly in enzymatic catalysis. However, we have found Rtt109-Vps75 activity in vitro is still sensitive to nanomolar concentrations of non-specific thiol-reactive electrophiles (Dahlin, Zhang, Walters, forthcoming results), as well as low micromolar concentrations of chemical aggregators (Dahlin et al., 2013).
HTS campaigns targeting yeast Rtt109
With the goal of identifying potential anti-fungal compounds, two HTS s to identify small-molecule inhibitors of Rtt109-catalyzed histone acetylation were recently reported (Dahlin et al., 2013; Lopes da Rosa et al., 2013). Both screens utilized a thiol-sensitive fluorescence probe, 7-diethylamino-3-(4′-maleimidyl-phenyl)-4-methylcoumarin (CPM), to quantify the amount of CoA by-product from the HAT reaction. Both screens utilized the Rtt109-Vps75 as the enzymatic component, though the screens differed in their substrates: yeast Asf1 complexed to full-length Drosophila H3-H4 versus an H3 peptide (and higher concentrations of acetyl-CoA). In the former screen, the majority of active compounds from the HTS were assay artifacts resulting from a combination of chemical aggregation, redox-activity, fluorescence quenching, compound-reagent interactions and non-specific thiol-reactivity (Dahlin et al., 2013). The few remaining active compounds could inhibit Rtt109-catalyzed histone acetylation in orthogonal assays for HAT activity, but are still undergoing further characterization to evaluate their selectivity, efficacy and mechanism(s) of inhibition (Dahlin, Walters, Zhang, unpublished results). In the other screen, performed using a different academic screening library, one small-molecule was reported to inhibit Rtt109-Vps75-catalyzed histone acetylation, but not the activity of related HATs p300 or Gcn5, at nanomolar compound concentrations in vitro (Figure 8C) (Lopes da Rosa et al., 2013). From a medicinal chemistry perspective, this hydantoin derivative does not contain any conspicuous structural alerts and is synthetically accessible. Surprisingly, for such a potent inhibitor, this compound did not affect cellular H3K56ac levels or increase genotoxic sensitivity in either S. cerevisiae or C. albicans. Additional close structural analogues of this compound were relatively inactive.
At first glance, this result may appear damning with respect to finding Rtt109 inhibitors that could be developed into useful therapeutics. However, we suggest caution when jumping to this conclusion. The lack of anti-fungal activity of this apparently potent and selective compound (Figure 8D) could be explained by several factors that are not directly related to the Rtt109-catalyzed histone acetylation, such as compound metabolism, cell permeability, compound efflux by membrane transporters, or even compound degradation in the cellular setting (i.e. lack of target engagement in vivo). Despite its potent in vitro activity (unusually potent for a non-optimized screening hit and the target class), and its apparent enzymatic selectivity, the lack of interpretable structure-activity relationship using close structural analogues, the relatively low standards reported for compound purity (>80%), and the absence of activity re-confirmation by HPLC-purified commercial samples and independent compound re-synthesis, raises questions about the validity of this reported inhibitor as a chemical probe (Baell, 2010; Dahlin & Walters, 2014). Inhibitory activity and apparent enzymatic selectivity may arise from variable intrinsic susceptibilities of enzymes to certain forms of non-specific enzymatic inhibition, such as thiol-reactive compounds or trace metal impurities. Until further studies are done on this compound and purified analogues, one should not exclude the possibility that this compound inhibits Rtt109 activity in vitro via a therapeutically uninteresting mechanism (Baell, 2010). For instance, an impurity such as a degradation product or a remnant from its chemical synthesis could disrupt enzymatic activity, though isolating and characterizing such impurities can be notoriously difficult (Beasley et al., 2003; Imbert et al., 2007).
Identifying compounds capable of inhibiting Rtt109-catalyzed histone acetylation, both in vitro and in vivo, is still an unmet need. Identifying HAT inhibitors may simply be an intrinsically difficult task with the current technology and screening libraries. For instance, a well-designed HTS versus a human MYST HAT only identified one small-molecule inhibitor, though its structure and any in vivo activity have not yet been reported due to intellectual property reasons (Falk et al., 2011; Laplante et al., 2013). While there are potent HDAC inhibitors (Carafa et al., 2013; Marson, 2009; Pan et al., 2012) and histone methyltransferase inhibitors available (Wagner & Jung, 2012), there is still an overall lack of bona fide HAT inhibitors reported in the scientific literature or patent applications (Furdas et al., 2011). Of course, the small numbers of reported HAT inhibitors could also result from the comparatively small amounts of academic and industrial resources directed towards HATs. Garcinol, a natural product with reported activity against several HATs (Balasubramanyam et al., 2004; Dahlin et al., 2013), can also inhibit Rtt109-Vps75 activity at low micromolar compound concentration in vitro (Dahlin et al., 2013). Several other reported HAT inhibitors showed weak activity against Rtt109 across several orthogonal assays (Dahlin, Walters, Zhang, unpublished results). However, many of these reported HAT inhibitors are likely promiscuous inhibitors, assay artifacts and/or therapeutically intractable chemotypes based on their chemical structures and available biological evaluations (Baell, 2010; Baell & Holloway, 2010). Protein-protein interactions like those found in the Rtt109-Vps75-Asf1 system may also be difficult to target using standard screening libraries, as their chemotypes may be biased towards more conventional drug discovery targets like GPCRs and protein kinases (Wells & McClendon, 2007). Therefore, targeting Rtt109-catalyzed histone acetylation with a small-molecule is likely a difficult endeavor given the size and composition of conventional academic screening libraries.
We can only speculate that success in identifying and optimizing a tractable therapeutic may require the use of a combination of different screening methodologies and alternative chemical matter. Given the propensity for CPM-based assays to select thiol-reactive compounds, along with lower signal-to-background ratios compared to other methods, any future cell-free assays targeting Rtt109 HATs should consider quantifying the modified protein product(s) rather than the CoA by-product. Such methods could include antibody-based assays like AlphaScreen and TR-FRET, or high-throughput electrophoresis (Caliper technology). Another concern we foresee with testing Rtt109 inhibitors for anti-fungal purposes is the cell-based readouts themselves. In most settings, cytotoxicty is a relatively non-specific readout, so candidate compounds will have to be rigorously evaluated for actual target engagement in a cellular context. In summary, the hypothesis that Rtt109 inhibitors can function as useful antifungal agents is still unresolved. HATs like Rtt109 may be difficult targets, but they represent a potential solution to an urgent, unmet clinical need.
Conclusions and future perspectives
Nucleosome assembly is a complex and highly regulated process in eukaryotes. Proper nucleosome disassembly and assembly is crucial for maintaining genomic integrity. Nucleosome assembly requires an intricate regulation of histone-modifying enzymes, histone chaperones and histone modifications. Many important discoveries pertaining to nucleosome assembly have originated initially using the SV40 DNA replication-coupled system. In recent years, yeast-based experiments have provided new insights into how assembly of new H3–H4 is regulated through histone modifications and chaperones. Not surprisingly for such a biologically critical process, there are many similarities between yeast and mammalian nucleosome assembly pathways, underscoring the continued utility of model organisms such as yeast. Despite these efforts, it remains largely unknown how parental H3–H4 are assembled into nucleosomes.
The HAT Rtt109 is an important fixture in fungal RCNA and genotoxic resistance. Rtt109 is the sole HAT responsible for H3K56ac in fungi. The roles of H3K56ac in mammalian systems are still being actively investigated, and it will be interesting to verify which features relating to Rtt109 and H3K56ac are conserved in mammalian cells, and which features are replaced or omitted. One of the main unanswered questions for Rtt109 HATs is how the histone chaperone Asf1 directs Rtt109 substrate specificity, particularly from a structural standpoint. Another remaining question with pro-found clinical relevance is whether potent and specific inhibitors of Rtt109-catalyzed histone acetylation can exploit the absence of Rtt109 HATs in metazoans to selectively inhibit fungal growth while sparing mammalian cells from cytotoxicity. Deeper understanding of the basic mechanisms of epigenetic processes such as nucleosome assembly will require continued investigations into histone-modifying enzymes, histone chaperones, histone PTMs and also patterns of molecular recognition (histone readers). This combined knowledge may have profound implications on human health, as epigenetic alterations have been associated with fungal virulence, cancers and aging (Box 3).
Box 3. Executive summary | Rtt109 HATs and chromatin assembly.
Nucleosomes must be disassembled to make way for replication machinery and then reassembled afterwards during DNA replication (replication-coupled nucleosome assembly, RCNA). Many of the same principles of RCNA apply to replication-independent (RINA) processes like histone exchange and transcription, as well as DNA damage and repair.
Proper RCNA is essential for maintaining genome stability, and dysfunctions in RCNA have been associated with aging as well as various human pathologies like cancers.
In yeast, the HAT Rtt109 is best known for its catalysis of H3K56ac, which is a PTM found abundantly on newly synthesized histones and is critical for proper RCNA.
H3K56ac enhances the Rtt101Mms1-catalyzed ubiquitination of these histones, which is thought to facilitate the formation of (H3–H4)2 tetramers and ultimately nascent nucleosome formation.
Rtt109 HATs represent a unique class of HATs that are conserved in fungi and are essential for the production of H3K56ac in fungi.
H3K56ac levels are less abundant in mammalian cells, and the roles of this modification in mammalian RCNA and the DNA damage response are still being characterized.
Rtt109 substrate specificity and enzymatic activity are subject to complex regulation by the histone chaperones Vps75 and Asf1 both in vitro and in vivo.
Inhibitors of Rtt109-catalyzed histone acetylation are hypothesized as minimally-toxic anti-fungal agents, but this therapeutic hypothesis is still unresolved.
Future work regarding Rtt109 HATs may focus on the structural determinants of substrate specificity and its roles in other fungi like opportunistic pathogens.
More work is needed in mammalian cells to clarify the role(s), if any, of H3K56ac in RCNA and RINA, especially with respect to DNA damage and repair.
Supplementary Material
Acknowledgments
The authors acknowledge (1) Mayo Clinic and the University of Minnesota librarians for assistance in obtaining scientific literature used for this review and (2) Drs. Rebecca Burgess, Barry Finzel, Andrew Limper and Georges Mer for helpful discussions.
Research in the Walters and Zhang labs has been supported by the Minnesota Partnership for Biotechnology and Medical Genomics (73–01 to M. A. W. and Z. Z.), the National Institutes of Health (NIH; GM72719 and GM81838 to Z. Z.), the Minnesota Supercomputing Institute and the Mayo Foundation for Medical Education and Research. J. L. D. was supported by the NIH Medical Scientist Training Program (T32 GM065841), an NIH pre-doctoral fellowship (F30 DK092026-01), a Pharmaceutical Research and Manufacturers of America Foundation pre-doctoral pharmacology/toxicology fellowship and the Mayo Foundation. X. C. was supported by the Mayo Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The opinions or assertions contained herein belong to the authors and are not necessarily the official views of the funders.
Footnotes
Declaration of interest: The authors report no declarations of interest.
References
- Abshiru N, Ippersiel K, Tang Y, et al. Chaperone-mediated acetylation of histones by Rtt109 identified by quantitative prote-omics. J Proteomics. 2012;81:80–90. doi: 10.1016/j.jprot.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S, Polo S, Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Adam S, Polo SE, Almouzni Gv. How to restore chromatin structure and function in response to DNA damage – let the chaperones play. FEBS J. 2014;281:2315–23. doi: 10.1111/febs.12793. [DOI] [PubMed] [Google Scholar]
- Adam S, Polo SE. Chromatin dynamics during nucleotide excision repair: histones on the move. Int J Mol Sci. 2012;13:11895–911. doi: 10.3390/ijms130911895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aherne GW, Rowlands MG, Stimson L, Workman P. Assays for the identification and evaluation of histone acetyltransferase inhibitors. Methods. 2002;26:245–53. doi: 10.1016/S1046-2023(02)00028-2. [DOI] [PubMed] [Google Scholar]
- Albaugh BN, Arnold KM, Lee S, Denu JM. Autoacetylation of the histone acetyltransferase Rtt109. J Biol Chem. 2011;286:24694–701. doi: 10.1074/jbc.M111.251579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh BN, Kolonko EM, Denu JM. Kinetic mechanism of the Rtt109-Vps75 histone acetyltransferase-chaperone complex. Biochemistry. 2010;49:6375–85. doi: 10.1021/bi100381y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez F, Munoz F, Schilcher P, et al. Sequential establishment of marks on soluble histones H3 and H4. J Biol Chem. 2011;286:17714–21. doi: 10.1074/jbc.M111.223453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AJ, Chen X, Zevin A, et al. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell. 2010;37:834–42. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antczak AJ, Tsubota T, Kaufman PD, Berger JM. Structure of the yeast histone H3-Asf1 interaction: implications for chaperone mechanism, species-specific interactions, and epigenetics. BMC Struct Biol. 2006;6:26. doi: 10.1186/1472-6807-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqeilan RI, Hassan MQ, Bruin Ad, et al. The WWOX tumor suppressor is essential for postnatal survival and normal bone metabolism. J Biol Chem. 2008;283:21629–39. doi: 10.1074/jbc.M800855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif M, Kumar GVP, Narayana C, Kundu TK. Autoacetylation induced specific structural changes in histone acetyltransferase domain of p300: probed by surface enhanced Raman spectroscopy. J Phys Chem B. 2007;111:11877–79. doi: 10.1021/jp0762931. [DOI] [PubMed] [Google Scholar]
- Ask K, Jasencakova Z, Menard P, et al. Codanin-1, mutated in the anaemic disease CDAI, regulates Asf1 function in S-phase histone supply. EMBO J. 2012;31:2013–23. doi: 10.1038/emboj.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, Ferrins L, Falk H, Nikolakopoulos G. PAINS: relevance to tool compound discovery and fragment-based screening. Aust J Chem. 2013;66:1483–94. [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–40. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Baell JB. Observations on screening-based research and some concerning trends in the literature. Future Med Chem. 2010;2:1529–46. doi: 10.4155/fmc.10.237. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Altaf M, Varier R, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279:33716–26. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K, Jiang L, Wang L, et al. Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. J Biol Chem. 2006;281:40292–301. doi: 10.1074/jbc.M608813200. [DOI] [PubMed] [Google Scholar]
- Barlev NA, Liu L, Chehab NH, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243–54. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Barman H, Takami Y, Ono T, et al. Histone acetyltransferase 1 is dispensable for replication-coupled chromatin assembly but contributes to recover DNA damages created following replication blockage in vertebrate cells. Biochem Biophys Res Commun. 2006;345:1547–57. doi: 10.1016/j.bbrc.2006.05.079. [DOI] [PubMed] [Google Scholar]
- Beasley JR, Dunn DA, Walker TL, et al. Evaluation of compound interference in immobilized metal ion affinity-based fluorescence polarization detection with a four million member compound collection. Assay Drug Dev Technol. 2003;1:455–59. doi: 10.1089/154065803322163768. [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko V, et al. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–93. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- Berndsen CE, Denu JM. Catalysis and substrate selection by histone/protein lysine acetyltransferases. Curr Opin Struc Biol. 2008;18:682–89. doi: 10.1016/j.sbi.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndsen CE, Tsubota T, Lindner SE, et al. Molecular functions of the histone acetyltransferase chaperone complex Rtt109-Vps75. Nat Struct Mol Biol. 2008;15:948–56. doi: 10.1038/nsmb.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower K, Napier C, Cole S, et al. Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLOS ONE. 2012;7:e55602. doi: 10.1371/journal.pone.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat Struct Mol Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RJ, Zhou H, Han J, Zhang Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell. 2010;37:469–80. doi: 10.1016/j.molcel.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafa V, Miceli M, Altucci L, Nebbioso A. Histone deacetylase inhibitors: a patent review (2009 – 2011) Expert Opin Ther Pat. 2013;23:1–17. doi: 10.1517/13543776.2013.736493. [DOI] [PubMed] [Google Scholar]
- Carmona EM, Limper AH. Update on the diagnosis and treatment of Pneumocystis pneumonia. Ther Adv Respir Dis. 2011;5:41–59. doi: 10.1177/1753465810380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchpool ED, Smallwood A, Joyce J, et al. Mutation analysis of H19 and NAP1L4 (hNAP2) candidate genes and IGF2 DMR2 in Beckwith–Wiedemann syndrome. J Med Genet. 2000;37:212–15. doi: 10.1136/jmg.37.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrat M, Kim CM, Thompson PR, et al. Synthesis and analysis of potential prodrugs of coenzyme A analogues for the inhibition of the histone acetyltransferase p300. Bioorg Med Chem. 2003;11:3307–13. doi: 10.1016/s0968-0896(03)00265-7. [DOI] [PubMed] [Google Scholar]
- Celic I, Masumoto H, Griffith WP, et al. The sirtuins Hst3 and Hst4p preserve genome integrity by controlling histone H3 lysine 56 deacetylation. Curr Biol. 2006;16:1280–9. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Celic I, Verreault A, Boeke JD. Histone H3 K56 hyperacetyla-tion perturbs replisomes and causes DNA damage. Genetics. 2008;179:1769–84. doi: 10.1534/genetics.108.088914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AL, Downs JA. The RSC and INO80 chromatin-remodeling complexes in DNA double-strand break repair. Prog Mol Biol Transl Sci. 2012;110:229–61. doi: 10.1016/B978-0-12-387665-2.00009-2. [DOI] [PubMed] [Google Scholar]
- Chan HM, Thangue NBL. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–73. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Chavez MS, Scorgie JK, Dennehey BK, et al. The conformational flexibility of the C-terminus of histone H4 promotes histone octamer and nucleosome stability and yeast viability. Epigenetics Chromatin. 2012;5:5. doi: 10.1186/1756-8935-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Carson JJ, Feser J, et al. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–43. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Fan M, Pfeffer LM, Laribee RN. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cissé OH, Pagni M, Hauser PM. De novo assembly of the Pneumocystis jirovecii genome from a single bronchoalveolar lavage fluid specimen from a patient. mBio. 2012;4:e00428–12. doi: 10.1128/mBio.00428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–97. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A, De Koning L, Toedling J, et al. Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J. 2011;30:480–93. doi: 10.1038/emboj.2010.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer RA, Rivera A, Hohl TM. Immune responses against Aspergillus fumigatus: what have we learned? Curr Opin Infect Dis. 2011;24:315–22. doi: 10.1097/QCO.0b013e328348b159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daganzo SM, Erzberger JP, Lam WM, et al. Structure and function of the conserved core of histone deposition protein Asf1. Curr Biol. 2003;13:2148–58. doi: 10.1016/j.cub.2003.11.027. [DOI] [PubMed] [Google Scholar]
- Dahlin JL, Kottom TJ, Han J, et al. Pneumocystis jirovecii expresses a functional ortholog of Pneumocystis carinii Rtt109. Mol Microbiol. 2013a in press. [Google Scholar]
- Dahlin JL, Sinville R, Solberg J, et al. A cell-free fluorometric high-throughput screen for inhibitors of Rtt109-catalyzed histone acetylation. PLOS ONE. 2013b;8:e78877. doi: 10.1371/journal.pone.0078877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin JL, Walters MA. The essential roles of chemistry in highthroughput screening triage. Future Med Chem, Ahead of Print. 2014 doi: 10.4155/fmc.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Antoni S, Mattina T, Di Mare P, et al. Altered replication timing of the HIRA/Tuple1 locus in the DiGeorge and Velocardiofacial syndromes. Gene. 2004;26:111–19. doi: 10.1016/j.gene.2004.02.029. [DOI] [PubMed] [Google Scholar]
- D’Arcy S, Luger K. Understanding histone acetyltransferase Rtt109 structure and function: how many chaperones does it take? Curr Opin Struc Biol. 2011;21:1–7. doi: 10.1016/j.sbi.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Lucia M, Hansen K, Tyler J. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–17. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Tyler JK. Histone exchange and histone modifications during transcription and aging. Biochem Biophys Acta. 2013;1819:332–42. doi: 10.1016/j.bbagrm.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennehey BK, Tyler J. Histone chaperones in the assembly and disassembly of chromatin. In: Workman JL, Abmayr SM, editors. Fundamentals of chromatin. New York: Springer; 2014. pp. 29–67. [Google Scholar]
- Di Tommaso P, Moretti S, Xenarios I, et al. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011;39:W13–17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315:649–52. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drogaris P, Villeneuve V, Pomiès C, et al. Histone deacetylase inhibitors globally enhance H3/H4 tail acetylation without affecting H3 lysine 56 acetylation. Sci Rep. 2012;2 doi: 10.1038/srep00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Ray S, Home P, et al. Regulation of angiogenesis by histone chaperone HIRA-mediated incorporation of lysine 56-acetylated histone H3.3 at chromatin domains of endothelial genes. J Biol Chem. 2010;285:41567–77. doi: 10.1074/jbc.M110.190025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlassi-Lassallette A, Mocquard E, Arnaud MC, Thiriet C. H4 replication-dependent diacetylation and Hat1 promote S-phase chromatin assembly in vivo. Mol Biol Cell. 2011;22:245–55. doi: 10.1091/mbc.E10-07-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English CM, Adkins MW, Carson JJ, et al. Structural basis for the histone chaperone activity of Asf1. Cell. 2006;127:495–508. doi: 10.1016/j.cell.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk H, Connor T, Yang H, et al. An efficient high-throughput screening method for MYST family acetyltransferases, a new class of epigenetic drug targets. J Biomol Screen. 2011;16:1196–205. doi: 10.1177/1087057111421631. [DOI] [PubMed] [Google Scholar]
- Fazly A, Li Q, Hu Q, et al. Histone chaperone Rtt106 promotes nucleosome formation using (H3-H4)2 tetramers. J Biol Chem. 2012;287:10735–60. doi: 10.1074/jbc.M112.347450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei M, Kim E, Sant C, et al. Predicting mortality from HIV-associated Pneumocystis pneumonia at illness presentation: an observational cohort study. Thorax. 2009;64:1070–76. doi: 10.1136/thx.2009.117846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feser J, Truong D, Das C, et al. Elevated histone expression promotes life span extension. Mol Cell. 2010;39:724–35. doi: 10.1016/j.molcel.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingham J, Recht J, Silva A, et al. Chaperone control of the activity and specificity of the histone H3 acetyltransferase Rtt109. Mol Cell Biol. 2008;28:4342–53. doi: 10.1128/MCB.00182-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller JC, Burgoyne NJ, Jackson RM. Predicting druggable binding sites at the protein-protein interface. Drug Discov Today. 2009 doi: 10.1016/j.drudis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Fulzele A, Malgundkar S, Govekar R, et al. Keratins in oral cancer: necessity of mass spectrometry for validation of antibody based identifications. J Proteomics. 2012;75:2404–16. doi: 10.1016/j.jprot.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Furdas SD, Kannan S, Sippl W, Jung M. Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Archiv der Pharmazie. 2011;345:7–21. doi: 10.1002/ardp.201100209. [DOI] [PubMed] [Google Scholar]
- Gashaw I, Ellinghaus P, Sommer A, Asadullah K. What makes a good drug target? Drug Discov Today. 2012;17 doi: 10.1016/j.drudis.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Girdwood D, Bumpass D, Vaughan OA, et al. P300 transcriptional repression is mediated by SUMO modification. Mol Cell. 2003;11:1043–54. doi: 10.1016/s1097-2765(03)00141-2. [DOI] [PubMed] [Google Scholar]
- Gkikopoulos T, Havas K, Dewar H, Owen-Hughes T. SWI/SNF and Asf1p cooperate to displace histones during induction of the saccharomyces cerevisiae HO promoter. Mol Cell Biol. 2009;29:4057–66. doi: 10.1128/MCB.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P, Robert X, Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–33. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Haldar D, Kamakaka RT. Schizosaccharomyces pombe Hst4 functions in DNA damage response by regulating histone H3 K56 acetylation. Eukaryotic Cell. 2008;7:800–13. doi: 10.1128/EC.00379-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhang H, Zhang H, et al. A Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell. 2013;155:817–29. doi: 10.1016/j.cell.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007a;315:653–5. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- Han J, Zhou H, Li Z, et al. The Rtt109-Vps75 histone acetyltransferase complex acetylates non-nucleosomal histone H3. J Biol Chem. 2007b;282:14158–64. doi: 10.1074/jbc.M700611200. [DOI] [PubMed] [Google Scholar]
- Han J, Zhou H, Li Z, et al. Acetylation of lysine 56 of histone H3 catalyzed by Rtt109 and regulated by Asf1 is required for replisome integrity. J Biol Chem. 2007c;282:28587–96. doi: 10.1074/jbc.M702496200. [DOI] [PubMed] [Google Scholar]
- Harris TK, Turner GJ. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- Heo K, Kim H, Choi S, et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- Hodawadekar S, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene. 2007;26:5528–40. doi: 10.1038/sj.onc.1210619. [DOI] [PubMed] [Google Scholar]
- Huang R, Holbert MA, Tarrant MK, et al. Site-specific introduction of an acetyl-lysine mimic into peptides and proteins by cysteine alkylation. J Am Chem Soc. 2010;132:9986–7. doi: 10.1021/ja103954u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide S, Saka K, Kobayashi T. Rtt109 prevents hyper-amplification of ribosomal RNA genes through histone modification in budding yeast. PLoS Genet. 2013;9:e1003410. doi: 10.1371/journal.pgen.1003410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbert P-E, Unterreiner V, Siebert D, et al. Recommendations for the reduction of compound artifacts in time-resolved fluorescence resonance energy transfer assays. Assay Drug Dev Technol. 2007;5:363–72. doi: 10.1089/adt.2007.073. [DOI] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, et al. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell. 1997;90:145–55. doi: 10.1016/s0092-8674(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamai A, Puglisi A, Strubin M. Histone chaperone spt16 promotes redeposition of the original H3-H4 histones evicted by elongating RNA polymerase. Mol Cell. 2009;35:377–83. doi: 10.1016/j.molcel.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Jasencakova Z, Scharf AND, Ask K, et al. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell. 2010;37:736–43. doi: 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- Johnston M, Hillier L, Riles L, et al. The nucleotide sequence of Saccharomyces cerevisiae chromosome XII. Nature. 1997;387:87–90. [PMC free article] [PubMed] [Google Scholar]
- Kadlec J, Hallacli E, Lipp M, et al. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat Struct Mol Biol. 2011;18:142–9. doi: 10.1038/nsmb.1960. [DOI] [PubMed] [Google Scholar]
- Kadyrova LY, Blanko ER, Kadyrov FA. Human CAF-1-dependent nucleosome assembly in a defined system. Cell Cycle. 2013;12:3286–97. doi: 10.4161/cc.26310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T, Liu C, Erkmann J, et al. Cell cycle-and chaperone-mediated regulation of H3K56ac incorporation in yeast. PLoS Genet. 2008;4:e1000270. doi: 10.1371/journal.pgen.1000270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanam B, Jiang L, Wang L, et al. Kinetic and mass spectrometric analysis of p300 histone acetyltransferase domain autoacetylation. J Biol Chem. 2006;281:40292–301. doi: 10.1074/jbc.M608813200. [DOI] [PubMed] [Google Scholar]
- Katan-Khaykovich Y, Struhl K. Splitting of H3-H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc Natl Acad Sci USA. 2011;108:1296–301. doi: 10.1073/pnas.1018308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck KM, Pemberton LF. Interaction with the histone chaperone Vps75 promotes nuclear localization and HAT activity of Rtt109 in vivo. Traffic. 2011;12:826–39. doi: 10.1111/j.1600-0854.2011.01202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenseth JR, Coldiron SJ. High-throughput characterization and quality control of small-molecule combinatorial libraries. Curr Opin Chem Biol. 2004;8:418–23. doi: 10.1016/j.cbpa.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Killela P, Reitman Z, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self renewal. Proc Natl Acad Sci USA. 2013;110:6021–6. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolonko EM, Albaugh BN, Lindner SE, et al. Catalytic activation of histone acetyltransferase Rtt109 by a histone chaperone. Proc Natl Acad Sci USA. 2010;107:20275–80. doi: 10.1073/pnas.1009860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottom TJ, Han J, Zhang Z, Limper AH. Pneumocystis carinii expresses an active Rtt109 histone acetyltransferase. Am J Resp Cell Mol Biol. 2011;44:768–76. doi: 10.1165/rcmb.2009-0443OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan N, Cagney G, Yu H, et al. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–43. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Krude T. Chromatin assembly during DNA replication in somatic cells. Eur J Biochem. 1999;263:1–5. doi: 10.1046/j.1432-1327.1999.00508.x. [DOI] [PubMed] [Google Scholar]
- Kubicek S, O’Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–81. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Laplante SR, Carson R, Gillard J, et al. Compound aggregation in drug discovery: implementing a practical NMR assay for medicinal chemists. J Med Chem. 2013;56:5142–50. doi: 10.1021/jm400535b. [DOI] [PubMed] [Google Scholar]
- Li B, Su T, Ferrari R, Li J-Y, Kurdistani SK. A unique epigenetic signature is associated with active DNA replication loci in human embryonic stem cells. Epigenetics. 2013;9:257–67. doi: 10.4161/epi.26870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhou H, Wurtele H, et al. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–55. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yuan YA. Structural insights into histone H3 lysine 56 acetylation by Rtt109. Structure. 2008;16:1503–10. doi: 10.1016/j.str.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Lin L, Schultz M. Promoter regulation by distinct mechanisms of functional interplay between lysine acetylase Rtt109 and histone chaperone Asf1. Proc Natl Acad Sci USA. 2011;108:19599–604. doi: 10.1073/pnas.1111501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden JWMvd, Snelders E, Kampinga GA, et al. Clinical implications of azole resistance in Aspergillus fumigatus, The Netherlands, 2007–2009. Emerging Infect Dis. 2011;17:1846–54. doi: 10.3201/eid1710.110226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang L, Zhao K, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451:846–50. doi: 10.1038/nature06546. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang D-L, Chen S, et al. Oncogene Ras/phosphatidy-linositol 3-kinase signaling targets histone H3 acetylation at lysine 56. J Biol Chem. 2012;287:41469–80. doi: 10.1074/jbc.M112.367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo KA, Bauchmann MK, Baumann AP, et al. Genome-wide profiling of H3K56 acetylation and transcription factor binding sites in human adipocytes. PLOS ONE. 2011;6:e19778. doi: 10.1371/journal.pone.0019778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Rosa J, Bajaj V, Spoonamore J, Kaufman PD. A small molecule inhibitor of fungal histone acetyltransferase Rtt109. Bioorg Med Chem Lett. 2013;23:2853–59. doi: 10.1016/j.bmcl.2013.03.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Rosa J, Boyartchuk VL, Zhu LJ, Kaufman PD. Histone acetyltransferase Rtt109 is required for Candida albicans pathogenesis. Proc Natl Acad Sci USA. 2010;107:1594–99. doi: 10.1073/pnas.0912427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola A, Tagami H, Bonaldi T, et al. The HP1alpha-CAF1-SetDB1-containing complex provides H3K9me1 for Suv39-mediated K9me3 in pericentric heterochromatin. EMBO Rep. 2009;10:769–75. doi: 10.1038/embor.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–19. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Malapeira J, Khaitova LC, Mas P. Ordered changes in histone modifications at the core of the Arabidopsis circadian clock. Proc Natl Acad Sci USA. 2012;109:21540–5. doi: 10.1073/pnas.1217022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning ET, Ikehara T, Ito T, et al. p300 forms a stable, template-committed complex with chromatin: role for the bromodo-main. Mol Cell Biol. 2001;21:3876–87. doi: 10.1128/MCB.21.12.3876-3887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochem Biophys Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr KA, Carter RA, Boeckh M, et al. Invasive aspergillosis in allogeneic stem cell transplant recipients: changes in epidemiology and risk factors. Blood. 2002;100:4358–66. doi: 10.1182/blood-2002-05-1496. [DOI] [PubMed] [Google Scholar]
- Marson C. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anticancer Agents Med Chem. 2009;9:661–92. doi: 10.2174/187152009788679976. [DOI] [PubMed] [Google Scholar]
- Maschmeyer G, Haas A, Cornely OA. Invasive aspergillosis: epidemiology, diagnosis and management in immunocompromised patients. Drugs. 2007;67:1567–601. doi: 10.2165/00003495-200767110-00004. [DOI] [PubMed] [Google Scholar]
- Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–8. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- May T, Yang J, Shoni M, et al. BRCA1 expression is epigenetically repressed in sporadic ovarian cancer cells by overexpression of C-terminal binding protein 2. Neoplasia. 2013;15:600–08. doi: 10.1593/neo.121674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight SL, Miller OL. Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophilia melano-gaster embryo. Cell. 1977;12:795–804. doi: 10.1016/0092-8674(77)90278-1. [DOI] [PubMed] [Google Scholar]
- McManus KJ, Hendzel MJ. Quantitative analysis of CBP-and P300-induced histone acetylations in vivo using native chromatin. Mol Cell Biol. 2003;23:7611–27. doi: 10.1128/MCB.23.21.7611-7627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Boxer LD, et al. Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle. 2009;8:2664–6. doi: 10.4161/cc.8.16.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–U15. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moggs J, Grandi P, Quivy J, et al. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol Cell Biol. 2000;20:1206–18. doi: 10.1128/mcb.20.4.1206-1218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A, Highland J, Krogan N, et al. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–75. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- Morrison A, Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol. 2009;10:373–84. doi: 10.1038/nrm2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosammaparast N, Ewart C, Pemberton L. A role for nucleosome assembly protein 1 in the nuclear transport of histones H2A and H2B. EMBO J. 2002;21:6527–38. doi: 10.1093/emboj/cdf647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan P, Ge Z, Sirbu B, et al. Histone acetyltransferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS Genet. 2013;9:e1003518. doi: 10.1371/journal.pgen.1003518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakan S, Stillman B, Horvitz HR. Replication-coupled chromatin assembly generates a neuronal bilateral asymmetry in C. elegans. Cell. 2011;147:1525–36. doi: 10.1016/j.cell.2011.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins D, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–9. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Padmanabhan B, Kataoka K, Umehara T, et al. Structural similarity between histone chaperone Cia1p/Asf1p and DNA-binding protein NF-kappaB. J Biochem. 2005;138:821–29. doi: 10.1093/jb/mvi182. [DOI] [PubMed] [Google Scholar]
- Pan H, Cao J, Xu W. Selective histone deacetylase inhibitors. Anticancer Agents Med Chem. 2012;12:247–70. doi: 10.2174/187152012800228814. [DOI] [PubMed] [Google Scholar]
- Park YJ, Sudhoff KB, Andrews AJ, et al. Histone chaperone specificity in Rtt109 activation. Nat Struct Mol Biol. 2008;15:957–64. doi: 10.1038/nsmb.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, Messer SA, Georgopapadakou N, et al. Activity of MGCD290, a Hos2 histone deacetylase inhibitor, in combination with azole antifungals against opportunistic fungal pathogens. J Clin Microbiol. 2009;47:3797–804. doi: 10.1128/JCM.00618-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Theocharis SE, Grandin L, et al. Clinical significance and prognostic value of chromatin assembly factor-1 overexpression in human solid tumours. Histopath. 2010;57:716–24. doi: 10.1111/j.1365-2559.2010.03681.x. [DOI] [PubMed] [Google Scholar]
- Pupaibool J, Kottom TJ, Bouchonville K, Limper AH. Characterization of the Pneumocystis carinii histone acetyltranferase chaperone proteins PcAsf1 and PcVps75. Infect Immun. 2013;81:2268–75. doi: 10.1128/IAI.01173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovani E, Cadorin M, Shams T, et al. The carboxyl terminus of Rtt109 functions in chaperone control of histone acetylation. Eukaryotic Cell. 2013;12:654–64. doi: 10.1128/EC.00291-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom M, Dennehey B, Tyler J. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–95. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht J, Tsubota T, Tanny JC, et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci USA. 2006;103:6988–93. doi: 10.1073/pnas.0601676103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Bader G. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013;9:1–17. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renella R, Roberts NA, Brown JM, et al. Codanin-1 mutations in congenital dyserythropoietic anemia type 1 affect HP1{alpha} localization in erythroblasts. Blood. 2011;117:6928–38. doi: 10.1182/blood-2010-09-308478. [DOI] [PubMed] [Google Scholar]
- Rishton G. Reactive compounds and in vitro false positives in HTS. Drug Discov Today. 1997;2:382–4. [Google Scholar]
- Roth S, Denu J. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Rufiange A, Jacques P, Bhat W, et al. Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell. 2007;27:393–405. doi: 10.1016/j.molcel.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Sagar V, Zheng W, Thompson PR, Cole PA. Bisubstrate analogue structure-activity relationships for p300 histone acetyltransferase inhibitors. Bioorg Med Chem. 2004;12:3383–90. doi: 10.1016/j.bmc.2004.03.070. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–41. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman N, Törün SH, Budan B, Somer A. Invasive aspergillosis in hematopoietic stem cell and solid organ transplantation. Exp Rev Ant-Infect Ther. 2011;9:307–15. doi: 10.1586/eri.11.13. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Valls E, Kouzarides T, Martínez-Balbás M. Mechanisms of P/CAF auto-acetylation. Nucleic Acids Res. 2003;31:4285–92. doi: 10.1093/nar/gkg655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem. 2011;80:703–32. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Schumacher B, Lombard DB, et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc Natl Acad Sci USA. 2010;107:21790–4. doi: 10.1073/pnas.1016306107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selth L, Svejstrup JQ. Vps75, a new yeast member of the NAP histone chaperone family. J Biol Chem. 2007;282:12358–62. doi: 10.1074/jbc.C700012200. [DOI] [PubMed] [Google Scholar]
- Singh N, Pursell KJ. Combination therapeutic approaches for the management of invasive aspergillosis in organ transplant recipients. Mycoses. 2008;51:99–108. doi: 10.1111/j.1439-0507.2007.01479.x. [DOI] [PubMed] [Google Scholar]
- Smerdon M. DNA repair and the role of chromatin structure. Curr Opin Cell Biol. 1991;3:422–8. doi: 10.1016/0955-0674(91)90069-b. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Whitehouse I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature. 2012;483:434–8. doi: 10.1038/nature10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Stillman B. Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 1991;10:971–80. doi: 10.1002/j.1460-2075.1991.tb08031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soekarman D, Lindern Mv, Daenen S, et al. The translocation (6;9) (p23;q34) shows consistent rearrangement of two genes and defines a myeloproliferative disorder with specific clinical features. Blood. 1992;79:2990–7. [PubMed] [Google Scholar]
- Stavropoulos P, Nagy V, Blobel G, Hoelz A. Molecular basis for the autoregulation of the protein acetyl transferase Rtt109. Proc Natl Acad Sci USA. 2008;105:12236–41. doi: 10.1073/pnas.0805813105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach WJ. Invasive aspergillosis in pediatric patients. Curr Med Res Opin. 2010;26:1779–87. doi: 10.1185/03007995.2010.487793. [DOI] [PubMed] [Google Scholar]
- Stillman B. Chromatin assembly during SV40 DNA replication in vitro. Cell. 1986;45:555–65. doi: 10.1016/0092-8674(86)90287-4. [DOI] [PubMed] [Google Scholar]
- Su D, Hu Q, Li Q, et al. Structural basis for recognition of H3K56-acetylated histone H3-H4 by the chaperone Rtt106. Nature. 2012;483:104–07. doi: 10.1038/nature10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D, Hu Q, Zhou H, et al. Structure and histone binding properties of the Vps75-Rtt109 chaperone-lysine acetyltransferase complex. J Biol Chem. 2011;286:15625–9. doi: 10.1074/jbc.C111.220715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Xue Y, Song C, Grunstein M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci USA. 2013;110:11493–8. doi: 10.1073/pnas.1309914110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Tanizawa H, Sriswasdi S, et al. Epigenetic regulation of condensin-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56 acetylation. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Holbert M, Wurtele H, et al. Fungal Rtt109 histone acetyltransferase is an unexpected structural homolog of metazoan p300/CBP. Nat Struct Mol Biol. 2008a;15:738–45. doi: 10.1038/nsmb.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Holbert MA, Delgoshaie N, et al. Structure of the Rtt109-AcCoA/Vps75 complex and implications for chaperone-mediated histone acetylation. Structure. 2011;19:221–31. doi: 10.1016/j.str.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Meeth K, Jiang E, et al. Structure of Vps75 and implications for histone chaperone function. Proc Natl Acad Sci USA. 2008b;105:12206–11. doi: 10.1073/pnas.0802393105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner K, Langer M, Kim Y, Denu J. Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J Biol Chem. 2000a;275:22048–55. doi: 10.1074/jbc.M002893200. [DOI] [PubMed] [Google Scholar]
- Tanner K, Trievel R, Kuo M, et al. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–60. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- Tanner KG, Langer MR, Denu JM. Kinetic mechanism of human histone acetyltransferase P/CAF. Biochemistry. 2000b;39:15652. doi: 10.1021/bi005121q. [DOI] [PubMed] [Google Scholar]
- Thaminy S, Newcomb B, Kim J, et al. Hst3 is regulated by Mec1-dependent proteolysis and controls the S phase checkpoint and sister chromatid cohesion by deacetylating histone H3 at lysine 56. J Biol Chem. 2007;282:37805–14. doi: 10.1074/jbc.M706384200. [DOI] [PubMed] [Google Scholar]
- Thomas CF, Jr, Limper AH. Current insights into the biology and pathogenesis of Pneumocystis pneumonia. Nat Rev Microbiol. 2007;5:298–308. doi: 10.1038/nrmicro1621. [DOI] [PubMed] [Google Scholar]
- Thompson PR, Wang D, Wang L, et al. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–15. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- Thornton CR. Detection of invasive aspergillosis. Adv App Microbiol. 2010;70:187–216. doi: 10.1016/S0065-2164(10)70006-X. [DOI] [PubMed] [Google Scholar]
- Tjeertes J, MIller K, Jackson S. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28:1878–89. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D, Erdel F, Bouazoune K, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell. 2013;51:454–68. doi: 10.1016/j.molcel.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomašić T, Mašič LP. Rhodanine as a scaffold in drug discovery: a critical review of its biological activities and mechanisms of target modulation. Exp Opin Drug Discov. 2012 doi: 10.1517/17460441.2012.688743. [DOI] [PubMed] [Google Scholar]
- Trievel RC, Rojas JR, Sterner DE, et al. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc Natl Acad Sci USA. 1999;96:8931–6. doi: 10.1073/pnas.96.16.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubota T, Berndsen CE, Erkmann JA, et al. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–12. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature. 2005;438:379–83. doi: 10.1038/nature04148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler JK, Adams CR, Chen SR, et al. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature. 1999;402:555–60. doi: 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- Valor L, Viosca J, Lopez-Atalaya J, Barco A. Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr Pharm Des. 2013;19:5051–64. doi: 10.2174/13816128113199990382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Beekum O, Kalkhoven E. Aberrant forms of histone acetyltransferases in human disease. Subcell Biochem. 2007;41:233–62. [PubMed] [Google Scholar]
- Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87:95–104. doi: 10.1016/s0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr Biol. 1998;8:96–108. doi: 10.1016/s0960-9822(98)70040-5. [DOI] [PubMed] [Google Scholar]
- Verreault A. De novo nucleosome assembly: new pieces in an old puzzle. Genes Dev. 2000;14:1430–8. [PubMed] [Google Scholar]
- Vetting MW, Carvalho LPSd, Yu M, et al. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–26. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Volkamer A, Kuhn D, Grombacher T, et al. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52:360–72. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- Volle C, Dalal Y. Histone variants: the tricksters of the chromatin world. Curr Opin Genet Dev. 2014;25:8–14. doi: 10.1016/j.gde.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Jung M. New lysine methyltransferase drug targets in cancer. Nat Biotechnol. 2012;30:622–3. doi: 10.1038/nbt.2300. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen J. SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. J Biol Chem. 2010;285:11458–64. doi: 10.1074/jbc.M109.087585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struc Biol. 2008;18:741–7. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–9. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Williams S, Truong D, Tyler J. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Prco Natl Acad Sci USA. 2008;105:9000–5. doi: 10.1073/pnas.0800057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Draper TM, Mintz-Cole RA, Morris TA, et al. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res. 2009;69:1792–9. doi: 10.1158/0008-5472.CAN-08-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtele H, Tsao S, Lépine G, et al. Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nat Med. 2010;16:774–80. doi: 10.1038/nm.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Song C, Young NL, et al. Histone H3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33:417–27. doi: 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang Q, Zhang K, et al. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, et al. Partitioning of histone H3–H4 tetramers during DNA replication-dependent chromatin assembly. Science. 2010;328:94–8. doi: 10.1126/science.1178994. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ayrapetov M, Xu C, et al. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell. 2012;48:723–33. doi: 10.1016/j.molcel.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Harper S, Speicher D, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self-acetylated intermediate. Nat Struct Biol. 2002;9:862–9. doi: 10.1038/nsb849. [DOI] [PubMed] [Google Scholar]
- Yang B, Miller A, Kirchmaier AL. HST3/HST4-dependent deacetylation of lysine 56 of histone H3 in silent chromatin. Mol Biol Cell. 2008;19:4993–5005. doi: 10.1091/mbc.E08-05-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zwaans BMM, Eckersdorff M, Lombard DB. The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle. 2009;8:2662–3. doi: 10.4161/cc.8.16.9329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Song JH, Cheng Y, et al. Long non-coding RNA HNF1A-AS1 regulates proliferation and migration in oesophageal adenocarcinoma cells. Gut. 2013 doi: 10.1136/gutjnl-2013-305266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Pu M, Zhang Z, Lou Z. Histone H3-K56 acetylation is important for genomic stability in mammals. Cell Cycle. 2009;8:1747–53. doi: 10.4161/cc.8.11.8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ouyang S, Kong X, et al. Catalytic mechanism of histone acetyltransferase p300: from the proton transfer to acetylation reaction. J Phys Chem B. 2014a;118:2009–19. doi: 10.1021/jp409778e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shibahara K, Stillman B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature. 2000;408:221–5. doi: 10.1038/35041601. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yang Q, Sun G, et al. Histone H3K56 acetylation is required for quelling-induced small RNA production through its role in homologous recombination. J Biol Chem. 2014b;289:9365–71. doi: 10.1074/jbc.M113.528521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yang F, Chen D-F, et al. Acetylation of chromatin-associated histone H3 lysine 56 inhibits the development of encysted Artemia embryos. PLOS ONE. 2013;8:e68374. doi: 10.1371/journal.pone.0068374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunder RM, Antczak AJ, Berger JM, Rine J. Two surfaces on the histone chaperone Rtt106 mediate histone binding, replication, and silencing. P Natl Acad Sci USA. 2012;109:E144–53. doi: 10.1073/pnas.1119095109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.