

Abstract
Purpose
The etiology of keratoconus (KC) and the factors governing its progression are not well understood. It has been suggested that this disease might be caused by biochemical alterations in the cornea; changes in the expression profiles of human aqueous humor (hAH) proteins have been observed in some diseases. To gain a new insight into the molecular mechanisms of KC pathology, we examined the hAH proteomes of those in the advanced stages of this disease. We used a high-throughput mass spectrometry approach to compare hAH protein expression in patients with KC and in control subjects.
Methods
Aqueous humor samples were acquired from five keratoconus patients during keratoplasty surgery and from five myopic control subjects during phakic intraocular lens implantation. Quantitative mass spectrometry analysis using spectral counting was performed to determine the relative amounts of hAH proteins in the samples from KC patients and control individuals.
Results
All KC patients included in the study presented severe keratoconus (K2 >52 D), and slit-lamp examination revealed microfolds in Descemet’s membrane, without clinical signs of hydrops. We found significant differences between the expression levels of 16 proteins in the two groups. In KC samples, seven proteins were overexpressed and nine were underexpressed in comparison with the control group. Gene ontology analysis revealed that these deregulated proteins are implicated in several biologic processes, such as the regulation of proteolysis, responses to hypoxia, and responses to hydrogen peroxide, among others.
Conclusions
The protein expression profiles in hAH from KC patients and myopic control subjects differ significantly. This result suggests that some components of the hAH proteome are involved in this disease. Further in-depth analysis of the hAH proteome should provide a better understanding of the mechanisms governing the pathophysiology of KC.
Introduction
Keratoconus (KC) is a pathological condition in which the cornea assumes a conical shape as a result of corneal thinning and central or paracentral conical protrusion [1]. The etiology and pathophysiology governing the progression of this disease are still poorly understood, although several risk factors have been described [1].
However, it has been reported that alterations in the activity of corneal collagenase leads to stromal thinning caused by collagen breakdown [2, 3]. In the early stages of KC, the cell membranes are disrupted, the basal cell layer disappears [4], and particulate material is deposited between the surface of basal epithelial cells and the Bowman’s layer [5]. Several studies of keratoconic corneas have demonstrated the loss of the corneal stroma caused by increased levels of proteases or by decreased levels of protease inhibitors, such as the α1-proteinase inhibitor and α2-macroglobulin [6]. Other reports have suggested that oxidative stress, producing reactive oxygen species (ROS), can cause apoptotic cell death and stromal mass loss [7, 8].
Human aqueous humor (hAH) contains a complex mixture of electrolytes, organic solutes, growth factors, cytokines, and other proteins involved in the metabolism of the avascular tissues of the anterior eye segment, such as the corneal endothelium [9]. Protein profiling has demonstrated the importance of hAH in the regulation of many functions of the eye. Several reports have shown a direct relationship between changes in the composition of this biologic fluid and ocular pathologies. Changes in hAH proteomes correlate with various stages of disease progression. One such example is an increase in the activity of transforming growth factor-beta2 in patients having keratoconus or glaucoma [10, 11]. Changes in the protein or ion concentrations in hAH might significantly affect the cellular functions and cell–matrix communication in the surrounding tissues. Nevertheless, the role of this biologic fluid in pathologies such as keratoconus has not been extensively studied. The relative scarcity of proteomic material in hAH is one reason for the lack of such studies; the other is invasive sample collection methods. To overcome these limitations, specialized and sensitive techniques have to be used. Using such specific techniques, we might provide new insights into the mechanisms of anterior segment homeostasis and the potential involvement of hAH proteins in KC disease.
We employed label-free liquid chromatography–tandem mass spectrometry analysis to identify alterations in the hAH protein expression in KC patients in comparison with age-matched control individuals. This sensitive proteomic approach should help to examine the role of hAH in the underlying pathophysiology of KC disease.
Methods
Subjects
Patients and control subjects were recruited in the Cornea and Ocular Surface Unit, Instituto de Oftalmología La Arruzafa (Cordoba, Spain), Instituto Barraquer (Barcelona, Spain), and Instituto Clínico-Quirúrgico de Oftalmología (Bilbao, Spain). The research was conducted by medically qualified personnel after approval by the Clinic Institutional Review Board and in compliance with the tenets of the Declaration of Helsinki. Informed consent was obtained from all patients after the nature and possible consequences of the study had been explained. Samples of hAH were obtained from five patients with severe KC during keratoplasty surgery and five myopic individuals (control subjects) during intraocular lens surgery to correct myopia. Best corrected distance visual acuity (CDVA: Snellen chart) measurements were employed in ophthalmic examinations. Slit-lamp biomicroscopy was used to identify stromal corneal thinning, Vogt’s striae, and Fleischer rings. Intraocular pressure (IOP) was also measured, and the fundus was examined with dilatation of the pupil. Corneal topography was performed using Orbscan (Bausch and Lomb, Rochester, NY). Topographic data were evaluated using Rabinowitz diagnostic criteria [1, 12]. The stage of KC was graded if there was at least one clinical sign of this disease: asymmetric bowtie, inferior-superior asymmetry greater than 1.4 D, central pachymetry giving the thickness of less than 490 μm, retinoscopic scissors-shadow, or corneal meridian (K2) curvature of more than 52 D.
Patients with a history of any systemic or ocular disorder or condition (including ocular surgery, trauma, and disease) were excluded. Current or recent use of topical ophthalmic or systemic medications was also a reason for exclusion from this study.
hAH sample collection and protein quantitation
Samples of hAH were collected from patients in the control group during the implantation of phakic intraocular lenses. The samples from KC patients were taken during keratoplasty procedures. To collect the samples, paracentesis was performed with a 15° ophthalmic slit knife. Then, a 30-gauge cannula was introduced into the anterior chamber, and an aliquot of 102 µl (mean) of hAH was aspirated. This procedure was performed at the beginning of the surgery and before the introduction of any substance into the anterior chamber. The protein samples were quantified using the EZQ Protein Quantification Kit (Invitrogen Dynal, Oslo, Norway) and stored at −80 °C until analysis.
Protein digestion and LC-MS/MS protein identification
Sample aliquots (6 µg) were dried in a Christ RVC 2–25 vacuum concentrator (Christ GmbH, Osterode, Germany) and resuspended in 16 µl of 6 M urea. Proteins were reduced and alkylated by incubation in dithiothreitol (200 mM DTT in 50 mM ammonium bicarbonate) for 45 min, followed by incubation in iodoacetamide (1 M IA in 50 mM ammonium bicarbonate) for another 45 min. Finally, the remaining IA was neutralized by adding 20 µl DTT and incubating for another 45 min. All incubations were performed at 25 °C. The samples were diluted to 100 µl with 50 mM ammonium bicarbonate, and trypsin (Trypsin Gold, Promega Corporation, Madison, WI, USA) was added to a final trypsin/protein ratio of 1:20. Samples were vortexed and incubated overnight at 37 °C. Peptides were dried in an RVC2 25 Speedvac concentrator (Christ), resuspended in 10 µl of 0.1% formic acid (FA), and sonicated for 5 min. Aliquots (2 µl) of each sample were desalted in C18 ZipTip pipette tips (Millipore) following the manufacturer’s protocol and dried in the vacuum concentrator. The samples were resuspended in 3 µl of 0.1% FA and sonicated for 5 min before mass spectrometry analysis.
Peptide mixtures obtained by digestion were separated (in triplicate) by online nanoLC and analyzed using electrospray tandem mass spectrometry. Peptide separation was performed on a nanoACQUITY UltraPerformance LC (UPLC) System (Waters, Manchester, UK) connected to an LTQ Orbitrap XL mass spectrometer (Thermo Electron, Bremen, Germany). An aliquot of each sample (3 µl) was diluted to a final volume of 5 µl in 0.1% FA and loaded onto a Symmetry 300 C18 UPLC Trap column, 180 µm × 20 mm, 5 µm (Waters). The precolumn was connected to a BEH130 C18 column, 75 μm x 200 mm, 1.7 μm (Waters), then equilibrated in 3% acetonitrile and 0.1% FA. Peptides were eluted directly into the nanoelectrospray capillary (Proxeon Biosystems, Odense, Denmark) at 300 nl/min with a 60 min linear gradient of 3%–50% acetonitrile.
The mass spectrometer automatically switched between MS and MS/MS in data-dependent acquisition mode. Full MS scan survey spectra (m/z 400–2000) were acquired in the Orbitrap with a mass resolution of 30,000 at m/z 400. The six most intense ions above 1,000 counts were sequentially subjected to collision-induced dissociation (CID) in the linear ion trap. Precursors with charge states of 2 and 3 were specifically selected for CID. Collision energy applied to each peptide was automatically normalized as a function of m/z and charge state. To identify less abundant peptides, a dynamic exclusion method was used; peptides were excluded from further analysis during 60 s.
Searches were performed using the Mascot search engine (Matrix Science, London, UK) with Proteome Discoverer 1.2 software (Thermo Electron, Bremen, Germany). Carbamidomethylation of cysteines was set as fixed modification, oxidation of methionines as variable modification; 5 ppm of peptide mass tolerance, 0.5 Da fragment mass tolerance, and two missed cleavages were allowed. Spectra were searched against the UniProtKB/Swiss-Prot database 2012_06 (536,489 sequences, 190,389 898 residues), using only Homo sapiens entries.
Quantitative protein analysis using spectral counting (APEX quantitation) and statistics
Protein quantitation was performed by label-free spectral counting using the APEX Quantitative Proteomics Tool v.1.1 as described previously [13]. Only Mascot-identified peptides with PeptideProphet probability ≥0.9 and proteins with ProteinProphet probability ≥0.95 were included in the analysis.
The data matrix obtained from APEX quantitation was used for multivariate statistical analysis. Before the statistical analysis, protein expression data were filtered by considering only the proteins identified in at least 50% of biologic samples for each condition. K-nearest neighbor data imputation was performed to complete the data matrix and manage the missing values. Data were then normalized using total spectral count normalization to avoid run-to-run variation. Significant differences between the groups were determined by the Significance Analysis of Microarrays (SAM) method [14]. After data filtering, the clustering of the samples was performed by principal component analysis (PCA) and agglomerative hierarchical clustering analysis using Tanagra statistical software v1.4.48 [15]. This procedure reduces the dimensionality of the data set and determines the extent of overlaps between the groups. The Mann–Whitney test was used to analyze differences in protein concentration between the groups. Statistical differences were set at p<0.05 level.
Functional interaction network analysis
To find network patterns related to diseases, the list of the most significantly deregulated proteins was loaded into Cytoscape version 2.8.3, using the Reactome FI Plugin [16, 17]. This plugin allows the construction of functional interaction (FI) sub-networks (modules) based on protein–protein interactions, gene coexpression, protein domain interactions, gene ontology (GO) annotations, and text-mined protein interactions. This procedure constructs an expression/interaction network in which the candidate protein biomarkers are interconnected in a direct or indirect manner by other interactions. It also performs functional enrichment and pathways analysis. For the selection of significant biologic processes, a false discovery rate (FDR)-based filtering was applied, with a cut-off value of 0.01.
Results
The mean age of KC patients was 34.16±9.62 years (1 female, 4 males) and the mean age of control individuals was 36±7.52 years (2 females, 3 males). All patients had severe KC (K2 >52 D) and microfolds in Descemet’s membrane revealed by slit-lamp examination.
Mean protein concentration was 0.82±1.40 μg/μl in the KC group and 0.14±0.04 μg/μl in the control group. There were no statistical differences between the total protein content in these groups (p=0.059; Mann–Whitney test). Protein concentrations are shown in Table 1 and the demographic data in Table 2.
Table 1. Protein concentration in hAH in KC group and in healthy subjects.
| Individuals | hAH total protein (μg) |
|---|---|
| CT 1 |
11.06 |
| CT 2 |
15.96 |
| CT 3 |
10.20 |
| CT 4 |
14.39 |
| CT 5 |
11.80 |
| KC 1 |
6.66 |
| KC 2 |
56.12* |
| KC 3 |
7.20 |
| KC 4 |
7.50 |
| KC 5 | 9.70 |
It should be emphasized the total protein concentration in a sample of hAH in a patient with KC is notably higher than each of the other hAH samples from KC patients (identified with an asterisk(*)). Because this sample had a higher concentration it consequently increased the standard deviation. CT=Control ; KC=Keratoconus
Table 2. Characteristics of healthy individuals and KC patients.
| Groups | Mean age | Sex (M/F) | Allergy | Rubbing |
|---|---|---|---|---|
| KC |
34.16±9.62 |
4/1 |
0.33 |
0.83 |
| CT | 36±7.52 | 3/2 | 0.5 | 0.75 |
CT=Control ; KC=Keratoconus
Label-free LC-MS/MS quantitative proteomics
The spectral counting-based label-free analysis identified 242 significantly distinct proteins. However, as only the proteins identified in at least 50% of the biologic samples for each condition were considered, this number was reduced to 137 proteins.
The data were processed using an APEX algorithms and subjected to data filtering by SAM analysis, with a cut-off value of 0.01; 16 proteins showed significant deregulation of their expression levels (Table 3). We found overexpression of hemoglobin subunit beta (HBB), haptoglobin (HP), plasma protease C1 inhibitor (SERPING1), alpha-2-HS-glycoprotein (AHSG), basement membrane-specific heparan sulfate proteoglycan core protein (HSPG2), hemoglobin subunit delta (HBD), and carbonic anhydrase 1 (CAI) in the KC samples (Figure 1A). There was also a group of proteins with reduced levels of expression in these samples. The group contained the following proteins: ceruloplasmin (CP), hemopexin (HPX), apolipoprotein A-II (APOA2), prostaglandin-H2 D-isomerase (PTGDS), actin cytoplasmic 2 (ACTG), semaphorin-7A (SEMA7A), alpha-1-acid glycoprotein 1 (ORM1), latent transforming growth factor beta-binding protein 2 (LTBP2), and Ig kappa chain V-I region EU (IGKC; Figure 1B). The largest protein expression changes were found for HBB (3.6-fold) and IGKC (−2.94-fold).
Table 3. List of proteins with significantly changed expression levels identified in the samples of hAH from KC patients (in comparison with healthy controls).
| Gene symbol | Protein | Fold | P value |
|---|---|---|---|
| HBB |
Hemoglobin subunit beta |
3.66 |
0.006 |
| HP |
Haptoglobin |
2.10 |
0.013 |
| SERPING1 |
Plasma protease C1 inhibitor |
2.06 |
0.004 |
| AHSG |
Alpha-2-HS-glycoprotein |
1.90 |
0.042 |
| HSPG2 |
Basement membrane-specific heparan sulfate proteoglycan core protein |
1.78 |
0.032 |
| HBD |
Hemoglobin subunit delta |
1.62 |
0.006 |
| CA1 |
Carbonic anhydrase 1 |
1.48 |
0.023 |
| CP |
Ceruloplasmin |
−1.60 |
0.022 |
| HPX |
Hemopexin |
−1.66 |
0.005 |
| APOA2 |
Apolipoprotein A-II |
−1.67 |
0.005 |
| PTGDS |
Prostaglandin-H2 D-isomerase |
−1.69 |
0.003 |
| ACTG |
Actin, cytoplasmic 2 |
−1.91 |
0.007 |
| SEMA7A |
Semaphorin-7A |
−1.97 |
0.029 |
| ORM1 |
Alpha-1-acid glycoprotein 1 |
−2.04 |
0.009 |
| LTBP2 |
Latent-transforming growth factor beta-binding protein 2 |
−2.63 |
0.015 |
| IGKC | Ig kappa chain V-I region EU | −2.94 | <0.001 |
Identification was achieved using LC-MS/MS and Mascot v.2.2.03 search engine.
Figure 1.

Whisker plot showing the mean expression values of proteins. A. Proteins upregulated in keratoconic corneas, listed in Table 3. B. Whisker plot showing the mean expression values of proteins downregulated in keratoconic corneas, listed in Table 3. KC=Keratoconus group; CT=control group.
We also evaluated the spatial separation for these proteins in the studied groups using data mining techniques such as PCA and hierarchical clustering. The results of PCA demonstrated a good separation between the KC and control groups, indicating that the analysis of hAH proteins might detect pathological changes associated with KC (Figure 2).
Figure 2.

Principal component analysis (PCA). Each symbol represents an individual, and the shape shows the group to which he/she belongs (circle: control - CT, triangle: keratoconus - KC).
Apart from data visualization using PCA, we also performed agglomerative hierarchical clustering analysis. This analysis detects close relationships between samples and establishes the degree of data homogeneity (or dispersion) within experimental groups. The results demonstrated that the samples were correctly assigned to their respective groups (Figure 3). These results corroborate the outcome of PCA, demonstrating that it is possible to capture the differences between pathological and normal expression profiles in hAH proteomes via LC-MS/MS.
Figure 3.
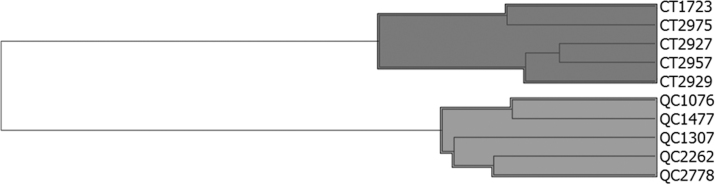
Dendrogram obtained by agglomerative hierarchical clustering analysis. Each branch represents an individual with their study code (CT: control group; QC: keratoconus group).
FI network analysis
The list of proteins whose expression differed between the experimental groups was used in FI network analysis, employing the Cytoscape program. The analysis produced an FI network in which these proteins were, directly or indirectly, interconnected (Figure 4). Functional enrichment analysis revealed significant involvement of these proteins in various biologic processes. These processes were: regulation of proteolysis, regulation of fibrinolysis, acute-phase response, platelet activation, regulation of ROS, response to hypoxia, response to calcium ions, cellular iron ion homeostasis, regulation of blood coagulation, response to retinoic acid, response to hydrogen peroxide, regulation of angiogenesis, response to osmotic stress, regulation of cell death, and response to vitamin D.
Figure 4.

Functional interaction network obtained using the FI Cytoscape plugin, which allowed construction of FI sub-networks (modules) based on protein–protein interactions and gene coexpression. The proteins were clustered into six modules numbered from 0 to 5, composed of 11, 9, 8, 5, 4, and 4 proteins. Proteins showing deregulation in our study are represented by circles, whereas newly added proteins or interconnectors are represented by diamonds. FIs extracted from pathways are shown as solid lines, while those predicted using NBC are shown as dashed lines. Extracted FIs involved in activation, expression regulation, or catalysis are shown with an arrowhead on the end of the line, and FIs involved in inhibition are shown with a “T” bar.
We also performed morphological analysis of the FI network and discovered six modules or groupings, as shown in Figure 4. GO analysis of the proteins in each module reveals that these modules are involved in a range of biologic functions, listed in Table 4. Thus, “Module 0” includes proteins associated with the response to retinoic acid, glucocorticoid stimulus, hypoxia, and regulation of proteolysis. “Module 1” is the largest group, including several proteins related mainly to apoptosis, angiogenesis, cell growth, and oxidative stress. “Module 2” includes the two proteins most overexpressed in the hAH of KC patients, HBB and HP. These proteins are involved in the regulation of cell death and the response to hydrogen peroxide. The regulation of endopeptidase activity and cellular iron ion homeostasis were among the processes enriched in the remaining modules.
Table 4. The results of GO analysis including all proteins of coexpression/interaction network, showing the proteins involved in each process and the significance of the results (FDR).
| Module | Biologic Process | FDR | Protein (Gene Symbol) |
|---|---|---|---|
| 0 |
Response to retinoic acid |
0.006 |
RBP4,EP300,SERPINF1 |
| 0 |
Response to glucocorticoid stimulus |
0.015 |
EP300,SERPINF1,PTGDS |
| 0 |
Regulation of proteolysis |
0.032 |
SERPINF1,SERPINA1 |
| 0 |
Response to hypoxia |
0.037 |
EP300,SERPINA1,USF1 |
| 1 |
Regulation of fibrinolysis |
<0.001 |
F2,HRG,THBS1 |
| 1 |
Regulation of reactive oxygen species |
<0.001 |
AGT,F2,THBS1 |
| 1 |
Regulation of blood coagulation |
0.006 |
F2,HRG |
| 1 |
Vitamin D metabolic process |
0.007 |
GC,LRP2 |
| 1 |
Regulation of cell growth |
0.008 |
AGT,HRG,AHSG |
| 1 |
Regulation of endothelial cell migration |
0.012 |
AGT,THBS1 |
| 1 |
Regulation of proteolysis |
0.011 |
AGT,SERPINC1 |
| 1 |
Acute-phase response |
0.011 |
F2,AHSG |
| 1 |
Induction of apoptosis |
0.011 |
AGT,THBS1,PLG |
| 1 |
Platelet activation |
0.011 |
F2,HRG,THBS1 |
| 1 |
Regulation of angiogenesis |
0.034 |
HRG,THBS1 |
| 2 |
Blood coagulation |
0.004 |
KCNMA1,TF,PPIA,ALB,HBB |
| 2 |
Cellular iron ion homeostasis |
0.004 |
TF,HPX,HP |
| 2 |
Regulation of cell death |
0.008 |
HP,HBB |
| 2 |
Platelet activation |
0.019 |
TF,PPIA,ALB |
| 2 |
Response to hydrogen peroxide |
0.017 |
HP,HBB |
| 4 |
Regulation of endopeptidase activity |
0.019 |
CSTB,CST3 |
| 4 |
Cellular iron ion homeostasis |
0.016 |
CP,SLC40A1 |
| 5 | Homeostasis of number of cells | 0.002 | PRDX2,IL7R |
*FDR=False discovery rate
Discussion
Different proteomic techniques that might be useful in the analysis of the pathophysiology of KC have been the subject of considerable interest in recent years [18–20]. This study is the first to use label-free LC-MS/MS quantitation to analyze the hAH proteome in KC disease. However, this technique has been used by Balaustraman et al. to analyze changes in the tear proteins in KC patients [21]. Recent advances in the field of proteomics have allowed the investigation of low-abundance and low-mass proteins or peptides. Fractionation strategies are essential for improving the isolation and enrichment of low-molecular-weight proteins or peptides from complex samples.
The sensitivity of this procedure overcomes the limitations of hAH analysis imposed by low protein content. The results of PCA and hierarchical clustering demonstrate that the procedure provides a good tool for discrimination between KC patients and control subjects.
We identified 16 proteins whose expression levels were altered in keratoconic hAH. These alterations might have important implications for the biologic processes associated with the disease (Figure 4, Table 4). Many of these processes might be directly involved in the events taking place in the cornea during the development of KC [7, 8, 22–26]. To obtain more details of biologic processes involved in the disease, we performed protein-network analysis. The results revealed several distinct modules formed by association of the identified proteins with specific processes. The results revealed several distinct modules formed by association of the identified proteins with specific processes (shown in Figure 4). The involvement of some of these processes in KC has been already reported [27–30].
The hAH provides nutrition to the avascular ocular tissues such as the cornea; changes in the composition of this liquid might affect the structures of these tissues. The endothelium, being in direct contact with the hAH, might be particularly sensitive to such changes. Several studies have reported that KC causes changes in the cornea at all levels. However, these changes might be not directly related to the alterations in the protein composition in keratoconus, but a consequence of preexisting proteomic variation [22–25].
Apoptosis and oxidative stress are the events most frequently reported in the keratoconic cornea [7, 8, 31, 32]. One of the most important, although often overlooked, functions of the cornea is to neutralize free radicals and oxidants that are typically formed by cellular metabolism and caused by exposure to ultraviolet light. High levels of ROS and reactive nitrogen species (RNS) damage the DNA, the mitochondrial respiratory chain, and denature proteins. They also cause lipid peroxidation, which further generates free radicals, initiating a vicious cycle of oxidation [33]. Keratocytes are particularly susceptible to oxidative stress, which may play an important role in the development and progression of keratoconus.
Kenney et al. [33] proposed a “cascade hypothesis of keratoconus,” in which the alterations in corneal protein expression could lead to oxidative damage (and ultimately to apoptosis), changes in signaling pathways, and increases in enzyme activities. Irreversible cell damage leads to apoptosis; the less damaged cells undergo wound healing or repair. During the wound-healing process, various degradative enzymes and wound healing factors are upregulated, which might lead to the formation of focal areas of corneal thinning and fibrosis.
Our results are consistent with such phenomena, and, therefore, could reflect the molecular alterations proposed in the cascade hypothesis of keratoconus.
Our study, evaluating the possible changes in the protein composition of the hAH in KC disease, was conducted with a relatively small number of patients. Once the existence of such changes is confirmed, the results should be validated in future studies with increased statistical power.
In conclusion, this work represents the first attempt to analyze the changes in the proteome composition in the hAH of KC patients. Clear spatial separation between pathological and control groups obtained using exploratory data techniques shows that the analysis of the hAH proteome by LC-MS/MS is a valuable tool in the studies of KC. The results presented here provide new insights into the underlying molecular mechanisms of this disease. Our results and observations should be further explored to generate essential data needed to understand the role of hAH in KC pathology.
Acknowledegments
Financial Support: This work was partially supported by the Centre for the Development of Industrial Technology (CDTI,) CENIT Program, grant CEN-20091021. The funding organizations had no role in the design or conduct of this research.
References
- 1.Rabinowitz YS. Keratoconus. Surv Ophthalmol. 1998;42:297–319. doi: 10.1016/s0039-6257(97)00119-7. [DOI] [PubMed] [Google Scholar]
- 2.Tuft SJ, Moodaley LC, Gregory WM, Davison CR, Buckley RJ. Prognostic factors for the progression of keratoconus. Ophthalmology. 1994;101:439–47. doi: 10.1016/s0161-6420(94)31313-3. [DOI] [PubMed] [Google Scholar]
- 3.Kenney MC, Nesburn AB, Burgeson RE, Butkowski RJ, Ljubimov AV. Abnormalities of the extracellular matrix in keratoconus corneas. Cornea. 1997;16:345–51. [PubMed] [Google Scholar]
- 4.Teng CC Electron microscope study of the pathology of keratoconus: I. Am J Ophthalmol. 1963;55:18–47. doi: 10.1016/0002-9394(63)91645-3. [DOI] [PubMed] [Google Scholar]
- 5.Iwamoto T, Devoe AG. Particulate structures in keratoconus. Arch Ophtalmol Rev Gen Ophtalmol. 1975;35:65–76. [PubMed] [Google Scholar]
- 6.Sawaguchi S, Twining SS, Yue BY, Wilson PM, Sugar J, Chan SK. Alpha-1 proteinase inhibitor levels in keratoconus. Exp Eye Res. 1990;50:549–54. doi: 10.1016/0014-4835(90)90044-u. [DOI] [PubMed] [Google Scholar]
- 7.Behndig A, Karlsson K, Johansson BO, Brännström T, Marklund SL. Superoxide dismutase isoenzymes in the normal and diseased human cornea. Invest Ophthalmol Vis Sci. 2001;42:2293–6. [PubMed] [Google Scholar]
- 8.Buddi R, Lin B, Atilano SR, Zorapapel NC, Kenney MC, Brown DJ. Evidence of oxidative stress in human corneal diseases. J Histochem Cytochem. 2002;50:341–51. doi: 10.1177/002215540205000306. [DOI] [PubMed] [Google Scholar]
- 9.To CH, Kong C, Chan C, Shahidullah M, Do C. The mechanism of aqueous humour formation. Clin Exp Optom. 2002;85:335–49. [PubMed] [Google Scholar]
- 10.Joseph R, Srivastava OP, Pfister RR. Differential epithelial and stromal protein profiles in keratoconus and normal human corneas. Exp Eye Res. 2011;92:282–98. doi: 10.1016/j.exer.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Ozcan AA, Ozdemir N, Canataroglu A. The aqueous levels of TGF-beta2 in patients with glaucoma. Int Ophthalmol. 2004;25:19–22. doi: 10.1023/b:inte.0000018524.48581.79. [DOI] [PubMed] [Google Scholar]
- 12.Rabinowitz YS. Videokeratographic indices to aid in screening for keratoconus. J Refract Surg. 1995;11:371–9. doi: 10.3928/1081-597X-19950901-14. [DOI] [PubMed] [Google Scholar]
- 13.Braisted JC, Kuntumalla S, Vogel C, Marcotte EM, Rodrigues AR, Wang R, Huang ST, Ferlanti ES, Saeed AI, Fleischmann RD, Peterson SN, Pieper R. The APEX Quantitative Proteomics Tool: generating protein quantitation estimates from LC-MS/MS proteomics results. BMC Bioinformatics. 2008;9:529. doi: 10.1186/1471-2105-9-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rakotomalala R. TANAGRA: un logiciel gratuit pour l’enseignement et la recherche. Actes EGC. 2005;2:697–702. [Google Scholar]
- 16.Smoot ME, Ono K, Ruscheinski J, Wang P-L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–2. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu G, Feng X, Stein L. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 2010;11:R53. doi: 10.1186/gb-2010-11-5-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lema I, Brea D, Rodríguez-González R, Díez-Feijoo E, Sobrino T. Proteomic analysis of the tear film in patients with keratoconus. Mol Vis. 2010;16:2055–61. [PMC free article] [PubMed] [Google Scholar]
- 19.Pannebaker C, Chandler HL, Nichols JJ. Tear proteomics in keratoconus. Mol Vis. 2010;16:1949–57. [PMC free article] [PubMed] [Google Scholar]
- 20.Acera A, Vecino E, Rodríguez-Agirretxe I, Aloria K, Arizmendi JM, Morales C, Durán JA. Changes in tear protein profile in keratoconus disease. Eye (Lond) 2011;25:1225–33. doi: 10.1038/eye.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balasubramanian SA, Wasinger VC, Pye DC, Willcox MDP. Preliminary identification of differentially expressed tear proteins in keratoconus. Mol Vis. 2013;19:2124–34. [PMC free article] [PubMed] [Google Scholar]
- 22.Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic Biol Med. 1998;25:392–403. doi: 10.1016/s0891-5849(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 23.Loh A, Hadziahmetovic M, Dunaief JL. Iron homeostasis and eye disease. Biochim Biophys Acta. 2009;1790:637–49. doi: 10.1016/j.bbagen.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yolton DP. Calcium: II. Role in keratoconus. J Am Optom Assoc. 1983;54:135–8. [PubMed] [Google Scholar]
- 25.Balasubramanian SA, Mohan S, Pye DC, Willcox MDP. Proteases, proteolysis and inflammatory molecules in the tears of people with keratoconus. Acta Ophthalmol (Copenh) 2012;90:e303–9. doi: 10.1111/j.1755-3768.2011.02369.x. [DOI] [PubMed] [Google Scholar]
- 26.Cheung IMY, McGhee CNJ, Sherwin T. A new perspective on the pathobiology of keratoconus: interplay of stromal wound healing and reactive species-associated processes. Clin Exp Optom. 2013;96:188–96. doi: 10.1111/cxo.12025. [DOI] [PubMed] [Google Scholar]
- 27.Arnal E, Peris-Martínez C, Menezo JL, Johnsen-Soriano S, Romero FJ. Oxidative stress in keratoconus? Invest Ophthalmol Vis Sci. 2011;52:8592–7. doi: 10.1167/iovs.11-7732. [DOI] [PubMed] [Google Scholar]
- 28.Brown DJ, Lin B, Chwa M, Atilano SR, Kim DW, Kenney MC. Elements of the nitric oxide pathway can degrade TIMP-1 and increase gelatinase activity. Mol Vis. 2004;10:281–8. [PubMed] [Google Scholar]
- 29.Radner W, Zehetmayer M, Skorpik C, Mallinger R. Altered organization of collagen in the apex of keratoconus corneas. Ophthalmic Res. 1998;30:327–32. doi: 10.1159/000055492. [DOI] [PubMed] [Google Scholar]
- 30.Wilson SE, He YG, Weng J, Li Q, McDowall AW, Vital M, Chwang EL. Epithelial injury induces keratocyte apoptosis: hypothesized role for the interleukin-1 system in the modulation of corneal tissue organization and wound healing. Exp Eye Res. 1996;62:325–7. doi: 10.1006/exer.1996.0038. [DOI] [PubMed] [Google Scholar]
- 31.Gondhowiardjo TD, van Haeringen NJ. Corneal aldehyde dehydrogenase, glutathione reductase, and glutathione S-transferase in pathologic corneas. Cornea. 1993;12:310–4. doi: 10.1097/00003226-199307000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Kenney MC, Brown DJ, Rajeev B. Everett Kinsey lecture. The elusive causes of keratoconus: a working hypothesis. CLAO J. 2000;26:10–3. [PubMed] [Google Scholar]
- 33.Kenney MC, Brown DJ. The cascade hypothesis of keratoconus. Cont Lens Anterior Eye. 2003;26:139–46. doi: 10.1016/S1367-0484(03)00022-5. [DOI] [PubMed] [Google Scholar]