

Abstract
The perimenopause is an aging transition unique to the female that leads to reproductive senescence which can be characterized by multiple neurological symptoms. To better understand potential underlying mechanisms of neurological symptoms of perimenopause, the current study determined genomic, biochemical, brain metabolic and electrophysiological transformations that occur during this transition using a rat model recapitulating fundamental characteristics of the human perimenopause. Gene expression analyses indicated two distinct aging programs: chronological and endocrine. A critical period emerged during the endocrine transition from regular to irregular cycling characterized by decline in bioenergetic gene expression, confirmed by deficits in FDG-PET brain metabolism, mitochondrial function, and long-term potentiation. Bioinformatic analysis predicted insulin/IGF1 and AMPK/PGC1α signaling pathways as upstream regulators. Onset of acyclicity was accompanied by a rise in genes required for fatty acid metabolism, inflammation, and mitochondrial function. Subsequent chronological aging resulted in decline of genes required for mitochondrial function and β-amyloid degradation. Emergence of glucose hypometabolism and impaired synaptic function in brain provide plausible mechanisms of neurological symptoms of perimenopause and may be predictive of later life vulnerability to hypometabolic conditions such as Alzheimer’s.
Keywords: Perimenopause, Female brain aging, Glucose metabolism, Mitochondria, Synaptic plasticity, Hypometabolism, Fatty acid metabolism, Long-term potentiation
1. INTRODUCTION
The perimenopause, unique to the female, is a midlife transition state that precedes and leads to reproductive senescence (Brinton, 2010, Butler and Santoro, 2011). Worldwide, there are currently over 850 million women aged 40–60 years of age, 88% of whom will transition through the perimenopause by age 55; all women who reach the age of 60 with reproductive organs intact will transition through the perimenopause to the menopause (Brinton, 2010, Harlow, et al., 2012). This final stage is associated with a near complete decrease in ovarian secretion of estrogen and progesterone hormones (Harlow, et al., 2012). Similar to the human, rodent and nonhuman primates share common features of the perimenopausal transition, including decline in follicles, irregular cycling, irregular fertility, steroid hormone fluctuations and insensitivity to estrogen (Brinton, 2012, Finch, 2014).
While the clinical definition of perimenopause focuses on functional changes in the reproductive system, the symptoms of the perimenopause are largely neurological in nature. The breadth of neurological symptoms coincident with the perimenopause is well documented in the clinical science literature (Genazzani, et al., 2005, Greendale, et al., 2010, Maki, et al., 2008, Nelson, 2008). These neurological symptoms including increased temperature, depression, insomnia, pain, and cognitive impairment during the perimenopausal transition are indicative of disruption in multiple systems whereas their emergence during the same transition is indicative of a common or a set of common underlying mechanisms. Substantial evidence indicates that ovarian and neural hormones including 17β-estradiol (E2) and progesterone (P4) regulate fundamental systems in brain at both the organizational and activational levels including neurogenesis, cell survival, brain metabolism, neuro-inflammation, and synaptic activity (Brinton, 2008, Brinton, 2009, Brinton, et al., 2008, McEwen, et al., 2012, Morrison, et al., 2006, Rettberg, et al., 2014, Simpkins, et al., 2010, Woolley, 2007, Yao, et al., 2011).
The present perimenopause model builds on prior studies in middle-aged rats during their transition from regular to irregular cycling (Adams, et al., 2001, Downs and Wise, 2009, Finch, et al., 1984, Gore, et al., 2000, Kermath and Gore, 2012). The transition to irregular cycles was associated with cognitive impairments and altered sex steroids in hippocampus and cerebral cortex (Paris, et al., 2011), while we found altered expression of estrogen receptor α(ERα) in astrocytes of cerebral cortex with altered neurotrophic activity in vitro (Arimoto, et al., 2013). To investigate potential underlying mechanisms of neurological dysfunction during the perimenopause, we conducted neural analyses in endocrine characterized rats spanning the transition from reproductively capable to reproductive senescent to aged. Using this model, we investigated the differential impact of endocrine aging versus chronological aging on hippocampal bioenergetic systems (glucose metabolism, inflammation, redox, lipid metabolism, and β-amyloid processing) and synaptic function (long-term potentiation). Findings from this study revealed bioenergetic transitions specific to endocrine aging during the perimenopause, that were coincident with significant decline in synaptic plasticity.
2. METHODS
Animals
Animal studies were performed following National Institutes of Health guidelines on use of laboratory animals; protocols were approved by the University of Southern California Institutional Animal Care and Use Committee. A total of 90 young or middle-aged female Sprague-Dawley rats were obtained from Harlan Laboratories. Regular 6 month group was cycled from 5 month of age using rats that had given birth to at least one litter. Rats for all other groups were aged from 8–9 month old retired breeders. One week after arrival, ovarian status were evaluated daily by the cytology of uterine cells obtained from lavage at 11am. The smear was morphologically characterized based on the four stages of the cycle: Estrus (E), Metestrus (M), Diestrus (D) and Proestrus (P). The regular 4-day estrus cycle is defined as the period between successive proestrus smears (P, E, M, D, P, E, M, D, P). In addition to regular cycling animals, selected groups included middle-aged rodents at defined stages of perimenopause (Fig. 1B). The irregular group was defined as 2 contiguous cycles of 5–8 days characterized by prolonged diestrus stages. The acyclic (constant estrus) group was defined as persistent vaginal cornification lasting > 9 days. Rats at designated age (6m, 9–10m or 16m) and cycling status were euthanized at estrus or constant estrus. Three sets of animals were used in this study. The first set (40 rats in total) included all 5 experimental groups (Reg-6m, Reg-9–10m, Irreg-9–10m, Acyc-9–10m and Acyc-16m). Of this set, N = 5–6 per group were used for analyses of E2/P4 levels, estrogen/progesterone receptors, and gene array (Figs. 1–3 and Tabs. 1, 2). A second set of rats containing only the Reg-9–10m and Irreg-9–10m groups (30 rats in total) were used for glucose tolerance test, brain glucose uptake (whole brain), protein expression (left hemisphere) and mitochondrial function (right hemisphere) analyses shown in Figs. 4, 5. A third set of rats containing only the Reg-9–10m and Irreg-9–10m groups (20 rats in total) was used for LTP (Fig. 6). Rats that did not meet the endocrine criteria for each group were excluded from analyses for this study.
Figure 1. The perimenopause rodent model.
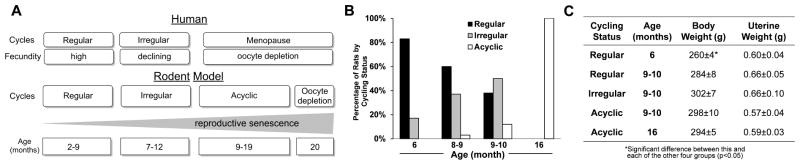
A. Comparison of main stages of the human perimenopause with rodent reproductive senescence. In humans and rodents, ovarian senescence from follicular depletion is associated with decreasing fecundity and increasing irregularity of estrus cycles (Finch, 2014). B. Transition of cycling stages with age: percentage of aging rats by cycling status from cohort of 90 rats. C. Body weight and uterine weight of animals with different age and cycling status. Data were presented as average ± SEM, *p ≤ 0.05, n = 6.
Figure 3. Bioenergetics-, inflammation- and AD pathology-related gene expression.

A. Heatmaps of standardized gene expression from selective functional groups. Each gene was normalized and colored based on its relative expression levels across 5 biological groups. This method allows genes that have different magnitude of signal intensity but belong to the same functional group and share similar expression patterns to be displayed on the same heatmap. B. The first principal component (PC1) scores of the bioenergetic gene category for each animal group. The middle line in the box is the median, the ends of the box are 25th and 75th percentiles; the ends of the dotted lines are the 25/75 percentile + 1.5 times the interquartile range. C. Mutual correlations between different gene categories (Bioenergetics, Redox, Inflammation, Lipid metabolism and AD pathology) based on PC1 scores from the PCA of gene expression data. The r-value and significance for each of the Pearson’s correlation was summarized in the table. n = 5, * p ≤ 0.05; ***p ≤ 0.001.
Table 1. Summary of significantly changed genes during each transition period.
Genes that showed statistically significant changes (p = 0.001 – 0.05) are grouped based on their primary function for each transition period. Results were based on pair-wise comparisons of the gene expression, either up-regulated or down-regulated.
| Comparison | Up-regulated | Down-regulated |
|---|---|---|
| Reg-9–10m vs Reg-6m | Redox regulation: Nfe2l2 Phospholipase: Pld1, Plcg2 Inflammation: C4a, Hrh1 Fatty acid metabolism: Hadh Cholesterol homeostasis: Abca7, Apoa1 Alzheimer’s related: Apeh, Clu Apoptosis: Casp8 |
Molecule Transport: Hspd1, Bnip1 Neurotransmission: Gad1, Gad2 Redox homeostasis: Grx2, Prx3 Neuronal survival: Cxcr4 |
| Irreg-9–10m vs Reg-9–10m | Mitochondrial function: Atpaf2, Esrra Stress/Inflammation: Nfkb2, Il1rl1, Mapk3 Phospholipase: Plcb3 Amyloid metabolism: Thop1 Microtubule stabilization: Map2 |
|
| Acyc-9–10m vs Irreg-9–10m | Mitochondrial function: Atpaf2, Esrrb Mitochondrial fatty acid uptake: Cpt2 Cholesterol homeostasis: Apoa1, Star Phospholipase: Pla2g2a Inflammation: Tnfaip2, Cxcl12, Cd40 Anti-apoptosis: Bcl2l1 Amyloid metabolism: Apba3 |
Amyloid metabolism: Apbb1 |
| Acyc-16m vs Acyc-9–10m | Glycolysis: Pfk1 Stress/Inflammation: Mapk3 |
Mitochondrial function: Atpaf2, Ndufs4, Ndufs8, Cox4i2, Pdp1, Timm23 Lipid metabolism: Pparg, Pla2g1b Amyloid metabolism: Apba3, Ide Anti-apoptosis: Bcl2l1 |
Table 2. Network nodes and upstream regulators identified by IPA.
Network nodes and upstream regulators identified by the IPA network- and pathway analyses for each transition period and each gene category were listed.
| Network Nodes | Upstream Regulators | |
|---|---|---|
| Bioenergetics | Insulin, IGF-1, IRS1, PI3K, Akt, Creb, JNK, ERK1/2, AMPK, NFκB, CD3 | Insulin, IGF1, INSR, IGF1R, IRS1, ESRRA, PPARGC1A, PPARG, PPARA, NRF1, CREB, NR4A3, Myc, TP53, NFE2L2, TNF |
| Inflammation | IL1, IL12, CD40, ERK1/2, TLR | TNF, CD40, IFNg, NFκB, IL1, IL1B, IL6, ICAM1 |
| AD Pathology | ERK1/2, MEK, CREB, IL1, AP1, Bcl2, BclXL, Bax, Bad, Casp8, Cyto c | IGF1, PI3K/Akt, PTEN, Myc, ERK1/2, NFκB |
Figure 4. Bioenergetic protein expression and related signaling pathway in hippocampus of 9.

–10 month regular- and irregular cycling rats.
A. Glucose transporter 3 (GLUT3) protein expression. B. PDH E1α subunit protein expression. C. ATPAF2 protein expression. D. Relative expression of p-IRS1Tyr608 (activated) to IRS1. E. Relative expression of p-Akt (activated) to Akt. F. Levels of phosphorylated (activated) ERK1 and ERK2. G. Levels of phosphorylated (activated) CREB. H. Relative expression of p-AMPK (activated) to AMPK. I. NFκB (p65) protein expression. J. Relative expression of phosphorylated I B to I B. Data were presented as average ± SEM, *p ≤0.05, n = 10–12. Representative blot images were shown under the bar graphs.
Figure 5. Brain metabolic function during regular-irregular transition.

A. Representative FDG-microPET/CT scan images in regular and irregular cycling rats. B. Relative glucose uptake in regular and irregular groups measured by FDG-microPET/CT. C. Glucose tolerance test of regular and irregular cycling rats and the quantification of the area under the curve (AUC). D. Respiratory control ratio of mitochondria isolated from whole brain. E. Complex IV activity of mitochondria isolated from whole brain. F. ATP synthase activity of mitochondria isolated from whole brain. G. Rate of H2O2 production of mitochondria isolated from whole brain. Data were presented as average ± SEM, *p ≤ 0.05, n = 10–12.
Figure 6. Synaptic plasticity in the regular and irregular cycling rats.
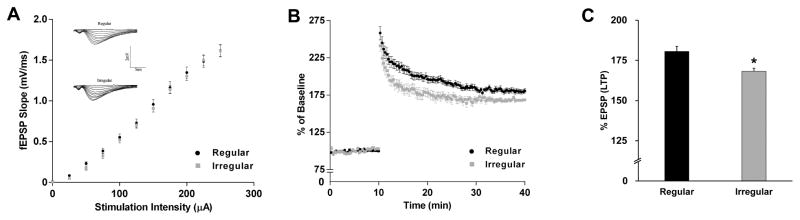
A. I/O relationships with increasing stimulation to the stratum radiatum of the CA1 region of the hippocampus. The inserts are the representative I/O recordings. B. Recorded fEPSP slopes normalized to the pre-tetanus baseline. Data analyzed by repeated measures ANOVA. C. LTP shown as %EPSP for the last 5 min of the response to TBS stimulation. Data were presented as average ± SEM, *p ≤ 0.05, n = 10–12.
Brain Tissue Collection
Rats were euthanized and the brains rapidly dissected on ice. Cerebellum and brainstem was removed from each brain and two hemispheres were separated. The cortical hemisphere was fully peeled laterally and hippocampus was then separated. Both cortex and hippocampus were harvested and frozen in −80°C for subsequent analyses.
Quantitative Analysis of Steroid Hormones by LC-MS/MS
Levels of P4 and E2 extracted from cerebral cortex and serum were measured as previously described (Caruso, et al., 2013). 2,2,4,6,6–17α,21,21,21-D9-PROG (D9-PROG) was obtained from Medical Isotopes, (Pelham, NH, USA); 2,3,4-13C3-17β-estradiol (13C3-17β-E) was obtained from Sigma-Aldrich. SPE cartridges (Discovery DS-C18 500 mg) were from Supelco, Italy. Cerebral cortex and serum were spiked with 13C3-17β-E (1 ng/sample) and D9-PROG (0.2 ng/sample), as internal standards and homogenized in 2 ml of MeOH/acetic acid (99:1 v/v). After an overnight extraction at 4°C, samples were centrifuged at 12000 rpm for 5 min and the pellet was extracted twice with 1 ml of MeOH/acetic acid (99:1 v/v). The organic phases were combined and evaporated to dryness. The organic residues were resuspended with 3 ml of MeOH/H20 (10:90 v/v) and passed through a SPE cartridges, previously activated with MeOH (5ml) and MeOH:H2O 1:9 v/v (5ml), the steroids were eluted in MeOH. Quantitative analysis was performed on the basis of calibration curves daily prepared and analyzed. Positive atmospheric pressure chemical ionization (APCI+) experiments were performed with a linear ion trap - mass spectrometer (LTQ, ThermoElectron Co, San Jose, CA, USA) using nitrogen as sheath, auxiliary and sweep gas. The mass spectrometer (MS) was employed in tandem mode (MS/MS) using helium as collision gas. The Hypersil Gold column (100×3 mm, 3 μm; ThermoElectron Co, San Jose, CA, USA) was maintained at 40 °C. Peaks of the LC-MS/MS were evaluated by means of the software Excalibur® release 2.0 SR2. The steroids data were analyzed by ANOVA followed by pairwise comparisons. The statistical significance was indicated by p ≤ 0.05.
Low Density Gene Expression Array
Low density gene expression array was performed as previously described (Zhao, et al., 2012). Briefly, customerized 384-well Taqman® low-density array (TLDA) cards (See Table S1 for complete list of genes) were manufactured by Invitrogen (Carlsbad, CA). Total RNA was isolated from rat hippocampal tissues using the PureLink RNA Mini Kit (Invitrogen). RNA quantity and quality were analyzed before cDNA synthesis using the High Capacity RNA-to-cDNA Master Mix (Applied Biosystems). Taqman real-time qPCR reactions were performed on 50 ng cDNA samples mixed with the TaqMan Universal PCR Master 2x Mix and fluorescence was detected on an ABI 7900HT Fast Real-Time PCR System (Applied Biosystems). Data were analyzed using the RQ Manager Version 1.2 and DataAssist Version 2.0. Relative gene expression levels or fold changes relative to the reference group were calculated by the comparative Ct (ΔΔCt) method, with Ct denoting threshold cycle. For each sample, ΔCt was calculated as the difference in average Ct of the target gene and the endogenous control gene. For each group, mean 2−ΔCt was calculated as the geometric mean of 2−ΔCt of all samples in the group. Fold change was then calculated as mean 2−ΔCt (comparison group)/mean 2−ΔCt (reference group). The 2−ΔCt values for each target gene were statistically analyzed by ANOVA followed by pairwise comparisons using Student’s t-test. The statistical significance was indicated by p ≤ 0.05; p-values were not adjusted for multiple testing as discussed previously (Zhao, et al., 2012).
Principal Component Analysis (PCA) of Gene Expression Data
As a data reduction method to obtain a summary of gene expression across a group of genes, PCA (Basilevsky, 1994) was used on groupings of gene array data. The specific gene groupings were: Bioenergetics, Inflammation, Redox, Lipid metabolism and Alzheimer’s pathology. As the transform explaining the largest degree of variance in gene expression, we extracted the first principal component (PC1) and calculated the PC1 factor scores for each animal and each gene group.
Bioinformatic Analysis by Ingenuity Pathway Analysis (IPA)
Expression data for genes that exhibited > 5% fold change (increase or decrease) AND with the p-value < 0.5 were analyzed by IPA core analysis composed of a network analysis and an upstream regulator analysis. We used these relaxed criteria to maximize the coverage of the gene array results in the bioinformatic analyses. The network analysis identified biological connectivity among molecules in the dataset that were up- or down-regulated in a comparison (focus molecules that serve as “seeds” for generating networks) and their interactions with other molecules present in the Ingenuity Knowledge Base. Focus molecules were combined into networks that maximized their specific connectivity. Additional molecules from the Ingenuity Knowledge Base (interacting molecules) were used to specifically connect two or more smaller networks to merge them into a larger one. A network was composed of direct and indirect interactions among focus molecules and interacting molecules, with a maximum of 70 molecules per network. Generated networks were ranked by the network score according to their degree of relevance to the network eligible molecules from the dataset. The network score was calculated with Fisher’s exact test, taking into account the number of network eligible molecules in the network and the size of the network, as well as the total number of network eligible molecules analyzed and the total number of molecules in the Ingenuity Knowledge Base that were included in the network. Higher network scores are associated with lower probability of finding the observed number of network eligible molecules in a given network by chance.
The Ingenuity’s Upstream Regulator Analysis in IPA is a tool that predicts upstream regulators from gene expression data based on the literature and compiled in the Ingenuity Knowledge Base. A Fisher’s exact test p-value was calculated to assess the significance of enrichment of the gene expression data for the genes downstream of an upstream regulator. A z-score was given to indicate the degree of consistent agreement or disagreement of the actual versus the expected direction of change among the downstream gene targets. A prediction about the state of the upstream regulator, either activated or inhibited, was made based on the z-score.
Brain Glucose Uptake
Brain glucose uptake (18-FDG-microPET/microCT) was determined as previously described (Ding, et al., 2013). Briefly, rats (n = 10–12 per group) were maintained under anesthesia during microPET and microCT scans with 2–2.5% isoflurane in oxygen. Scans were performed in an imaging chamber equipped with a nose cone for anesthesia delivery and heating pad for body temperature control as previously described. MicroPET imaging was performed with a microPET R4 rodent model scanner (Concorde Microsystems Inc, Knoxville, TN) and micro CT imaging was performed on MicroCAT II tomography (Siemens Preclinical Solutions, Knoxville, TN). Rats were injected intravenously via the tail vein with radiotracer [18F] Fluoro-2-deoxy-2-D- glucose (FDG, 400 μCi, 100 uL). Radioactive dose was determined prior to injection by radioisotope dose calibrator (Capintec, CRC-712M). At 40 min post-injection of FDG, each rat was positioned in the microPET scanner in the center of the 10.8 cm transaxial and 8 cm axial field of view (FOV). Brain microPET data were collected for 10 min followed by a 10 min microCT scan for the purpose of co-registration. Co-registration of microPET and microCT data was performed using the AMIDE software package (http://amide.sourceforge.net/). After co-registration of the PET and CT images, ROI (region of interest) was defined and used to measure the radioactivity concentration in brains. Decay correction was used to adjust the actual radioactivity dosage injected (Actual radioactivity dosage at time of injection , T = minutes between injected time point and initial time point). Data were analyzed using Student’s t-test.
Glucose Tolerance Test
Rats were fasted overnight and micro-volume blood samples were collected by puncturing tail vein. Baseline blood glucose level was measured before injection with D-glucose (2g/kg, intraperitoneally). After glucose injection, micro-volume blood samples were collected at 5, 15, 30, 60, 90, and 120 minutes and glucose levels were determined using a glucometer (Abbott Diabetes Care, Alameda, CA). The area under the curve for each group based on their averaged glucose levels at different time points were calculated.
Protein Expression Analysis
Rats were euthanized and the brains were rapidly dissected on ice. Hippocampal tissue was processed for protein extraction using Tissue Protein Extraction Reagent (Thermo Scientific, Rockford, IL) with phosphatase- and protease inhibitors (Sigma, St. Louis, MO). Protein concentrations were determined with the Bio-Rad Bradford assay. Equal amounts of protein (20 μg/well) were loaded to SDS PAGE Criterion gel (Bio-Rad, Hercules, CA) and electrophoresed with Tris/glycine running buffer (pH 8.3). Proteins were transferred to 0.45 mm pore size polyvinylidene difluoride (PVDF) membranes and probed with primary antibodies against GLUT3, PDH-E1α, ERβ (Abcam, Cambridge, MA), and ERα and ATPAF2 (Proteintech, Chicago, IL). All other primary antibodies were obtained from Cell Signaling Technology (Beverly, MA). HRP-conjugated anti-rabbit antibody and HRP-conjugated anti-mouse antibody (Vector Laboratories, Burlingame, CA) were used as secondary antibodies. Immunoreactive bands were visualized with Pierce SuperSignal Chemiluminescent Substrates (Thermo Scientific, Waltham, MA) and captured by Molecular Imager ChemiDoc XRS System (Bio-Rad Laboratories, Hercules, CA). All band intensities were quantified using the Un-Scan-it (version 6.0, Silk Scientific, Orem, UT) software and normalized to the expression of β-actin.
Brain Mitochondria Isolation
Rats were euthanized and the brains were rapidly dissected on ice. Cerebellum and brain stem were excluded for mitochondria isolation. Whole brain mitochondria were isolated as previously described (Lam, et al., 2009, Yao, et al., 2009). Briefly, the brain was rapidly minced and homogenized at 4°C in mitochondrial isolation buffer (MIB) (PH 7.4), containing sucrose (320 mM), EDTA (1 mM), Tris-HCl (10mM), and Calbiochem’s Protease Inhibitor Cocktail Set I (AEBSF-HCl 500μM, aprotonin 150 nM, E-64 1μM, EDTA disodium 500μM, leupeptin hemisulfate 1μM). Single-brain homogenates were then centrifuged at 1500g for 5 min. The pellet was resuspended in MIB, rehomogenized, and centrifuged again at 1500g for 5 min. The postnuclear supernatants from both centrifugations were combined, and crude mitochondria were pelleted by centrifugation at 21000g for 10 min. The resulting mitochondrial pellet was resuspended in 15% Percoll made in MIB, layered over a preformed 23%/40% Percoll discontinuous gradient, and centrifuged 31000g for 10 min. The purified mitochondria were collected at the 23%/40% interface and washed with 10 ml MIB by centrifugation at 16700g for 13 min. The loose pellet was collected and transferred to a microcentrifuge tube and washed in MIB by centrifugation at 9000 g for 8 min. The resulting mitochondrial pellet was resuspended in MIB to an approximate concentration of 1mg/ml. The resulting mitochondrial samples were used immediately for respiratory measurements and hydrogen peroxide production. During mitochondrial purification, aliquots were collected for confirmation of mitochondrial purity and integrity following a previously established protocol (Irwin, et al., 2008).
Mitochondrial Respiration
Mitochondrial respiration was measured using the Seahorse XF-96 metabolic analyzer following established protocol (Rogers, et al., 2011). Briefly, protein concentration of purified mitochondria was determined for protein concentration and resuspended into 1X MAS solution (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5mM MgCl2, 2 mM HEPES, 1 mM EGTA and 0.5% (w/v) fatty acid free BSA, pH7.2 at 37°C) 4μg of mitochondrial samples in 25μl 1X MAS buffer were seeded into each assay well. Mitochondrial samples were spun down at 2000g for 10 minutes and then supplemented with 75ul 1X MAS buffer with substrate (glutamate/malate 5 mM). Metabolic flux cartridges were loaded with the following reagents in 1X MAS: Port A: ADP (20 mM); Port B: Oligomycin (30 μg/ml); Port C: FCCP (28 μM); and Port D: Rotenone (16 μM) + Antimycin (70 μM). Injection volumes for all ports were 25μl. Mitochondrial respiration was measured sequentially in a coupled state with substrate present (basal respiration, state 4 respiration), followed by ADP stimulated state 3 respiration after port A injection, (phosphorylating respiration in the presence of ADP and substrates). Injection of oligomycin in port B induced state 40 respiration and the subsequent injection of FCCP in port C induced uncoupler-stimulated respiration state 3u while the final injection of rotenone and antimycin in port D resulted in non-oxidative phosphorylation related residual oxygen consumption OCRresidual. Respiratory control ratio value was calculated as the ratio of oxygen consumption rate (OCR) at state 3 respiration over OCR at state 4 respiration: RCR= OCRstate4/OCRstate3.
H2O2 Production Assay
The rate of hydrogen peroxide production by fresh isolated mitochondria (20 μg) was determined by the Amplex Red Hydrogen Peroxide/Peroxidase Assay kit (Invitrogen) as previously described (Yin, et al., 2012) with the presence of 5 mM glutamate/malate but not ADP (state 4).
Enzyme Activity Assays
Complex I, complex IV and ATP synthase activity were assessed in isolated mitochondria (20 μg) using Rapid Microplate Assay kit for rat Complex I, Complex IV, and ATP synthase Activity respectively (Mitosciences, Eugene, OR) following the manufacturer’s instructions.
Long-Term Potentiation and I/O Curves
Input/output (I/O) responses and long-term potentiation of hippocampal region was determined electrophysiologically as previously described (Sancheti, et al., 2013). Briefly, each animal was decapitated after deep isoflurane anesthesia and slices processed in sucrose-modified artificial cerebrospinal fluid containing: 105 mM sucrose; 62 mM NaCl, 3 mM KCl, 4 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3 and 10 mM glucose. Field EPSPs (fEPSPs) were recorded from stratum radiatum of CA1 using a glass pipette filled with 2M NaCl (yielding a resistance of 2–3 MΩ) in response to orthodromic stimulation (twisted nichrome wires, 50 μm) of the Schaffer collateral-commissural projections in CA1 stratum radiatum. Pulses of 0.1 ms duration were delivered to the stimulating electrode every 20 seconds. The responses were amplified with Axoclamp 2A DC amplifier (Axon Instruments, Foster City, CA), filtered at 6 kHz and digitized at 20 kHz. Data acquisition was controlled by Clampex 9.0 software. Input/output (I/O) curves were generated using stimulus intensities from 100–350 μA in increments of 50 μA. Baseline fEPSP were evoked at 30–50% of maximal fEPSP in 20 sec intervals. LTP was induced at baseline intensity using Theta Burst Stimulation (TBS) consisting of ten trains of five 100Hz stimulation repeated at 5 Hz. Recording continued for at least 30 min following TBS. fEPSP slope magnitude was calculated as the difference between two cursors, separated by 1 ms, and placed on the middle portion of the ascending phase of the fEPSP. Three consecutive responses separated with 20 sec intervals were averaged and presented as a single point to reduce deviations. LTP were expressed as a percentage of the average slope from the baseline recordings. Comparison of theta burst-induced plasticity was performed between groups using repeated measures ANOVA (across all post-theta burst time points). This analysis was followed by a post-hoc t-test performed between groups for the average change in fEPSP amplitude recorded during the final 5 minutes of recording (30 min post theta burst stimulation).
3. RESULTS
The Perimenopause Animal Model (PAM)
9–10 month-old female Sprague-Dawley rats were stratified into groups according to the stage of ovarian senescence that followed the classification of the human perimenopause-menopause transition as per Stages of Reproductive Aging Workshop (STRAW) (Finch, 2014, Harlow, et al., 2012). The endocrine aging groups included regular cyclers (4–5 day cycles), irregular cyclers (5–8 day cycles), and acyclic (no cycling within 9 days) at 9–10 months. 6 month-old regular cycling rats and 16 month-old acyclic rats were included to distinguish endocrine aging from chronological aging (Brinton, 2013) (Fig. 1A). From 6 months to 9–10 months, rats exhibited the greatest breadth of endocrine aging phenotypes (Fig 1B): at 6 months, 82% of females were regular cyclers and 18% were irregular cyclers; at 8–9 months, 60% were regular cyclers and the percentage of irregular cyclers doubled to 37%, with the first appearance of acyclicity in 3%; at 9–10 months, the percent of regular cyclers declined to 38%, the percent of irregular cyclers increased to 50%, and the remaining 12% were acyclic; at 16 months, all rats were acyclic. Increase in body weight was observed between the 6 month- and the 9–10 month-old regular cyclers (p = 0.023) and was constant for all subsequent stages (Fig. 1C). There were no differences in body weight among 9–10 month-old animals at different endocrine stages (p = 0.33). Uterine weight did not differ across the 5 endocrine phenotypes (p = 0.69) and was consistent with all animals being euthanized at estrus or constant estrus (Fig. 1C).
E2 and P4 levels were quantified by LC-MS/MS in serum and cerebral cortex collected at estrus across all groups. The pattern of serum and cortical levels of E2 across all groups differed inasmuch as serum E2 was not correlated with cortex E2 at the same endocrine stage (Fig. 2A–C). While serum E2 levels were highest in the 9–10 month regular and irregular cyclers and declined thereafter (Fig. 2A), the most robust E2 change was a collapse in cortex levels from 6 month to 9–10 month rats with negligible brain E2 observed in acyclic rats (Fig. 2B). In contrast to E2, serum P4 levels were correlated with cortical levels (Fig. 2F). In serum, P4 level was greatest in the 9–10 month irregular cyclers and declined significantly with the transition into 9–10 month acyclic, and remained low in the 16 month acyclic group (Fig. 2D). In cortex, there was a trend towards decreased P4 level from 6 month regular to 9–10 month regular and irregular cyclers, which was statistically significant when animals transitioned into acyclicity at the same age (Fig. 2E).
Figure 2. Steroids levels and the expression of steroid receptors in PAM.

A. Serum levels of 17β-estradiol (E2). B. Cortical levels of E2. C. Correlation of serum- and cortical levels of E2 with r-value and p-value shown. D. Serum levels of progesterone (P4). E. Cortical levels of P4. F. Correlation of serum- and cortical levels of P4 with r-value and p-value shown. G–K. Expression of estrogen- and progesterone receptors in the hippocampus: G. ERα; H. ERβ; I. PR-A; J. PR-B; K. PGRMC1. All samples were collected on estrus day. Data were presented as average ± SEM, *p ≤ 0.05, n = 6.
The impact of chronological and endocrine aging on ovarian hormone receptors in hippocampus was assessed by the protein expressions of the estrogen receptors (ERα and ERβ) in the hippocampus, the progesterone receptors A (PR-A) and B (PR-B), and the progesterone receptor membrane component 1 (PGRMC) (Brinton, 2009, Brinton, et al., 2008, Singh, et al., 2013). ERα expression was unaffected by either chronological or endocrine aging (Fig. 2G, p = 0.22). Conversely, ERβ expression declined in the 9–10 month irregular cyclers (p = 0.05) and rebounded with the onset of acyclicity at the same age (p = 0.004) (Fig. 2H). Compared to 6 month regular cyclers, the 9–10 month irregular cyclers had an increased expression of both PR-A (Fig. 2I, p = 0.001) and PR-B (Fig. 2J, p < 0.001). The major difference in PR-A expression was between the 6 month- and 9–10 month regular cyclers (p < 0.001) whereas that in PR-B expression was between the 9 month regular- and irregular cyclers (p = 0.04) (Fig. 2I, J). The expression of PGRMC1 did not change across groups (Fig. 2K, p = 0.83).
Bioenergetics-, Inflammation- and Alzheimer’s Pathology-Related Gene Expression in the Hippocampus
Gene expression profiles in the hippocampus derived from the PAM rats were determined with emphasis on energy metabolism, inflammation, and Alzheimer’s disease (AD) pathology. The AD-pathology related gene group is a panel related to brain aging and AD that emphasizes targets/pathways implicated in estrogen-mediated protection. Conceptually, estrogen protects the brain against AD in part by (i) reducing β-amyloid production and enhancing its clearance, (ii) protecting against apoptosis, (iii) supporting neuronal function and signaling. This panel assesses these three established estrogen actions. Differences in gene expression were analyzed by ANOVA followed by pairwise t-tests for each transition period. Heatmaps of gene expression clustered by their function were generated (Fig. 3A) and significant differences are summarized in Table 1 (p = 0.001–0.05).
Three sets of comparisons of the gene expression profiles were conducted to distinguish age-related versus endocrine-related changes:
a) Comparison between 6 month- and 9–10 month regular cyclers
This comparison represents the effect of chronological aging prior to the perimenopausal transition (Fig. 3A, Table 1). The 9–10 month regular cyclers showed a significant decline in the expression of genes involved in molecule transport (heat shock 60 kDa protein 1 (HSPD1) and Bcl2/adenovirus E1B interacting protein 1 (Bnip1)); mitochondrial redox enzymes glutaredoxin 2 (Glrx2) and peroxiredoxin 3 (Prx3); the glutamate decarboxylases (Gad1 and Gad2, which generate the neurotransmitter GABA), and a neuronal survival gene (chemokine receptor Cxrcr4) (Table 1). Genes significantly upregulated included those involved in redox-sensitive transcription (nuclear factor erythroid 2-like 2, Nfe2l2), lipid metabolism (hydroxyacyl-CoA dehydrogenase (Hadh), phospholipases C gamma 2 (Plcg2) and D1 (Plcd1)), cholesterol homeostasis and trafficking (apolipoprotein A1 (Apoa1) and ATP-binding cassette sub-family A member7 (Abca7)), inflammation (complement component 4A (C4a)), amyloid processing (N-acylaminoacyl-peptide hydrolase (Apeh)), and apoptosis (caspase 8 (Casp8) and clusterin (Clu)) (Table 1). Consistent with these significant differences, functional group analyses (Fig. 3A) indicated a trend towards decline in genes involved in substrate transport, TCA cycle, electron transfer chain and mitochondrial biogenesis/dynamics and an increase in genes involved in inflammation, apoptosis, and cholesterol homeostasis.
b) Comparison among endocrine status groups in 9–10 month rats
Comparisons among 9–10 month regular cyclers, irregular cyclers, and acyclic animals represent the impact of endocrine status independent of chronological age. Compared to regular cyclers, the irregular cyclers exhibited a significant decline in the expression of genes involved in bioenergetics (ATP synthase F1 complex assembly factor 2 (Atpaf2) and estrogen-related receptor alpha (Esrra, a regulator of mitochondrial biogenesis)), lipid metabolism (phospholipase C, beta 3 (Plcb3)), inflammation (interleukin 1 receptor-like 1 (Il1rl1), the downstream Mapk3 (Erk1) and nuclear factor of kappa light polypeptide gene enhancer in B-Cells 2 (Nfkb2)), and amyloid processing (thimet oligopeptidase (Thop1) and microtubule-associated protein 2 (Map2)) (Table 1). Compared to the 9–10 month irregular cyclers, the acyclic animals exhibited higher expression of genes involved in fatty acid uptake by the mitochondria (carnitine palmitoyltransferase 2 (Cpt2) and phospholipase A2 group 2A (Pla2g2a)), mitochondrial biogenesis and ATP generation (Atpaf2 and estrogen-related receptor β (Esrrb)), inflammation (C-X-C motif chemokine 12 (Cxcl12), Cd40, TNF-alpha-induced protein 2 (Tnfaip2)), cholesterol homeostasis (Apoa1 and steroidogenic acute regulatory (Star)), and amyloid and apoptosis related (Apba3 and Bcl2l1) (Table 1). The expression of APP-Binding, Family B, Member 1 (Apbb1), decreased in acyclic animals (Table 1). Functional group analyses revealed a decline in irregular cyclers relative to regular cyclers in genes involved in glucose transport, glycolysis, mitochondrial energy-transducing systems (TCA cycle, electron transfer chain, oxidative phosphorylation, mitochondrial biogenesis and dynamics), cellular redox homeostasis, inflammation (adhesion, chemokines, interleukins, phospholipases), Aβ clearance and degradation, apoptosis, and synaptic plasticity (Fig. 3A). Functional group analyses between the 9–10 month irregular cyclers and acyclic animals revealed that the deficits in glycolytic genes persisted into the acyclic condition. In contrast, mitochondrial genes were increased with the transition into acyclicity and those involved in synaptic plasticity were partially recovered (Fig. 3A).
c) Comparison between 9–10 month acyclic and 16 month acyclic groups
This transition primarily reflects the impact of chronological aging on gene expression profiles. Compared to the 9–10 month acyclic group, the 16 month acyclic group exhibited lower expression of genes involved in bioenergetics (translocase of inner mitochondrial membrane 23 homolog (Timm23), pyruvate dehydrogenase phosphatase catalytic subunit 1 (Pdp1), NADH dehydrogenase (ubiquinone) iron-sulfur protein 4 and 8 (Ndufs4 and Ndufs8), cytochrome oxidase subunit IV isoform 2 (Cox4i2), and Atpaf2), lipid metabolism (phospholipase A2 group 1B (Pla2g1b) and peroxisome proliferator-activated receptor gamma (Pparg)), amyloid processing (Apba3, and insulin-degrading enzyme (Ide)), apoptosis (Bcl2l1) (Table 1). In contrast, the glycolytic gene phosphofructokinase 1 (Pfk1) and the cell signaling gene Erk1 were significantly increased in the 16 month acyclic group (Table 1). Functional group analyses indicated down-regulation of genes involved in glucose metabolism, mitochondrial biogenesis and dynamics, Aβ clearance/degradation, and apoptosis (Fig. 3A).
Changes in gene expression patterns across all age and endocrine states were quantified by principal component analysis (PCA) for the 5 major functional gene groups; the first principal component (PC1) scores of individual animals were calculated for each gene group. The bioenergetic gene group (encompassing glucose metabolism and mitochondrial function) was the only gene group that showed a significant difference in PC1 score across the 5 animal groups (Fig. 3B, F = 5.04, p < 0.01). The other gene groups (redox, inflammation, lipid metabolism, and AD) did not show statistically significant differences across groups (data not shown). Pair-wise comparison of the PC1 scores of bioenergetic genes showed a significant decline from 6 month regular cyclers to 9–10 month irregular cyclers and from the 9–10 month acyclic to 16 month acyclic. The 9–10 month acyclic group showed a significant increase in bioenergetic PC1 scores compared to the 9–10 month irregular cyclers (Fig. 3B). Moreover, the changes in bioenergetic gene expression profile were independent of other functional group (ie, redox-, inflammation-, lipid metabolism-, and AD-pathology related genes), at the transcription level (Fig. 3C). In contrast, changes in redox-, inflammation- and lipid metabolism-related genes were tightly correlated with each other, indicating that these three functional groups may either be under a common regulatory pathway or share a common intermediating pathway. The changes in AD-pathology related genes were correlated with the inflammatory gene group, indicating that prolonged inflammation may contribute to the AD pathogenic pathway (Fig. 3C).
Network and Pathway Analyses Identified Gene Nodes and Upstream Regulators
Hippocampal gene expression data were further analyzed by the Ingenuity Pathway Analysis to identify networks of genes and their upstream regulators (Table 2). The network analysis revealed known interactions among the genes in the dataset and predicted interactions from the knowledge base and identified nodes within the network. Analyses for upstream regulators identified transcriptional networks predictive of the observed patterns of gene expression.
The network analysis of bioenergetic genes identified key nodes that unified gene changes across both chronological and endocrine aging including insulin, IGF-1, IRS1, PI3K, Akt, CREB, JNK, ERK1/2, AMPK, NFκB, and CD3. Analyses of predictive upstream regulators indicated that differences in gene expression patterns during chronological and endocrine aging was primarily related to the insulin/IGF1-IRS-PI3K/Akt-ERK signaling pathway and the AMPK-PGC1α-NRF1 transcriptional pathway (Table 2). Insulin/IGF1 signaling is primarily involved in regulation of bioenergetic functions whereas the AMP-PGC1α pathway senses energy needs and drives mitochondrial biogenesis.
Key nodes within the inflammatory network include IL1, IL12, CD40, ERK1/2 and toll-like receptors (TLRs) and upstream regulator analyses indicated that differences in patterns of inflammatory gene expression could be ascribed to the IL1/TNF-MAPK-NFκB pathway (Table 2). Key nodes of the Alzheimer’s-related genes included ERK1/2, MEK, CREB, IL1, AP1, Bcl2, BclxL, Bax, Bad, Casp8 and Cyto c. Upstream regulator analyses indicated that differences in Alzheimer’s related gene expression patterns could be attributed to the IGF1-PI3K/Akt-ERK1/2 signaling pathway and the NFκB pathway (Table 2).
Decline in Bioenergetic Enzyme Expression Was Accompanied by Inhibition of the Insulin/IGF1 and AMPK Signaling Pathways in Hippocampus during the Regular-Irregular Transition
Gene expression analyses indicated that changes in the brain bioenergetic profile were among the earliest indicators associated with the perimenopausal transition in the hippocampus; these changes were prominently represented by the difference between 9–10 month regular- and irregular cyclers. The endocrine transition state manifested by irregular cycling thus emerged as a critical period for bioenergetic function. We therefore further characterized the irregular cycling group regarding their bioenergetic and synaptic function using regular cycling rats of the same age as the reference group. Consistent with the gene array data, the expression of glucose transporter 3 (GLUT3, p = 0.03), pyruvate dehydrogenase E1α subunit (PDH-E1α, p = 0.03), and ATPAF2 (p = 0.04) were decreased in the hippocampus of irregular cyclers (Fig. 4A–C). Cytosolic signaling components and transcriptional pathways identified by bioinformatic analyses were assessed in the hippocampus, with results shown in Fig. 4D–J. The insulin/IGF1 signaling cascade from IRS-1 (Fig. 4D; p-IRS1Tyr608/IRS1, p < 0.001), to Akt (Fig. 4E; p-Akt/Akt, p = 0.02) and ERK1/2 (Fig. 4F; p-ERK1, p = 0.009; p-ERK2, p = 0.03) were substantially decreased in the irregular cyclers; this pattern was accompanied by the decline in transcriptional pathways CREB (Fig. 4G; p-CREB, p = 0.005) and AMPK (Fig. 4H; p-AMPK, p < 0.001). The expression of PGC1α did not differ between regular and irregular cycling groups (data not shown). The expression and activation of the master regulator of cellular inflammatory responses, NFκB, was also decreased in the irregular cyclers (Fig. 4I, NFκB, p = 0.02; Fig. 4J, p-IκB/IκB, p = 0.04).
Brain Metabolism Declines during Regular-Irregular Transition
18-FDG-microPET co-registered with microCT imaging revealed a 15% decrease in whole brain glucose uptake in the 9–10 month irregular cyclers relative to regular cyclers (Fig. 5A, B, p = 0.03); this decrease correlated with a decrease in systemic glucose tolerance (Fig. 5C, p = 0.01 for AUC). The glucose-metabolizing machinery in the whole brain (not including cerebellum and brain stem) of 9–10 month irregular cyclers was also substantially diminished in terms of mitochondrial respiratory control ratios (Fig. 5D, p = 0.04) and the activities of mitochondrial complexes IV (Fig. 5E, p = 0.03) and V (Fig. 5F, p = 0.03). Activity of the mitochondrial complex I remained unchanged (data not shown). These metabolic changes were accompanied by a substantial increase in mitochondrial H2O2 production (Fig. 5G, p = 0.03), thus substantiating a compromised mitochondrial energy-redox axis (Yin, et al., 2014).
Decline of Synaptic Plasticity in Irregular Cyclers
Deficits in bioenergetic gene and protein expression, brain metabolism, mitochondrial function and the inactivation of ERK pathway are expected to lead to a decline in synaptic transmission. LTP observed in the hippocampal CA1 region has been considered critical for learning and memory. Analysis of input/output (I/O) responses in the CA1 region indicated that the machinery required for synaptic transmission remained intact as there was no difference in the I/O curves between the regular and irregular cyclers (Fig. 6A, p = 0.13). In contrast, long-term potentiation (LTP) in the CA1 region of irregular cyclers was significantly lower than that of regular cyclers: responses following high frequency stimulation (relative to baseline) were lower in the irregular cyclers (Fig. 6B) (F = 366.2, p < 0.001); the LTP magnitude of regular cyclers was 180.6% EPSP, whereas that of the irregular cyclers was 168.2% EPSP (Fig. 6C, p = 0.007). These data indicate that the machinery necessary to establish synaptic potentials is functional in both regular and irregular cyclers; however, the ability to sustain high frequency synaptic activity was significantly compromised in the irregular cyclers consistent with their deficits in bioenergetic gene expression profile (Fig. 3B), decreased glucose uptake (Fig. 5A, B), and mitochondrial energy-conservation systems and their regulators (Figs. 4, 5).
4. DISCUSSION
Findings from this investigation support the PAM model as a physiologically relevant rodent model of human perimenopause and provide mechanistic insights regarding the transitions within bioenergetic systems inherent in chronological and endocrine aging of the female brain and the associated decline in synaptic plasticity during endocrine aging. In the perimenopausal rodent model, two distinct aging programs were identified: endocrine and chronological. Endocrine aging refers to changes that were coincident with changes in rat cycling phenotype independent of chronological age, as age was held constant at 9–10 months for comparison between regular, irregular and acyclic groups. In contrast, chronological aging refers to those changes that occurred in animals at different ages but the same cycling status (e.g., Regular-6m vs. Regular-9m; Acyclic-9–10m vs. Acyclic-16m).
The PAM in this study is a physiologically-relevant rodent model of the human perimenopausal transition that–from an endocrine perspective– recapitulates key aspects of perimenopausal physiology: the 6 month regular cyclers correspond to STRAW stage 4 of regular cycles and peak fecundability; the 9–10 month irregular cyclers with prolonged cycles correspond to STRAW stage 3b of declining fecundability with increased cycle irregularity; 9–10 month acyclic may correspond to STRAW Stage 2 with increased anovulatory cycles, both hypo- and hyper estrogenic; and the 16 month acyclic rodents correspond to postmenopausal STRAW stage 0 (Finch, 2014). Similar to humans, the PAM exhibited individual variability in both age of onset and rate of perimenopausal transition.
With respect to the number of statistical tests performed, the potential for false positives is of concern. To avoid interpretation system level responses based on a single indicator, we interrogated system level functions across multiple domains including peripheral and neural steroid hormone concentration in tandem with their cognate receptors, gene expression, protein expression, FDG-PET whole brain glucose metabolism, peripheral glucose tolerance, mitochondrial function, and long-term potentiation (synaptic plasticity). The consistency of systems level indicators across multiple systems/platforms support our conclusions and allay concerns regarding false positive findings.
The perimenopausal transition was associated with complex profiles in steroid levels in brain and serum. How the perimenopausal transition affects brain levels of steroid hormones in human and rodents is poorly understood, although decreased brain estrogens are associated with AD in postmenopausal women (Rosario, et al., 2011) and in an AD mouse model (Yue, et al., 2005). Recent evidence indicates good agreement between plasma and brain levels of E2 and P4 across the ovarian cycle in young female rats, although higher than expected brain levels suggest significant brain neurosteroidogenesis (Kato, et al., 2013). However, the data herein indicate that correlations evident in young animals do not generalize to the brain in transition as peripheral levels of steroids were not predictive of brain levels, particularly for E2.
Of note, changes in ERβ expression (Fig. 2H) temporally correlated with those of the bioenergetic gene expression profile during perimenopause (Fig. 3B); these changes may be ascribed to nuclear and/or extra-nuclear (mitochondrial and mitochondria-associated endoplasmic reticulum) ERβ localization (Alvarez-Delgado, et al., 2010, Milner, et al., 2005, Yang, et al., 2004). It is noteworthy that the ERβ selective agonist, DPN, as well as an ERβ targeting phytoSERM (selective estrogen receptor modulator) formulation potentiates brain mitochondrial function (Irwin, et al., 2012, Yao, et al., 2013). These data indicate a regulatory role of ERβ in brain bioenergetics such that the endocrine-associated alterations in ERβ expression may contribute to the changes in brain metabolism. Unaltered ERα level during perimenopause is consistent with previous reports that E2 treatment had minimal effect on ERα expression in the female brains (Adams, et al., 2002, Naugle, et al., 2014, Wang, et al., 2010).
Our hypothesis-driven gene array focused on 5 functional domains revealed perimenopausal transformations that occur in female brain aging. These functional domains were selected to test the hypothesis that the perimenopausal transition is composed of bioenergetic, redox, lipid, and inflammatory responses that could be predictive of AD pathology. Overall, the outcomes of the gene array analyses indicate that: (a) the transition from regular to irregular cycling exhibited profound changes in gene expression; (b) the bioenergetic category was the most affected cluster (Fig. 3B); (c) the expression profiles of gene groups including redox-, inflammation- and lipid metabolism genes are tightly correlated and appear to be independent of that of the bioenergetic genes (Fig. 3C). The impact of endocrine aging can be sustained in vitro as evidenced by astrocytes cultured from irregular cycling rats exhibited impaired glial neurotrophic activity relative to same aged rats with regular cycles (Arimoto, et al., 2013).
The endocrine transition manifested by irregular cycling thus emerged as a critical period for bioenergetic function characterized by a significant decline in the expression of genes required for glucose metabolism and mitochondrial function.
Further analyses of the gene expression data defined the metabolic phenotypes of the perimenopausal transition. The sequence from 6 month regular cyclers to 9–10 month regular cyclers to 9–10 month irregular cyclers represents an interconnected system of responses towards decreased expression of bioenergetic genes; this is likely due to an additive effect of chronological aging (6 month- to 9–10 month regular cyclers) and endocrine aging (9–10 month regular- to irregular cyclers), in which the latter proceeded on the background of the former and together induced a significant decrease in gene expression. Of note, the similarities in the bioenergetic gene profiles of the 9–10 month irregular cyclers and 16 month acyclic rats are consistent with the pattern of gene expression observed in earlier studies of aged male rats, aged female non-human primates and of AD patients (Blalock, et al., 2011, Blalock, et al., 2004, Kadish, et al., 2009). The reduced expression of glucose-dependent metabolic genes in female rats transitioning from 6 month regular to 9 month regular and irregular cycling stages was also seen in male rats starting at 3–6 month-of-age (Kadish, et al., 2009, Rowe, et al., 2007). Secondly, the transition from 9–10 month irregular to 9–10 month acyclic indicated that the bioenergetic phenotype that characterized the irregular cyclers was stage-limited: the acyclic animals exhibited a recovery of components of mitochondria oxidative phosphorylation capacity and fatty acid metabolism to a level comparable to age-matched regular cyclers whereas other components of the same system such as glycolysis were not recovered. This switch to fatty acid/ketone metabolism is consistent with the upregulation of fatty acid metabolism genes shown in male rats at 9–12 months of age (Kadish, et al., 2009), although in male rats, the shift starts as early as 3–6 months and there was not a stage when both the glucose and fatty acid metabolic systems were downregulated as seen in irregular cycling females. Lastly, the transition from 9–10 month acyclic to 16 month acyclic is a direct chronological aging effect that is independent of the endocrine status and resulted in an overall decline in genes involved in bioenergetics.
The endocrine transition from 9–10 month regular to irregular cyclers was associated with a decrease in inflammatory genes (Nfkb2, Il1rl1, and Erk1); these changes were confirmed at the protein level (Fig. 4). It is intriguing that endocrine aging does not activate pro-inflammatory genes in hippocampus, although further investigation is warranted given that the inflammatory response is usually region and cell-type-specific in the brain. Notably, an upregulation of genes involved in inflammatory and complement pathways from 6 month to 9 month regular cycling females was also observed in male Fischer 344 rats between 6- and 9 month of age (Kadish, et al., 2009); the continued activation of inflammatory genes when male rats aged further to 12 months was only seen in the acyclic but not the irregular cycling female rats in our study.
Analysis of genes related to amyloid processing showed an early age-related increase in Clu, a stress-induced chaperone linked to AD pathogenesis by numerous pathways (Yu and Tan, 2012). In contrast, the perimenopausal transition is associated with a down-regulation of Thop1 that protects against Aβ toxicity (Pollio, et al., 2008), and more advanced chronological aging was marked by reduced expression of Ide. Although we did not observe significant changes in Ide expression across the different stages of endocrine aging, our prior work demonstrated that ovariectomy-induced hormone depletion lowers mRNA and protein levels of Ide, effects that are reversed by E2 treatment (Jayaraman, et al., 2012, Zhao, et al., 2011).
Because endocrine and chronological aging coincide during middle age, we expect that these gene changes not only have additive downstream functional consequences but common upstream regulators. The insulin/IGF1 signaling was identified as the upstream pathway by both aging programs, as confirmed by the diminished activation of IRS-1 and Akt in irregular cyclers (Fig. 4D, E). This is also consistent with previous studies showing impaired insulin/IGF1 signaling during brain aging and in AD animal models and patients (Craft, et al., 2013, Sancheti, et al., 2013, Yin, et al., 2013). The identification of the AMPK-PGC1α-NRF1 signaling/transcriptional pathway –involved in regulation of mitochondrial biogenesis– was confirmed by decreased AMPK activation in 9–10 irregular cyclers (Fig. 4H), consistent with reports of AMPK inhibition in rodent aging- and AD brains (Jiang, et al., 2013, Pedros, et al., 2014).
The deficits in bioenergetic gene expression during endocrine aging were further corroborated by a panel of functional indicators, including diminished brain glucose uptake, decreased mitochondrial respiration and compromised bioenergetic enzyme activity (Figs. 4,5). This phenotype of bioenergetic decline is consistent with studies demonstrating that loss of estrogen regulation is associated with systematic deficits in brain metabolism, ranging from glucose uptake to aerobic glycolysis to mitochondrial function and morphology (Hara, et al., 2014, Nilsen, et al., 2007, Yao, et al., 2010, Yao, et al., 2012, Yao, et al., 2009). It was also shown that the decline in brain glucose uptake is associated with a decrease in neuronal glucose transporter GLUT3 (Fig. 4A) as reported in a previous study on female brain aging (Ding, et al., 2013). Further, the estrogen-controlled glucose metabolism appears to be replaced by ketone body metabolism when the rats transitioned from irregular cycling to acyclic, as suggested by increased expression of fatty acid metabolism-related gene (Cpt2) and mitochondria-related genes (Atpaf2 and Esrra) but unaltered glycolytic genes. This could represent an adaptive response of the midlife brain to enhance fatty acid metabolism and to utilize ketone bodies as alternative substrates due to declined glucose metabolism (Kadish, et al., 2009); the adaptive response was transient and lost with further chronological aging to 16 months.
The energy-dependent synaptic transmission is susceptible to mitochondria dysfunction (Cheng, et al., 2010), and the expression of genes required for synaptic function decreases in AD (Blalock, et al., 2011). Compromised LTP in the irregular cyclers (Fig. 6) can thus be correlated to the decline in glucose metabolism and mitochondrial function (Fig. 5). Moreover, one of the major transcriptional pathways implicated in hippocampal LTP, the CREB-CRE transcriptional pathway, is co-regulated by ERK and activated by E2 (Brinton, 2008, Brinton, 2009, Foy, et al., 1999, Thomas and Huganir, 2004). Therefore, the decreased activation (phosphorylation) of both ERK1/2 and CREB (Fig. 4) in the irregular cyclers can also contribute to the impairment of LTP. The deficit in LTP of irregular cyclers is consistent with previous findings that female Long-Evans rats with irregular cycling exhibited poorer cognitive performance compared to rats with regular cycles (Paris, et al., 2011). In rhesus monkeys, peri/post-menopausal animals exhibited significant impairment in recognition memory which was paralleled by a decline in perforated synapses (Hara, et al., 2012). These changes were relative to premenopausal performance and synaptic morphology and were independent of chronological age (Hara, et al., 2012). Collectively, these data are consistent with the hypothesis that mechanisms underlying select cognitive functions are part of an endocrine aging program rather than a chronological aging process (Hara, et al., 2012).
Our preclinical findings are consistent with clinical observations that menopausal women exhibit decreased brain glucose uptake in the posterior cingulate and prefrontal cortex of the brain (Rasgon, et al., 2005). In humans, cognitive deficits during the perimenopausal transition are increasingly documented (Maki and Resnick, 2001) and interestingly, these deficits were found to be improved following the transition into menopause (Greendale, et al., 2009). These clinical findings are consistent with outcomes of gene and protein expression, metabolic function and LTP, which indicate that cognitive deficits evident during the perimenopause (irregular cycling) are associated with the bioenergetic deficits in glucose transport and metabolism whereas the recovery of cognitive function upon menopause would be mechanistically linked to the utilization of ketone bodies as an adaptive and transitory response to impaired glucose metabolism. Bioenergetic deficits in the irregular cyclers were paralleled by systemic glucose intolerance, which confirmed earlier studies of the correlation between dysregulated peripheral metabolism (encompassing insulin resistance and dyslipidemia) and brain mitochondrial function (Blalock, et al., 2010, Rettberg, et al., 2014). Consistent with these preclinical studies, findings from the Early versus Late Intervention Trial with Estradiol (ELITE) study indicate that a subgroup of menopausal women with dysregulated glucose metabolism exhibit poorer cognitive performance (Rettberg, et al., 2013). Systemic glucose intolerance indicates an early stage of compromised insulin signaling, corroborating the bioinformatic prediction of decreased insulin/IGF-1 signaling at this stage. Together with clinical data documenting deficits in glucose metabolism in prodromal AD (Ewers, et al., 2014, Mosconi, 2013, Protas, et al., 2013), our findings suggest that the bioenergetic transition of the perimenopause could have implications for development of a metabolic phenotype of risk for AD and may be relevant to the disproportionate burden of AD pathology in females and their two-fold greater lifetime risk of AD (Alzheimer’s, 2014, Barnes, et al., 2005). Although these data do not directly address the contributions of the perimenopause transition to the development of AD, they provide important and novel insights into understanding an early bioenergetic transition in brain that is associated with profile of risk. Specifically, these data indicate that endocrine aging is not a linear trajectory of overall decline but a selective adaptive systems biology response that results in a shift in the bioenergetic system of the brain that includes substrate utilization and mitochondrial function. This bioenergetic phenotype is reminiscent of early stage of AD (Yao, et al., 2011), and suggest that bioenergetic changes that occur early in the female aging brain contributes to an increased risk for AD in women. Collectively these findings suggest a critical window for earliest detection of bioenergetic vulnerability of brain for later life development of Alzheimer’s disease in those most affected, postmenopausal women.
Supplementary Material
Table S1. List of Gene Symbols and Names in the Low Density Array
HIGHLIGHTS.
Bioenergetic transformations characterize the perimenopausal aging transition of female brain.
A rat model recapitulating fundamental characteristics of human perimenopause was developed.
Gene expression analyses indicated two distinct aging programs: chronological and endocrine.
A critical period emerged during the endocrine transition from regular to irregular cycling characterized by declines in bioenergetic gene expression, glucose uptake, mitochondrial respiration, and synaptic plasticity.
The onset of acyclicity was characterized by a rise in the expression of genes required for fatty acid metabolism, mitochondrial function and inflammation.
Acknowledgments
This research was supported by the National Institute on Aging P01AG026572 to RDB. We thank Jennifer Mao and Eric Hernandez for their exceptional technical assistance.
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MM, Fink SE, Shah RA, Janssen WG, Hayashi S, Milner TA, McEwen BS, Morrison JH. Estrogen and aging affect the subcellular distribution of estrogen receptor-alpha in the hippocampus of female rats. J Neurosci. 2002;22(9):3608–14. doi: 10.1523/JNEUROSCI.22-09-03608.2002. 20026275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MM, Morrison JH, Gore AC. N-methyl-D-aspartate receptor mRNA levels change during reproductive senescence in the hippocampus of female rats. Exp Neurol. 2001;170(1):171–9. doi: 10.1006/exnr.2001.7687. [DOI] [PubMed] [Google Scholar]
- Alvarez-Delgado C, Mendoza-Rodriguez CA, Picazo O, Cerbon M. Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V. Exp Gerontol. 2010;45(7–8):580–5. doi: 10.1016/j.exger.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s A. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10(2):e47–92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Arimoto JM, Wong A, Rozovsky I, Lin SW, Morgan TE, Finch CE. Age increase of estrogen receptor-alpha (ERalpha) in cortical astrocytes impairs neurotrophic support in male and female rats. Endocrinology. 2013;154(6):2101–13. doi: 10.1210/en.2012-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005;62(6):685–91. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- Basilevsky A. Statistical Factor Analysis and Related Methods, Theory and Applications. John Wiley & Sons; New York, NY: 1994. [Google Scholar]
- Blalock EM, Buechel HM, Popovic J, Geddes JW, Landfield PW. Microarray analyses of laser-captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer’s disease. J Chem Neuroanat. 2011;42(2):118–26. doi: 10.1016/j.jchemneu.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101(7):2173–8. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Grondin R, Chen KC, Thibault O, Thibault V, Pandya JD, Dowling A, Zhang Z, Sullivan P, Porter NM, Landfield PW. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci. 2010;30(17):6058–71. doi: 10.1523/JNEUROSCI.3956-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008;31(10):529–37. doi: 10.1016/j.tins.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD. Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol Sci. 2009;30(4):212–22. doi: 10.1016/j.tips.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD. Neuroendocrinology of Aging. In: Fillit HM, Rockwood K, Woodhouse K, editors. Brockelhurst’s Textbook of Geriatric Medicine and Gerontology. Saunders Elsevier; 2010. pp. 163–71. [Google Scholar]
- Brinton RD. Minireview: translational animal models of human menopause: challenges and emerging opportunities. Endocrinology. 2012;153(8):3571–8. doi: 10.1210/en.2012-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD. Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol. 2013;9(4):241–50. doi: 10.1038/nrendo.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton RD, Thompson RF, Foy MR, Baudry M, Wang J, Finch CE, Morgan TE, Pike CJ, Mack WJ, Stanczyk FZ, Nilsen J. Progesterone receptors: form and function in brain. Front Neuroendocrinol. 2008;29(2):313–39. doi: 10.1016/j.yfrne.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler L, Santoro N. The reproductive endocrinology of the menopausal transition. Steroids. 2011;76(7):627–35. doi: 10.1016/j.steroids.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso D, Pesaresi M, Abbiati F, Calabrese D, Giatti S, Garcia-Segura LM, Melcangi RC. Comparison of plasma and cerebrospinal fluid levels of neuroactive steroids with their brain, spinal cord and peripheral nerve levels in male and female rats. Psychoneuroendocrinology. 2013;38(10):2278–90. doi: 10.1016/j.psyneuen.2013.04.016. [DOI] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN neuro. 2010;2(5):e00045. doi: 10.1042/AN20100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft S, Cholerton B, Baker LD. Insulin and Alzheimer’s disease: untangling the web. J Alzheimers Dis. 2013;33(Suppl 1):S26–75. doi: 10.3233/JAD-2012-129042. [DOI] [PubMed] [Google Scholar]
- Ding F, Yao J, Rettberg JR, Chen S, Brinton RD. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One. 2013;8(11):e79977. doi: 10.1371/journal.pone.0079977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JL, Wise PM. The role of the brain in female reproductive aging. Mol Cell Endocrinol. 2009;299(1):32–8. doi: 10.1016/j.mce.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Brendel M, Rizk-Jackson A, Rominger A, Bartenstein P, Schuff N, Weiner MW Alzheimer’s Disease Neuroimaging I. Reduced FDG-PET brain metabolism and executive function predict clinical progression in elderly healthy subjects. NeuroImage Clinical. 2014;4:45–52. doi: 10.1016/j.nicl.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch CE. The menopause and aging, a comparative perspective. J Steroid Biochem Mol Biol. 2014;142:132–41. doi: 10.1016/j.jsbmb.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch CE, Felicio LS, Mobbs CV, Nelson JF. Ovarian and steroidal influences on neuroendocrine aging processes in female rodents. Endocr Rev. 1984;5(4):467–97. doi: 10.1210/edrv-5-4-467. [DOI] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81(2):925–9. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Genazzani AR, Bernardi F, Pluchino N, Begliuomini S, Lenzi E, Casarosa E, Luisi M. Endocrinology of menopausal transition and its brain implications. CNS spectrums. 2005;10(6):449–57. doi: 10.1017/s1092852900023142. [DOI] [PubMed] [Google Scholar]
- Gore AC, Oung T, Yung S, Flagg RA, Woller MJ. Neuroendocrine mechanisms for reproductive senescence in the female rat: gonadotropin-releasing hormone neurons. Endocrine. 2000;13(3):315–23. doi: 10.1385/ENDO:13:3:315. [DOI] [PubMed] [Google Scholar]
- Greendale GA, Huang MH, Wight RG, Seeman T, Luetters C, Avis NE, Johnston J, Karlamangla AS. Effects of the menopause transition and hormone use on cognitive performance in midlife women. Neurology. 2009;72(21):1850–7. doi: 10.1212/WNL.0b013e3181a71193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greendale GA, Wight RG, Huang MH, Avis N, Gold EB, Joffe H, Seeman T, Vuge M, Karlamangla AS. Menopause-associated symptoms and cognitive performance: results from the study of women’s health across the nation. Am J Epidemiol. 2010;171(11):1214–24. doi: 10.1093/aje/kwq067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Park CS, Janssen WG, Roberts MT, Morrison JH, Rapp PR. Synaptic correlates of memory and menopause in the hippocampal dentate gyrus in rhesus monkeys. Neurobiol Aging. 2012;33(2):421.e17–28. doi: 10.1016/j.neurobiolaging.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci U S A. 2014;111(1):486–91. doi: 10.1073/pnas.1311310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow SD, Gass M, Hall JE, Lobo R, Maki P, Rebar RW, Sherman S, Sluss PM, de Villiers TJ, Group SC. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. J Clin Endocrinol Metab. 2012;97(4):1159–68. doi: 10.1210/jc.2011-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin RW, Yao J, Hamilton RT, Cadenas E, Brinton RD, Nilsen J. Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology. 2008;149(6):3167–75. doi: 10.1210/en.2007-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin RW, Yao J, To J, Hamilton RT, Cadenas E, Brinton RD. Selective oestrogen receptor modulators differentially potentiate brain mitochondrial function. J Neuroendocrinol. 2012;24(1):236–48. doi: 10.1111/j.1365-2826.2011.02251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman A, Carroll JC, Morgan TE, Lin S, Zhao L, Arimoto JM, Murphy MP, Beckett TL, Finch CE, Brinton RD, Pike CJ. 17beta-estradiol and progesterone regulate expression of beta-amyloid clearance factors in primary neuron cultures and female rat brain. Endocrinology. 2012;153(11):5467–79. doi: 10.1210/en.2012-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Yin F, Yao J, Brinton RD, Cadenas E. Lipoic acid restores age-associated impairment of brain energy metabolism through the modulation of Akt/JNK signaling and PGC1a transcriptional pathway. Aging Cell. 2013;12:1021–31. doi: 10.1111/acel.12127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I, Thibault O, Blalock EM, Chen KC, Gant JC, Porter NM, Landfield PW. Hippocampal and cognitive aging across the lifespan: a bioenergetic shift precedes and increased cholesterol trafficking parallels memory impairment. J Neurosci. 2009;29(6):1805–16. doi: 10.1523/JNEUROSCI.4599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Hojo Y, Higo S, Komatsuzaki Y, Murakami G, Yoshino H, Uebayashi M, Kawato S. Female hippocampal estrogens have a significant correlation with cyclic fluctuation of hippocampal spines. Frontiers in neural circuits. 2013;7:149. doi: 10.3389/fncir.2013.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermath BA, Gore AC. Neuroendocrine control of the transition to reproductive senescence: lessons learned from the female rodent model. Neuroendocrinology. 2012;96(1):1–12. doi: 10.1159/000335994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam PY, Yin F, Hamilton RT, Boveris A, Cadenas E. Elevated neuronal nitric oxide synthase expression during ageing and mitochondrial energy production. Free Radic Res. 2009;43(5):431–9. doi: 10.1080/10715760902849813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki PM, Drogos LL, Rubin LH, Banuvar S, Shulman LP, Geller SE. Objective hot flashes are negatively related to verbal memory performance in midlife women. Menopause. 2008;15(5):848–56. doi: 10.1097/gme.0b013e31816d815e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki PM, Resnick SM. Effects of estrogen on patterns of brain activity at rest and during cognitive activity: a review of neuroimaging studies. Neuroimage. 2001;14(4):789–801. doi: 10.1006/nimg.2001.0887. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Akama KT, Spencer-Segal JL, Milner TA, Waters EM. Estrogen effects on the brain: actions beyond the hypothalamus via novel mechanisms. Behav Neurosci. 2012;126(1):4–16. doi: 10.1037/a0026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491(2):81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Brinton RD, Schmidt PJ, Gore AC. Estrogen, menopause, and the aging brain: how basic neuroscience can inform hormone therapy in women. J Neurosci. 2006;26(41):10332–48. doi: 10.1523/JNEUROSCI.3369-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clinical and translational imaging : reviews in nuclear medicine and molecular imaging. 2013;1(4) doi: 10.1007/s40336-013-0026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugle MM, Nguyen LT, Merceron TK, Filardo E, Janssen WG, Morrison JH, Rapp PR, Gore AC. G-protein coupled estrogen receptor, estrogen receptor alpha, and progesterone receptor immunohistochemistry in the hypothalamus of aging female rhesus macaques given long-term estradiol treatment. Journal of experimental zoology Part A, Ecological genetics and physiology. 2014;321(7):399–414. doi: 10.1002/jez.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson HD. Menopause. Lancet. 2008;371(9614):760–70. doi: 10.1016/S0140-6736(08)60346-3. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Irwin RW, Gallaher TK, Brinton RD. Estradiol in vivo regulation of brain mitochondrial proteome. J Neurosci. 2007;27(51):14069–77. doi: 10.1523/JNEUROSCI.4391-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, Walf AA, Frye CA. II. Cognitive performance of middle-aged female rats is influenced by capacity to metabolize progesterone in the prefrontal cortex and hippocampus. Brain Res. 2011;1379:149–63. doi: 10.1016/j.brainres.2010.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedros I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, Auladell C, Pallas M, Vazquez-Carrera M, Casadesus G, Folch J, Camins A. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(9):1556–66. doi: 10.1016/j.bbadis.2014.05.025. [DOI] [PubMed] [Google Scholar]
- Pollio G, Hoozemans JJ, Andersen CA, Roncarati R, Rosi MC, van Haastert ES, Seredenina T, Diamanti D, Gotta S, Fiorentini A, Magnoni L, Raggiaschi R, Rozemuller AJ, Casamenti F, Caricasole A, Terstappen GC. Increased expression of the oligopeptidase THOP1 is a neuroprotective response to Abeta toxicity. Neurobiol Dis. 2008;31(1):145–58. doi: 10.1016/j.nbd.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Protas HD, Chen K, Langbaum JB, Fleisher AS, Alexander GE, Lee W, Bandy D, de Leon MJ, Mosconi L, Buckley S, Truran-Sacrey D, Schuff N, Weiner MW, Caselli RJ, Reiman EM. Posterior cingulate glucose metabolism, hippocampal glucose metabolism, and hippocampal volume in cognitively normal, late-middle-aged persons at 3 levels of genetic risk for Alzheimer disease. JAMA neurology. 2013;70(3):320–5. doi: 10.1001/2013.jamaneurol.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasgon NL, Silverman D, Siddarth P, Miller K, Ercoli LM, Elman S, Lavretsky H, Huang SC, Phelps ME, Small GW. Estrogen use and brain metabolic change in postmenopausal women. Neurobiol Aging. 2005;26(2):229–35. doi: 10.1016/j.neurobiolaging.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Rettberg JR, Dang H, Hodis H, Stanczyk F, Brinton RD, Mack WJ. Development of metabolic and physiological biomarker profiles for early detection of women with an at-risk for Alzheimer’s disease phenotype. Society for Neuroscience Annual Meeting; San Diego, CA. 2013. [Google Scholar]
- Rettberg JR, Yao J, Brinton RD. Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol. 2014;35(1):8–30. doi: 10.1016/j.yfrne.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6(7):e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Chang L, Head EH, Stanczyk FZ, Pike CJ. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiol Aging. 2011;32(4):604–13. doi: 10.1016/j.neurobiolaging.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe WB, Blalock EM, Chen KC, Kadish I, Wang D, Barrett JE, Thibault O, Porter NM, Rose GM, Landfield PW. Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. J Neurosci. 2007;27(12):3098–110. doi: 10.1523/JNEUROSCI.4163-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancheti H, Akopian G, Yin F, Brinton RD, Walsh JP, Cadenas E. Age-dependent modulation of synaptic plasticity and insulin mimetic effect of lipoic acid on a mouse model of Alzheimer’s disease. PLoS One. 2013;8(7):e69830. doi: 10.1371/journal.pone.0069830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochim Biophys Acta. 2010;1800(10):1113–20. doi: 10.1016/j.bbagen.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Su C, Ng S. Non-genomic mechanisms of progesterone action in the brain. Front Neurosci. 2013;7:159. doi: 10.3389/fnins.2013.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5(3):173–83. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Wang AC, Hara Y, Janssen WG, Rapp PR, Morrison JH. Synaptic estrogen receptor-alpha levels in prefrontal cortex in female rhesus monkeys and their correlation with cognitive performance. J Neurosci. 2010;30(38):12770–6. doi: 10.1523/JNEUROSCI.3192-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley CS. Acute effects of estrogen on neuronal physiology. Annu Rev Pharmacol Toxicol. 2007;47:657–80. doi: 10.1146/annurev.pharmtox.47.120505.105219. [DOI] [PubMed] [Google Scholar]
- Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci U S A. 2004;101(12):4130–5. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Hamilton RT, Cadenas E, Brinton RD. Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim Biophys Acta. 2010;1800:1121–6. doi: 10.1016/j.bbagen.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Irwin R, Chen S, Hamilton R, Cadenas E, Brinton RD. Ovarian hormone loss induces bioenergetic deficits and mitochondrial beta-amyloid. Neurobiology of Aging. 2012;33(8):1507–21. doi: 10.1016/j.neurobiolaging.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106(34):14670–5. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD. Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol Aspects Med. 2011;32(4–6):247–57. doi: 10.1016/j.mam.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Zhao L, Mao Z, Chen S, Wong KC, To J, Brinton RD. Potentiation of brain mitochondrial function by S-equol and R/S-equol estrogen receptor beta-selective phytoSERM treatments. Brain Res. 2013;1514:128–41. doi: 10.1016/j.brainres.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Boveris A, Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal. 2014;20(2):353–71. doi: 10.1089/ars.2012.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Jiang T, Cadenas E. Metabolic triad in brain aging: mitochondria, insulin/IGF-1 signalling, and JNK signalling. Biochem Soc Trans. 2013;41:101–5. doi: 10.1042/BST20120260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Sancheti H, Cadenas E. Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim Biophys Acta. 2012;1817(3):401–9. doi: 10.1016/j.bbabio.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JT, Tan L. The role of clusterin in Alzheimer’s disease: pathways, pathogenesis, and therapy. Mol Neurobiol. 2012;45(2):314–26. doi: 10.1007/s12035-012-8237-1. [DOI] [PubMed] [Google Scholar]
- Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci U S A. 2005;102(52):19198–203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Morgan TE, Mao Z, Lin S, Cadenas E, Finch CE, Pike CJ, Mack WJ, Brinton RD. Continuous versus cyclic progesterone exposure differentially regulates hippocampal gene expression and functional profiles. PLoS One. 2012;7(2):e31267. doi: 10.1371/journal.pone.0031267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Yao J, Mao Z, Chen S, Wang Y, Brinton RD. 17beta-Estradiol regulates insulin-degrading enzyme expression via an ERbeta/PI3-K pathway in hippocampus: relevance to Alzheimer’s prevention. Neurobiol Aging. 2011;32(11):1949–63. doi: 10.1016/j.neurobiolaging.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Gene Symbols and Names in the Low Density Array