

Abstract
Unnatural amino acids with bioorthogonal reactive groups have the potential to provide a rapid and specific mechanism for covalently inhibiting a protein of interest. Here, we use unnatural mutagenesis to insert an azide (Z) into the target protein at positions such that a “click” reaction with an alkyne modulator (X) will alter protein function. This bioorthogonal reactive pair can engender specificity of X for the Z-containing protein even if the target is otherwise identical to another protein, allowing for rapid target validation in living cells. We demonstrate our method using inhibition of the E. coli enzyme aminoacyl transferase by both active site occlusion and allosteric mechanisms. This “clickable magic bullet” strategy should be generally applicable to protein inhibition, within the limits of unnatural amino acid mutagenesis.
Keywords: chemical genetics, bioorthogonal chemistry, unnatural amino acids, covalent inhibitor, drug discovery
Most pharmaceuticals are designed to function as molecular entities acting on specific biological targets, what Erlich referred to as “magic bullets” (MBs).[1] A key early event in the development of any pharmaceutical is target-validation: confirmation that modulation of a particular gene or protein will improve a disease phenotype.[2] In modern drug discovery efforts, validating a target often begins with genetic deletion of the protein product. This can be accomplished either with RNA interference (RNAi) or the breeding of knock out animal models.[3] However, there are limitations to these two technologies, and in many cases greater understanding of biological pathways can be attained with small molecules of high target specificity, even if they do not ultimately become drugs.[4] The field of Chemical Genetics encompasses an array of strategies to link genotypes and phenotypes using small molecules in model systems.[5] Here, we present a method designed to attain small molecule specificity rapidly and simply using a combination of genetic manipulation and molecular design.
Our strategy is simple: use unnatural amino acid mutagenesis to insert bioorthogonal functional groups (Z in Figure 1) into a “magic bullet” target (MBT) protein at positions such that reaction with a small molecule partner bearing a complementary reactive group (X in Figure 1) will alter protein function. In this work, we will use azidophenylalanine (Azf) as the Z group and dibenzocyclooctynes (DBCOs) as X groups, taking advantage of the strain-promoted cycloaddition of Azf and DBCO to rapidly and specifically attach X to the MBT protein.[6] We note that our general strategy is equally applicable to other bioorthogonal “click” chemistry reactive pairs such as cycloalkene/tetrazine.[7] Protein modulation will be specific, provided that the reactions are truly chemoselective and only the MBT protein contains the targeting group Z. The position of the Z targeting moiety can be easily scanned throughout the protein sequence to identify positions for which the MBT protein has wild type (WT) activity prior to reaction with an X molecule. These MBT mutants will be taken forward to studies in which they are reacted with X molecules and the phenotype of the modified protein is evaluated.
Figure 1.
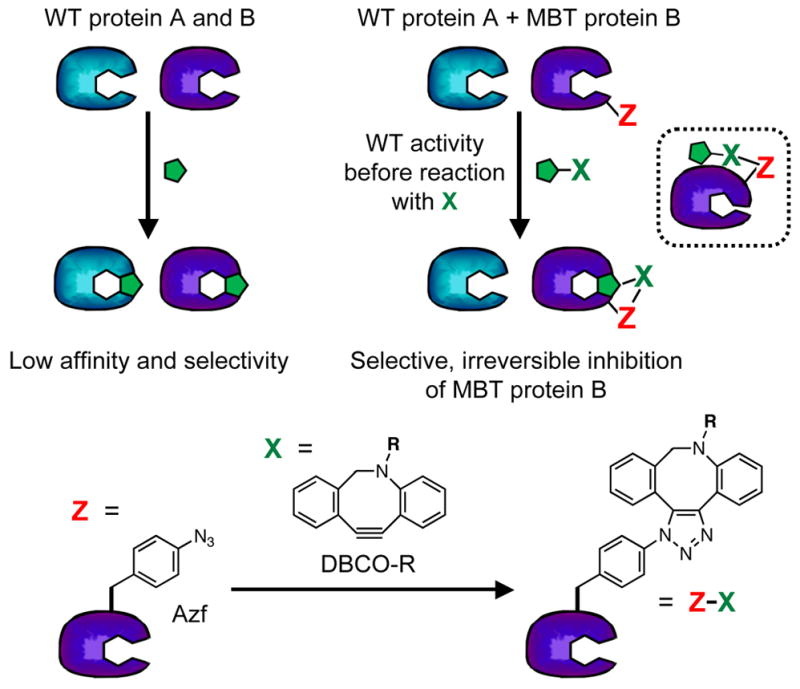
Clickable Magic Bullet Strategy. Top: Replacing WT protein B with MBT protein B enables selective inhibition by reaction of Z with X. Inset: Allosteric inhibition through ZX reaction. Bottom: Cycloaddition of azidophenylalanine (Azf, Z) with dibenzocyclooctyne derivative (DBCO-R, X).
The two previous Chemical Genetics experiments most relevant to our work are the “bump-and-hole” strategy employed by Schreiber, Shokat, and others; and the tethering approach of Erlanson and Wells.[8] In the “bump-and-hole” method, a bump must be added to the small molecule in a way that precisely matches a hole generated in the binding pocket of the target enzyme.[9] To be implemented well, this strategy generally requires prior structural knowledge and a deep binding pocket like an active site. Tethering uses reaction with an exogenous Cys thiol to covalently attach small molecules to the surface of proteins.[8a, 10] These molecules can act as allosteric modulators, so they do not need to reside in active sites.[11] Moreover, the Cys thiol can be scanned across the surface of a protein in a screening protocol, so that careful, structure-based design is not necessary. However, since many proteins contain Cys, specificity derived from tethering is not easily retained in lysates or in vivo. Our method shares many features with these two strategies, but the ability to scan Z through a protein sequence should make it much more broadly applicable than the bump-and-hole strategy and the chemoselectivity of “click” reactions should allow it to be applied in vivo, unlike the tethering strategy. Strategies similar to our own using tetracysteine motifs and bisarsenical compounds have been described, but these methods have not been broadly applied.[12] Finally, it should be noted that unnatural amino acids, including bioorthogonal reactive handles, have been used extensively to probe protein structure and function, particularly in membrane protein work by Sakmar, Wang, and Dougherty.[13] However, none of these previous studies made use of the reactive handle as a general tool for perturbing function as we do here.
To incorporate Azf into the MBT proteins site-specifically, we can employ unnatural amino acid mutagenesis techniques developed by Schultz and coworkers.[14] These methods allow one to insert an unnatural amino acid by using a “21st” aminoacyl tRNA synthetase (aaRS) which charges a tRNACUA that recognizes the UAG stop codon. Two plasmids are transformed into E. coli: pAzfRS (previously referred to as pDULE2Azf), encoding Azf aaRS and tRNACUA, and a plasmid encoding the MBT protein with the UAG mutation.[15] After growth in the presence of Azf, the MBT protein is either purified for in vitro studies, or reacted with DBCO reagents in vivo.
To show proof-of-concept, we have used the MB strategy to inhibit the E. coli aminoacyl transferase (AaT) protein.[16] This protein functions in the N-end rule pathway by tagging proteins bearing N-terminal Lys or Arg with Leu, Phe, or Met using aminoacyl tRNA as a substrate.[17] Addition of the hydrophobic amino acid constitutes a recognition motif for ClpS, which targets the tagged protein for degradation by ClpAP. We and others have previously shown that AaT can accept a variety of hydrophobic unnatural amino acids, which can be used to report on AaT activity.[18]
Using existing AaT crystal structures, we chose six locations for incorporation of Azf.[19] These included three positions right in the active site, and three positions near the exterior of AaT that were not expected to directly alter activity. In all cases, we mutated aromatic amino acids because we expected replacement of these amino acids with Azf to be minimally perturbing to protein function prior to reaction with Azf. We expressed and purified His-tagged versions of each AaT mutant and assessed the ability to react them with X molecules using DBCO rhodamine derivatives (DBCO545 or DBCOTMR, Supporting Information, Fig. S1). MALDI MS and PAGE gel data showed that for those positions taken forward to additional study, labeling was quantitative within the limits of MS detection. (Supporting Information, Table S1 and Fig. S5) Next, we tested the activity of these Azf mutants prior to reaction with DBCO545. Five mutants – Z47, Z59, Z68, Z81, and Z135 – were found to have activities within 4-fold of WT. The positions of these amino acids are shown in Figure 2. All five mutants were taken forward to inhibition assays. While it is surprising that Azf replacement of nearly isosteric Phe at position 81 should cause such a large change in activity, this is consistent with previous reports of the sensitivity of AaT to mutation.[19a] The reduction in activity on mutation of Y42 is less surprising as this makes a hydrogen bond with the Lys or Arg of the peptide substrate.[19b]
Figure 2.

Analysis of AaT Inhibition by DBCO545 Reaction. Top: Transferase activity assay uses tRNA aminoacylated in situ with Phe (for detection by HPLC). AaT activity is evaluated before and after reaction of Azf mutants with DBCO545. Middle: Structures of seven AaT Azf mutants showing the position of adenosine donor (gray), peptide product (black), and Azf substitution sites. Structures with aminoacyl adenosine (PDB ID: 2Z3K, orange) or peptide product (PDB ID: 2Z3N, cream) are overlayed.[19b] Bottom: Activity before (dark colors) and after (light colors) reaction with DBCO545 evaluated based on conversion of LysAlaAcm in an HPLC assay (see Supporting Information).
All five mutants were inhibited by DBCO545 treatment. The levels of inhibition that we observed varied from 52% for Z135 (relative to unmodified protein) to 98% for Z47. It is perhaps not surprising that Z47 was most inhibited, since the mutation occurred in the active site. Inhibition presumably occurs by simply occluding access to the active site. Z59 modification would also block the active site. Inhibition of Z68, Z81, or Z135, on the other hand, probably occurs through an allosteric mechanism. Using crystal structures of either the acyl nucleotide-bound or peptide product-bound states of AaT, we made simple computational models of the DBCO adducts.[19b] (Supporting Information, Fig. S9) The DBCO545 and DBCOTMR adducts should not be able to reach the active site in either state of the enzyme. In WT AaT, W135 occurs at a crystallographic dimer interface, and some weak dimerization has been observed in gel analysis of purified AaT. However, our analysis of WT AaT indicates that activity varies linearly with concentration. (Supporting Information, Table S2) Therefore, we believe that AaT is monomeric in the concentration range used, and that Z68, Z81, and Z135 inhibition occurs through allostery. It should be noted that we observed some reactivity between WT AaT and the DBCO reagent, presumably at one or more of the 6 Cys residues. Blocking with iodoacetamide can be used to eliminate this reaction (Supporting Information, Fig. S6), but this strategy is not viable in cells. Moreover, the differences in inhibition seen for the Azf mutants give us confidence that high levels of inhibition are only achieved upon DBCO reaction with a precisely placed Azf residue.
We also wished to determine whether inhibition was in fact dose-dependent. The cycloaddition is essentially irreversible, therefore we must carry out the reaction under conditions where the DBCO inhibitor is present at much higher concentrations than the enzyme so that pseudo-first-order kinetic analysis can be applied.[20] Timepoints must also be selected so that the irreversible reaction has not been allowed to reach completion. Under the conditions of our assay (1 μM AaT, 30 min), DBCO545 has an apparent IC50 of 8.5 μM for the Z47 mutant. (Supporting Information, Fig. S8) While a more rigorous analysis would be required to determine a true IC50, this is sufficient to show that inhibition is dose-dependent, provided that the assay time is comparable to the half-life for the DBCO545 cycloaddition (hours).
Initially, we used DBCO545 as an inhibitor because we could assess the levels of protein modification using the chromophore. However, modeling studies implied that for Z47, the rhodamine moiety was unnecessary since the DBCO moiety occupied the binding pocket and rhodamine extended into solution (Supporting Information, Fig. S9). Therefore, we also tested inhibition of the DBCO amine (DBCOA) alone and saw 99% inhibition, similar to that obtained with DBCO545. For the Z68 and Z135 mutants, DBCOA actually gave higher levels of inhibition than DBCO545, 87% and 94%, respectively (Supporting Information, Table S3). If necessary, the DBCO moiety can also be functionalized with other groups (R in Figure 1), so that a variety of inhibitors can be made. In the cell studies below, we used a fluorescent R group in order to confirm reaction with AaT in lysates and in cells.
Next, we wished to demonstrate that our method could be used to inhibit AaT selectively in complex mixtures. Therefore, we carried out labeling reactions using a reporter protein derived from α-synuclein (αS) in lysates of TS351G E. coli cells, which lack endogenous AaT activity.[17b] Here, we determined the activity of AaT based on its ability to transfer Azf to the reporter protein from Azf-acylated tRNA generated in situ by a mutant aaRS. Transfer of Azf was then imaged using a copper-catalyzed “click” reaction to append a fluorescein derivative. Note: Although azide-alkyne chemistry is being used for both inhibition and the AaT assay, no cross-reactivity of the click reagents is expected because of the order of addition. To make appropriate calculations of inhibition levels, we carried out control experiments in which we either withheld ATP (necessary for acylation of the tRNA with Azf) from the transfer reaction or added DMSO vehicle only. Since no Azf should be transferred in the “-ATP” controls, this control allowed us to determine that the level of non-specific fluorescein binding was very low. Therefore, we calculated inhibition by comparing the levels of fluorescence in DBCO545 treated lanes to those treated with DMSO. We chose three mutants for further study: Z47, Z68, Z135. Z47 showed almost complete inhibition by active site occlusion, and Z68 and Z135 showed the highest levels of presumed allosteric modulation. In all cases, we observed inhibition levels comparable to those observed with the small reporter peptide in purified reactions: > 99% for Z47, 66% for Z68, and 73% for Z135. (Supporting Information, Fig. S12) This shows that we can effectively inhibit AaT in the presence of the other proteins in the TS351G lysates using 200 μM DBCO545.
Finally, in order to demonstrate that we can inhibit AaT mutants in living cells, we reacted DBCOTMR (which we expected to be more cell permeable than DBCO545 or DBCOA) with AaT in intact E. coli cells, and then analyzed activity in crude lysates as described above. Our protocol is outlined in Figure 3. We transformed BL21(DE3) cells with pAzfRS and plasmids encoding the AaT mutants. After growth in the presence of Azf and IPTG, the cells were pelleted, washed repeatedly to remove unincorporated Azf, and then treated with a DBCOTMR solution or DMSO vehicle. The cells were washed, resuspended, and lysed. The levels of AaT activity were analyzed by Azf transfer and reaction with fluorescein alkyne as described above. Note that Azf is used both to introduce the reactive handle for inhibition in vivo and then to determine AaT activity. However, because of the order of our steps, no crosstalk is expected. Note also that BL21(DE3) cells were used here since we observed little endogenous AaT activity in these cells as compared to overexpressed AaT (data not shown) and we obtained low yields of unnatural amino acid protein in TS351G cells.
Figure 3.

In Vivo Assay Protocol. Cells are transformed with an AzfRS plasmid and a WT AaT plasmid or a mutant AaT (e.g. Z47) plasmid and subjected to the following conditions: a) AaT production is induced with the addition of Azf and IPTG; b) Cells are pelleted, washed, and reacted with DBCOTMR or DMSO vehicle; c) Cells are grown again after resuspension; d) Cells are lysed and the lysates subjected to an Azf transfer assay using the αS reporter protein, E. coli AzfRS, and AaT; e) Azf transfer levels determined by Cu-catalyzed reaction with a fluorescein alkyne probe. WT control experiments determine the level of transfer activity that should be expected from uninhibited AaT as well as the levels of non-specific binding of the DBCOTMR reagent. Vehicle control experiments determine the level of transfer activity that should be expected from uninhibited AaT mutants (i.e., What is the effect of the Azf mutation alone?). Inhibition by the DBCOTMR reaction is determined by comparing the levels of labeling in the final step to the levels seen in the vehicle control.
As one can see in Figure 4, we successfully inhibited all three Azf-tagged AaT proteins through in vivo treatment with DBCOTMR. Since we used a tetramethylrhodamine label on our inhibitor and a fluorescein probe for AaT activity, the red channel reports on reaction of the Azf residue in AaT with the DBCOTMR inhibitor, while the green channel reports on the levels of Azf reporter transferred in the activity assay. Once again, trials in which ATP was withheld from the transfer reaction served as controls for non-specific fluorescein binding. Although we observed labelling of other proteins (possibly endogenous AaT substrates) with the fluorescein alkyne probe, analysis of the αS bands allowed us to assay AaT activity.
Figure 4.
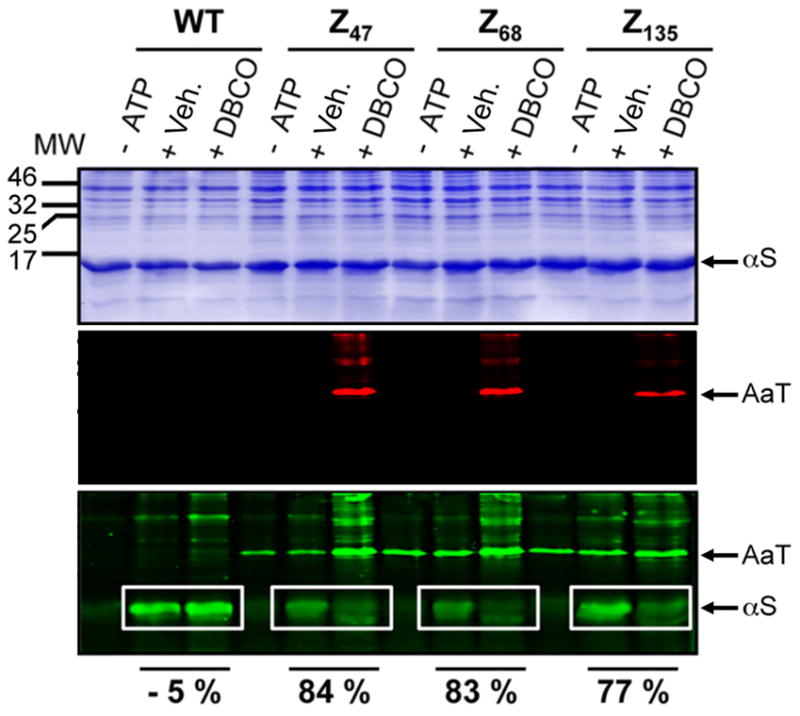
AaT Inhibition in Cells. BL21(DE3) cells expressing AaT WT and mutants were subjected to one of three conditions, + DBCO: AaT-expressing cells were treated with DBCOTMR in DMSO, then lysates of these cells were used to transfer Azf to the αS reporter protein; + Veh.: AaT-expressing cells were treated with DMSO only, then lysates of these cells were used to transfer Azf to the αS reporter protein; − ATP: AaT-expressing cells were treated with DMSO only, then lysates of these cells were used in a mock transfer assay in which ATP was withheld to prevent Azf transfer. In all cases, Azf transfer was detected with a fluorescein alkyne. Transfer yields calculated from the fluorescence intensity of αS bands were used to determine percent inhibition (relative to the corresponding + Veh. lane in the white box). All three images depict the same gel, stained with Coomassie dye (top), imaged using 532 nm excitation (middle), or using 473 nm excitation (bottom). MW: Molecular weight markers (kDa). The protocols for the + DBCO and + Veh. experiments are shown in Figure 3.
As one can see by comparing the “+ DBCO” and “+ Veh.” lanes, significant inhibition of AaT activity was observed in all cases: 84 % for Z47, 83 % for Z68, and 77 % for Z135. This shows that we can effectively inhibit AaT by dosing DBCOTMR in intact E. coli cells. Moreover, we can track the selectivity of the DBCO reagent by analyzing the red fluorescence channel. We see relatively low levels of non-specific binding for WT AaT, indicating that the bands seen for the Azf mutants may be due to off-target incorporation of Azf or association of AaT with other proteins. One feature of note is the green bands observed in the bottom gel, corresponding to fluorescein labeling of unreacted Azf in AaT mutants that had not been treated with DBCOTMR. This shows that Azf was present in the active site of these AaT samples (“+ DBCO” lanes appear to have higher background due to emission from TMR excited with 473 nm light). Thus, we see specific, DBCO-dependent inhibition of Azf mutants of AaT at levels that are comparable to or greater than those seen for the purified enzyme. However, it must be noted that there are still steps to be taken to make this a truly in vivo technology. While the DBCO reaction was carried out in cells, the assay was carried out in lysates, as is common for AaT, which lacks a clear activity-dependent phenotype (even the TS351G AaT-deletion cell line does not exhibit an obvious phenotype).[13, 17b, 21] A discussion of potential improvements to this method is given below.
We have shown, for the first time, that unnatural amino acids and bioorthogonal chemical reactions can be used as part of a general strategy to connect genotype to phenotype. We believe that our method could be a valuable complement to RNAi for target validation. While it is more complex to implement than RNAi, it offers validation in the context of a dosable small molecule. Since many microRNAs (miRNAs) act catalytically, controlling dosage can be hard.[22] A few additional advantages of our method are worth noting. Firstly, RNAi prevents translation of the protein of interest, which may lead to phenotypes resulting from the absence of all of its protein interactions rather than inhibition of one specific activity. These changes would be hard to replicate with a small molecule or protein therapeutic, which typically bind to one face of the target protein. Second, although we have only found examples of inhibition in this initial study of AaT, the method can conceivably also be used to find activators. RNAi can only reduce the activity of its protein target, not enhance it.
While we are excited about the many possible applications of this method, several considerations are worth noting:
Washout of unincorporated Azf is essential. Azf can act as a buffer, consuming DBCO-R and preventing modification of Azf in the MBT protein.
The ability to remove the endogenous copy of the protein, at least from cell lysates, is essential. This can be done with a chromosomal deletion as in this work, or using miRNA in appropriate cell lines. Judicious codon usage can prevent the miRNA from targeting the MBT as well as the WT protein.[23] However, deleting the endogenous copy must either have a very mild phenotype (as AaT deletion does here), or the knockdown of the endogenous gene must be carefully timed with expression of the replacement MBT protein.
The UAG stop codon, though rare (~ 5 %), is present in many natural proteins (~ 300).[24] The presence of AzfRS could lead many of these codons to be read through, yielding proteins with aberrant activity and/or susceptibility to inhibition by reaction with DBCO reagents. These proteins may be misfolded and degraded, but they may also contribute to the observed phenotype.
If the tRNACUA does not efficiently deliver the unnatural amino acid at the UAG stop codon, high levels of truncated protein may be synthesized, resulting in a dominant negative phenotype that would mask the effect of any DBCO-based modulation of the MBT protein.
Click chemistry is imperfect and the reactions used here may not be optimal for this application. Although the fact that we are able to achieve nearly 100 % inactivation of the Z47, Z68, and Z135 mutants implies that the cycloaddition goes to completion with respect to the protein, this will not be true for many residues in many proteins.[21c] Also, the cyclooctyne probes used here are relatively hydrophobic and may be prone to non-specific binding. Newer, faster bioorthogonal reactions are continually being developed, and a great variety of R groups and X/Z reactive partners are possible, so there are many options, including more polar reactive groups or meta-stable linkages (e.g. oxime) to make reversible inhibitors.[7a]
The ultimate goal of connecting genotype to phenotype requires that the phenotype be either intrinsically-discernable or that a reporter construct be used to assess the phenotype. In the latter case, this could mean transformation of a third plasmid and maintenance of the cell line in the presence of three antibiotics, which could introduce intolerable levels of stress to the cells (as we observed in attempts to perform an in vivo assay of AaT activity with a GFP reporter). Chromosomal insertion of the synthetase and tRNA or the use of multi-gene vectors like pET-DUET would reduce this burden and may be necessary for phenotypic characterization.
In spite of these potential complications, the ability to rapidly generate a protein that is selectively-targetable by simple small molecules could have tremendous value in the drug discovery process. The fact that we can do this through a single amino acid substitution is a significant advantage relative to other similar technologies that require a greater perturbation to confer selectivity.[25] Although we use an enzyme with a crystal structure in this proof-of-principle experiment, this is not required. One could scan the unnatural mutation through a protein sequence as one does in an Ala scan and simply rely on an activity assay to determine which positions are non-perturbing prior to reaction with the small molecule. The demonstration of our method in this manuscript is limited to a single-celled model organism, E. coli. However, unnatural amino acid mutagenesis has been demonstrated in mammalian cells as well as multicellular organisms such as C. elegans and Drosophila melanogaster.[26] Moreover, alternate coding strategies have been employed leading to efficient incorporation at four base codons, which may address issues 3 and 4 above.[26a, 27] Schultz, Church, Isaacs, and coworkers have also generated a strain of E. coli with the TAG stop codons replaced by TAA codons.[28] We will explore these applications and continue to improve our method. We have begun to apply our method to other systems including membrane transporters implicated in drug resistance and aggregating proteins like α-synuclein. These types of gain-of-function systems may be more ammenable to our methods, as deletion of the endogenous copy of the protein may be less important and there are clear phenotypes. The generality of our approach should make it equally applicable to the discovery of competitive inhibitors, allosteric modulators, or modulators of protein interactions with DNA or other proteins.
Experimental Section
Wild type AaT expression and purification
His10-tagged E. coli AaT was expressed from the pAaT-WT plasmid (previously referred to as pEG6) in E. coli BL21-Gold (DE3) cells using a procedure modified from Graciet et al.[17b] Briefly, E. coli were grown in a primary culture of 4 mL LB at 37 °C to OD600 of 0.5 which was diluted into a secondary culture of 500 mL LB and grown to OD600 of 0.6 – 0.8. AaT expression was induced using 0.1 mM isopropyl β-D-thiogalactoside (IPTG) and cells were grown at 25 °C for 16 hours. Cells were pelleted at 6, 000 rpm using a GS3 rotor and Sorvall RC-5 centrifuge. Cell pellets were resuspended in the Ni-NTA binding buffer (50 mM Tris, 10 mM imidazole, and 300 mM KCl, pH 8.0) that included protease inhibitor cocktail, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 units/mL DNAse1–Grade II. Following resuspension, the cells were lysed using sonication. Soluble proteins were collected via centrifugation at 13, 200 rpm for 15 minutes. Collected soluble protein was gently shaken for 1 hour on ice with Ni-NTA Superflow (Qiagen). Protein was purified by rinsing with Ni-NTA binding buffer and then washing with Ni-NTA wash buffer (50 mM Tris, 50 mM imidazole, and 300 mM KCl, pH 8.0). The proteins were eluted with elution buffer (50 mM Tris, 250 mM imidazole, and 300 mM KCl, pH 8.0). Fractions containing E. coli AaT were dialyzed overnight in transferase buffer consisteing of 50 mM HEPES, 30 % glycerol, 120 mM (NH4)2SO4, 1 μM β-mercaptoethanol (β-ME), pH 8.0. The dialyzed enzymes were stored at −80 °C.
Unnatural amino acid incorporation into AaT
Site-directed mutagenesis was used to insert TAG stop codons into the pAaT-WT plasmid in order to introduce an unnatural amino acid at position XX (pAaT-ZXX). The sequences and mutagenic primers are shown in Figure S3 (Supporting Information). BL21-Gold (DE3) cells were transformed with the pAaT-ZXX plasmid and a pAzfRS (previously referred to as pDULE2Azf) plasmid containing an M. jannaschii mutant tRNA synthetase and tRNACUA pair. Cells were selected using ampicillin (100 mg/L) and streptomycin (100 mg/L). Azf was incorporated into AaT as described above with the addition of 1.0 mM Azf (solubilized by dropwise addition of 1 M NaOH) to the growth media. When working with Azf, cultures and purified proteins were kept shielded from light.
AaT cyclooctyne labeling
Ni-NTA-purified AaT and AaT-ZXX mutants (0.20 mg/mL) were incubated with 200 μM (final concentration) DBCOA, DBCO545, or DBCOTMR from a DMSO stock for 2 hours at 37 °C. As a vehicle control, AaT and AaT-ZXX mutants were also treated with an equal volume dimethyl sulfoxide. Excess DBCO reagent was removed by dialysis twice in transferase buffer (50 mM Tris, 30 % glycerol, 120 mM (NH4)2SO4, 1 μM β-ME, pH 8.0). Labeled proteins were then analyzed by PAGE gel or trypsin digest and MALDI MS as described above and stored at −80 °C.
Activity of AaT using LysAlaAcm ligation assay
The activity of DMSO- and DBCOTMR-treated purified AaT and AaT-ZXX mutants were assayed by transfer of Phe to the N-terminus of a LysAlaAcm peptide (Bachem; Torrance, CA, USA) as previously described.[18b] Each ligation reaction, 125 μL total volume, contained the following reagents: Phe (1 mM), yeast Phe tRNA (12.5 μg), Adenosine-5′-triphosphate (1.25 mM, ATP), yeast Phe tRNA aminoacyl synthetase (yPheRS, 2.5 μg), AaT (1 μM), and LysAlaAcm (100 μM) in the AaT Ligation Buffer (50 mm HEPES pH 8.0, 150 mM KCl, 10 mM MgCl2, 50 mM Tris, pH 8.0; 30 % glycerol; 120 mM ammonium sulfate). The reaction mixtures were incubated at 37 °C for 4 hours and quenched with 1 % acetic acid. Proteins were removed from the peptide using four volumes of acetone and cooled at −20 °C for 1 hour. The reactions were centrifuged at 13, 200 rpm at 4 °C for 20 minutes to separate the peptide from precipitated protein. The supernatant was transferred to a 1.5 mL centrifuge tube and the acetone was allowed to evaporate overnight at room temperature. Residual acetone was removed by drying in a Speedvac (Savant, Thermo Scientific, Fisher Inc.) for 30 minutes. The resulting reaction volume was dissolved in a total volume of 1 mL of 1:9 0.1 % TFA in acetonitrile:water and analyzed by HPLC (see Supporting Information for details) to determine ligation efficiency by integration of separated PheLysAlaAcm and LysAlaAcm peak intensities monitored at 325 nm.
In vivo AaT labeling activity assay
E. coli BL21(DE3) cells were grown with pAzfRS and either pAaT-WT or pAaT-ZXX plasmids using ampicillin and streptomycin. A 4 mL primary culture was used to inoculate a 100 mL secondary culture. Secondary cultures were grown to an OD600 of 0.6 and induced with 1.0 mM Azf and 0.1 mM IPTG. Protein expression was induced for 16 hours at 25 °C. Cell pellets were harvested at 6,000 rpm for 10 min using a GS3 rotor and Sorvall RC-5 centrifuge. Cell pellets were washed three times with wash buffer (50 mM Tris and 300 mM KCl, pH 8.0) to remove excess Azf. Washed cell pellets were resuspended in 2 mL wash buffer and incubated with either DMSO or 500 μM DBCOTMR at 37 °C for 2 h with 250 rpm shaking. Cells were then washed three times with wash buffer containing 1% DMSO and then washed once with wash buffer. Cell pellets were then resuspended in 1 mL wash buffer and sonicated (one minute on then one minute off) three times. Lysed cells were stored on ice until further use. αS2-140K2 (31.3 μg) was modified with Azf (0.85 mM) in the E. coli lysate using E. coli total tRNA (125 μg), ATP (2.1 mM), and E. coli AzfRS (2.5 μg) in a reaction volume of 73.2 μL in modified AaT buffer (50 mm HEPES pH 8.0, 150 mM KCl, 10 mM MgCl2) and AaT (1.25 μg). ATP was omitted and replaced with an equivalent amount of MilliQ water for negative controls. The reaction mixture was incubated at 37 °C for 2 hours. The crude reaction (12.5 μL) was then labeled with fluorescein-alkyne (33 μM) using THPTA (3.546 mM), CuSO4 (681 μM), and sodium ascorbate (5.4 mM) at 37 °C for 1 hour. The reaction was boiled with gel loading dye LDS (5 μL, Pierce) for ten minutes at 95 °C and analyzed by SDS-PAGE. Fluorescence images were obtained with a Typhoon FLA 7000. Fluorescein was imaged using 473 nm excitation and an Y520 filter. TMR was imaged using 532 nm excitation and an O580 filter. Images were collected using a 100 μM pixel size. Total protein content was visualized using Coomassie Brilliant Blue.
Supplementary Material
Acknowledgments
This work was supported by funding from the University of Pennsylvania and the Searle Scholars Program (10-SSP-214 to EJP). We thank Rakesh Kohli for assistance with HRMS (NIH RR-023444) and MALDI-MS (supported by NSF MRI-0820996). We also thank Dr. Christopher Lanci of the UPenn Biological Chemistry Resource Center for assistance with gel imager. JBW thanks George Furst and Jun Guo for assistance with NMR data acquisition (instrument supported by NIH RR-022442).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Strebhardt K, Ullrich A. Nat Rev Cancer. 2008;8:473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 2.a) Lindsay MA. Nat Rev Drug Disc. 2003;2:831–838. doi: 10.1038/nrd1202. [DOI] [PubMed] [Google Scholar]; b) Sams-Dodd F. Drug Disc Today. 2005;10:139–147. doi: 10.1016/S1359-6446(04)03316-1. [DOI] [PubMed] [Google Scholar]
- 3.a) Zambrowicz BP, Sands AT. Nat Rev Drug Disc. 2003;2:38–51. doi: 10.1038/nrd987. [DOI] [PubMed] [Google Scholar]; b) Sharpless NE, DePinho RA. Nat Rev Drug Disc. 2006;5:741–754. doi: 10.1038/nrd2110. [DOI] [PubMed] [Google Scholar]; c) Iorns E, Lord CJ, Turner N, Ashworth A. Nat Rev Drug Disc. 2007;6:556–568. doi: 10.1038/nrd2355. [DOI] [PubMed] [Google Scholar]; d) Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 4.a) Bunnage ME, Chekler ELP, Jones LH. Nat Chem Biol. 2013;9:195–199. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]; b) Lee J, Bogyo M. Curr Opin Chem Biol. 2013;17:118–126. doi: 10.1016/j.cbpa.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Stockwell BR. Nat Rev Genetics. 2000;1:116–125. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walsh DP, Chang YT. Chem Rev. 2006;106:2476–2530. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]
- 6.a) Jewett JC, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Debets MF, van Berkel SS, Schoffelen S, Rutjes F, van Hest JCM, van Delft FL. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]; c) Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG. J Am Chem Soc. 2002;124:9026–9027. doi: 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- 7.a) Sletten EM, Bertozzi CR. Angew Chem, Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seitchik JL, Peeler JC, Taylor MT, Blackman ML, Rhoads TW, Cooley RB, Refakis C, Fox JM, Mehl RA. J Am Chem Soc. 2012;134:2898–2901. doi: 10.1021/ja2109745. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Devaraj NK, Weissleder R, Hilderbrand SA. Bioconj Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Erlanson DA, Wells JA, Braisted AC. Ann Rev Biophys Biomol Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]; b) Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]; c) Bishop A, Buzko O, Heyeck-Dumas S, Jung I, Kraybill B, Liu Y, Shah K, Ulrich S, Witucki L, Yang F, Zhang C, Shokat KM. Ann Rev Biophys Biomol Struct. 2000;29:577–606. doi: 10.1146/annurev.biophys.29.1.577. [DOI] [PubMed] [Google Scholar]
- 9.a) Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]; b) Bishop AC, Buzko O, Shokat KM. Trends Cell Biol. 2001;11:167–172. doi: 10.1016/s0962-8924(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 10.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Proc Natl Acad Sci USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardy JA, Wells JA. Curr Opin Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 12.a) Zhang XY, Bishop AC. J Am Chem Soc. 2007;129:3812–3813. doi: 10.1021/ja069098t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Erster O, Eisenstein M, Liscovitch M. Nat Methods. 2007;4:393–395. doi: 10.1038/nmeth1046. [DOI] [PubMed] [Google Scholar]
- 13.a) Ebhardt HA, Xu Z, Fung AW, Fahlman RP. Anal Chem. 2009;81:1937–1943. doi: 10.1021/ac802423d. [DOI] [PubMed] [Google Scholar]; b) Ninnis RL, Spall SK, Talbo GH, Truscott KN, Dougan DA. EMBO J. 2009;28:1732–1744. doi: 10.1038/emboj.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Schultz PG. Angew Chem, Int Ed. 2005;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 15.Miyake-Stoner SJ, Miller AM, Hammill JT, Peeler JC, Hess KR, Mehl RA, Brewer SH. Biochemistry. 2009;48:5953–5962. doi: 10.1021/bi900426d. [DOI] [PubMed] [Google Scholar]
- 16.Leibowitz MJ, Soffer RL. Biochem Biophys Res Commun. 1969;36:47–53. doi: 10.1016/0006-291x(69)90647-0. [DOI] [PubMed] [Google Scholar]
- 17.a) Abramochkin G, Shrader TE. J Biol Chem. 1996;271:22901–22907. doi: 10.1074/jbc.271.37.22901. [DOI] [PubMed] [Google Scholar]; b) Graciet E, Hu RG, Piatkov K, Rhee JH, Schwarz EM, Varshavsky A. Proc Natl Acad Sci USA. 2006;103:3078–3083. doi: 10.1073/pnas.0511224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Taki M, Kuno A, Matoba S, Kobayashi Y, Futami J, Murakami H, Suga H, Taira K, Hasegawa T, Sisido M. Chem Bio Chem. 2006;7:1676–1679. doi: 10.1002/cbic.200600181. [DOI] [PubMed] [Google Scholar]; b) Wagner AM, Fegley MW, Warner JB, Grindley CLJ, Marotta NP, Petersson EJ. J Am Chem Soc. 2011;133:15139–15147. doi: 10.1021/ja2055098. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Connor RE, Piatkov K, Varshavsky A, Tirrell DA. Chem Bio Chem. 2008;9:366–369. doi: 10.1002/cbic.200700605. [DOI] [PubMed] [Google Scholar]
- 19.a) Suto K, Shimizu Y, Watanabe K, Ueda T, Fukai S, Nureki O, Tomita K. EMBO J. 2006;25:5942–5950. doi: 10.1038/sj.emboj.7601433. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Watanabe K, Toh Y, Suto K, Shimizu Y, Oka N, Wada T, Tomita K. Nature. 2007;449:867–871. doi: 10.1038/nature06167. [DOI] [PubMed] [Google Scholar]
- 20.Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 21.a) Shrader TE, Tobias JW, Varshavsky A. J Bacteriol. 1993;175:4364–4374. doi: 10.1128/jb.175.14.4364-4374.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hoskins JR, Yanagihara K, Mizuuchi K, Wickner S. Proc Natl Acad Sci USA. 2002;99:11037–11042. doi: 10.1073/pnas.172378899. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Humbard MA, Surkov S, De Donatis GM, Jenkins LM, Maurizi MR. J Biol Chem. 2013;288:28913–28924. doi: 10.1074/jbc.M113.492108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valencia-Sanchez MA, Liu JD, Hannon GJ, Parker R. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 23.Arduini RM, Li ZF, Rapoza A, Gronke R, Hess DM, Wen DY, Miatkowski K, Coots C, Kaffashan A, Viseux N, Delaney J, Domon B, Young CN, Boynton R, Chen LL, Chen LQ, Betzenhauser M, Miller S, Gill A, Pepinsky RB, Hochman PS, Baker DP. Protein Exp Purification. 2004;34:229–242. doi: 10.1016/j.pep.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Blattner FR, Plunkett G, Bloch A, Perna NT, Burland V, Riley M, ColladoVides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 25.a) Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Goreshnik I, Maly DJ. J Am Chem Soc. 2010;132:938–940. doi: 10.1021/ja907886v. [DOI] [PubMed] [Google Scholar]
- 26.a) Parrish AR, She XY, Xiang Z, Coin I, Shen ZX, Briggs SP, Dillin A, Wang L. ACS Chem Biol. 2012;7:1292–1302. doi: 10.1021/cb200542j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bianco A, Townsley FM, Greiss S, Lang K, Chin JW. Nat Chem Biol. 2012;8:748–750. doi: 10.1038/nchembio.1043. [DOI] [PubMed] [Google Scholar]; c) Greiss S, Chin JW. J Am Chem Soc. 2011;133:14196–14199. doi: 10.1021/ja2054034. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sakamoto K, Hayashi A, Sakamoto A, Kiga D, Nakayama H, Soma A, Kobayashi T, Kitabatake M, Takio K, Saito K, Shirouzu M, Hirao I, Yokoyama S. Nucleic Acids Res. 2002;30:4692–4699. doi: 10.1093/nar/gkf589. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Liu WS, Brock A, Chen S, Chen SB, Schultz PG. Nat Methods. 2007;4:239–244. doi: 10.1038/nmeth1016. [DOI] [PubMed] [Google Scholar]
- 27.a) Neumann H, Wang KH, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]; b) Magliery TJ, Anderson JC, Schultz PG. J Mol Biol. 2001;307:755–769. doi: 10.1006/jmbi.2001.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hohsaka T, Ashizuka Y, Taira H, Murakami H, Sisido M. Biochemistry. 2001;40:11060–11064. doi: 10.1021/bi0108204. [DOI] [PubMed] [Google Scholar]
- 28.a) Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, Tolonen AC, Gianoulis TA, Goodman DB, Reppas NB, Emig CJ, Bang D, Hwang SJ, Jewett MC, Jacobson JM, Church GM. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, Isaacs FJ. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.