

Abstract
Background
Quercetinases are metal-dependent dioxygenases of the cupin superfamily. While fungal quercetinases are copper proteins, recombinant Streptomyces quercetinase (QueD) was previously described to be capable of incorporating Ni2+ and some other divalent metal ions. This raises the questions of which factors determine metal selection, and which metal ion is physiologically relevant.
Results
Metal occupancies of heterologously produced QueD proteins followed the order Ni > Co > Fe > Mn. Iron, in contrast to the other metals, does not support catalytic activity. QueD isolated from the wild-type Streptomyces sp. strain FLA contained mainly nickel and zinc. In vitro synthesis of QueD in a cell-free transcription-translation system yielded catalytically active protein when Ni2+ was present, and comparison of the circular dichroism spectra of in vitro produced proteins suggested that Ni2+ ions support correct folding. Replacement of individual amino acids of the 3His/1Glu metal binding motif by alanine drastically reduced or abolished quercetinase activity and affected its structural integrity. Only substitution of the glutamate ligand (E76) by histidine resulted in Ni- and Co-QueD variants that retained the native fold and showed residual catalytic activity.
Conclusions
Heterologous formation of catalytically active, native QueD holoenzyme requires Ni2+, Co2+ or Mn2+, i.e., metal ions that prefer an octahedral coordination geometry, and an intact 3His/1Glu motif or a 4His environment of the metal. The observed metal occupancies suggest that metal incorporation into QueD is governed by the relative stability of the resulting metal complexes, rather than by metal abundance. Ni2+ most likely is the physiologically relevant cofactor of QueD of Streptomyces sp. FLA.
Electronic supplementary material
The online version of this article (doi:10.1186/s12858-015-0039-4) contains supplementary material, which is available to authorized users.
Keywords: Dioxygenase, Flavonol, Metalloprotein, Nickel, Coordination geometry, Cell-free protein synthesis
Background
Quercetinase (flavonol dioxygenase, EC 1.13.11.24) catalyzes the 2,4-dioxygenolytic cleavage of quercetin, a flavonol that is produced by numerous plants, to form carbon monoxide and the depside (diphenolic ester) 2-protocatechuoylphoroglucinol carboxylic acid (Scheme 1). The bacterial and fungal quercetinases that have been characterized to date all belong to the cupin superfamily [1-4]. The cupin domain is characterized by a β-barrel fold which comprises two conserved motifs with the consensus sequences G(X)5HXH(X)3,4E(X)6G and G(X)5PXG(X)2H(X)3N. The conserved glutamate and the three histidine residues provide the ligands for a divalent metal ion in the active center of the enzyme [5] (Figure 1). Among the fungal quercetinases, the enzyme of Aspergillus japonicus has been characterized most intensively [1,6-9]. It is an extracellular glycoprotein with a bicupin scaffold, i.e., each subunit consists of two cupin domains. Two examples of bacterial quercetinases have been reported; both are cytoplasmic proteins. The enzyme of Bacillus subtilis consists of bicupin subunits that form a homodimer [2], whereas the enzyme of Streptomyces sp. FLA, which shows 35.9% sequence identity with the C-terminal cupin domain of Bacillus quercetinase, is a dimer of monocupin subunits [3,10].
Scheme 1.
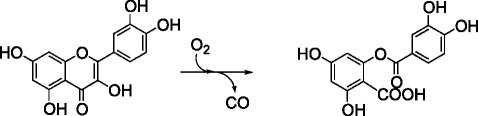
Conversion of quercetin to 2-protocatechuoylphloroglucinol carboxylic acid, catalyzed by quercetinase (flavonol 2,4-dioxygenase).
Figure 1.
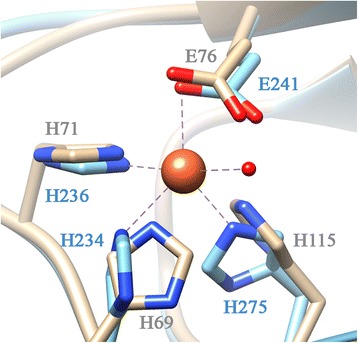
Model of the QueD metal center. The iron-containing Bacillus subtilis quercetinase [2] (PDB code 1y3t (chain B); residues indicated in blue) served as template for homology modelling of Streptomyces QueD (gray).
The majority of dioxygenases require a metal cofactor for catalysis, which is most often a nonheme iron [11]. Interestingly, all the extracellular fungal quercetinases isolated so far rely on a mononuclear copper center for activity [1,4,12-14], whereas the cytoplasmic quercetinases of B. subtilis and Streptomyces sp. FLA appear to be promiscuous enzymes capable of using different metal ions for catalysis. Bacillus quercetinase has originally been purified from a recombinant E. coli host grown in LB (Lysogeny Broth) as an iron protein [2,15,16]. Reconstitution experiments suggested that Co, Cu, and Mn also support catalysis [2], and characterization of the kinetic parameters of the Mn- and Co-forms revealed that the catalytic efficiency was highest for the Mn-enzyme [17]. In contrast to Bacillus QueD, Streptomyces QueD, purified from recombinant E. coli cells grown with various metal ions, was found to be most active with Ni2+, which is highly unusual for oxygenases [3]. However, in this as well as in other studies on bacterial quercetinases, the proteins were heterologously produced in E. coli. As pointed out in a recent review, when using a high-expression heterologous system for the synthesis of Ni-QueD, “the relevance to metal speciation of protein in the native host remains unclear” [18].
Streptomyces sp. strain FLA grows very poorly on quercetin as sole carbon source [10]. Transcription of the queD gene of strain FLA is induced by quercetin, however, the regulatory mechanism remained elusive. An effect of Ni2+ on queD expression was not observed [19]. Because quercetin shows antibacterial activity, acting on multiple cellular targets [20-23], it is conceivable that a major physiological role of quercetinase is to detoxify the flavonol. The quercetin O-methylation, hydroxylation, or glycosylation reactions mediated by various Streptomyces spp. [24,25] may serve the same function.
In this study, we prepared different metal forms of Streptomyces QueD by in vivo and in vitro approaches, to identify the physiologically relevant metal cofactor(s), and to find out which factors determine metal selection. Replacement of individual residues of the 3His/1Glu motif gave insight into the significance of the amino acid ligands for metal occupancy, protein folding, and function.
Results
Metal selectivity and catalytic activity of recombinant QueD
To enable a systematic study of the metal selectivity of QueD, as well as of the effects of different transition metal ions on quercetinase activity, we produced the enzyme recombinantly in E. coli which was grown in media supplemented with an excess of the metal ion of interest. Preparations of C-terminally Strep-tagged QueD, purified to electrophoretic homogeneity from cells grown in Ni2+-supplemented medium (Additional file 1: Figure S1), generally contained about 0.6 − 0.8 equivalents of nickel per protein monomer. Cobalt contents of QueD isolated from cells grown in CoCl2-supplemented medium varied significantly between the batches (0.5 to >1 equivalent per protein monomer). QueD protein produced by recombinant E. coli grown in Fe2+-supplemented medium showed varying iron contents in the range of 0.3 to 0.8 equivalents of iron per protein monomer. When QueD was purified from cells grown in medium supplemented with 30 μM MnCl2, the proteins contained only between 0.2 to 0.5 equivalents of Mn and varying amounts of other metals, predominantly Ni, but also Fe and Zn. Increasing the concentration of MnCl2 in the cultivation medium to 1 mM did not increase the Mn occupancy of the recombinant protein. To assess the effect of the metal ion on the overall structure of the protein, the CD spectra of Ni-, Co-, Fe- and Mn-QueD produced in vivo were recorded. The spectra were very similar and characteristic of proteins that are predominantly composed of β-sheets (Figure 2A).
Figure 2.
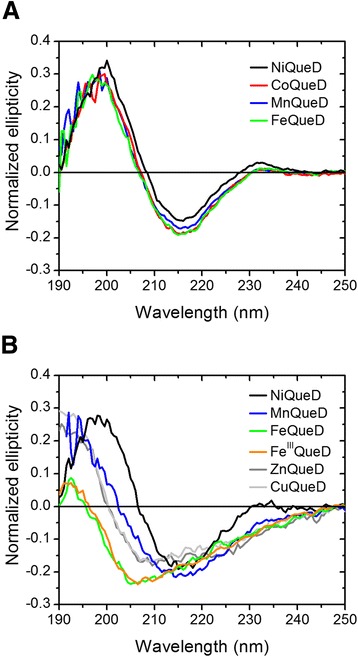
Far-UV CD spectra of QueD proteins produced in vivo and in vitro. (A), Proteins purified from recombinant E. coli cells, and (B), proteins synthesized in vitro by cell-free protein synthesis in presence of 1 mM Ni2+, Mn2+, or iron salts, or 0.2 mM Cu2+ or Zn2+. Protein samples in 10 mM potassium phosphate buffer (pH 8.0) were adjusted to ~10 μM (based on measuring absorbance at 280 nm). Spectra were recorded at 25°C in a 1 mm path length cell; bandwidth: 0.1 nm (A) or 0.5 nm (B).
The catalytic activity of the recombinant proteins decreased in the order Ni-QueD > Co-QueD > Mn-QueD (Table 1). An estimation of the metal-related specific activities of QueD forms, i.e., the specific activities if QueD were fully occupied with the metal of interest (Additional file 2: Table S1), suggests that the iron form of QueD is not catalytically competent.
Table 1.
Metal contents and specific activities of selected batches of recombinant QueD proteins
| Protein a | Metal content (equivalents per protein monomer) | Specific activity (U mg −1 ) |
|---|---|---|
| Ni-QueD | 0.70 Ni, 0.20 Fe, 0.03 Co, 0.06 Zn, 0.01 Mn | 137 |
| Co-QueD | 0.51 Co, 0.07 Ni, 0.05 Fe, 0.02 Zn, 0.01 Mn, 0.01 Cu | 30 |
| Mn-QueD | 0.44 Mn, 0.12 Zn, 0.08 Fe, 0.05 Ni, 0.01 Co, 0.01 Cu | 18 |
| Fe-QueD | 0.72 Fe, 0.05 Ni, 0.02 Zn, 0.02 Cu, 0.01 Mn | 8 |
aThe designations of the QueD metal forms refer to the metal of interest and do not imply exclusive occupancy of the protein with this metal.
Metal ions which contribute to ≥0.01 equivalents per protein monomer are listed.
Characterization of a Zn-form of QueD would be very interesting from a mechanistic point of view, because the Zn2+ ion is redox-inert in biological systems. However, attempts to produce Zn-QueD by the in vivo approach resulted in protein preparations that contained nickel rather than zinc (up to 0.67 equivalents of Ni and between 0.1 and 0.26 equivalents of Zn per protein monomer).
Since fungal quercetinases depend on a Cu2+ center, it also would be interesting to characterize a Cu-form of the bacterial QueD. However, in the reducing environment of the E. coli cytoplasm, copper ions are in the Cu1+ state, and attempts to produce Cu-QueD by the standard expression system were not successful. Fusing QueD to PelB- or DsbA-leader peptides in order to secrete the recombinant QueD to the E. coli periplasm resulted in formation of intracellular rather than periplasmic protein, precluding the production of Cu-QueD by periplasmic Cu2+ incorporation.
In summary, the order of metal ions according to occupancy of recombinant QueD is Ni ≈ Co > Fe > Mn, while in terms of activity we observe the ranking Ni > Co > Mn.
QueD from the wild-type strain, Streptomyces sp. FLA
The bacterial quercetinases described to date were all produced heterologously in E. coli. They were observed to be promiscuous with respect to metal ion incorporation, thus their natural cofactor(s) incorporated by the original wild-type strain remained unassigned [2,3,10,15-17]. Using a four-step protocol, QueD from Streptomyces sp. FLA was purified to approximately 91% electrophoretic homogeneity, as deduced from densitometric analysis of the Coomassie-stained gel (Additional file 1: Figure S1). According to ICP-MS (inductively coupled plasma – mass spectrometry) analysis, the protein preparation contained 0.33, 0.28, 0.16 and 0.02 equivalents of Ni, Zn, Fe and Cu, respectively, per monomer. We cannot exclude the possibility that the purification protocol enriches distinct metal forms of QueD, however, QueD activity eluted as distinct peaks from the columns. Because heterologously produced QueD can incorporate cobalt or manganese besides nickel and iron (see above), it is remarkable that the content of these metals was below detection, especially as the mineral salts medium used for growth of Streptomyces contained CoCl2 (8.4 nM) and MnCl2 (1.5 nM) besides NiCl2 (0.8 nM). The relatively high contents of zinc in the protein preparation were surprising. The growth medium was supplemented with 3.5 nM ZnSO4, but as cells accumulate metal ions to a different extent, zinc may be concentrated by a high factor. The protein preparation, which contained minor amounts of other proteins, showed a specific activity of 49 U mg−1 (Additional file 3: Table S2). Considering that recombinant QueD with a nickel occupancy of 33% should have a specific activity of about 60 U mg−1 (cf. Additional file 2: Table S1), the catalytic activity of wild-type QueD is likely mediated by a Ni2+ center.
Cell-free protein synthesis (CFPS) of QueD in the presence of metal ions
From the analysis of QueD proteins produced in vivo, it is difficult to assess to which extent metal ion availability and/or intrinsic preferences of the protein determine metal incorporation. To exclude effects caused by the limited in vivo availability of free metal ions, we aimed at producing QueD using in vitro approaches. However, in vitro reconstitution experiments, following protocols used for other cupin proteins [2,26], resulted in precipitation of QueD. Therefore, we tested an E. coli cell-free coupled transcription-translation system for production of different metal forms of the QueD protein. There are relatively few studies on cell-free synthesis of metalloproteins in the literature. For example, successful synthesis by in vitro transcription-translation systems was reported for manganese peroxidase (a heme protein) [27], a ferredoxin [28], [FeFe] hydrogenase [29], and Cu,Zn-superoxide dismutase [30]. The yields reported were in the range of about 30 − 50 μg ml−1 [27,29,30] or lower and seldom reached several 100 μg of protein from 1 ml reaction [28]. Often only part of the total protein was functional [27,29].
In order to identify suitable conditions for CFPS in the presence of metal ions, we first used a QueD-EGFP fusion protein as reporter, and determined the relative protein level by measuring EGFP fluorescence during CFPS. At concentrations below or equal to 1 mM of Ni2+, Fe2+, or Mn2+, the relative EGFP fluorescence intensities in the coupled transcription-translation reactions were similar to those observed in CFPS reactions performed in the absence of additional metal ions (Additional file 4: Figure S2 A − C, F), suggesting that these metals do not significantly affect translational efficiency in general. In case of CFPS reactions in the presence of Ni2+ or Mn2+, the quercetinase activity in the samples increased with increasing metal concentration in the reaction mixtures (except for the reaction that contained 3 mM Ni2+) (Additional file 4: Table S3). Unfortunately, zinc and copper ions had an inhibitory effect on the efficiency of QueD-EGFP synthesis (Additional file 4: Figure S2 D − E), as also reported in CFPS studies on other metalloproteins [30,31]). The absence of quercetinase activity in the Cu2+- and Zn2+-CFPS samples (Additional file 4: Table S3) suggested that Cu2+ and Zn2+ are either not incorporated into the QueD-EGFP fusion protein, or do not support catalysis.
To purify (Strep-tagged) QueD proteins synthesized by CFPS, reactions were performed on a 1 ml scale in the presence of Ni2+, Mn2+, or Fe2+/3+, or Zn2+ or Cu2+, and the proteins were prepared by Strep-Tactin affinity chromatography. The isolation of QueD proteins was verified in Western blots of denaturing polyacrylamide gels (Additional file 5: Figure S4). Far-UV CD spectra were recorded to find out whether the proteins synthesized in vitro have adopted the same overall fold as those produced in vivo (Figure 2). The spectrum of QueD produced in Ni2+-supplemented CFPS reactions retained the characteristics of that of the Ni-QueD formed in vivo, with a negative band at about 215 nm and a positive band at 198 nm, indicating that QueD in the presence of nickel ions is able to fold to its native state in vitro. The protein produced in the presence of 1 mM Mn2+ retained a high β-sheet content. In contrast, the CD spectra of the proteins obtained from CFPS in presence of Cu2+ (0.2 mM), Zn2+ (0.2 mM), Fe2+ or Fe3+ (1 mM) differ significantly from that of the nickel enzyme. Thus, the metal ions tested can be ordered as Ni > Mn > > Zn, Fe, Cu according to their effect on secondary structure of QueD. Altogether, the findings suggest that while the efficiency of CFPS is similar in the presence of Ni2+, Mn2+ or Fe2+, in the in vitro situation only nickel ions support QueD folding to its native state.
QueD purified from CFPS reactions supplemented with Ni2+ and Mn2+ showed quercetinase activities of 21.3 U mg−1 and 6.6 U mg−1, respectively. For QueD isolated from CFPS-reactions supplemented with Cu2+, Zn2+, Fe2+ or Fe3+, the activity was approximately 0.1 U mg−1. Control experiments without metal ion supplementation indicated that such residual activity cannot be attributed to the supplemented metals, however, traces of Mn2+ and/or Ni2+ ions present in the E. coli extracts may confer activity to a subpopulation of the in vitro produced proteins. In terms of catalytic activity of QueD metal forms, we therefore obtain the ranking Ni > Mn.
Replacement of the 3His/1Glu ligand amino acids of Ni-QueD
To analyze the role of the metal-ligating amino acids for the structural integrity and function of Ni-QueD, the individual residues of the 3His/1Glu motif (Figure 1) were replaced by site-directed mutagenesis, and the QueD proteins produced by the respective E. coli strain grown in Ni2+-supplemented medium were purified and characterized with respect to metal contents, catalytic activity, and ability to bind quercetin (Table 2). All variants, with the exception of the H69A substitution, showed a decrease in nickel contents compared to the wild-type protein, and a relative increase in the zinc contents pointing towards a somewhat relaxed metal specificity. The KD values of the protein-quercetin complexes show that all protein variants retained a high affinity for the organic substrate. Replacement of the histidine residues at position 69 or 115 with alanine resulted in catalytically inactive protein variants, whereas the QueD-H71A and -E76D proteins retained marginal activity (Table 2). The CD spectra of the protein variants showed an increased intensity of the negative band (at approx. 215 nm) and a hypsochromic (blue) shift of the positive band (<200 nm) compared to the wild type protein (Figure 3A). Thus, the loss of activity could be due to perturbation of the secondary structure.
Table 2.
Metal contents and activities of QueD proteins carrying ligand replacements, and K D values of protein-quercetin complexes
| Protein a | Metal content (equivalents per protein monomer) | Spec. activity (U mg −1 ) | K D (μM) |
|---|---|---|---|
| Ni-QueD-H69A | 0.88 Ni, 0.07 Fe | b.d.b | 3.1 (0.9) |
| Ni-QueD-H115A | 0.34 Ni, 0.29 Zn, 0.07 Fe, 0.02 Co, 0.01 Mn | b.d. | 2.6 (0.6) |
| Ni-QueD-H71A | 0.18 Ni, 0.16 Zn, 0.06 Fe | 0.90 (0.04) | 10.4 (2.9) |
| Ni-QueD-E76D | 0.28 Ni, 0.24 Zn, 0.05 Fe | 1.32 (0.01) | 4.0 (1.0) |
| Ni-QueD-E76H | 0.54 Ni, 0.03 Cu, 0.02 Fe, 0.01 Zn | 3.44 (0.13) | 1.3 (0.1) |
| Co-QueD-E76H | 0.6 Co, 0.03 Cu, 0.02 Fe, 0.01 Zn, 0.02 Ni | 0.19 (0.01) | n.d.c |
aThe designations of the QueD metal forms refer to the metal of interest and do not imply exclusive occupancy of the protein with this metal.
bb.d., below detection.
cn.d., not determined.
Metal ions which contribute to ≥0.01 equivalents per protein monomer are listed. For the specific activities and K D values, the average of 3 experiments is given with the standard deviations in brackets. The K D value of the complex of Ni-QueD with quercetin, determined for the preparation specified in Table 1, was 10.1 ± 2.9 μM.
Figure 3.
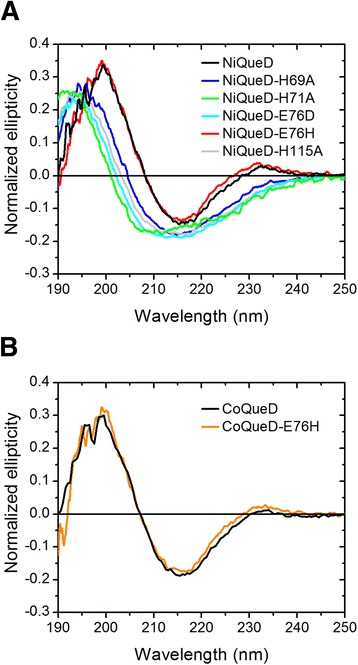
Far-UV CD spectra of QueD variants carrying amino acid replacements. (A), Ni-QueD proteins with amino acid substitutions in the 3His/1Glu motif proposed to ligate the metal ion, and (B) Co-QueD with a substitution of the glutamate ligand by histidine. Protein samples in 10 mM potassium phosphate buffer (pH 8.0) were adjusted to ~10 μM (based on measuring absorbance at 280 nm). Spectra were recorded at 25°C in a 1 mm path length cell; bandwidth: 0.1 nm.
Replacement of E76 by histidine resulted in a protein that showed about 2% of the activity of the wild-type protein and an about 8-fold decreased KD value compared to that of the quercetin complex with Ni-QueD (Table 2). Interestingly, the CD spectrum of the Ni-QueD-E76H protein was nearly superimposable to that of Ni-QueD (Figure 3A), demonstrating that, in contrast to the other protein variants tested, the E76H variant retained the wild-type conformation. This was also observed for the cobalt form of this protein (Figure 3B). To verify that the mutant protein catalyzes the same reaction as wild-type QueD, the organic products of quercetin conversion by Co-QueD and Co-QueD-E76H were extracted and subjected to LC/MS analysis. The LC elution profiles were identical and both showed a main peak at a retention time of 18.2 min. ESI mass spectrometry of the corresponding peaks revealed ions at m/z 324.1 (100%) and 307.0 (22%) for [M + NH4]+ and [M + H]+, respectively. This corresponds to the previously observed mass for the molecular ion of the product of quercetin conversion by recombinant Ni- and Co-QueD and is consistent with formation of the depside 2-protocatechuoylphloroglucinol carboxylic acid (C14H10O8) [3]. The possibility that the same product could also result from a 2,3-dioxygenolytic side reaction to form an α-oxo acid, and subsequent hydrolysis to formate and the depside, was examined by determining the concentration of formate in enzyme assays. Previous analysis of the reaction of (His6-tagged) Ni- and Co-QueD had revealed an amount of formate corresponding to about 1.3% of the quercetin converted [3]. In the reaction of Strep-tagged Co-QueD, the formate detected corresponded to 2.3% (±0.2%) of substrate converted, whereas 3.0% (±0.8%) of side product was observed in the reaction of the Co-QueD-E76H protein. Thus, both the wild-type and the mutant Co protein mediate this side reaction to a similar extent.
The observation that Ni- and Co-QueD-E76H were the only proteins in the series of variants that retained the wild-type fold may suggest that the metal ion is coordinated by four amino acid ligands in these variants, i.e., in a 4His motif instead of the canonical 3His/1Glu motif. The HSAB concept predicts a higher stability of the Ni/His complex compared to a Ni/Glu complex, because the ligating nitrogen of His is softer than the ligating oxygen in Glu, leading to a larger stability of complexes with the soft Ni ion (or the Co ion). The E76H variant shows an increase in affinity for its electron-rich organic substrate, indicated by the low KD value (Table 2). This effect might be due to the altered charge distribution around the metal, since a carboxylate ligand (as in E76) transfers more charge to the metal ion than an imidazole nitrogen. Alternatively, or additionally, subtle rearrangements of all the ligands around the metal may account for the decrease of the KD value of Ni-QueD-E76H compared to that of the Ni-QueD-quercetin complex.
Discussion
Bacterial quercetinases have been described to be able to use a defined set of different metal ions for catalysis, but the molecular basis of such metal selection is largely unclear. When using a recombinant high-expression system in E. coli for the production of Streptomyces QueD, the metal contents of the purified proteins suggested that QueD binds Mn2+, Fe2+, Co2+, and Ni2+ in the order Mn < Fe < Co ≈ Ni. This ranking is similar to the Irving-Williams series, which describes the stability of divalent transition-metal complexes as Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+ [32]. Interestingly, QueD purified from the wild-type Streptomyces sp. strain contained mainly nickel and zinc. However, quantitative metal analyses cannot distinguish between metal ions unspecifically adsorbed to the protein surface or those coordinated to the conserved metal-binding 3His/1Glu motif of the cupin protein.
Because the concentrations of “free” transition metal ions are thought to be in the 10−7 to 10−15 M range [33,34], metal incorporation into QueD in the E. coli expression system may be limited by metal ion availability. In contrast, the cell-free transcription-translation system allows for supplementation with defined metal ion concentrations. To outcompete divalent ions possibly brought in by the highly concentrated E. coli extracts used in the CFPS reactions, we added metal ions in excess, at concentrations which still supported protein synthesis (cf. Additional file 4). The CD spectra and activity data of the QueD forms produced in vitro indicated that intrinsic properties of the protein rather than metal ion availability govern the formation of native metalloenzyme, with Ni2+ as the strongly preferred ion.
The coordination geometry of the metal center should play a significant role in determining the preference of QueD for its active-site ion. Ni2+ centers with metal binding by a 3His/1Glu motif show a preference for octahedral geometry [35,36]. Co2+ and Mn2+ centers are usually also hexa-coordinated with an octahedral geometry [37]. Zn2+ typically has coordination numbers of four or five in active sites of enzymes, with a preference for tetrahedral geometry [37]. Cu2+ bound to proteins is typically coordinated by four ligands in a square planar geometry [37,38], or with an additional water ligand positioned to form a trigonal bipyramidal geometry [39]. Fe2+ complexes in proteins are either penta- or hexa-coordinated, with penta-coordination as most common [40]. Possibly, formation of native, catalytically competent (Ni-, Co-, Mn-) QueD correlates with the propensity of the metal ions to form an octahedral complex geometry, as dictated by the ligands that constitute the metal binding site of the protein. If so, the metal center of Streptomyces QueD would comprise two additional ligands (such as water molecules) besides the four amino acid ligands and thus would be different from that of copper-dependent A. japonicus quercetinase, which shows a penta-coordinated metal center with 3 His, 1 Glu and one water molecule [1]. Interestingly, an octahedral coordination sphere was also proposed for the catalytically highly active Mn-QueD of B. subtilis [17], whereas its less active Fe-form, which has been crystallized, is penta-coordinate with a square pyramidal or distorted trigonal bipyramidal geometry [2]. Thus, the geometry of the nickel center of Streptomyces QueD may be different from that shown in Figure 1 which is based on homology modelling using the crystal structure of quercetinase of B. subtilis as template.
The CD spectra of QueD proteins carrying replacements of individual residues of the 3His/1Glu motif suggested that all four residues are important for structural integrity of QueD, although Glu76 can be exchanged to histidine without affecting the secondary structure of the protein. The important structural role of both metal and ligands in QueD is in contrast to several other cupin proteins, which tolerate the substitution of individual metal-coordinating amino acid residues or which even retain their structure upon in vitro metal depletion [26,41,42].
Conclusions
Streptomyces quercetinase (QueD), a metal dependent flavonol dioxygenase with a cupin fold, is rather promiscuous with respect to the incorporated metal ion. This study shows that QueD produced in a heterologous host requires Ni2+, Co2+ or Mn2+, i.e. metal ions that prefer an octahedral coordination geometry, for native secondary structure content and catalytic activity. Ni2+ ions support the formation of native QueD in a cell-free transcription/translation system. The in vivo and in vitro data suggest that metal ion specificity of QueD is determined by both chemical stability (the Irving-Williams series) and intrinsic geometric constraints dictated by the metalloprotein. Taking all criteria together, Ni appears to be the physiologically optimal ion for Streptomyces quercetinase. Crystal structures of QueD, as well as of QueD-E76H protein and enzyme-substrate complexes, would be most interesting to compare this bacterial quercetinase to the fungal copper quercetinase with atomic resolution.
Methods
Research performed in this study does not involve human subjects, human material, or human data.
Materials
Chemicals were purchased from Sigma-Aldrich, except for tRNAs from E. coli strain MRE 600 which were obtained from Roche Applied Science. Restriction enzymes, T4 DNA ligase, and other reagents for molecular biology were purchased from Thermo Scientific. DNA primers, listed in Table 3, were obtained from Microsynth AG (Balgach, Switzerland) or MWG Biotech AG (Ebersberg, Germany).
Table 3.
Oligonucleotides used for gene amplification and site-directed mutagenesis
| Name | Nucleotide sequence (5′ → 3′) | Application |
|---|---|---|
| quedfor | TGACCATCGAATACGCCACCCGTCACC | Amplification of queD |
| quedrev | GACGTCGTACTGCTCGGGCACGGTC | |
| strepfor | ATAAGAATGCGGCCGCATTAGAATGGAGCCACCCGCAGTTCGAAAAATGAGATCCGGCTGCTAACAAAGCCCGAAAGG | Exchange of His6-tag for StrepII-tag |
| streprev | ATAAGAATGCGGCCGCAAGCTTCCTTCCCTCGATACTCCCGGTGTGC | |
| gfpfor | TATAAGCTTGCCGGTGCGGAAAATC | Amplification of the EGFP gene |
| gfprev | TATAGCGGCCGCCTGGTACAGTTCATCCATG | |
| H69Afor | CCTCGCACGCGGACACCTACG | Exchange of His69 for Ala |
| H69Arev | CGGCGGGGATCACCTCGCCCTTG | |
| H71Afor | CGCCCACTCGGCCGCGGACACCTACGAGGTCTTC | Exchange of His71 for Ala |
| H71Arev | GGGATCACCTCGCCCTTGGGGCCCTCGCAG | |
| H115Afor | ATGGAACGCCACCACTCGCAGGTCG | Exchange of His115 for Ala |
| H115Arev | GCGGTAGGCGGCCACGCAGTTCTTCGGTACGAAGC | |
| E76Hfor/E76Dfor | CGTCTTCTACATCACCCAGGGC | Forward primer for the exchange of Glu76 for His or Asp |
| E76Hrev | TGGTAGGTGTCCGCGTGCGAGTGG | Reverse primer for the exchange of Glu76 for His |
| E76Drev | TCGTAGGTGTCCGCGTGCGAG | Reverse primer for the exchange of Glu76 for Asp |
Recognition sites for restriction endonucleases are italicized.
DNA techniques
Isolation of plasmid DNA from recombinant E. coli strains, extraction of DNA fragments from agarose gels, and purification of PCR products were carried out by the innuPREP kit series (Analytik Jena AG, Jena, Germany). Standard protocols were used for agarose gel electrophoresis, restriction digestion, and DNA ligation. Exchange of the His6-tag sequence in pET23a-queD [10] for a StrepII-tag sequence was performed by amplification of the plasmid with the primer pair strepfor/streprev (Table 3), restriction with DpnI and NotI, and circularisation with T4 DNA ligase. For fusion of enhanced green fluorescent protein to QueD, the egfp gene was amplified by PCR, using the primer pair gfpfor/gfprev (Table 3), and plasmid pET27b-egfp harboring a synthetic gene (GenScript) for EGFP (F64L/S65T/Q80R/K238Q) as template. The PCR product and pET23a-queD-Strep were restricted with HindIII and NotI, purified with the innuPREP Doublepure kit (Analytik Jena AG), and ligated, resulting in pET23a-queD-egfp-Strep. Transformation of E. coli strains DH5α and BL21 (DE3) [pLysS] with the constructed plasmid was performed as described by Hanahan [43]. Site-directed mutagenesis of the queD gene (GenBank accession number AM234612) was performed according to the protocol of the Phusion™ Site-Directed Mutagenesis Kit (Thermo Scientific) using pET23a-queD-Strep as template, Phusion™ High-Fidelity DNA polymerase for amplification, and the primers listed in Table 3. Confirmatory DNA sequencing of the insert and flanking regions of the plasmids was performed by GATC Biotech AG (Konstanz, Germany) or MWG Biotech AG.
Cell-free protein synthesis (CFPS)
Cell-free protein synthesis was conducted at 30°C using the PANOx-SP system [44], except that instead of S30 cell extract, S12 extract from E. coli strain BL21 (DE3) was used, which was prepared as described by Kim et al. [45]. Crude extract of E. coli strain BL21 (DE3) containing T7 RNA polymerase was prepared separately as described in ref. [44]. In order to quantify the relative amounts of protein produced by CFPS, the plasmid pET23a-queD-egfp-Strep was used as template for in vitro transcription-translation reactions supplemented with metal salts (0.05 − 3 mM). The relative amount of EGFP present in the samples (as QueD-EGFP fusion) was determined by measuring the average fluorescence emission at 510 − 570 nm upon excitation at 490 nm. Cell-free synthesis of QueD-Strep, using pET23a-queD-Strep as template, was performed in the presence of 1 mM NiCl2, 1 mM MnCl2, 0.5 mM FeCl2 or 1 mM (NH4)2Fe(SO4)2 (optionally supplemented with 1 mM DTT), 1 mM FeCl3, 1 mM or 0.2 mM CuCl2, and 1 mM or 0.2 mM ZnCl2.
Expression and purification of C-terminally Strep-tagged QueD proteins
E. coli BL21 (DE3) [pLysS, pET23a-queD-Strep] as well as E. coli strains containing a mutant pET23a-queD-Strep plasmid were grown at 37°C to an OD600 of 0.5 in M9 minimal medium [46] containing ampicillin (100 μg ml−1) and chloramphenicol (34 μg ml−1). Subsequently, metal salts (MnCl2, FeCl2, CoCl2, NiCl2, or ZnCl2) were added to the cultures to final concentrations of 10 μM (in case of Ni2+ and Co2+), 30 μM (for Mn2+, Fe2+, and Zn2+) or 1 mM (for Mn2+), and expression of queD was induced with 1 mM IPTG. The cultivation temperature was reduced to 25°C for 3 h and then shifted to 20°C for another 15 h. The cells were centrifuged (4°C, 10 min at 12,000 × g) and the cell pellets were stored at −80°C. For the preparation of cell extracts, cells were resuspended in 100 mM Tris/HCl buffer (pH 8) containing 300 mM NaCl and 1 mM MgCl2, supplemented with 12.5 units of Benzonase (Novagen). After incubation on ice for 1 h, the suspension was sonicated and then centrifuged (4°C, 40 min at 38,360 × g). Recombinant QueD proteins were purified from cell extract supernatant by affinity chromatography on a 2 ml Strep-Tactin® column (IBA, Göttingen, Germany) using an FPLC system. QueD proteins synthesized by in vitro transcription/translation were isolated using a 0.5 ml Strep-Tactin® gravity column. After removing unbound proteins with 100 mM Tris/HCl buffer (pH 8.0) containing 300 mM NaCl, Strep-tagII fusion proteins were eluted with 2.5 mM desthiobiotin in the same buffer. Fractions containing the protein of interest were pooled, concentrated by ultrafiltration in a Vivaspin 20 concentrator (Sartorius Stedim Biotech), frozen in liquid nitrogen and stored at –80°C.
Growth of Streptomyces sp. FLA and purification of QueD protein
Growth of Streptomyces sp. strain FLA in complex medium, followed by resuspension of the biomass in mineral salts medium supplemented with quercetin, was performed as described previously [10]. The growth medium contains ammonium ferric citrate (1 μM) and 100 μl/l of SL-6 trace element solution according to Pfennig [47], resulting in final concentrations of Cu2+, Ni2+, Mn2+, Zn2+ and Co2+ salts of 0.6 nM, 0.8 nM, 1.5 nM, 3.5 nM, and 8.4 nM, respectively. For the preparation of cell extracts, Streptomyces biomass was suspended in 50 mM Tris/HCl buffer (pH 7.0) and disrupted by sonication. After a centrifugation step (38,360 × g, 90 min) to remove cell debris, (NH4)2SO4 was gradually added to the cell extract supernatant to a final concentration of 0.4 M, and precipitated proteins were removed by centrifugation at 38,360 × g for 40 minutes at 4°C. The supernatant was applied to a 20 ml Phenyl-Sepharose CL-4B column (GE Healthcare) that was equilibrated in 50 mM Tris/HCl, pH 7.0, containing 0.4 M (NH4)2SO4. Quercetinase was eluted by stepwise reducing the concentration of (NH4)2SO4 (0.4 to 0 M) in the buffer. The protein fractions with quercetinase activity were pooled and dialyzed against 50 mM Tris/HCl buffer (pH 8.0). The protein pool was applied to a 6 ml Q Sepharose Fast Flow column (GE Healthcare) equilibrated in 50 mM Tris/HCl buffer (pH 8). Protein fractions with quercetinase activity were eluted with 0.3 M NaCl and dialyzed as described above. A second anion exchange step was performed on a 2 ml Source 15Q (GE Healthcare) column. Quercetinase was eluted with 0.2 M NaCl, concentrated by ultrafiltration, and stored in 50 mM Tris/HC (pH 8) at −80°C.
Protein analysis
Molecular graphics was performed with the UCSF Chimera package, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311) [48]. Protein concentrations in cell-free extracts and CFPS reactions were estimated using the Bradford method as modified by Zor and Selinger [49]. Concentrations of electrophoretically pure QueD-Strep proteins were estimated from their absorbance at 280 nm, using the theoretical molar extinction coefficient of ε280nm = 21,555 M−1 cm−1, calculated according to Pace et al. [50]. Metal contents of QueD preparations and buffer blanks were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES), or by ICP-MS, performed by the Chemical Analysis Laboratory, Center for Applied Isotope Studies, University of Georgia (Athens, GA, USA). To prepare samples for metal analysis, the proteins were washed 3 times with 10-fold volumes of 50 mM Tris/HCl buffer (pH 8.0). Before use, the buffer was treated with Serdolit® Chelite® CHE (SERVA Electrophoresis, Heidelberg, Germany) to remove divalent cations. QueD activity was determined spectrophotometrically at 30°C by measuring quercetin consumption as described previously [3]. One unit (U) of QueD is defined as the amount of enzyme that catalyzes the conversion of 1 μmol of quercetin per minute at 30°C. Assays for the determination of apparent steady-state parameters of QueD for quercetin were performed in air-saturated buffer (50 mM Tris/HCl, pH 8.0) [3].
Identification of organic reaction products
Samples for identifying the organic products of quercetin conversion by Co-QueD-E76H and Co-QueD were prepared and extracted as described previously [3]. LC/MS analysis of the extracts was performed on a Dionex™ Ultimate™ 3000 UHPLC system (Thermo Scientific), coupled to an amaZon speed ion trap mass spectrometer (Bruker). Products were separated with an Eurospher II 100-5 C18 column (Knauer GmbH, Berlin, Germany), using a linear gradient of 20% to 100% (over 45 min) methanol in 10 mM aqueous ammonium acetate, adjusted to pH 4.0 with acidic acid, at a flow rate of 0.4 ml min−1.
To analyze whether formate is formed from quercetin in a possible side reaction, 0.1 U ml−1 of Co-QueD or Co-QueD-E76H were incubated with 200 μM quercetin in 20 mM potassium phosphate buffer (pH 7.0) for 90 min. Subsequently, Candida boidinii formate dehydrogenase (Fluka, ~50 U ml−1) and NAD+ (40 mM) were added to final concentrations of 0.5 U ml−1 and 2 mM, respectively. Formation of NADH was monitored at 340 nm over 180 min. NAD+ solutions were always prepared freshly in 20 mM phosphate buffer (pH 7.0). For background correction, reference samples without formate dehydrogenase containing the QueD-quercetin reaction mixtures and NAD+ were monitored as well. Under these assay conditions, the detection limit of formic acid is in the μM range: 10 μM and 1 μM formic acid were detected with percentage deviations of ±12% and ±77%, respectively.
Fluorescence titration
Dissociation constants KD of enzyme-quercetin complexes were determined by fluorescence titrations, measuring intrinsic protein fluorescence in a Jasco FP-6500 spectrofluorometer. In an anaerobic glove box, degassed anoxic solutions of QueD protein were diluted in an anaerobic cuvette with N2-saturated buffer (50 mM Tris/HCl, pH 8.0, treated with Serdolit® Chelite® CHE) to a final concentration of approximately 1 μM. Small amounts of anoxic quercetin solution were added stepwise, mixed gently, and allowed to equilibrate at 30°C for 15 − 20 minutes. Upon excitation at 280 nm, the fluorescence intensities at λmax = 340 nm were recorded after each titration step and plotted against the quercetin concentration. KD values were determined from analysis of binding curves using the equation for a quadratic binding curve [51].
Circular Dichroism (CD) spectroscopy
Far-UV CD spectra of proteins were recorded from 190 to 250 nm at 25°C with a J-815 circular dichroism spectrometer (Jasco), using a 0.1-cm path length cell, with a digital time integration of 1 s and a scan speed of 50 − 100 nm/min. Spectra were accumulated 7 − 10-fold. Protein samples were in 10 mM potassium phosphate buffer (pH 8.0). The protein concentration in the cuvette was adjusted to a final concentration of ~10 μM. All protein spectra were corrected by subtracting the CD spectrum of the buffer. To correct for errors such as slight differences in protein concentrations, spectra were scaled by normalizing each spectrum by ‖Θ‖ = (∫Θ(λ)2dλ)1/2 [52].
Acknowledgments
We thank Prof. Dr. Simone König (Integrated Functional Genomics, University of Münster, Germany) for MALDI-MS analysis. We also thank Steffen Drees for the synthetic EGFP template and Almut Kappius for expert technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (grant no. FE 383/18-1 to S.F.). We also acknowledge support by the Open Access Publication Fund of University of Münster.
Abbreviations
- CD
Circular dichroism
- CFPS
Cell-free protein synthesis
- DTT
Dithiothreitol
- FPLC
Fast protein liquid chromatography
- HSAB
Hard-soft acids and bases
- ICP-MS
Inductively coupled plasma mass spectroscopy
- ICP-OES
Inductively coupled plasma optical emission spectroscopy
- IPTG
Isopropyl-β-D-thiogalactopyranoside
- LB
Lysogeny broth
- LC/MS
Liquid chromatography coupled with mass spectroscopy
- NADH
Nicotinamide adenine dinucleotide
- QueD
Quercetinase of Streptomyces sp. FLA
- U
Enzyme units
- UV
Ultraviolet
Additional files
Electrophoretic analysis of QueD proteins.
Metal-related catalytic activity of QueD.
Purification of QueD from Streptomyces sp. FLA.
Production of QueD-EGFP fusion protein by cell-free protein synthesis (CFPS), and quercetinase activity of QueD-EGFP in the CFPS reactions.
Verification of QueD synthesis by the CFPS system.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DN carried out the molecular genetic experiments, the cell-free synthesis of QueD, and characterized the biochemical properties of the QueD variants. ST identified the organic products of the reaction catalyzed by QueD, and performed statistical analyses of data. DN, LS and PL designed and performed circular dichroism experiments. PL and DK analyzed CD data. DK contributed to manuscript writing. SF conceived of the study and wrote the manuscript draft. All authors contributed to the final version of the manuscript, and all authors read and approved the final manuscript.
Contributor Information
Dimitrios Nianios, Email: d_nian01@uni-muenster.de.
Sven Thierbach, Email: s.thierbach@uni-muenster.de.
Lenz Steimer, Email: lenz.steimer@gmx.de.
Pavel Lulchev, Email: plulc_01@uni-muenster.de.
Dagmar Klostermeier, Email: dagmar.klostermeier@uni-muenster.de.
Susanne Fetzner, Email: fetzner@uni-muenster.de.
References
- 1.Fusetti F, Schröter KH, Steiner RA, van Noort PI, Pijning T, Rozeboom HJ, et al. Crystal structure of the copper-containing quercetin 2,3-dioxygenase from Aspergillus japonicus. Structure. 2002;10:259–68. doi: 10.1016/S0969-2126(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 2.Gopal B, Madan LL, Betz SF, Kossiakoff AA. The crystal structure of a quercetin 2,3-dioxygenase from Bacillus subtilis suggests modulation of enzyme activity by a change in the metal ion at the active site(s) Biochemistry. 2005;44:193–201. doi: 10.1021/bi0484421. [DOI] [PubMed] [Google Scholar]
- 3.Merkens H, Kappl R, Jakob RP, Schmid FX, Fetzner S. Quercetinase QueD of Streptomyces sp. FLA, a monocupin dioxygenase with a preference for nickel and cobalt. Biochemistry. 2008;47:12185–96. doi: 10.1021/bi801398x. [DOI] [PubMed] [Google Scholar]
- 4.Tranchimand S, Ertel G, Gaydou V, Gaudin C, Tron T, Iacazio G. Biochemical and molecular characterization of a quercetinase from Penicillium olsonii. Biochimie. 2008;90:781–9. doi: 10.1016/j.biochi.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Dunwell JM, Purvis A, Khuri S. Cupins: the most functionally diverse protein superfamily? Phytochemistry. 2004;65:7–17. doi: 10.1016/j.phytochem.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 6.Steiner RA, Kalk KH, Dijkstra BW. Anaerobic enzyme-substrate structures provide insight into the reaction mechanism of the copper-dependent quercetin 2,3-dioxygenase. Proc Natl Acad Sci U S A. 2002;99:16625–30. doi: 10.1073/pnas.262506299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kooter IM, Steiner RA, Dijkstra BW, van Noort PI, Egmond MR, Huber M. EPR characterization of the mononuclear Cu-containing Aspergillus japonicus quercetin 2,3-dioxygenase reveals dramatic changes upon anaerobic binding of substrates. Eur J Biochem. 2002;269:2971–9. doi: 10.1046/j.1432-1033.2002.02973.x. [DOI] [PubMed] [Google Scholar]
- 8.Steiner RA, Kooter IM, Dijkstra BW. Functional analysis of the copper-dependent quercetin 2,3-dioxygenase. 1. Ligand-induced coordination changes probed by X-ray crystallography: inhibition, ordering effect, and mechanistic insights. Biochemistry. 2002;41:7955–62. doi: 10.1021/bi0159736. [DOI] [PubMed] [Google Scholar]
- 9.Steiner RA, Meyer Klaucke W, Dijkstra BW. Functional analysis of the copper-dependent quercetin 2,3-dioxygenase. 2. X-ray absorption studies of native enzyme and anaerobic complexes with the substrates quercetin and myricetin. Biochemistry. 2002;41:7963–8. doi: 10.1021/bi015974y. [DOI] [PubMed] [Google Scholar]
- 10.Merkens H, Sielker S, Rose K, Fetzner S. A new monocupin quercetinase of Streptomyces sp. FLA: identification and heterologous expression of the queD gene and activity of the recombinant enzyme towards different flavonols. Arch Microbiol. 2007;187:475–87. doi: 10.1007/s00203-007-0215-z. [DOI] [PubMed] [Google Scholar]
- 11.Bugg TDH. Dioxygenase enzymes: catalytic mechanisms and chemical models. Tetrahedron. 2003;59:7075–101. doi: 10.1016/S0040-4020(03)00944-X. [DOI] [Google Scholar]
- 12.Oka T, Simpson FJ. Quercetinase, a dioxygenase containing copper. Biochem Biophys Res Commun. 1971;43:1–5. doi: 10.1016/S0006-291X(71)80076-1. [DOI] [PubMed] [Google Scholar]
- 13.Oka T, Simpson FJ, Krishnamurty HG. Degradation of rutin by Aspergillus flavus. Studies on specificity, inhibition, and possible reaction mechanism of quercetinase. Can J Microbiol. 1972;18:493–508. doi: 10.1139/m72-076. [DOI] [PubMed] [Google Scholar]
- 14.Hund HK, Breuer J, Lingens F, Hüttermann J, Kappl R, Fetzner S. Flavonol 2,4-dioxygenase from Aspergillus niger DSM 821, a type 2 CuII-containing glycoprotein. Eur J Biochem. 1999;263:871–8. doi: 10.1046/j.1432-1327.1999.00574.x. [DOI] [PubMed] [Google Scholar]
- 15.Barney BM, Schaab MR, LoBrutto R, Francisco WA. Evidence for a new metal in a known active site: purification and characterization of an iron-containing quercetin 2,3-dioxygenase from Bacillus subtilis. Protein Expr Purif. 2004;35:131–41. doi: 10.1016/j.pep.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Bowater L, Fairhurst SA, Just VJ, Bornemann S. Bacillus subtilis YxaG is a novel Fe-containing quercetin 2,3-dioxygenase. FEBS Lett. 2004;557:45–8. doi: 10.1016/S0014-5793(03)01439-X. [DOI] [PubMed] [Google Scholar]
- 17.Schaab MR, Barney BM, Francisco WA. Kinetic and spectroscopic studies on the quercetin 2,3-dioxygenase from Bacillus subtilis. Biochemistry. 2006;45:1009–16. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- 18.Boer JL, Mulrooney SB, Hausinger RP. Nickel-dependent metalloenzymes. Arch Biochem Biophys. 2013;544:142–52. doi: 10.1016/j.abb.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merkens H, Fetzner S. Transcriptional analysis of the queD gene coding for quercetinase of Streptomyces sp. FLA. FEMS Microbiol Lett. 2008;287:100–7. doi: 10.1111/j.1574-6968.2008.01296.x. [DOI] [PubMed] [Google Scholar]
- 20.Mirzoewa OK, Grishanin RN, Calder PC. Antimicrobial action of propolis and some of its components: the effects on growth, membrane potential and motility of bacteria. Microbiol Res. 1997;152:239–46. doi: 10.1016/S0944-5013(97)80034-1. [DOI] [PubMed] [Google Scholar]
- 21.Plaper A, Golob M, Hafner I, Oblak M, Solmajer T, Jerala R. Characterization of quercetin binding site on DNA gyrase. Biochem Biophys Res Commun. 2003;306:530–6. doi: 10.1016/S0006-291X(03)01006-4. [DOI] [PubMed] [Google Scholar]
- 22.Wu D, Kong Y, Han C, Chen J, Hu L, Jiang H, et al. D-Alanine:D-alanine ligase as a new target for the flavonoids quercetin and apigenin. Int J Antimicrob Agents. 2008;32:421–6. doi: 10.1016/j.ijantimicag.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Cushnie TPT, Lamb AJ. Recent advances in understanding the antibacterial properties of flavonoids. Int J Antimicrob Agents. 2011;38:99–107. doi: 10.1016/j.ijantimicag.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 24.Hosny M, Dhar K, Rosazza JPN. Hydroxylations and methylations of quercetin, fiestin, and catechin by Streptomyces griseus. J Nat Prod. 2001;64:462–5. doi: 10.1021/np000457m. [DOI] [PubMed] [Google Scholar]
- 25.Ma B, Zeng J, Shao L, Zhan J. Efficient bioconversion of quercetin into a novel glycoside by Streptomyces rimosus subsp. rimosus ATCC 10970. J Biosci Bioeng. 2013;115:24–6. doi: 10.1016/j.jbiosc.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moomaw EW, Angerhofer A, Moussatche P, Ozarowski A, García-Rubio I, Richards NGJ. Metal dependence of oxalate decarboxylase activity. Biochemistry. 2009;48:6116–25. doi: 10.1021/bi801856k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazaki-Imamura C, Oohira K, Kitagawa R, Nakano H, Yamane T, Takahashi H. Improvement of H2O2 stability of manganese peroxidase by combinatorial mutagenesis and high-throughput screening using in vitro expression with protein disulfide isomerase. Protein Eng. 2003;16:423–8. doi: 10.1093/protein/gzg054. [DOI] [PubMed] [Google Scholar]
- 28.Boyer ME, Wang CW, Swartz JR. Simultaneous expression and maturation of the iron-sulfur protein ferredoxin in a cell-free system. Biotechnol Bioeng. 2006;94:128–38. doi: 10.1002/bit.20830. [DOI] [PubMed] [Google Scholar]
- 29.Boyer ME, Stapleton JA, Kuchenreuther JM, Wang CW, Swartz JR. Cell-free synthesis and maturation of [FeFe] hydrogenases. Biotechnol Bioeng. 2008;99:59–67. doi: 10.1002/bit.21511. [DOI] [PubMed] [Google Scholar]
- 30.Ezure T, Suzuki T, Ando E, Nishimura O, Tsunasawa S. Expression of human Cu, Zn-superoxide dismutase in an insect cell-free system and its structural analysis by MALDI-TOF MS. J Biotechnol. 2009;144:287–92. doi: 10.1016/j.jbiotec.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 31.Matsuda T, Kigawa T, Koshiba S, Inoue M, Aoki M, Yamasaki K, et al. Cell-free synthesis of zinc-binding proteins. J Struct Funct Genomics. 2006;7:93–100. doi: 10.1007/s10969-006-9012-1. [DOI] [PubMed] [Google Scholar]
- 32.Irving H, Williams RJP. The stability of transition-metal complexes. J Chem Soc. 1953;3192−210. doi:10.1039/jr9530003192.
- 33.Outten CE, O’Halloran TV. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science. 2001;292:2488–92. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 34.Williams RJP. Chemical selection of elements by cells. Coord Chem Rev. 2001;216–217:583–95. doi: 10.1016/S0010-8545(00)00398-2. [DOI] [Google Scholar]
- 35.He MM, Clugston SL, Honek JF, Matthews BW. Determination of the structure of Escherichia coli glyoxalase I suggests a structural basis for differential metal activation. Biochemistry. 2000;39:8719–27. doi: 10.1021/bi000856g. [DOI] [PubMed] [Google Scholar]
- 36.Chai SC, Ju TT, Dang M, Goldsmith RB, Maroney MJ, Pochapsky TC. Characterization of metal binding in the active sites of acireductone dioxygenase isoforms from Klebsiella ATCC 8724. Biochemistry. 2008;47:2428–38. doi: 10.1021/bi7004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudev T, Lim C. Competition among metal ions for protein binding sites: determinants of metal ion selectivity in proteins. Chem Rev. 2014;114:538–56. doi: 10.1021/cr4004665. [DOI] [PubMed] [Google Scholar]
- 38.Rulíšek L, Vondrášek J. Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J Inorg Biochem. 1998;71:115–27. doi: 10.1016/S0162-0134(98)10042-9. [DOI] [PubMed] [Google Scholar]
- 39.McCall KA, Fierke CA. Probing determinants of the metal ion selectivity in carbonic anhydrase using mutagenesis. Biochemistry. 2004;43:3979–86. doi: 10.1021/bi0498914. [DOI] [PubMed] [Google Scholar]
- 40.Harding MM. Geometry of metal-ligand interactions in proteins. Acta Cryst. 2001;D57:401–11. doi: 10.1107/s0907444900019168. [DOI] [PubMed] [Google Scholar]
- 41.Leitgeb S, Straganz GD, Nidetzky B. Biochemical characterization and mutational analysis of the mononuclear non-haem Fe2+ site in Dke1, a cupin-type dioxygenase from Acinetobacter johnsonii. Biochem J. 2009;418:403–11. doi: 10.1042/BJ20081161. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Li W, Wang M, Zhu G, Liu D, Sun F, et al. Crystal structure and mutagenic analysis of GDOsp, a gentisate 1,2-dioxygenase from Silicibacter pomeroyi. Protein Sci. 2008;17:1362–73. doi: 10.1110/ps.035881.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–80. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 44.Yang WC, Patel KG, Wong HE, Swartz JR. Simplifying and streamlining Escherichia coli-based cell-free synthesis. Biotechnol Prog. 2012;28:413–20. doi: 10.1002/btpr.1509. [DOI] [PubMed] [Google Scholar]
- 45.Kim TW, Keum JW, Oh IS, Choi CY, Park CG, Kim DM. Simple procedures for the construction of a robust and cost-effective cell-free protein synthesis system. J Biotechnol. 2006;126:554–61. doi: 10.1016/j.jbiotec.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch EF, Maniatis T. In: Molecular cloning: a laboratory manual 2nd ed. Cold Spring Harbor NY, editor. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 47.Pfennig N. Rhodopseudomonas globiformis, sp. n., a new species of the Rhodospirillaceae. Arch Microbiol. 1974;100:197–206. doi: 10.1007/BF00446317. [DOI] [Google Scholar]
- 48.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 49.Zor T, Selinger Z. Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal Biochem. 1996;236:302–8. doi: 10.1006/abio.1996.0171. [DOI] [PubMed] [Google Scholar]
- 50.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–23. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Groemping Y, Hellmann N. Spectroscopic Methods for the Determination of Protein Interactions. Curr Protoc Protein Sci. 2005;39:20.8:20.8.1–20.8.27. doi: 10.1002/0471140864.ps2008s39. [DOI] [PubMed]
- 52.Randolph TW. Scale-based normalization of spectral data. Cancer Biomark. 2006;2:135–44. doi: 10.3233/cbm-2006-23-405. [DOI] [PubMed] [Google Scholar]