

Background: Rap1 is a general regulator of transcription and a component of the telomere.
Results: Rap1 binds DNA in at least two binding modes.
Conclusion: Access to binding modes in vitro is affected by different regions of the protein.
Significance: The ability of Rap1 to access different binding modes may impact its functions.
Keywords: Analytical Ultracentrifugation, Fluorescence Anisotropy, Isothermal Titration Calorimetry (ITC), Telomere, Transcription, Rap1, Binding Modes, Transcription
Abstract
Budding yeast Rap1 is a specific double-stranded DNA-binding protein involved in repression and activation of gene transcription and in the establishment of the nucleoprotein complex formed at telomeres. The DNA-binding domain (DBD) of Rap1 forms a high affinity complex with DNA where both Myb-like domains bind to the recognition site. However, we recently showed that the DBD can also access an alternative, lower affinity DNA-binding mode where a single Myb-like domain binds. This results in Rap1-DNA complexes with stoichiometry higher than previously anticipated. In this work, we show that the ability of the DBD to form higher stoichiometry complexes on DNA is maintained also in larger Rap1 constructs. This indicates that transition between at least two DNA-binding modes is a general property of the protein and not a specific feature of the DBD in isolation. The transition between binding modes is modulated by the C-terminal wrapping loop within the DBD, consistent with the proposed model in which the transient opening of this region allows a switch between binding modes. Finally, we provide evidence that the Rap1 C terminus interacts with the DNA-binding domain, suggesting a complex network of interactions that couples changes in conformation of the protein to the binding of its DNA recognition sequence.
Introduction
Saccharomyces cerevisiae Rap1 (repressor activator protein 1) is a multifunctioning protein acting as a key regulator of gene transcription and chromosomal integrity. Rap1 was first discovered as a repressor of the HMR/HML silent mating-type loci and an activator of gene expression for ribosomal protein genes and glycolytic enzymes (1–3), hence earning its designation as both a repressor and activator. Rap1 is also a critical regulator of telomere homeostasis responsible for proper maintenance of telomere length and integrity (4, 5). Found in abundance at telomeres (6), Rap1 is recruited to the highly repetitive, TG-rich regions to form the large nucleoprotein complexes that “cap” the chromosome ends. Rap1 binds directly to telomeric DNA (7, 8) and is responsible for recruitment of other proteins like the Rap1 interacting factors, Rif1 and Rif2, and Sir2, Sir3, and Sir4 (9–12). Together with Rif1 and Rif2, Rap1 forms the core of the yeast shelterin-like complex, which protects the chromosomal ends from DNA damage response processing (reviewed in Refs. 13–15).
Rap1 is a modular protein composed of at least three major domains (16). The N terminus contains a non-essential BRCA1 C-terminal (BRCT) domain whose function is not well understood but may be important for interactions with Gcr-1 (17, 18) and possibly DNA bending (19, 20). The DNA-binding domain (DBD)3 of Rap1, responsible for the specific interaction with the different recognition sequences (1, 21, 22), is centrally located within the 827-amino acid sequence, from residues 358–601. The DBD consists of two tandem Myb-like motifs, followed by a C-terminal wrapping loop (23–25). In the most recent crystal structure of the DBD bound to a telomeric recognition sequence, this loop interacts with the N-terminal Myb-like domain, forming a closed complex on DNA (25). Within the C-terminal wrapping loop, residues 591–597 were recently identified as being important for cell viability (25). Following the C-terminal wrapping loop is the tox region, identified as being responsible for toxicity upon overexpression of Rap1 (26), and the “act” domain, responsible for transcriptional activation (27). Finally, the Rap1 C-terminal (RCT) domain (residues 672–827) contains the region required for recruitment of the known Rap1-interacting proteins (9, 10, 28, 29).
We recently showed that in addition to the known high affinity complex observed in the crystal structures, the DBD forms, with lower affinity, complexes with higher stoichiometry (30). Access to these complexes is independent of the presence of a Rap1 recognition sequence. We proposed that access to these higher stoichiometry complexes is due to the ability of the DBD to transition between at least two DNA-binding modes. In the high affinity mode, both Myb-like domains bind to the hemisites within the recognition sequence. In the alternative lower affinity mode, only one Myb-like domain binds to DNA. One basic feature of this model proposes that the transient opening of the C-terminal wrapping loop within the DBD modulates the ability of Rap1 to access the alternative binding mode. In this work, we used different DBD constructs with varying lengths of the wrapping loop and different deletions of this region within larger Rap1 constructs to test the proposed model. We show that the residues spanning positions 591–597 of the DBD contain a region important for “latching” the C-terminal wrapping loop into the high affinity state. Deletion of this domain releases the “lock” of the 1:1 complex, favoring access to the alternate binding modes and thus formation of higher stoichiometric complexes. Moreover, we show that the ability to access higher stoichiometry on dsDNA (i.e. multiple binding modes) is not an exclusive property of the DBD in isolation but rather a feature maintained in the full-length protein. Also, we provide evidence that the RCT domain affects the maximum stoichiometry achievable on these model substrates and that complex conformational changes couple Rap1 to DNA binding.
EXPERIMENTAL PROCEDURES
Reagents and Buffers
All chemicals used were reagent grade. All solutions were prepared with distilled and deionized Milli-Q water (18 megaohms at 25 °C). Oligonucleotides were purchased from Integrated DNA Technology (Coralville, IA). Annealed duplex dsDNAs were prepared by mixing equimolar concentrations of each oligonucleotide strand in 20 mm HEPES (pH 7.4), 50 mm NaCl, 10% (v/v) glycerol, 2 mm MgCl2 and incubated in a preheated 95 °C water bath, followed by slow cooling to room temperature. The “top” strands of the oligonucleotides used are as follows (for DNAs with 6-carboxyfluorescein (FAM) or Cy3 the position is indicated, and the hemisites are underlined): TeloA, 5′-CCGCACACCCACACACCAGTG (5′- FAM, 3′-FAM, 5′-Cy3); RPG, 5′-CCGCACACCCATACATTAGTG (5′-FAM); HMRE, 5′-CCGCAAACCCATCAACCAGTG (5′-FAM, 3′-FAM, 5′-Cy3); RND, 5′-CCGCCGCGGAACTTATTAGTG (5′-FAM, 3′-FAM, 5′-Cy3); TeloS, 5′-CCGCACACCCACACCAGTG (5′-FAM, 5′-Cy3).
Cloning and Overexpression of Rap1 Constructs
Full-length Rap1 was initially cloned from S. cerevisiae strain W303 and provided in pET30a (Recombinant DNA Laboratory, University of Texas Medical Branch, Galveston, TX). All of the subsequent Rap1 constructs, including full-length Rap1, were generated with standard molecular biology techniques and inserted into pGEX-6p-1 at EcoRI and XhoI restriction sites. The RCT domain (residues 672–827) bearing the single C741S or C757S mutation was also cloned in pGEX-6p-1. The proteins were overexpressed from Rosetta2(DE3)pLysS by growing cells in LB-Miller broth at 37 °C until the A600 measured 0.6–0.8 and then quickly chilled and induced with 0.7 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C for overnight expression. Harvested cell pellets were stored at −80 °C for later use.
Purification of Rap1 Constructs
The DNA-binding domains of Rap1 comprising residues 358–579 (DBD579), 358–590 (DBD590), and 358–615 (DBD615) were purified with a two-column purification strategy as described recently (30). Briefly, after cell lysis, the clarified supernatant was incubated with 0.3% (v/v) polyethyleneimine, recovered in the supernatant, and incubated overnight with glutathione-Sepharose 4 Fast Flow GST affinity resin (GE Healthcare). After on-column cleavage of the GST tag, the DBD constructs were directly loaded on a Poros 50 HE Heparin (Life Technologies, Inc.) and eluted at 600 mm NaCl. The construct comprising residues 1–601 (Rap1(1–601)) and 440–601 (Myb-C) followed the same procedure. Rap1 constructs comprising residues 358–827 (DBD827, DBD + linker + RCT domain) and its internal deletion constructs were eluted from Poros 50 HE Heparin with 300 mm NaCl. Full-length Rap1 and residues 440–827 (Myb-C827) were dialyzed overnight against Buffer D50 (20 mm Tris-HCl (pH 8.3 at 4 °C), 50 mm NaCl, 10% (v/v) glycerol, 1 mm DTT, 0.5 mm EDTA, and 0.1 mm PMSF) before loading onto heparin and then eluted with Buffer D containing 250 mm NaCl. The RCT domain variants (residues 672–827) were purified with glutathione-Sepharose 4 Fast Flow GST affinity resin after on-column cleavage. Finally, all of the protein constructs were dialyzed overnight against Storage Buffer (20 mm HEPES (pH 7.4), 400 mm NaCl, 40% (v/v) glycerol, 1 mm DTT, and 0.5 mm EDTA) and stored at −80 °C for later use. Unless otherwise indicated, prior to experiments, the proteins were dialyzed against Buffer HN50 (20 mm HEPES, pH 7.4, 50 mm NaCl, 2 mm MgCl2, 10% (v/v) glycerol), including 1 mm tris(2-carboxyethyl)phosphine for cysteine-containing constructs. All proteins were quantified spectrophotometrically using calculated extinction coefficients (31, 32).
The RCT domains containing a single cysteine were labeled with 6-carboxyfluorescein maleimide (Invitrogen) by incubating overnight with a 10-fold excess of dye in 20 mm sodium phosphate (pH 7.5), 400 mm NaCl, 1 mm EDTA, 10% (v/v) glycerol, and 50 μm tris(2-carboxyethyl)phosphine). Unincorporated dye was removed by size exclusion using Bio-Rad P6 matrix. The labeled protein was concentrated and dialyzed overnight against Storage Buffer before storing at −80 °C. Before use, the labeled RCT domain (FAMRCT) was dialyzed in Buffer HN50 and quantitated using the extinction coefficient of FAM at 495 nm (78,000 liters mol−1 cm−1). The different Rap1 constructs and their abbreviations are shown in Fig. 1b.
FIGURE 1.

a, model for how the DNA-binding domain of Rap1 can access different binding modes on dsDNA, thus forming high stoichiometry complexes. b, schematic representation of Rap1 domain organization and list of the different Rap1 constructs used.
Analytical Ultracentrifugation
All sedimentation experiments were collected on an Optima XL-A analytical ultracentrifuge using a An60Ti rotor (Beckman Coulter, Brea, CA). Sedimentation equilibrium experiments were performed using Epon charcoal-filled six-sector centerpieces at the appropriate rpm with 0.001-cm spacing, scanned every 4 h, averaged from 10 replicates, and recorded at 545 nm to monitor the Cy3-labeled DNA. The data were processed and analyzed with SedFit/SedPhat (Peter Schuck) (33–36). The apparent molecular weights of the complexes were determined as described (30). Sedimentation velocity experiments with the labeled RCT domain were performed using Epon charcoal-filled two-sector centerpieces and scanning at 500 nm, where the unlabeled proteins do not contribute to the signal.
Electrophoretic Mobility Shift Assays (EMSAs)
EMSAs with labeled or unlabeled dsDNAs were performed on 6 or 8% acrylamide/bisacrylamide 1× TBE minigels with running buffer prechilled at 4 °C. Gels were scanned using a Typhoon 9400 variable mode imager (GE Healthcare). EMSAs with unlabeled DNAs were stained with GelRed (Phenix Research, Candler, NC) and scanned on an Alpha Imager HP imager (Protein Simple, Santa Clara, CA).
Equilibrium Fluorescence Titrations
All fluorescence titrations were performed with an L-format PC1 spectrofluorimeter (ISS, Champaign, IL) equipped with Glenn-Thompson polarizers. Measurements of the anisotropy and total fluorescence intensity of FAM-labeled DNA were recorded using excitation and emission wavelengths of 480 and 530 nm, respectively, as described (30).
Isothermal Titration Calorimetry (ITC)
The experiments were performed using a VP-ITC calorimeter (Microcal, GE Healthcare) after extensive dialysis of both the protein and the DNA in Buffer HN50. Titrations were carried out with 10-μl injections of 20 μm titrant into 2 μm samples containing either protein or duplex DNA, at either 20 or 30 °C. Reference titrations to account for the heat of dilution of each titrant were performed by titrating into sample cells loaded with the appropriate reaction buffer and equilibrated at the appropriate temperature.
RESULTS
C-terminal Wrapping Loop of the DBD Modulates Access to Higher Stoichiometry on DNA and Deletion of the “Latch” Is Sufficient to Favor Transition in Binding Modes
Fig. 1a shows the model we recently proposed to explain the ability of the DBD of Rap1 to access different binding modes on dsDNA (30). One prediction of this model is that deletion of the C-terminal wrapping loop of the DBD should favor the transition between binding modes. To test this prediction, we generated constructs where the C-terminal wrapping loop has been partially or entirely deleted (Fig. 1b). The first DBD variant comprises residues 358–590 (DBD590), which removes the latch that makes direct interaction with the N-terminal Myb-like domain of the DBD (25). The second DBD variant comprises residues 358–579 (DBD579), which remove the entire C-terminal wrapping loop as defined by the crystal structure of the DBD bound to DNA (23–25). As a control, we also generated an extended version of the DBD that comprises residues 358–615 (DBD615) to include part of the toxicity region shown to relieve the growth inhibition phenotype observed by overexpression of Rap1 (26). We performed EMSAs at different DNA concentrations to target either formation of the high affinity, 1:1 complex or the lower affinity, higher stoichiometry complexes. Fig. 2, a and b, shows EMSAs at 30 nm TeloA or HMRE (labeled at the 5′-end with FAM) as a function of the concentration of the different DBD constructs. For TeloA, containing the high affinity Rap1 recognition sequence found at telomeres (7, 37, 38), all of the DBDs bind to the substrate, with little apparent effect of deleting the C-terminal wrapping loop. However, for HMRE, which contains the lower affinity recognition sequence found at the HM locus (2), DBD579 does not bind to the substrate at this concentration. Even for DBD590, which is missing the latch region, the affinity for HMRE appears to be reduced. This suggests that the C-terminal wrapping loop and potentially just the latch region contribute significantly to the stabilization of the 1:1 complex formed on a moderate affinity recognition sequence.
FIGURE 2.

The C-terminal wrapping loop regulates access to binding modes. a, EMSA as a function of -fold excess of the different DBD constructs for 30 nm TeloA, labeled at the 5′-end of the top strand with FAM (see “Experimental Procedures”). b, same experiments as in a but for HMRE. c and d, EMSA as a function of -fold excess of the different DBD constructs targeting the protein-DNA complexes formed with 2 μm unlabeled TeloA in c and HMRE in d. e, change in fluorescence anisotropy of dsDNAs FAM-labeled at the 5′-end of the top strand. The experiments were performed with 255 nm TeloA (black circles), RND (gray circles), or HMRE (open circles). Shown are the data for DBD615 (left), DBD590 (middle), and DBD579 (right).
The stoichiometry (Table 1) of the protein-DNA complexes formed with the different DBD variants at saturating protein concentration was determined by equilibrium analytical ultracentrifugation experiments with TeloA, HMRE, RND (a dsDNA of random composition), and TeloS (a 19-bp dsDNA that contains two direct repeats as in TeloA but spaced by only 1 bp) (see “Experimental Procedures”). Independent of the dsDNA sequence, all of the tested DBD constructs form a 3:1 complex at saturation, indicating that the changes in the C-terminal wrapping loop do not affect the maximum achievable stoichiometry.
TABLE 1.
Molecular mass of protein-DNA complexes for different DBD constructs determined by equilibrium analytical sedimentation monitoring the absorbance of Cy3 at 550 nm
| DBD615 |
DBD590 |
DBD579 |
||||
|---|---|---|---|---|---|---|
| Mass (observed) | P/D | Mass (observed) | P/D | Mass (observed) | P/D | |
| kDa | kDa | kDa | ||||
| Cy3-TeloA | ND | ND | 97.2 ± 0.8 | 2.98 ± 0.03 | 85.8 ± 0.2 | 2.67 ± 0.07 |
| Cy3-HMRE | 95.0 ± 0.3 | 2.63 ± 0.01 | 103.6 ± 0.4 | 3.20 ± 0.02 | 104.7 ± 0.6 | 3.37 ± 0.02 |
| Cy3-RND | ND | ND | ND | ND | 101.3 ± 0.3 | 3.25 ± 0.09 |
| Cy3-TeloS | 93.3 ± 0.7 | 2.61 ± 0.03 | 91.5 ± 0.1 | 2.8 ± 0.04 | 94.0 ± 0.2 | 3.02 ± 0.07 |
Next, we examined the propensity of the different DBD constructs to access these higher stoichiometry complexes. Fig. 2, c and d, shows EMSAs targeting the singly (PD) and multiply bound (PnD) species formed at 2 μm TeloA or HMRE. For TeloA, the three examined DBD constructs show very similar behavior. However, for HMRE, the data show that as the C-terminal wrapping loop is shortened, the DBD becomes more prone to access the higher stoichiometry complexes. Indeed, for DBD579, formation of PD is not clearly detectable, and at a 2-fold excess of protein, the complex is already in the multiply bound state (PnD). This suggests that removal of the C-terminal wrapping loop in the DBD favors transition to higher stoichiometry (i.e. transition between binding modes).
Binding of the different DBD variants was also monitored by the change in anisotropy of FAM-labeled DNA (Fig. 2e). As recently reported for the DBD construct terminated at residue 601 (30), the observed change in anisotropy depends on whether or not the DNA contains a Rap1 recognition sequence. For 5′-FAM-labeled dsDNA containing a Rap1 recognition sequence (TeloA, HMRE), the fluorescence anisotropy is insensitive to the binding of the first DBD molecule, with major contributions to the signal arising from the binding of the second and third molecule. However, for a dsDNA of random sequence composition (RND), the signal is sensitive to the binding of the first DBD molecule. Extending the wrapping loop to residue 615 (DBD615) to include part of the toxicity region does not have any significant effect on the different fluorescence signals as compared with DBD601 (30). Deletion of the latch (DBD590) or the entire wrapping loop (DBD579) affects the anisotropy response for HMRE, whereas it does not affect TeloA (Fig. 2e, second and third columns). For HMRE, the signal response becomes almost indistinguishable from that of the random DNA.
The differences in signal between the RND substrate and those containing the recognition sequence are consistent with the formation of different complexes on DNA. Moreover, binding to a DNA of random composition (or a lower affinity Rap1 recognition sequence) appears to favor transition of the DBD between binding modes (30). The data in Fig. 2 show that it is sufficient to delete the latch region of the wrapping loop to generate a DBD construct that, on a lower affinity substrate (HMRE), is less sensitive to the presence of the Rap1 recognition sequence. These data strongly support the conclusion that the wrapping loop is a crucial modulator of both the 1:1 complex (high affinity and proper conformation) and the access to different binding modes.
Access to Higher Stoichiometry Complexes on DNA Is Maintained in Larger Rap1 Constructs
Above, we showed that the ability of the DBD to populate the 3:1 complex on DNA (i.e. switch between binding modes) is modulated by the presence of the C-terminal wrapping loop, consistent with the model in Fig. 1a. We next asked whether the N-terminal region or the RCT domain affects the ability of Rap1 to access higher stoichiometry (i.e. binding modes). In electromobility shift assays, a construct comprising amino acids 1–601, which contains the entire N-terminal region including the BRCT domain, behaves qualitatively similar to the DBD (not shown). Access to the observed higher stoichiometry and the accompanying change in binding modes is not hampered by the presence of the N-terminal region of Rap1. This is consistent with the reported lack of preferential interactions of this region with the rest of the protein (25).
Next, we examined the effect of the C-terminal region of Rap1 on the ability to access high stoichiometry and change in binding modes. For this, we expressed a region of Rap1 comprising residues 358–827 (DBD827) (25). In addition to the DBD, this construct contains the latch region, the “tox” region, and the entire RCT domain. EMSAs targeting the protein-DNA complexes formed at 2 μm unlabeled TeloA, RPG, HMRE, or TeloS show a clear presence of supershifts at the highest concentrations of DBD827, consistent with the formation of higher stoichiometry complexes on DNA (Fig. 3a). Equilibrium analytical ultracentrifugation shows that approximately two molecules of DBD827 can bind at saturation on the three different substrates containing a Rap1 recognition sequence (Table 2). These data strongly suggest that DBD827 can still access higher stoichiometry on DNA and thus transition between binding modes.
FIGURE 3.
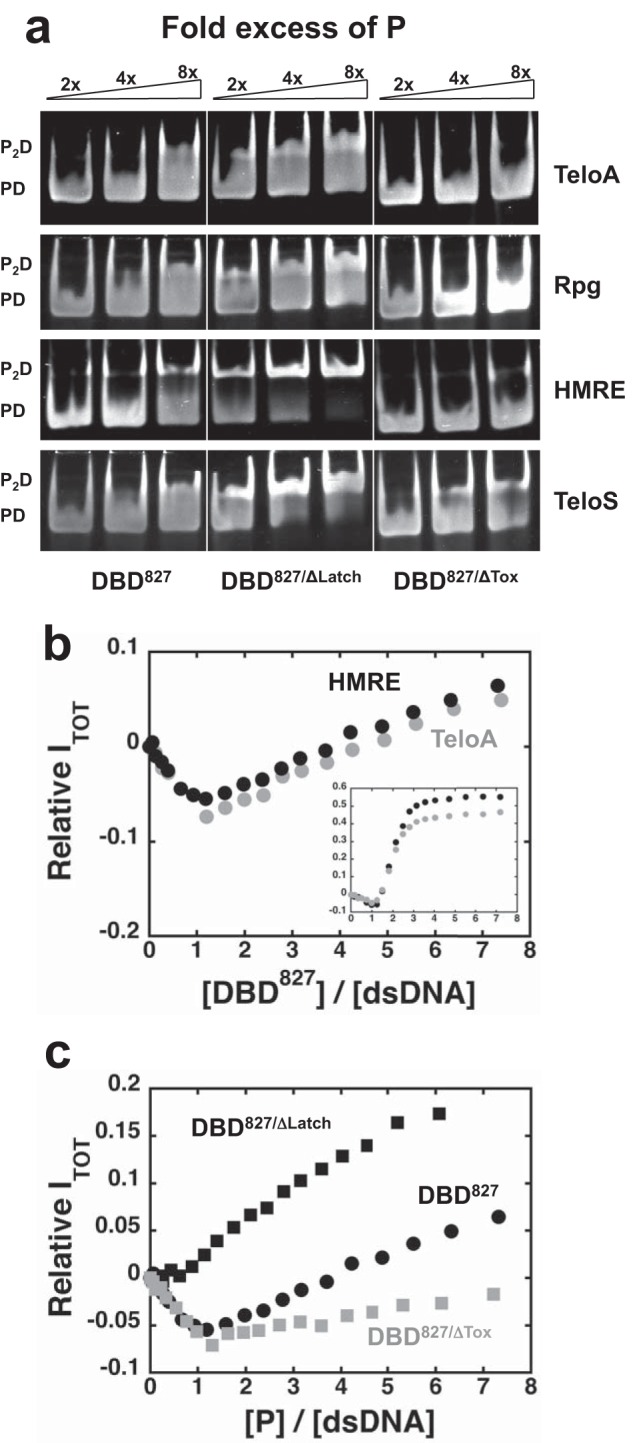
DBD827 can access higher stoichiometry on DNA and it is regulated by the latch and tox regions. a, EMSA targeting the protein-DNA complexes as a function of -fold excess of the DBD827, DBD827/ΔLatch, and DBD827/ΔTox for 2 μm unlabeled TeloA, RPG, HMRE, and TeloS. b, change in relative total fluorescence intensity as a function of DBD827/DNA ratio for 255 nm TeloA (gray) or HMRE (black) FAM-labeled at the 3′-end. Inset, same data for the DBD615 construct. c, change in relative total fluorescence intensity for 255 nm HMRE (FAM-labeled at the 3′-end) for the three different DBD827 constructs.
TABLE 2.
Molecular mass of protein-DNA complexes for different DBD827 constructs determined by equilibrium analytical sedimentation
| DBD827 |
DBD827/ΔTox |
DBD827/ΔLatch |
||||
|---|---|---|---|---|---|---|
| Mass (observed) | P/D | Mass (observed) | P/D | Mass (observed) | P/D | |
| kDa | kDa | kDa | ||||
| Cy3-TeloA | 103.7 ± 0.3 | 1.64 ± 0.02 | 101.4 ± 0.3 | 1.66 ± 0.02 | 128.3 ± 0.3 | 2.12 ± 0.02 |
| Cy3-HMRE | 113.2 ± 0.4 | 1.82 ± 0.02 | 119.1 ± 0.2 | 2.0 ± 0.01 | 130.8 ± 0.3 | 2.17 ± 0.01 |
| Cy3-TeloS | 107.5 ± 0.3 | 1.74 ± 0.02 | 105.0 ± 0.2 | 1.75 ± 0.01 | 132.6 ± 0.2 | 2.23 ± 0.01 |
Interestingly, we found that all of the spectroscopic signals observed upon binding to labeled substrates of the different variants containing only the DBD are dampened with this larger Rap1 construct. This suggests that the ability of a third molecule of the DBDs to bind to the dsDNA contributes significantly to the observed signals. Fig. 3b shows the change in relative total fluorescence intensity of TeloA and HMRE with FAM at the 3′-end (see “Experimental Procedures”). With the label at this position, binding of the first molecule of DBD615 (Fig. 3b, inset) leads to ∼7–9% quenching followed by a large ∼50–60% fluorescence increase upon binding of the second and third molecule. For DBD827, binding of the first protein molecule is accompanied by a similar quenching of fluorescence followed by only a small, yet detectable, fluorescence increase. Notably, the presence of fluorescence changes of opposite sign is a clear indication that more than one molecule of DBD827 binds to the DNA.
The Latch and Tox Regions in the C-terminal Wrapping Loop Affect the Ability of DBD827 to Transition between Binding Modes
The data in Fig. 2 strongly suggest that removal of the latch in the C-terminal wrapping loop of the DBD (DBD590) is sufficient to favor transition to higher stoichiometry complexes. Also, the addition of the part of the tox region to the DBD (DBD615) does not appear to affect the ability to transition between binding modes. Next, we deleted the region 591–597 (DBD827/ΔLatch) or 598–616 (DBD827/ΔTox) in the context of the DBD827 construct.
Equilibrium analytical ultracentrifugation showed that at saturating protein concentration, these constructs form a 2:1 complex with the dsDNA studied (Table 2), indicating that deletion of the latch or tox region does not prevent transition between binding modes. Similar to what was observed for the DBD constructs, EMSAs with the high affinity TeloA or RPG substrate did not show substantial differences between the different DBD827 constructs (Fig. 3a). However, EMSAs at 2 μm HMRE, the lower affinity Rap1 recognition sequence, or TeloS, where the intersite spacing was reduced from 3 to 1 bp, showed that for DBD827/ΔLatch, supershifts appear at lower concentrations than for DBD827. Also, for DBD827/ΔTox, the EMSAs at this DNA concentration did not reveal significant supershifts on HMRE, despite the fact that equilibrium analytical ultracentrifugation shows formation of a 2:1 complex. This is possibly due to some dissociation of the complexes during electrophoresis, suggesting that the 2:1 complex has lower affinity. Instead, for this construct, clear supershifts were observed with the TeloS sequence, strongly suggesting that removal of the tox region does not completely abolish the ability of Rap1 to access higher stoichiometry complexes. A similar trend is also observed when monitoring the change in total fluorescence of HMRE with FAM at the 3′-end (Fig. 3c). Compared with DBD827, for DBD827/ΔLatch, binding of the first protein molecule does not lead to an initial fluorescence quenching but rather to an immediate fluorescence increase. For DBD827/ΔTox, binding of the first molecule is accompanied by a similar fluorescence quenching followed by a smaller fluorescence increase. Consistent with what we showed for DBD590, the data strongly suggest that for DBD827, removal of the latch also favors transition to higher stoichiometry and a switch in binding modes. However, deletion of the tox region stabilizes the 1:1 complex, thus disfavoring transition in binding modes.
ITC Shows a Complex Coupling between DNA Binding and Conformational Changes
We previously showed that for the DBD601 asymmetrical ITC isotherms are obtained, depending on whether protein is titrated into DNA (P into D, P/D molar ratio) or vice versa (D into P, D/P molar ratio), consistent with a complex behavior originating from more than one protein molecule binding to the DNA (30). We used the same strategy to study DBD827 binding to dsDNA and the effect of deleting the latch or tox regions. Fig. 4a shows the isotherms obtained for DBD827 and TeloA at 20 °C in Buffer HN50. The two isotherms are moderately asymmetrical, and the midpoint of the P into D isotherm is lower than expected for binding of two DBD827 molecules (Table 2). It is possible that for TeloA, the observed molar heat is dominated by the ΔH of the high affinity binding of the first Rap1 molecule, with binding of the second, lower affinity molecule contributing little to the overall signal.
FIGURE 4.

ITC of different DBD827 constructs binding DNA. a, isotherms in buffer HN50 at 20 °C for 20 μm protein titrated into 2 μm TeloA (black, P/D) or vice versa (gray, D/P, DNA into protein). b, same experiments as in a but at 30 °C. Open circles, P into D isotherm for DBD827 in buffer HN50 devoid of MgCl2. Gray triangles, P into D isotherm for DBD615. c, isotherms in buffer HN50 at 30 °C for the HMRE sequence. d, same experiments as in b but in buffer HN150. e and f, isotherms in buffer HN50 at 30 °C for DBD827/ΔLatch (e) and DBD827/ΔTox (f).
With the idea to reduce the affinity for TeloA, next we performed the same experiments at higher temperature (30 °C; Fig. 4b). At this higher temperature, the two isotherms are highly asymmetrical. For the D into P isotherm, the increase in temperature leads to a gain of ∼−10 kcal/mol in ΔH. However, the midpoint of the transition is not affected. To our surprise, the P into D isotherm is dramatically changed compared with titrations at 20 °C. The midpoint of the transition observed at higher P/D ratio is significantly shifted to the right, suggesting that under these conditions, the binding of the second Rap1 molecule is now being detected. Moreover, the maximum change in ΔH (plateau at ∼−17–18 kcal/mol) is almost a factor of 2 lower than for the D into P isotherm (∼−35 kcal/mol). Strikingly, at lower P/D ratios, the data show an increasing molar heat rather than a plateau. This behavior is independent of the presence of Mg2+ in solution (Fig. 4b, open circles). Moreover, similar results are obtained with the lower affinity HMRE substrate (Fig. 4c), suggesting that the observed behavior of the P into D isotherm is independent of the nature of the Rap1 recognition sequence. However, for HMRE, binding of DBD827 is accompanied by an overall decrease in the observed ΔH, as expected by formation of different protein-DNA contacts (24). Furthermore, under these conditions, a qualitatively similar behavior is also observed for DBD615 that is missing the RCT domain (Fig. 4b, gray triangles). ITC experiments at 30 °C were also performed at higher NaCl concentration (150 mm) with the TeloA substrate (Fig. 4d). At this higher NaCl concentration, the asymmetry in the ΔH for the experiments performed in either direction is reduced. However, asymmetry is still present for the midpoint of the transition, suggesting that even at this higher NaCl concentration, at least one additional DBD827 molecule can bind to the substrate, albeit with lower affinity compared with the lower NaCl concentration. Moreover, for the P into D titration at this higher NaCl concentration, the initial increase in ΔH is not present, suggesting that either the ΔH for this step is minimal, or it does not occur at this higher NaCl concentration.
In order to test the effect of the internal deletions within the C-terminal wrapping loop in the DBD827 construct, ITC experiments were also performed for DBD827/ΔLatch and DBD827/ΔTox binding to TeloA. At 30 °C in Buffer HN50 for DBD827/ΔLatch, the asymmetrical behavior of the isotherms is still present (Fig. 4e), consistent with the binding of at least two Rap1 molecules (Table 2). However, for this construct, removal of the latch leads to a decrease in the overall ΔH and, as compared with DBD827, a much smaller difference in the maximum molar heat between the two isotherms. Also, the increase in ΔH at lower P/D ratios observed for DBD827 is not as pronounced, if present at all. Similar results were obtained with DBD827/ΔTox (Fig. 4f). For this construct, the difference in midpoint between the two isotherms is even lower. Again, this suggests that the lower affinity binding of a second Rap1 molecule is masked by the formation of the high affinity 1:1 complex. Also, for the D into P titration, when the tox region is removed, the overall ΔH is not strongly affected. However, despite the fact that this construct contains the latch region, for the P into D titration, the initial decrease in ΔH is not very pronounced, and also the observed ΔH at the plateau (∼−25 kcal/mol) is higher than for the wild-type construct (∼−17–18 kcal/mol). The removal of different regions in the C-terminal wrapping loop affects differently the overall ΔH, and this suggests the presence of additional interactions within the protein.
The RCT Domain Interacts with the DBD
Above, we showed that the ability of the DBD to access higher stoichiometry on DNA is maintained also within the larger Rap1 constructs containing the linker and RCT domain. However, the data also suggest that the presence of the RCT domain affects the ability to access these complexes. In addition, formation of the 1:1 complex on DNA is accompanied by a different behavior when monitored by ITC. Matot et al. (25) showed that binding of Rap1 to DNA induces a large conformational change of the RCT domain and that the N-terminal region of Rap1 does not interact with the rest of the protein. Therefore, next we asked whether the RCT domain behaves as an independent domain or whether it can interact with any other region of Rap1. For this, we generated a RCT domain variant where either one of the two natural cysteines was mutated to serine, and the other one was labeled with FAM. Similar results were obtained with either labeled RCT domain construct, and the data are shown for the C741S variant. The distribution of sedimentation coefficients for FAMRCT in Fig. 5a shows a single peak consistent with the presence of a monomer in solution (24). Fig. 5a also shows the distribution of sedimentation coefficients of the labeled FAMRCT in the presence of a 3-fold excess of DBD615, DBD601, and DBD579. Independent of the degree of change, the increase in sedimentation coefficient of the RCT domain in the presence of the different DBD constructs strongly suggests that the RCT domain can interact with the DBD. Moreover, a small yet detectable change in the sedimentation coefficient of the RCT domain is also observed in the presence of a Rap1 construct spanning amino acids 440–601 (Myb-C), containing the C-terminal Myb-like domain and the wrapping loop (Fig. 5a). This suggests that although the N-terminal Myb-like domain might contribute to the interaction, it is not required. Because of the reported large conformational change of the RCT domain upon Rap1 binding to DNA (25), next we tested whether binding of the DBD to a DNA containing its recognition sequence would affect its ability to interact with the RCT domain in trans. Fig. 5b shows the distribution of sedimentation coefficients of the RCT domain in the presence of a 3-fold excess of DBD615 in a 1:1 complex with either TeloA or HMRE. Independent of the recognition sequence when the DBD is bound to DNA the sedimentation coefficient of the RCT domain is little affected, suggesting that now the DBD can no longer interact.
FIGURE 5.
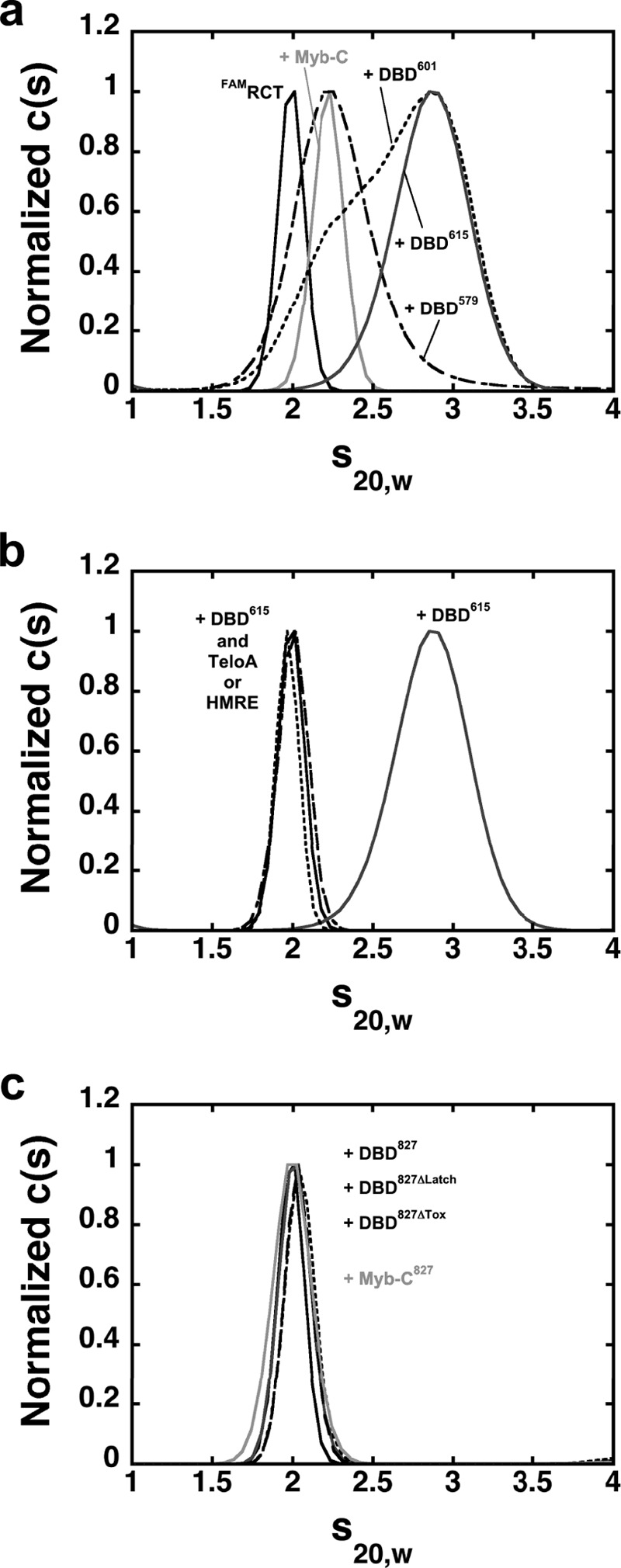
The RCT domain interacts with the DBD. a, distribution of sedimentation coefficients for FAMRCT alone (solid) or in the presence of excess DBD590 (dash), DBD601 (dotted), DBD615 (dark gray), or Myb-C (light gray). b, distribution of sedimentation coefficients of FAMRCT alone (solid) or in the presence of excess DBD615 and TeloA (dotted) or HMRE (dash). The data for FAMRCT + DBD615 (dark gray) are shown for reference. c, distribution of sedimentation coefficients of FAMRCT alone (solid) and in the presence of excess DBD827 (dark gray), DBD827/ΔLatch (dotted), DBD827/ΔTox (dash), or Myb-C827 (light gray).
Next we asked whether the RCT domain could still bind in trans to the DBD within the DBD827 constructs, where the RCT domain is already present in cis. Fig. 5c shows the distribution of sedimentation coefficients of the RCT domain in the presence of a 2-fold excess of DBD827, DBD827/ΔLatch, or DBD827/ΔTox. The lack of any significant change in the sedimentation coefficient strongly suggests that the RCT domain cannot bind in trans to the DBD when a RCT domain is already present in cis. At the concentration of proteins used, the data show that within the Rap1 protein, binding of the RCT domain to the DBD cannot be competed by the addition of the RCT domain in trans, pointing to an interaction that is much stronger than the one observed for the binding of this domain in trans. Also, neither DBD827/ΔLatch nor DBD827/ΔTox can bind the RCT domain in trans, showing that the interaction in cis of this domain with the DBD is independent of this region. Finally, we also generated a Rap1 construct spanning amino acids 440–827 (Myb-C827) that contains only the C-terminal Myb-like domain, the linker region, and the RCT domain. The sedimentation coefficient of the FAMRCT in the presence of a 10-fold excess of this construct is little affected (Fig. 5c), strongly suggesting that the binding in trans of the labeled RCT domain is blocked by the presence of the RCT domain in cis. Consistent with the ability of the RCT domain to interact with the C-terminal Myb-like domain in trans (Fig. 5a), this suggests that this single Myb-like domain of the DBD is sufficient for the RCT domain to bind in cis.
Access to Higher Stoichiometry Is Maintained in Full-length Rap1
We showed that transition to higher stoichiometries on DNA (i.e. binding mode switch) is a clear property of the DBD, and it is maintained in the larger construct containing the entire region C-terminal to the DBD, including the RCT domain. Next, we asked whether the ability to transition between binding modes is maintained in full-length Rap1 and whether the latch region is involved in regulating access to the higher stoichiometry complexes. EMSAs at 300 nm FAM-labeled HMRE as a function of increasing Rap1 concentration in Fig. 6a (left) show that on this substrate, Rap1 forms a clear supershift, consistent with the binding of more than one Rap1 protein. Also, the ability of Rap1 to form a supershift is independent of the presence of a recognition sequence (Fig. 6a, center). Rather, on this dsDNA of random composition, the higher stoichiometry complex is formed at lower Rap1 concentration, strongly suggesting that transition between binding modes is favored. Finally, the right panel of Fig. 6a shows an EMSA of FAM-labeled HMRE for a Rap1 construct missing amino acids 591–597 (Rap1ΔLatch). For this construct, formation of supershifts occurs at lower protein concentration as compared with the full-length protein, suggesting that deletion of the latch favors the binding of at least an additional protein molecule (i.e. switch in binding modes).
FIGURE 6.

The ability to access higher stoichiometry on DNA is maintained in full-length Rap1. a, EMSAs with 5′-FAM-labeled 300 nm HMRE (left) or RND (center) as a function of -fold excess of Rap1 and HMRE with Rap1ΔLatch (right). b, ITC isotherms in buffer HN50 at 30 °C for 20 μm Rap1 titrated into 2 μm TeloA (black, P/D) or vice versa (gray, D/P). c, isotherms for Rap1 binding to HMRE. d, isotherms for Rap1ΔLatch binding to TeloA.
ITC experiments in Fig. 4 showed clear evidence of a complex coupling between multiple DBD827 binding and protein conformational changes. Next, we performed the same experiments at 30 °C with full-length Rap1 and Rap1ΔLatch. Fig. 6b shows the two isotherms for Rap1 binding to TeloA. For this high affinity substrate, the isotherms are only slightly asymmetrical, suggesting that if a second Rap1 molecule binds, it is not clearly detectable. However, for the lower affinity HMRE substrate, the two isotherms are highly asymmetrical, providing clear evidence that at least a second Rap1 molecule can bind. For Rap1ΔLatch, the isotherms with TeloA are more asymmetrical as compared with Rap1. This suggests that removal of the latch may favor binding of a second Rap1 molecule, consistent with the EMSAs. However, for this construct, removal of the latch has a significant effect on the overall ΔH, similar to what was observed for DBD827.
DISCUSSION
We recently showed that in addition to forming a high affinity 1:1 complex with dsDNAs containing its recognition sequence, the DNA-binding domain of S. cerevisiae Rap1 can also form, with lower affinity, higher stoichiometry complexes (30). We proposed that the ability of the DBD to access these complexes is due to a change in DNA binding modes (Fig. 1a). In the high affinity binding mode, both Myb-like domains are bound to the two hemisites of the recognition sequence. In the alternative, lower affinity binding mode, only a single Myb-like domain binds to the dsDNA. The data in this work showed that the ability to transition between the two binding modes (i.e. access to higher stoichiometry complexes) is not an exclusive property of the DBD in isolation; rather it is a general feature maintained in larger Rap1 constructs. Both full-length Rap1 and DBD constructs containing either the N-terminal (residues 1–601) or C-terminal (residues 358–827) regions can form higher stoichiometry complexes on dsDNA, independent of a Rap1 recognition sequence. However, the data showed that the presence of the C-terminal region in the DBD827 construct has an effect on the maximum stoichiometry achievable on these short dsDNA substrates. Although all of the DBD constructs studied can form a 3:1 complex with the dsDNA at saturation, only two molecules of DBD827 can bind under the same conditions. The presence of the C-terminal region past the DBD effectively abolishes binding of the third molecule, observed for the different DBD constructs. This could be due to a lowering of the affinity for the DBD bound in the second binding mode (e.g. single Myb-like domain interacting) with the result that binding of the third molecule, if it happens at all, is so weak that is not detectable. Alternatively, this might result from a steric exclusion on the DNA due to the larger size of these protein constructs.
The model in Fig. 1a proposes that the first step for the DBD to transition between binding modes is the transient opening of the C-terminal wrapping loop, shown in the crystal structure to interact with the N-terminal Myb-like domain to form a closed complex on dsDNA (23–25). A direct prediction of this model is that removal of the C-terminal wrapping loop should favor the transition to the second, lower affinity binding mode. This would result in easier access to the higher stoichiometry complexes formed on DNA. Indeed, the data indicate that when the wrapping loop is deleted in the DBD, these complexes are formed at lower protein concentration, consistent with the prediction of the model. Furthermore, the data also suggest that within the DBD, it is sufficient to remove merely the latch region of the wrapping loop to favor the transition between binding modes. This is further confirmed by the observation that deletion of this same region in either the full-length or DBD827 likewise favors formation of the higher stoichiometry complexes. In addition, for both full-length and DBD827, deletion of the latch region results in a significant decrease in ΔH of the interaction (Figs. 4c and 6d). This strongly suggests that when the latch is removed, protein-protein and protein-DNA contacts are being lost. A similar effect on ΔH was observed by Matot et al. when performing ITC experiments titrating DNA into protein (25). Interestingly, the authors reported isotherms with ΔH of the opposite sign compared with the ones in this work. It is possible that differences in solution conditions contribute to this discrepancy. Nonetheless, our results reinforce the proposal that the latch region changes the interaction of Rap1 with DNA. Moreover, our data show that its removal indeed favors transition between binding modes, and possibly this provides a reason why this construct cannot fully complement wild-type Rap1 (25).
Removal of the wrapping loop also affects formation of the 1:1 complex. For the lower affinity HMRE substrate, deletion of the wrapping loop has a significant effect on the stability of this complex (Fig. 2b). This indicates that the closure of the loop and possibly the latch region has a major contribution to the formation of the high affinity complex on dsDNA observed in the crystal structures (23–25). It is interesting to speculate that the affinity (i.e. specificity) for different Rap1 sequences does not originate exclusively from the specific base recognition within the Myb-like domains but also from the wrapping loop region and/or the latch and the ability of different sequences to allow for an optimal closed state on DNA. Interestingly, we found that although removal of part of the tox region in the DBD827 construct still allows for a 2:1 complex, its formation is hindered on DNAs containing a high affinity recognition sequence. For this construct, a 2:1 complex becomes more evident on TeloS, where the spacing between the half-sites has been reduced. It has been shown that this region in Rap1 is a major determinant for the toxic phenotype observed upon overexpression of the protein (26). The ability of any Rap1 construct tested to access higher stoichiometry complexes as a function of protein concentration shows that this property is not exclusive to the DNA-binding domain. Although Rap1 is an abundant protein (4390 molecules/cell (39)), it remains to be determined whether the higher stoichiometries can be accessed in vivo. However, under overexpression conditions, where Rap1 becomes toxic, it is interesting to speculate that at least in part, the ability to form high stoichiometry complexes on DNA might provide a reason for its toxicity. The observation that removal of the tox region makes DBD827 less prone to form the 2:1 complex may possibly be a means to bypass the toxic effect upon overexpression. In addition, the ability to access alternative binding modes could play a role at telomeres, where the heterogeneity of Rap1 sites could lead to a heterogeneous nucleoprotein complex and/or where binding of Rap1-interacting factors could modulate its DNA binding mode.
ITC experiments with DBD827 at 30 °C showed an unexpected behavior at the lower P/D ratios, with the ΔH increasing. For the P into D isotherm, at these P/D ratios, the DNA is in excess relative to protein, and thus the 1:1 complex is populated. Intuitively, this behavior might be expected if binding of the protein to the DNA is coupled to a conformational change of the protein, with a ΔH of the opposite sign. For example, this could arise from a change in oligomeric state of the protein coupled to DNA binding. For Rap1, this does not appear to be the case; for all constructs examined, analytical sedimentation velocity experiments performed at the same protein concentration as for ITC (20 μm) indicate that the proteins are well behaved monomers in solution (not shown). One other possible origin of this behavior may be due to a contribution to the ΔH from closure of the wrapping loop/latch to form a closed complex on DNA. This is supported by the fact that a qualitatively similar behavior is also observed with the DBD615 construct (Fig. 4b). Moreover, deletion of either the latch or tox residues in DBD827 appear to affect this step, suggesting that this region contributes to the observed increase of the ΔH at low P/D ratios. However, we note that this behavior is absent in the full-length Rap1 (Fig. 6, b and c). In this case, it is possible that the presence of the N-terminal region either affects this step or contributes a compensatory ΔH to the interaction. Indeed, for full-length Rap1, the P into D isotherm has a maximum ΔH larger than for either DBD615 or DBD827. The complex behavior observed in ITC experiments with the different Rap1 constructs suggests that in addition to DNA binding, conformational transitions of the protein might contribute to the signal.
To our surprise, the data in Fig. 5 show that in the absence of DNA, the RCT domain can bind in trans to the DBD and that this interaction is blocked when the RCT domain is already present in cis within the larger Rap1 constructs. These data strongly suggest that the RCT domain does not behave as an independent domain, but rather in the larger Rap1 construct, it interacts with the DBD, possibly with the single C-terminal Myb-like domain. However, binding of DNA to the DBD releases this interaction. Interestingly, Matot et al. (25) also showed by small angle x-ray scattering that Rap1 binding to DNA is accompanied by a large reorientation of the C-terminal domain. Our data strongly suggest that the DNA binding of Rap1 is coupled to a complex network of conformational changes that include both reorientation and release of the C-terminal domain from the DBD closure of the wrapping loop.
In summary, we showed that access to higher stoichiometry on DNA is an intrinsic property of Rap1 and is internally modulated by different regions of the protein. This ability of the DBD may impact both the Rap1 transcriptional activities and its function at telomeres, where it may allow Rap1 to form a nucleoprotein complex with a heterogeneous distribution of bound states and/or affect its interaction with other binding partners.
Acknowledgments
We thank Prof. Timothy Lohman for suggestions and critical comments and Dr. Alex Kozlov for help with the ITC experiments.
This work was supported, in whole or in part, by National Institutes of Health Grant GM098509 (to R. G.).
- DBD
- DNA-binding domain
- ITC
- isothermal titration calorimetry
- FAM
- 6-carboxyfluorescein
- RCT and BRCT domains
- Rap1 and BRCA1 C-terminal domains, respectively
- FAMRCT
- FAM-labeled RCT domain.
REFERENCES
- 1. Lieb J. D., Liu X., Botstein D., Brown P. O. (2001) Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28, 327–334 [DOI] [PubMed] [Google Scholar]
- 2. Shore D., Nasmyth K. (1987) Purification and cloning of a DNA binding protein from yeast that binds to both silencer and activator elements. Cell 51, 721–732 [DOI] [PubMed] [Google Scholar]
- 3. Kurtz S., Shore D. (1991) RAP1 protein activates and silences transcription of mating-type genes in yeast. Genes Dev. 5, 616–628 [DOI] [PubMed] [Google Scholar]
- 4. Conrad M. N., Wright J. H., Wolf A. J., Zakian V. A. (1990) RAP1 protein interacts with yeast telomeres in vivo: overproduction alters telomere structure and decreases chromosome stability. Cell 63, 739–750 [DOI] [PubMed] [Google Scholar]
- 5. Lustig A. J., Kurtz S., Shore D. (1990) Involvement of the silencer and UAS binding-protein Rap1 in regulation of telomere length. Science 250, 549–553 [DOI] [PubMed] [Google Scholar]
- 6. Wright J. H., Zakian V. A. (1995) Protein-DNA interactions in soluble telosomes from Saccharomyces cerevisiae. Nucleic Acids Res. 23, 1454–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchman A. R., Kimmerly W. J., Rine J., Kornberg R. D. (1988) Two DNA-binding factors recognize specific sequences at silencers, upstream activating sequences, autonomously replicating sequences, and telomeres in Saccharomyces cerevisiae. Mol. Cell. Biol. 8, 210–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang S. S., Zakian V. A. (1990) Sequencing of Saccharomyces telomeres cloned using T4-DNA polymerase reveals 2 domains. Mol. Cell. Biol. 10, 4415–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wotton D., Shore D. (1997) A novel Rap1p-interacting factor, Rif2p, cooperates with Rif1p to regulate telomere length in Saccharomyces cerevisiae. Genes Dev. 11, 748–760 [DOI] [PubMed] [Google Scholar]
- 10. Moretti P., Freeman K., Coodly L., Shore D. (1994) Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 8, 2257–2269 [DOI] [PubMed] [Google Scholar]
- 11. Palladino F., Laroche T., Gilson E., Axelrod A., Pillus L., Gasser S. M. (1993) Sir3 and Sir4 proteins are required for the positioning and integrity of yeast telomeres. Cell 75, 543–555 [DOI] [PubMed] [Google Scholar]
- 12. Kyrion G., Liu K., Liu C., Lustig A. J. (1993) Rap1 and telomere structure regulate telomere position effects in Saccharomyces cerevisiae. Genes Dev. 7, 1146–1159 [DOI] [PubMed] [Google Scholar]
- 13. de Lange T. (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110 [DOI] [PubMed] [Google Scholar]
- 14. Palm W., de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 15. de Lange T. (2009) How telomeres solve the end-protection problem. Science 326, 948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morse R. H. (2000) RAP, RAP, open up! New wrinkles for RAP1 in yeast. Trends Genet. 16, 51–53 [DOI] [PubMed] [Google Scholar]
- 17. Mizuno T., Kishimoto T., Shinzato T., Haw R., Chambers A., Wood J., Sinclair D., Uemura H. (2004) Role of the N-terminal region of RapIp in the transcriptional activation of glycolytic genes in Saccharomyces cerevisiae. Yeast 21, 851–866 [DOI] [PubMed] [Google Scholar]
- 18. López M. C., Smerage J. B., Baker H. V. (1998) Multiple domains of repressor activator protein 1 contribute to facilitated binding of glycolysis regulatory protein 1. Proc. Natl. Acad. Sci. U.S.A. 95, 14112–14117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vignais M. L., Sentenac A. (1989) Asymmetric DNA bending induced by the yeast multifunctional factor Tuf. J. Biol. Chem. 264, 8463–8466 [PubMed] [Google Scholar]
- 20. Müller T., Gilson E., Schmidt R., Giraldo R., Sogo J., Gross H., Gasser S. M. (1994) Imaging the asymmetrical DNA bend induced by repressor activator protein 1 with scanning tunneling microscopy. J. Struct. Biol. 113, 1–12 [DOI] [PubMed] [Google Scholar]
- 21. Wahlin J., Cohn M. (2000) Saccharomyces cerevisiae RAP1 binds to telomeric sequences with spatial flexibility. Nucleic Acids Res. 28, 2292–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee S., Berger M. F., Jona G., Wang X. S., Muzzey D., Snyder M., Young R. A., Bulyk M. L. (2004) Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat. Genet. 36, 1331–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Konig P., Giraldo R., Chapman L., Rhodes D. (1996) The crystal structure of the DNA-binding domain of yeast RAP1 in complex with telomeric DNA. Cell 85, 125–136 [DOI] [PubMed] [Google Scholar]
- 24. Taylor H. O., O'Reilly M., Leslie A. G., Rhodes D. (2000) How the multifunctional yeast Rap1p discriminates between DNA target sites: a crystallographic analysis. J. Mol. Biol. 303, 693–707 [DOI] [PubMed] [Google Scholar]
- 25. Matot B., Le Bihan Y. V., Lescasse R., Pérez J., Miron S., David G., Castaing B., Weber P., Raynal B., Zinn-Justin S., Gasparini S., Le Du M. H. (2012) The orientation of the C-terminal domain of the Saccharomyces cerevisiae Rap1 protein is determined by its binding to DNA. Nucleic Acids Res. 40, 3197–3207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Freeman K., Gwadz M., Shore D. (1995) Molecular and genetic-analysis of the toxic effect of Rap1 overexpression in yeast. Genetics 141, 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hardy C. F. J., Balderes D., Shore D. (1992) Dissection of a carboxy-terminal region of the yeast regulatory protein Rap1 with effects on both transcriptional activation and silencing. Mol. Cell. Biol. 12, 1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moretti P., Shore D. (2001) Multiple interactions in sir protein recruitment by Rap1p at silencers and telomeres in yeast. Mol. Cell. Biol. 21, 8082–8094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kyrion G., Boakye K. A., Lustig A. J. (1992) C-terminal truncation of Rap1 results in the deregulation of telomere size, stability, and function in Saccharomyces cerevisiae. Mol. Cell. Biol. 12, 5159–5173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feldmann E. A., Galletto R. (2014) The DNA-binding domain of yeast Rap1 interacts with double-stranded DNA in multiple binding modes. Biochemistry 53, 7471–7483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino-acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 32. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) How to measure and predict the molar absorption-coefficient of a protein. Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 [DOI] [PubMed] [Google Scholar]
- 34. Balbo A., Brown P. H., Braswell E. H., Schuck P. (2007) Measuring protein-protein interactions by equilibrium sedimentation. Curr. Protoc. Immunol. 10.1002/0471142735.im1808s79 [DOI] [PubMed] [Google Scholar]
- 35. Schuck P. (1998) Sedimentation analysis of noninteracting and self-associating solutes using numerical solutions to the Lamm equation. Biophys. J. 75, 1503–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dam J., Velikovsky C. A., Mariuzza R. A., Urbanke C., Schuck P. (2005) Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s). Biophys. J. 89, 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vignais M. L., Huet J., Buhler J. M., Sentenac A. (1990) Contacts between the factor Tuf and Rpg sequences. J. Biol. Chem. 265, 14669–14674 [PubMed] [Google Scholar]
- 38. Graham I. R., Chambers A. (1994) Use of a selection technique to identify the diversity of binding sites for the yeast RAP1 transcription factor. Nucleic Acids Res. 22, 124–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]