

Abstract
NEMO (NF-κB essential modulator) associates with the catalytic subunits IKKα and IKKβ to form the IκB kinase (IKK) complex, and is a key regulator of NF-κB pathway signaling. Biochemical and structural characterization of NEMO has been challenging, however, leading to conflicting data on basic biochemical properties such as the oligomeric state of active NEMO and its binding affinity for IKKβ. We show that up to seven of NEMO’s 11 cysteine residues can be mutated to generate recombinant full-length NEMO that is highly soluble and active. Using a fluorescence anisotropy binding assay we show that full-length NEMO binds a 44-mer peptide encompassing residues 701-745 of IKKβ with KD = 2.2 ± 0.8 nM. The IKKβ binding affinities of mutants with five and seven Cys-to-Ala substitutions are indistinguishable from that of wild-type NEMO. Moreover, when expressed in NEMO −/− fibroblasts, the 5xAla and 7xAla NEMO mutants can interact with cellular IKKβ and restore NF-κB signaling to provide protection against TNFα-induced cell death. Treatment of the NEMO-reconstituted cells with H2O2 led to formation of covalent dimers for wild-type NEMO and the 5xAla mutant, but not for the 7xAla mutant, confirming that Cys54 and/or Cys347 can mediate inter-chain disulfide bonding. However, the IKKβ binding affinity of NEMO is unaffected by the presence or absence of inter-chain disulfide bonding at Cys54 – which lies within the IKKβ binding domain of NEMO – or at Cys347, indicating that NEMO exists as a noncovalent dimer independent of the redox state of its cysteines. This conclusion was corroborated by the observation that the secondary structure content of NEMO and its thermal stability were independent of the presence or absence of inter-chain disulfide bonds.
Keywords: IKKγ, IKK, bacterial expression, cysteine mutations
The transcription factor NF-κB regulates a large number of genes that control key cellular processes including cell proliferation and survival, and plays important roles in cancer, immunity and inflammation (1). The canonical pathway for activation of NF-κB is regulated by the multi-component cytoplasmic signaling kinase IκB kinase (IKK), which comprises two catalytic subunits, IKKα and IKKβ, plus the regulatory subunit NEMO (NF-κB Essential Modulator, also known as IKKγ) (2–4). Activation of the IKK complex in response to upstream signals leads IKK to phosphorylate the NF-κB inhibitor IκB, which forms a latent, inactive complex with NF-κB (5). Phosphorylation of IκB triggers its degradation by the proteasome, thereby releasing NF-κB to translocate to the nucleus where it affects gene transcription. NEMO is essential for activation of the NF-κB signaling cascade (2–4, 6); cells and mice in which the NEMO gene has been knocked out cannot activate NF-κB in response to external stimuli such as TNF or lipopolysaccharide (7, 8). In addition to scaffolding the IKK complex, NEMO bridges the interaction between the catalytic subunits of IKK and IκB (9); and also serves to recruit IKK to the cell membrane through ubiquitin-mediated interactions with upstream receptors (10–14). Because of its key role in the regulation of NF-κB signaling, as well as the occurrence of mutations in the NEMO gene in certain human immunodeficiences (15), there is great interest in understanding the structural, biochemical and functional properties of the NEMO protein.
The 419 amino acid NEMO protein contains multiple domains, including an N-terminal domain that can bind to IKKα or -β (16), a central, ubiquitin-binding domain (17, 18), and a C-terminal zinc finger domain (Figure 1) (19). In addition, NEMO can be post-translationally modified by ubiquitination, phosphorylation and SUMOylation depending on cell type and stimulus (20, 21). Previous studies aiming to establish the basic biochemical properties of NEMO, such as its functional oligomeric state and its interaction affinity for its binding partners, have generally used only NEMO fragments or truncated constructs (9, 10, 13, 22–33) and, perhaps for this reason, have often given conflicting results. For example, using size exclusion chromatography with in-line multi-angle lightscattering (SEC-MALS), Lo et al. reported that a truncated NEMO(1-196) protein existed in a variety of oligomeric states containing 1, 2, 3 or 5 NEMO subunits, as well as a much larger aggregate, whereas inclusion of a fragment of IKKβ comprising residues 680-756 gave mostly 2:2 complexes with a smaller fraction of a 4:4 species (28). Agou et al. reported that truncated NEMO constructs encompassing the coiled-coil 2 (CC2) and leucine zipper (LZ) domains form trimers in solution (22, 34). More recently, Ivins et al. showed by both SEC-MALS and analytical ultracentrifugation that a NEMO(1-355) construct containing a C54S mutation existed in a dimer-tetramer equilibrium with no detectible monomer (13). Attempts to establish the interaction affinity between NEMO and IKKβ have also given inconsistent results. Binding studies using similar IKKβ(701-745) peptides but a range of truncated NEMO constructs have reported KD values ranging from single digit nanomolar to micromolar (28, 30, 35, 36). The structural characterization of NEMO has also been challenging. Although X-ray crystal structures have been reported for several fragments of NEMO (11, 14, 19, 26, 30, 33, 37), the structure of the full-length protein is not known.
Figure 1.

Schematic representation of the domain structure of NEMO (59), showing the approximate locations of the 11 cysteine residues. CC1 and CC2 are the first and second coiled-coil regions, LZ is the leucine zipper region, and ZF is the zinc finger domain. Below the scheme is shown the X-ray co-crystal structure of NEMO(44-111) in complex with IKKβ(701-745) (30). Two molecules of NEMO(44-111) (blue) form a coiled coil that binds one molecule of IKKβ(701-745) (green) at each face. The locations of NEMO residues Cys54 (left), Cys76 (middle) and Cys95 (right) are highlighted in red.
NEMO contains 11 cysteine (Cys) residues. Four of these – at positions 396, 397, 400 and 417 – lie within the C-terminal zinc finger domain of NEMO, of which Cys397, 400, and 417 directly chelate the Zn ion. Mutation of Cys417 in humans has been shown to cause ectodermal dysplasia with immunodeficiency (19), and mutation of the residues in mouse NEMO corresponding to human NEMO Cys397 and Cys400 in the zinc finger domain abolished the ability of IKK to phosphorylate IκB though without affecting the interaction of NEMO with IKKβ (9). However, the functional importance of NEMO’s other Cys residues, several of which are highly conserved (Figure S1, Supporting Information), remains unclear. Previous work using full-length NEMO mutants transfected into mammalian cells has shown that cysteines 54 and 347 can form intermolecular disulfide bonds in the NEMO dimer, especially when cells are treated with hydrogen peroxide (38). But the extent to which the presence or oxidation state of these cysteines, or those at positions 11, 76, 95, 131 or 167, affect the oligomeric state or IKKβ binding properties of NEMO is unclear.
A significant obstacle to the rigorous biochemical characterization of NEMO is that full-length recombinant NEMO is highly prone to form high molecular weight aggregates (24, 28). We show here that the simultaneous mutation of multiple Cys residues N-terminal to the Zn finger domain of NEMO enables the generation of homogeneous and highly soluble recombinant full-length NEMO, and we use these well-behaved mutants to characterize the oligomeric state of full-length NEMO and its binding activity towards IKKβ. We show that all seven of these cysteines can be simultaneously mutated to Ala without affecting the binding affinity of NEMO for IKKβ, and that the resulting NEMO Cys mutants are functional with respect to IKKβ binding in vitro, and can restore NF-κB activation in NEMO −/− mouse fibroblasts. Finally, we show that Cys54 and Cys347 can mediate inter-chain disulfide bond formation in NEMO; however, the functional form of NEMO is a constitutive dimer independent of the redox state of these two Cys residues.
EXPERIMENTAL PROCEDURES
Plasmids and Site-directed Mutagenesis
Site-directed mutations in NEMO were generated by overlapping polymerase chain reaction-based mutagenesis and were then subcloned into the relevant vectors. The genes for the Cys variants were created using the QuickChange mutagenesis method starting with the 7xAla base construct. The 5xAla construct was synthesized with codon optimization to increase protein yield for E. coli expression (Genscript, Piscataway, NJ). For recombinant expression, each NEMO construct was subcloned into the NdeI and XhoI sites of the pET24b(+) vector (Novagen). Retroviral vectors for expression of wild-type NEMO or NEMO Cys mutants in mouse cells were created in pBABE-puro, as described previously (38). All constructs were confirmed by DNA sequencing. Details of retroviral vector constructions can be found at www.nf-kb.org.
E. coli Expression and Purification
The wild-type and NEMO mutant pET24b(+) constructs were transformed into Rosetta 2(DE3)pLysS competent cells (Novagen), except for the 5xAla pET24b(+), which was transformed into T7 Express Iq Competent E. coli (New England Biolabs, Ipswich, MA) to take advantage of the codon optimization of this construct. NEMO-transfected E. coli were grown overnight in LB broth at 37 °C. A new culture was seeded from the overnight culture at a 1:200 dilution, and grown to OD600 ~0.4. Protein expression was induced by 1 mM isopropyl β-D-thiogalactoside (IPTG), and the cells were grown for an additional 4 h. Cells were pelleted and stored at −80°C. In the standard protocol, cells were lysed by adding Bacterial Permeabilization Reagent II (B-PER II, Pierce, Rockford, IL) at 2 mL/g of cell pellet together with 5 mg/mL lysosome (Pierce), 10 mg/mL streptomycin sulfate (Gold Bio, St. Louis, MO), 10 μL/mL Halt Protease Inhibitor Cocktail (Thermo Scientific, Rockford, IL), and 0.4% w/v DNase I (Pierce, Rockford, IL). The lysate was incubated for 20 min at room temperature, then 8 M urea was added to solubilize inclusion bodies, and the lysate was incubated for a further 1 h at 37 °C. The lysate was clarified by centrifugation at 38,000 rpm for 30 min, and was then filtered through a 0.8 μm filter. For large-scale preparations, the prepared lysate was applied to a 5 mL HisTrap FF crude column (GE Healthcare, Piscataway, NJ), followed by 5 column volumes of 20 mM sodium phosphate, 500 mM NaCl, 40 mM imidazole, 6 M urea at pH 7.4. The bound protein was refolded on the column using a 20 column volume urea gradient from 6 M to zero. The refolded protein was eluted using a step gradient of increasing concentration of 20 mM sodium phosphate 500 mM NaCl, 500 mM imidazole at pH 7.4. This denaturation and refolding step was necessary to remove an impurity, as described in the text, and did not appear to affect the solubility of the final expressed protein product. In some preparations of wild-type NEMO a modified protocol was developed in which lysate and buffers were supplemented with 5 mM TCEP to reduce disulfide-mediated misfolding and aggregation. In both the standard and modified protocols, NEMO eluted from the Nickel-NTA column was concentrated and applied to a HiPrep 26/60 Sephacryl S-300 HR column (GE Healthcare) equilibrated with 20 mM sodium phosphate, 500 mM NaCl at pH 7.4. Purified NEMO was concentrated from the fractions eluting between the void volume and 160 min, aliquoted, and stored at −80°C. The six NEMO Cys addback mutants were purified using a modified protocol that did not include a gel-filtration step. Protein concentrations were calculated from the absorbance at 280 nm (NanoDrop, Thermo Scientific) using an extinction coefficient of 14,815 M−1cm−1 calculated from the protein sequence.
SDS-PAGE
Recombinant NEMO variants were separated using 12% polyacrylamide gels. To measure the extent of disulfide crosslinking, 20 μM protein was incubated in non-reducing SDS sample buffer for 5 min without boiling prior to analysis. Fully reduced protein was analyzed by incubating a sample of the same protein in SDS sample buffer containing 100 mM DTT with boiling for 5 min.
Analytical Gel Filtration Chromatography
The oligomeric state of recombinant NEMO preparations was determined using analytical gel filtration. A 40 μM sample of each NEMO preparation was pre-incubated for 1 h in 20 mM sodium phosphate, 500 mM NaCl at pH 7.4 containing 0, 1, or 10 mM DTT. A 100 μL sample of the treated protein was then loaded onto a Superose 6 10/300 GL analytical gel-filtration column (GE Healthcare), and eluted over 1.5 column volumes of 20 mM sodium phosphate, 500 mM NaCl at pH 7.4 run at 0.4 mL/min.
Fluorescence Anisotropy Binding Studies
FITC-labeled IKKβ(701-745) peptide was synthesized commercially (Genscript) and then further purified on a RESOURCE RPC reversed phase FPLC column (GE Healthcare), using a buffer system of 0.125% ammonium hydroxide and 0.125% ammonium hydroxide in 100% methanol. The concentration of the purified FITC-IKKβ peptide was determined from its UV absorbance at 280 and 493 nm, by Nanodrop using the formula (39):
Fluorescence Anisotropy assays were performed in 96-well polypropylene black plates (Corning, Corning, NY) in triplicate, using an assay volume of 200 μL and a buffer of 50 mM Tris pH 7.4, 200 mM NaCl, 0.01% v/v Triton X-100, 1 mM DTT, unless stated otherwise. Samples were incubated for 1 h at 25 °C, which control experiments showed was sufficient time to reach equilibrium at all concentrations (data not shown). Direct binding experiments were performed by first pre-incubating NEMO for 1 hour at 4 °C with either no addition to the buffer, 1 or 10 mM DTT, or 5% v/v H2O2. The concentration of FITC-IKKβ or FITC-IKKβ(C716S) was held constant (typically at 15 nM), and the NEMO protein concentration was varied. Competitive binding experiments were performed by holding FITC-IKKβ and NEMO concentrations constant, and varying the concentration of the unlabeled IKKβ(701-745) competitor. Assay plates were read using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA), using excitation and emission wavelengths of 488 and 520 nm, respectively. Anisotropy values were calculated by measuring the intensity of the parallel (I||) and perpendicular (I⊥) components of the fluorescence emission in each well, subtracting the corresponding parallel or perpendicular reading measured in control wells containing only assay buffer, and then calculating anisotropy (r) using the following equation:
Note that anisotropy differs from the related quantity of fluorescence polarization, which is calculated in a similar manner but using an equation that has (I|| + I⊥) rather than (I|| + 2I⊥) in the denominator of the equation shown above (40). Anisotropy is the preferred measure for the quantitative analysis of binding behavior, for the reason given in Results (41). The anisotropy dose-response curves for the direct binding experiments were fitted to a quadratic binding equation, modified to include an offset, r0, to account for the non-zero anisotropy observed in the absence of NEMO, and also a linear term:
In this formula, [L]T and [R]T are the total concentrations of ligand (FITC-IKKβ) and receptor (NEMO), KD is the dissociation constant for their interaction, and m is the slope of the linear term. A quadratic binding equation is appropriate to fit the data from the direct binding experiments because the fixed component in the assay, FITC-IKKβ, is present at a concentration that exceeds its KD for binding to NEMO. Consequently, the condition required for hyperbolic binding – that the equilibrium concentration of the varied component can be approximated by its total concentration – is not satisfied (42). The origin of the linear term, which is needed to fit a small positive slope that was observed in all direct binding experiments at high NEMO concentrations, is unclear at present. It may derive from low affinity (KD ≫ 1 μM) self-association of NEMO dimers to form tetramers, as reported by Ivins et al., that results in a gradual concentration-dependent increase in anisotropy at high [NEMO] (13). Data for the competition experiments, involving inhibition of the interaction between 5xAla NEMO and FITC-IKKβ (15 nM each) by various concentrations of unlabeled IKKβ(701-745), were analyzed by fitting the inhibition dose-response curves to the following competitive binding model using the numerical nonlinear regression software DynaFit 4 (43), holding [FITC-IKKβ], [NEMO] and KD at their fixed or independently-determined values so that KI was the only variable parameter (see Supporting Information for the complete DynaFit script):
Circular Dichroism Spectroscopy
A 2 mL sample of wild-type, 5xAla, and 7xAla NEMO was dialyzed overnight against 2 L of 20 mM sodium phosphate pH 7.4, 150 mM NaCl, 2.5 mM TCEP (CD Buffer). After dialysis, the protein was diluted to ~10 μM in CD buffer. The CD spectrum of a 300 μL sample of each variant was taken in a 0.1 cm cuvette at 20 °C from 195 nm to 260 nm. The thermal stability of each variant was measured using the same protein preparation by monitoring the increase in CD signal at 222 nm as the temperature was increased from 10–70 °C at a rate of 1 °C/min. To determine that thermal unfolding was independent of the temperature ramping rate, melting was measured for 5xAla at ramping rates of 0.5 °C/min and 0.33 °C/min, and the resulting melting temperatures were found to be identical to that measured at 1 °C/min. The measured circular dichroism signals (D, in units of millidegrees) were normalized to the concentration of the protein to give the mean molar residue ellipticity (θ, in units of deg.cm2.dmol−1), using the following equation (44):
where C is the concentration of protein in units of M, L is the path length in mm, and N is the number of residues. The secondary structure content was analyzed using the SOMCD algorithm. The CD signals of the wild-type, 5xAla, and 7xAla spectra from 200–240 nm were analyzed by the algorithm (45).
Mammalian Cell Culture and Transfection
NEMO-deficient mouse fibroblasts were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Biologos, Montgomery, IL) as described (38, 46). Virus stocks for the expression of NEMO proteins were prepared by co-transfecting BOSC23 packaging cells in a 60-mm dish with 10 μg of a pBABE-puro-based plasmid and 10 μg of the pCL101a helper plasmid using 60 μg of polyethylenimine (PEI) as described (47). Virus stocks were then used to infect NEMO-deficient fibroblasts, and selection was performed using 2.5 μg/mL puromycin (Sigma, St, Louis, MO) for approximately 1–2 weeks (47).
Western Blotting and Co-immunoprecipitation
Western blotting was performed essentially as described (38). When analyzing NEMO following H2O2 treatment, whole-cell extracts from transduced NEMO-deficient fibroblasts were prepared by heating cell pellets in SDS sample buffer lacking β-mercaptoethanol (0.625 M Tris, pH 6.8, 2.3% w/v SDS, 10% w/v glycerol). For analyzing phosphorylation of IκBα, cells were treated with 20 ng/mL TNFα (R&D Systems, Minneapolis, MN) for 10 min and then lysed directly in SDS sample buffer containing 5% β-mercaptoethanol. In all cases, samples containing approximately equal amounts of protein were separated on SDS-polyacrylamide gels, proteins were transferred to nitrocellulose membranes, and filters were incubated overnight at 4 °C with anti-NEMO antiserum (#2685, Cell Signaling Technology, Danvers, MA; 1:1000 dilution) or anti-phospho-IκBα (#9246, Cell Signaling Technology; 1:1000 dilution). Horseradish peroxidase-labeled secondary antiserum was added, and immunoreactive proteins were detected by Supersignal Dura West chemiluminescence (Thermo Scientific).
For co-immunoprecipitation experiments, retrovirally transduced NEMO −/− cells in confluent 100 mm tissue culture dishes were lysed in 500 μL of AT buffer (20 mM HEPES, pH 7.9, 1 mM EDTA, 1 mM EGTA, 20 mM Na4P2O7, 1 mM DTT, 1% v/v Triton X-100, 20% w/v glycerol, 1 mM Na3VO4, 1 μg/mL PMSF, 1 μg/mL leupeptin, 1 μg/mL pepstatin). An aliquot (20 μL) was saved as the input sample. To the remainder of the lysate, 30 μL of anti-NEMO agarose beads (sc-8330 AC; Santa Cruz Biotechnology, Santa Cruz, CA) was added, and samples were incubated for 2 h with rocking at 4 °C. The beads were washed several times with AT buffer, and then bound proteins were removed by heating the samples at 90 °C in SDS sample buffer containing β-mercaptoethanol. Samples (input or immunoprecipitates) were then analyzed by reducing SDS-PAGE followed by Western blotting with anti-IKKβ antibody (sc-7607; Santa Cruz Biotechnology) or anti-NEMO antiserum (#2685; Cell Signaling Technology) as described above.
TNFα-induced Cell Death Assays
NEMO-deficient mouse fibroblasts reconstituted with wild-type or mutant NEMO proteins were treated with 50 ng/mL TNFα (R&D Systems) for 18 h, and the number of live cells was determined using a crystal violet-based cell viability assay, as described previously (38, 47).
RESULTS
Design and Preparation of NEMO Cysteine Mutants
To develop recombinant full-length NEMO protein for detailed biophysical and structural characterization, we generated constructs of wild-type NEMO and also two mutants in which either five or seven of NEMO’s 11 Cys residues were mutated to Ala (Figure 1). In the 7xAla construct, all Cys residues except for the four in the C-terminal zinc finger domain were substituted (19). In the 5xAla construct, Cys54 and Cys347, which have been shown to be capable of inter-chain disulfide bond formation (38), were additionally retained, to test whether these two residues contribute to the stability of the protein or are important for achieving full IKKβ binding activity. The potential for disulfide bonding at Cys54 to affect IKKβ binding was of particular interest, because the published structure of NEMO fragment 44-111 bound to IKKβ 701-745 suggests that formation of an inter-chain disulfide at Cys54 is compatible with the active conformation of the protein (Figure 1), though the structure shows the protein in a reduced state (30). The wild-type and mutant NEMO cDNAs were subcloned into the pET24b(+) vector, which appends a C-terminal 6xHis tag to the protein. The bacterially expressed proteins were purified by nickel-NTA affinity chromatography followed by gel filtration chromatography. When initially developing the purification protocol we found that an approximately 70 kDa protein, which we suspected to be the bacterial chaperone protein DnaK that was observed as a contaminant in previously reported preparations of a truncated form of NEMO (22), co-eluted with NEMO from the nickel column (data not shown). We eliminated this contaminant by loading the crude protein onto the nickel-NTA column in 8 M urea, and then refolding the bound NEMO directly on the column by applying a urea gradient, before finally eluting the folded NEMO with imidazole (48).
Bacterially Expressed NEMO is Highly Prone to Disulfide-Mediated Aggregation
Analysis of the three purified NEMO constructs by SDS-PAGE (Figure 2A) showed that, even after boiling with SDS, under non-reducing conditions little wild-type NEMO protein entered the gel, and the small amount that did so appeared as a smear of high molecular-weight species. In contrast, the 5xAla NEMO gave two major bands, both corresponding to some form of NEMO dimer, while the 7xAla-NEMO gave predominantly a mixture of NEMO dimer and monomer. Western blotting with anti-NEMO and anti-6xHis-tag antibodies showed that all visible bands contained His-tagged NEMO (data not shown). Boiling with DTT and SDS reduced all three preparations to the molecular weight expected for monomeric NEMO, showing that the NEMO dimers and oligomers observed under non-reducing conditions were covalently linked through disulfide bonds (Figure 2A). Analytical gel-filtration of wild-type NEMO after the nickel purification step yielded a chromatogram that indicated extensive heterogeneity (Figure 2B), which we again attributed to improper folding and/or covalent aggregation involving Cys residues. Incubation of wild-type NEMO prepared in this way for 1 h with up to 10 mM DTT eliminated only a fraction of the aggregated forms (data not shown). The aggregation observed with wild-type NEMO was so extensive that the overall yield of the protein, ~0.3 mg/L of bacterial cell culture, was too low to be useful for further experiments. In contrast, the 5xAla and 7xAla constructs typically gave purified yields of several mg/L. SDS-PAGE analysis of crude E. coli lysates under reducing conditions showed that the expression levels of wild-type, 7xAla and 5xAla NEMO were roughly equal (data not shown), indicating that the poor yield obtained with wild-type NEMO was due to loss of material during purification, presumably resulting from oxidation and aggregation during or after cell lysis. The yield and homogeneity of wild-type NEMO could be somewhat improved by including 5 mM tris(2-carboxyethyl)phosphine (TCEP) in all post-lysis steps of the purification up to and including elution from the Ni/NTA column. However, even when purified under these stringently reducing conditions the yield of wild-type NEMO remained poor. The wild-type NEMO used in the further characterizations described below was purified under these reducing conditions, which were not needed for the 5xAla and 7xAla mutants.
Figure 2.
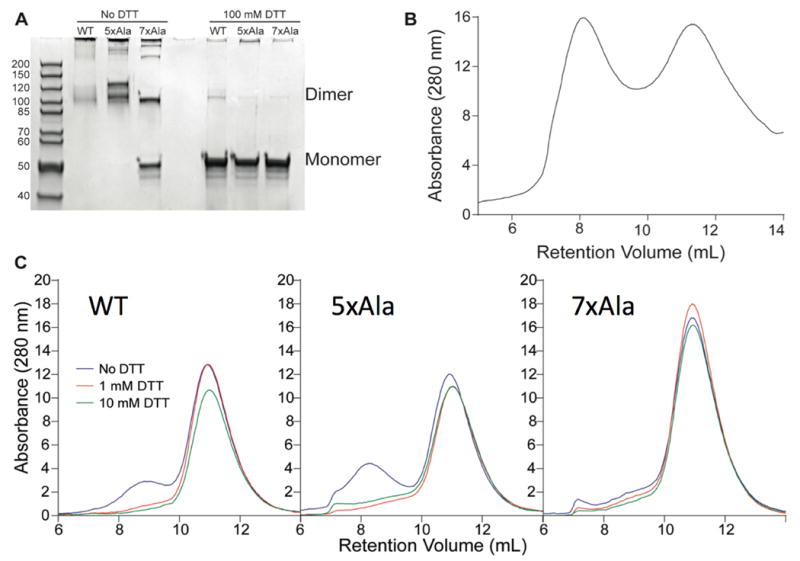
Aggregation states of wild-type, 5xAla and 7xAla NEMO proteins. (A) Reducing and non-reducing SDS-PAGE of wild-type NEMO and the 5xAla and 7xAla mutants after purification by Nickel-NTA affinity chromatography using the standard protocol (i.e., without inclusion of 5 mM TCEP). (B) Analytical gel filtration analysis of wild-type NEMO purified under these standard conditions, in 20 mM sodium phosphate, 500 mM NaCl, at pH 7.4. (C) Analytical gel filtration analysis of wild-type NEMO (left), 5xAla (middle) and 7xAla (right) in 20 mM sodium phosphate, 500 mM NaCl, at pH 7.4 after incubation for 1 h with 0 mM (black line), 1 mM (blue line) or 10 mM (red line) DTT. Wild-type NEMO in this experiment was purified under stringently reducing conditions involving inclusion of 5 mM TCEP in all steps, while the 5xAla and 7xAla NEMO were purified using the standard protocol that does not include TCEP.
The homogeneity and oligomerization state of wild-type NEMO purified in the presence of 5 mM TCEP were compared to those of 5xAla and 7xAla NEMO, both purified under the standard (no TCEP) conditions, by analytical gel-filtration of freshly prepared protein after incubation for 1 h with 0, 1, or 10 mM DTT. In the absence of DTT, wild-type and 5xAla NEMO both showed a major peak eluting at about 11 mL, together with a smaller peak that eluted with the void volume of the column and corresponds to aggregated protein (Figure 2C). The slower migrating peak corresponds to an apparent molecular weight of >450 kDa, as compared to the elution times of globular molecular weight standards. However, as previously reported, NEMO migrates anomalously on gel-filtration columns (13), which has been attributed to the protein having an elongated rather than a globular structure, and so the true size of the species eluting at 11 mL cannot be inferred from its gel filtration retention time alone. For both wild-type and 5xAla NEMO, the aggregated component present in these preparations was largely eliminated by treatment with 1 mM DTT (Figure 2C). In contrast to the other two constructs, 7xAla NEMO appeared relatively homogeneous by gel filtration, with only small amounts of higher molecular weight material that was not eliminated by inclusion of DTT. These results suggest that even when wild-type NEMO is purified under stringently reducing conditions it retains a high propensity to form disulfide-linked aggregates. The soluble aggregates observable by gel filtration appear to be primarily mediated by Cys54 and/or Cys347, in that replacement of the five Cys residues at positions 11, 76, 95, 131 and 167 with Ala in the 5xAla mutant did not reduce the observed level of these aggregates. However, the greatly improved yield of 5xAla NEMO compared to the wild-type protein suggests that elimination of these five cysteine residues substantially reduces the occurrence of larger, insoluble aggregates that lead to loss of the wild-type protein through aggregation and precipitation during the purification process. In the case of the 5xAla and 7xAla mutants, we were able to concentrate the purified proteins at concentrations of up to 35–40 μM in buffers containing no DTT or TCEP; moreover, in the presence of 2.5 mM TCEP both constructs could be concentrated to concentrations of 50 μM (~2.5 mg/ml) with little or no precipitation.
The 5xAla and 7xAla NEMO Mutants Retain Full Binding Affinity for IKKβ
To assess whether the multiple Cys mutations in the 5xAla and 7xAla constructs affected the biochemical properties of NEMO, we tested the ability of each protein to bind to a peptide derived from the C-terminal domain of IKKβ (30). IKKβ(701-745) binds to the N-terminal domain of NEMO, occupying the groove that is formed when two NEMO molecules associate into a dimer to form an α-helical coiled-coil at the N-terminal IKKβ-binding region (30). The NEMO dimer contains two IKKβ binding sites, one on each face of CC1, leading to a 2:2 binding stoichiometry with IKKβ (Figure 1) (28, 30). As shown in Figure 1, each IKKβ molecule makes extensive contact with the region of NEMO extending between roughly residues 44 and 111. This region of NEMO contains three cysteines, at positions 54, 76 and 95, with additional Cys residues flanking the IKKβ binding region in both the N-terminal (Cys11) and C-terminal (Cys131, Cys167) directions (Figure 1). The published structure of NEMO(44-111) in complex with IKKβ(701-745) shows that the Cys54 residues appear to be in a position to form an inter-chain disulfide bond in the active conformation of the protein, whereas Cys76 and Cys95 are not (Figure 1) (30). Comparison of the 7xAla and 5xAla forms of NEMO, which in the vicinity of the IKKβ binding region differ only by the presence or absence of Cys54, therefore provides a means to assess whether formation of a disulfide bond at this position is important for IKKβ binding.
To measure the binding of NEMO to IKKβ(701-745) we developed a fluorescence anisotropy (FA) binding assay. As tracer probe we used a synthetic IKKβ(701-745) peptide labeled at the N-terminus with fluorescein isothiocyanate (FITC-IKKβ). FA works on the principle that when a fluorescently labeled ligand of low molecular weight is excited with plane polarized light, due to the ligand’s rapid tumbling in solution the excited state dipole will undergo substantial reoriention before decaying with the release of a photon, and thus the polarization observed in the emitted light will be small. However, if the labeled ligand becomes bound as part of a much larger complex its tumbling will be slowed, leading to retention of more polarization in the emitted light. Thus, the degree of polarization in the emitted fluorescence provides a measure of what fraction of tracer probe is bound versus unbound (40, 41). In the following experiments we quantified the degree of polarization in terms of fluorescence anisotropy rather than fluorescence polarization per se (see Experimental Procedures for the distinction between these measures). We did this because total measured anisotropy is simply the linear sum of the contributions of the different bound and free forms of the tracer probe, allowing straightforward quantitative analysis of the binding data by fitting to the appropriate binding equation, whereas for fluorescence polarization the relationship of total measured signal to the contributions of individual components is more complex (40, 41). Representing the measured signal in terms of anisotropy is thus preferred for applications where quantitative fitting of the data to binding models is desired (41).
Figure 3A (inset) shows that inclusion of 15 nM 5xAla NEMO substantially increased the anisotropy observed with 15 nM FITC-IKKβ. Moreover, the anisotropy could be returned to baseline in a dose-dependent manner upon addition of unlabeled IKKβ(701-745) as a competitive inhibitor (Figure 3A, main plot). Fitting the data from inhibition experiments such as that shown in Figure 3A to a competitive binding model, as described in Experimental Procedures, gave a value for the binding affinity of unlabeled IKKβ(701-745) to 5xAla NEMO of KD = 2.2 ± 0.8 nM (n = 4). We used the FA binding assay to compare the IKKβ binding affinity of wild-type NEMO with that of the 5xAla and 7xAla mutants. These binding measurements were performed after pre-incubation of the NEMO for 1 h with 10 mM DTT. Preincubation with 10 mM DTT resulted in the elimination of most high molecular weight aggregates from the wild-type and 5xAla constructs (Figure 2C), and was sufficient to convert the 5xAla and 7xAla protein predominantly to a reduced state (Figure 3C; and Figure S2, Supporting Information). As shown in Figure 3B, incubation of 15 nM FITC-IKKβ with various concentrations of NEMO for 1 h gave a dose-dependent increase in anisotropy. Fitting the data for each NEMO construct to a quadratic binding equation (see Experimental Methods) showed, over multiple independent experiments, that the binding affinity of FITC-IKKβ for wild-type, 5xAla, and 7xAla NEMO were indistinguishable within experimental error, giving values of KD = 7.4 ± 2.1 nM (n = 3), 2.8 ± 1.1 nM (n = 3) and 4.9 ± 0.6 nM (n = 3), respectively, where the uncertainty limits represent one standard deviation. These results show that the binding affinity of NEMO for IKKβ is unaffected by the presence or absence of Cys54, Cys347 or the five other Cys residues outside the C-terminal zinc-finger domain, when the protein is tested under reducing conditions.
Figure 3.

Measurement of IKKβ binding activity by fluorescence anisotropy. (A) Inclusion of 15 nM 5xAla NEMO increases the anisotropy signal observed for 15 nM FITC-IKKβ (inset plot), and this signal can be reduced back to baseline in a dose-dependent fashion by addition of unlabeled IKKβ(701-745) as a competitive inhibitor (50 mM Tris pH 7.4, 200 mM NaCl, 0.01% v/v Triton X-100, 1 mM DTT). Error bars represent the variation between triplicate measurements on the same assay plate. The solid line represents the best fit to a competitive binding mechanism, performed by numerical nonlinear regression analysis as described in Experimental Procedures. The horizontal dashed lines at top and bottom represent the mean values for the high and low controls on the assay plate (i.e. 15 nM FITC-IKKβ plus 15 nM NEMO, or 15 nM FITC-IKKβ alone), plus and minus the standard deviation among these control wells. The data shown are representative of four independent experiments. (B) Direct binding of 15 nM FITC-IKKβ to wild-type NEMO (circles), 5xAla (squares) and 7xAla (triangles), after pre-treatment for 1 h with 10 mM DTT (assay buffer: 50 mM Tris pH 7.4, 200 mM NaCl, 0.01% v/v Triton X-100, 1 mM DTT). The solid lines represent the best fits to a modified quadratic binding equation, as described in Experimental Procedures. Error bars represent the variation between triplicate measurements on the same assay plate. Data shown are representative of at least three independent experiments. (C) Non-reducing SDS-PAGE analysis showing that 5xAla NEMO that is incubated for 1 h in buffer with no reducing agent, or containing 5% H2O2, forms a covalent dimer, while treatment with 10 mM DTT results in protein that like the 7xAla protein is predominantly covalent monomer. The samples were not boiled prior to running the gel, to more accurately reflect the proportion of monomers versus dimers existing in the fluorescence anisotropy assay after pretreatment of the NEMO protein under the specified conditions. (D) Direct binding to the variant tracer probe FITC-IKKβ(C716S) for 5xAla NEMO that was pre-incubated for 1 h in buffer containing no reducing agent (squares), with 10 mM DTT (triangles) or with 5% H2O2 (circles), compared to 7xAla NEMO (lozenges). The solid lines represent the best fits to a modified quadratic binding equation, as described in Experimental Procedures. Error bars represent the variation between triplicate measurements on the same assay plate. Data shown are representative of at least three independent experiments.
The binding affinities of IKKβ peptides encompassing residues 701-745 for truncated forms of NEMO have previously been reported as ranging from single digit nanomolar to 1 μM, depending on the NEMO construct and the measurement method used (28, 30, 35). Our results show that our recombinant full-length NEMO constructs give IKKβ binding affinities comparable to the highest affinities previously reported for various fragments or truncated forms of NEMO, establishing that this interaction occurs with low nM KD.
Binding Affinity for IKKβ is Unaffected by the Presence or Absence of Inter-Chain Disulfide Bonds at Cys54/Cys347
To test whether formation of a covalent NEMO dimer through inter-chain disulfide bonding involving Cys54 and/or Cys347 affects IKKβ binding, we compared the interaction affinity with 5xAla NEMO measured in the presence of 10 mM DTT with that seen in the absence of reducing agent, or after oxidation with H2O2 to force the formation of covalent dimers (Figure 3C). For these binding experiments we used a variant of the FITC-IKKβ peptide containing a Cys-to-Ser mutation at the position corresponding to IKKβ residue 716, to allow its use under non-reducing conditions without concern that the tracer probe itself might oxidize via this cysteine. This C716S mutation in IKKβ weakens its binding to NEMO by about 20-fold (Figure 3D). To generate 5xAla NEMO containing inter-chain disulfides at Cys54 and Cys347 we oxidized the protein in two ways: by air oxidation, through incubating the protein in assay buffer without DTT, and also by treating the protein for 1 h with 5% H2O2. Figure 3C shows that these oxidizing treatments converted the protein to covalent dimers, whereas 5xAla NEMO incubated with 10 mM DTT, like 7xAla NEMO, is predominantly monomeric. Figure 3D shows the results of FA binding assays using the FITC-IKKβ(C716S), in which we compared the activity of 5xAla NEMO that had been oxidized by air oxidation or by treatment with 5% H2O2 with that seen for 5xAla NEMO under reducing conditions and for 7xAla NEMO. The results show that the air-oxidized 5xAla, the reduced 5xAla and the 7xAla NEMO gave binding affinities for FITC-IKKβ(C716S) that were identical, with KD values of 60 ± 16 nM (n = 3), 54 ± 11 nM (n = 3) and 52 ± 11 nM (n = 3), respectively. The H2O2-treated 5xAla NEMO appeared to bind FITC-IKKβ(C716S) with a somewhat higher KD of 260 ± 30 nM (n = 3). The results obtained for air-oxidized 5xAla NEMO compared to reduced 5xAla NEMO or with 7xAla NEMO show that the binding affinity of 5xAla NEMO for IKKβ is essentially unaffected by whether the protein exists as a covalent dimer, or whether the protein is fully reduced. The roughly 4-fold decrease in binding affinity observed after H2O2 treatment probably results from partial inactivation of the NEMO under these strongly oxidizing conditions, through formation of non-native oxidized forms of the protein. Indeed, Figure 3C shows that the dimer generated by treatment with H2O2 migrates more slowly on the SDS gel than does the dimer that is observed after air oxidation, indicating that the protein oxidized with H2O2 has a different covalent structure.
To further understand the role of different cysteine residues on the disulfide-mediated dimerization of NEMO, we created a larger series of NEMO Cys mutants. Starting from the 7xAla construct, the seven cysteines upstream of NEMO residue 395 were individually mutated from alanine back to cysteine, to generate seven 6xAla NEMO constructs (Figure 4A). Figure 4B shows that, as expected, the presence of Cys54 causes a substantial increase in the tendency of NEMO to form covalent dimers under non-reducing conditions. A tendency to form disulfide-linked dimers was also seen for the 6xAla constructs containing cysteines at positions 11, 76 and 347. No significant dimer was observed for the constructs containing cysteines at positions 95, 131 or 167. Interestingly, the dimers involving disulfides at Cys 76 or 347 migrate slightly higher on the nonreducing gel than the dimers involving Cys11 or Cys54, reminiscent of the slow-migrating dimer seen in Figure 3C for 5xAla NEMO after oxidation with H2O2. This finding accounts for the observation of a doublet in the non-reducing SDS-PAGE analysis of some preparations of 5xAla NEMO (e.g., in Figure 2A) which, when observed, presumably reflects the presence of a mixture of dimeric forms, some involving disulfide bonds at Cys54 and others at Cys347. The X-ray crystal structure of NEMO(44-111) in complex with IKKβ(701-745) (30) shows that inter-chain disulfide bonding at Cys54 is compatible with the active structure of the NEMO dimer (Figure 4C, left panel), while disulfide bonding at Cys76 is not (Figure 4C, right panel). The slower-migrating dimers seen in Figure 3C for the 6xAla construct with a disulfide bond at Cys76, and also that with a disulfide at Cys347, therefore presumably reflect non-native dimers, consistent with the reduced IKKβ binding activity seen for 5xAla NEMO after oxidation with H2O2. In contrast, the faster migrating dimer seen in Figure 4B for the 6xAla construct containing a disulfide at Cys54 reflects a native dimer, as shown by the unperturbed binding activity observed for air-oxidized 5xAla NEMO in Figure 3D.
Figure 4.
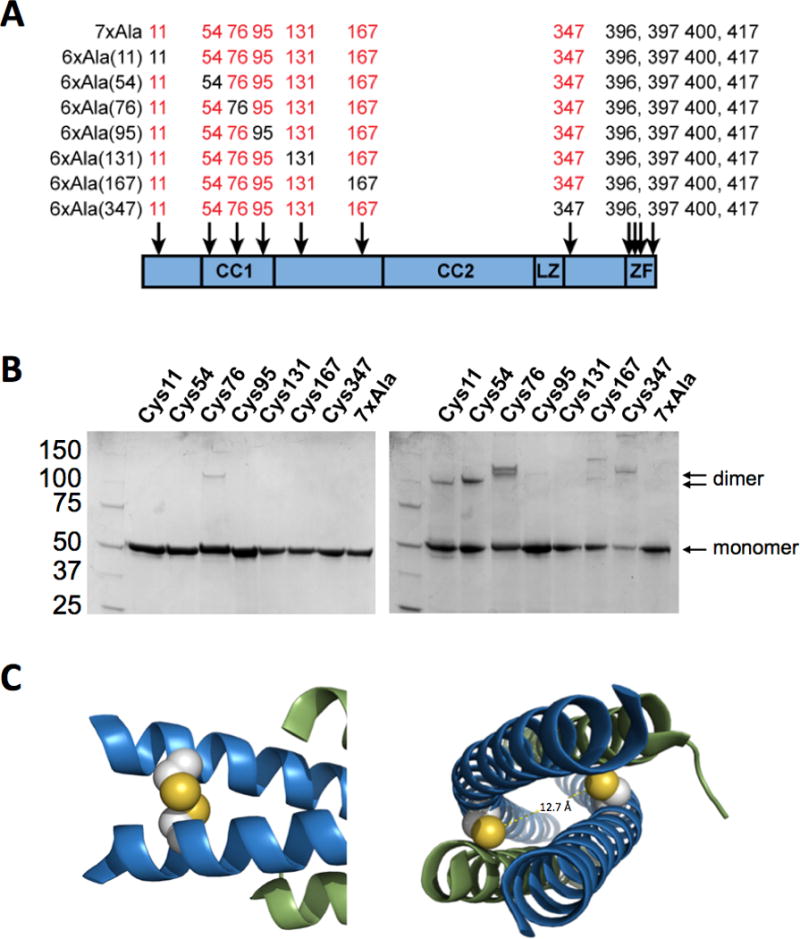
Role of individual cysteine residues in NEMO dimerization and aggregation. (A) Panel of 6xAla NEMO mutants in which each of the cysteines in the region of NEMO sequence between residue 1 and 395 has been individually added back into the 7xAla construct. (B) SDS-PAGE analysis of the 6xAla mutant panel under reducing (left) and non-reducing (right) conditions, showing that the mutants containing cysteines at positions 11, 54, 76 and 347 show a propensity to form disulfide-linked covalent dimers. (C) Images of the published co-crystal structure of NEMO(44-111) with IKKβ(701-745) (30), showing that the cysteines at position 54 (left) are well-positioned to form an inter-chain disulfide bond in the active conformation of the protein, while the cysteines at position 76 are not as their sulfur atoms are separated by 12.7 Å.
The greater propensity of 5xAla compared to 7xAla to form disulfide-mediated dimers and oligomers, highlighted in Figure 2A, supports previous work suggesting that Cys54 and Cys347 can mediate inter-chain disulfide bonding when NEMO is expressed in cells (38). The above results suggest that the binding of NEMO to IKKβ is not affected by inter-chain disulfide bonding involving Cys54, which lies in the IKKβ binding region of NEMO, though disulfide bonding at Cys347 and perhaps also at other positions can lead to formation of non-native dimers that in some cases might have reduced activity.
Active Site Titration shows that the 5xAla NEMO Preparation is Fully Active for Binding to IKKβ
To determine what fraction of the protein is active with respect to its ability to bind IKKβ, we used the FA binding assay to perform an active site titration. Active site titrations are well-established for quantifying the amount of active protein present in enzyme preparations (49), but the use of equivalent methods to characterize non-catalytic proteins is less common. By performing a binding assay under conditions where the fixed concentration component is present at levels well above the KD for interaction, the resulting binding curve becomes quadratic rather than hyperbolic, and can be analyzed to determine the stoichiometric end-point of binding. In cases where the concentrations of active ligand and receptor are accurately known, this approach can be used to determine the stoichiometry of complex formation (42, 50, 51), Alternatively, if the interaction stoichiometry is known, as is the case here for NEMO with IKKβ (28, 30), the method can be used to determine the concentration of active material in a preparation of one binding component by titrating it against a solution of its binding partner of known concentration. We performed such a titration for 5xAla NEMO by incubating various concentrations of the protein with several fixed, high concentrations of FITC-IKKβ ranging from 31.25–500 nM. FITC-IKKβ concentrations were standardized based on the absorbance of the labeled peptide measured at both 280 and 493 nm (Experimental Procedures). Figure 5A shows that each titration gave a well-defined titration end-point, and that the position of the end-point was approximately proportional to the concentration of FITC-IKKβ present in the experiment (inset plot). The slope of the inset plot in Figure 5A, in which the titration end-point is plotted against the nominal concentration of FITC-IKKβ, has a value close to unity. X-ray crystallography and other methods have shown that NEMO binds IKKβ in a 2:2 complex (28, 30). This result therefore indicates that essentially 100% of the NEMO protein present in the 5xAla preparation is active with respect to its ability to bind to IKKβ.
Figure 5.

Structure and stability of 5xAla NEMO. (A) Titration of 5xAla NEMO against various fixed concentrations of FITC-IKKβ peptide. Solid lines represent the best fit to a modified quadratic binding equation, as described in Experimental Procedures. Inset shows the titration equivalence point plotted as a function of the nominal concentration of FITC-IKKβ present in each titration, with the dashed line indicating the relationship titration end-point = [IKKβ] expected for 100% active protein. Data shown are representative of at least three independent experiments. (B) Circular dichroism spectra of wild-type NEMO, 5xAla and 7xAla NEMO after overnight dialysis against 2.5 mM TCEP. (C) Thermal melting of wild-type NEMO, 5xAla and 7xAla from 10–70 °C, as monitored by the change in molar ellipticity at 222 nm measured by circular dichroism spectroscopy. Inset plot shows the first derivative of each melting curve as a function of temperature, showing that all three variants show an unfolding transition at the same temperature of ~40 °C.
NEMO Secondary Structure, Stability and Oligomerization State
Circular dichroism spectroscopy was performed on NEMO to elucidate the secondary structure of the full-length protein and to probe the relationship between covalent dimerization and stability. Previous studies have suggested that different regions of NEMO can self-associate through formation of regions of α-helical coiled-coil (14, 26, 30, 33), though the stoichiometry of interaction has variously been proposed to involve formation of NEMO dimers, trimers, tetramers, pentamers and larger oligomers (2, 4, 13, 22, 34, 38, 52–54). Moreover, based on NMR data obtained using a fragment of NEMO encompassing residues 44-111, Rushe et al. suggested that the N-terminal domain of NEMO is disordered in the absence of bound IKKβ (30). To determine the secondary structure content of the NEMO variants, the CD spectra of wild-type NEMO, 5xAla and 7xAla NEMO were measured in the presence of 2.5 mM of reducing agent TCEP. This condition was shown to reduce non-specific aggregates while maintaining stable inter-chain disulfide bonds, resulting in wild-type NEMO that was largely a covalent dimer, 5xAla that was largely covalently monomeric but with a small fraction of covalent dimer, and 7xAla that was exclusively monomeric (Figure S3, Supporting Information). The CD spectra for all three NEMO preparations appeared identical, with strong minima at 208 and 222 nm that are characteristic of a highly α-helical protein (Figure 5B). Analysis of these spectra using the SOMCD algorithm for secondary structure estimation (45) gave helical percentages of 89.4 ± 7.3%, 89.4 ± 7.3%, and 96.3 ± 7.3% for wild-type, 5xAla, and 7xAla, respectively. The stability of the proteins was explored by performing thermal melting experiments, involving incrementally heating the samples from 10–70 °C and monitoring the loss of helical structure through the change in the CD signal at 222 nm (Figure 5C) (55). The melting temperature of the 5xAla NEMO was shown to be independent of the temperature ramping rate, and the melting was largely reversible, as shown by the fact that the CD spectrum observed after cooling back down to 10 °C was identical to the original spectrum except for a minimal (<20%) loss in signal intensity (Figure S4A, Supporting Information). All three NEMO variants showed identical melting temperatures of 41 ± 3 °C (n = 3–5), determined from the peak in the first derivative plot of the melting curve (Figure 5C, inset). Importantly, treatment of 5xAla NEMO with 0.5% H2O2, to fully convert the protein into a covalent dimer, had no measurable effect on the melting temperature of the protein compared to a sample of 5xAla that had been treated with 10 mM TCEP to ensure that the NEMO was essentially fully reduced (Figure S4B, Supporting Information). These results suggest that the presence or absence of disulfide crosslinks within a NEMO dimer at positions Cys54 and Cys347 does not significantly affect the structure or stability of the NEMO protein.
5xAla and 7xAla NEMO Mutants Are Active in Cells
NEMO is essential for NF-κB pathway signaling in response to extracellular stimuli such as TNFα. Consequently, treatment of NEMO-deficient mouse fibroblasts with TNFα induces extensive cell death as these cells cannot activate an NF-κB-dependent survival pathway (7). To test whether the 5xAla and 7xAla NEMO mutants can recapitulate the signaling effects of wild-type NEMO in mammalian cells, we retrovirally transduced NEMO-deficient fibroblasts with full-length wild-type, 5xAla, or 7xAla NEMO. Each of these NEMO proteins was able to bind to endogenous IKKβ, as shown by immunoprecipitation of NEMO followed by detection of IKKβ, whereas no IKKβ was detected when the procedure was performed using NEMO-deficient cells transduced with the empty vector (Figure 6A). Cells expressing all three forms of NEMO showed an increase in phospho-IκBα in response to treatment with TNFα, whereas cells transduced with the empty vector did not (Figure 6B). Figure 6C shows that retroviral transduction of all NEMO proteins restores the ability of NEMO-deficient cells to survive when challenged with TNFα, whereas transduction with an empty retroviral vector does not. Taken together, these results demonstrate that the 5xAla and 7xAla NEMO mutants retain the ability to interact with IKKβ to form a functional IKK signaling complex that can activate a downstream NF-κB dependent response in mammalian cells.
Figure 6.

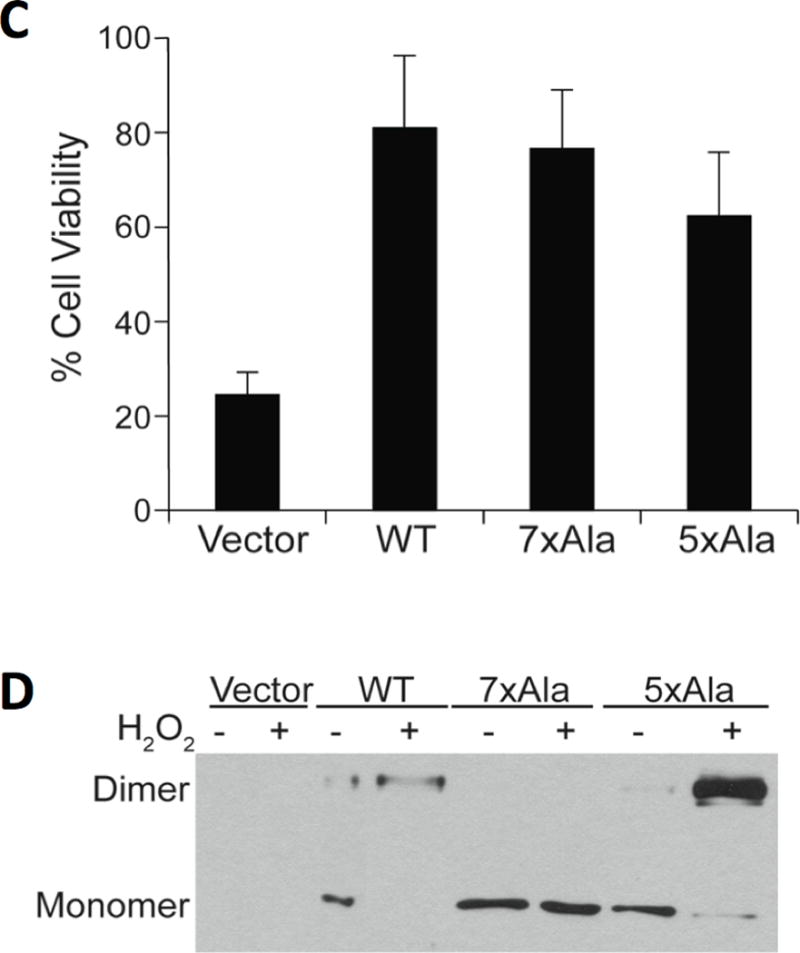
Wild-type, 5xAla and 7xAla NEMO are functionally active in cells. (A) Lysates from NEMO −/− mouse fibroblasts transduced with retroviral vectors containing no cDNA (pBABE vector), wild-type NEMO, 5xAla or 7xAla NEMO were immunoprecipitated with anti-NEMO antiserum, separated by SDS-PAGE, and immunoblotted for IKKβ (Co-IP). Panels labeled “Total” represent Western blots of 4% of the amount of lysate used for immunoprecipitation. (B) The transduced cells expressing the indicated NEMO proteins were lysed directly (−) or were treated with TNFα for 10 min before lysis (+). All lysates were then immunobloted for phospho-IκBα or for NEMO. (C) The transduced cells were treated with TNFα for 18 h, and cell viability was determined using crystal violet staining. Values (with standard deviation) are the averages of three experiments each performed with triplicate samples. For each cell type, % cell viability is relative to the value for untreated cultures (100%) measured in the same experiment. (D) Cell lines expressing the indicated NEMO proteins were treated with 200 μM H2O2 for 10 min, as described previously (38). Whole-cell extracts were prepared (in the absence of reducing agent), separated by SDS-PAGE under non-reducing conditions, and immunoblotted for NEMO. The positions of the NEMO monomer and dimer are indicated to the left of the panel.
Finally, we used the transduced fibroblasts to assess the ability of the 5xAla and 7xAla mutants to form disulfide-linked covalent dimers in cells. Through the analysis of a limited number of NEMO Cys-to-Ala mutants, we previously showed that treatment of cells with H2O2 can induce the formation of NEMO dimers that are covalently linked via homotypic intermolecular disulfide bonds involving Cys54 and Cys347 (38). To assess the effect of H2O2 on the 5xAla and 7xAla mutants, we treated retrovirally transduced NEMO-deficient cells with 200 μM H2O2 and then subjected the protein extracts to anti-NEMO Western blotting after separation by non-reducing SDS-PAGE. H2O2 treatment of cells expressing wild-type or 5xAla NEMO induced nearly complete NEMO dimer formation, whereas H2O2 treatment failed to induce covalent NEMO dimer formation in cells expressing the 7xAla NEMO mutant (Figure 6D). Thus, Cys54 and 347 are sufficient for the formation of a covalent NEMO dimer when cells are exposed to H2O2, but these disulfides are not required for NEMO to be functional in NF-κB signaling.
DISCUSSION
Despite strong interest in NEMO as a central mediator of NF-κB signaling, both the functional oligomeric state of the protein and its affinity for binding IKKβ have been subjects of widely conflicting reports in the literature (2, 4, 13, 22, 28, 30, 34, 35, 38, 53, 54). One obstacle to characterizing the biochemical properties of NEMO has been the difficulty in obtaining high quality, full-length NEMO protein. Consequently, previous reports describing the structural and biochemical analysis of NEMO have primarily used NEMO fragments or truncated forms (9, 10, 13, 14, 22–33), potentially limiting their applicability to the full-length protein. In the present study we show that mutation of five or seven of the 11 Cys residues in NEMO yields a full-length recombinant protein that is highly soluble and homogeneous, and has a binding affinity for IKKβ(701-745) that matches or exceeds previously reported values. Moreover, when expressed in NEMO −/− cells, both the 5xAla and 7xAla mutants can bind endogenous IKKβ and restore NF-κB signaling to provide protection against TNFα-induced cell death. Biochemical characterization of these well-behaved full-length NEMO constructs allowed us to resolve discrepancies in the literature concerning the affinity of NEMO-IKKβ binding and the quaternary structure of NEMO in its unbound state.
Even by maintaining stringent reducing conditions (5 mM TCEP) throughout the purification procedure we were able to obtain only low yields of bacterially expressed wild-type NEMO. Moreover, the small amount of NEMO that we obtained showed a high propensity to form large, disulfide-linked insoluble aggregates which could not be fully reduced under non-denaturing conditions, greatly limiting utility of this material for further study. These large aggregates presumably involve oxidation of multiple cysteines per NEMO monomer, as mutation of cysteines 11, 76, 95, 131 and 167 gave the much better behaved 5xAla NEMO, but additional mutation of Cys54 and Cys347 was required to fully eradicate the tendency of bacterially expressed NEMO to form small amounts of aggregate. Nevertheless, for 5xAla these aggregates were generally soluble and easily reducible.
Consistent with our previous work (38), we found that Cys54 and Cys347 can mediate inter-chain disulfide bonds to form a covalent NEMO dimer, both in solution and – under oxidizing conditions – in cells. Remarkably given that Cys54 lies within the region of the NEMO N-terminal domain that binds IKKβ, the binding affinity of full-length 5xAla NEMO for IKKβ(701-745) was not measurably affected by the presence or absence of an inter-chain disulfide bond at this position. The published X-ray crystal structure of NEMO fragment 44-111 in complex with IKKβ(701-745) (30), shown in Figures 1 and 4C, indicates that the Cys54 thiol groups are well-positioned to form an inter-chain disulfide bond in the bound complex. The finding that disulfide bonding at Cys54 has no discernible effect on the binding affinity of NEMO for IKKβ(701-745) suggests that the preorganization of the two Cys54 residues for disulfide bond formation across the NEMO dimer is essentially perfect. Analysis of the set of mutants in which each cysteine from residue 1-395 was separately added back into the 7xAla construct confirmed that cysteines at positions 11 and 76 can also mediate covalent dimer formation when these mutants are expressed in bacteria.
It is somewhat surprising that none of the seven cysteines in the region of NEMO encompassing residues 1-395 is required for formation of a high affinity, functional complex with IKKβ. A large number of single amino acid mutations scattered along the entire NEMO protein have been identified as disease-causing mutations in humans and have been shown to compromise NEMO’s ability to function in activating NF-κB (15). Moreover, Cys54 is strictly conserved across the 20 terrestrial mammals for which NEMO sequences are available in the NCBI database, and cysteines 76, 131 and 347 are also highly conserved across these species (Figure S1, Supporting Information). Our demonstration that 7xAla is active for IKKβ binding and IKKβ-dependent activation of NF-κB signaling establishes that inter-chain disulfide bonding involving Cys54 or Cys347 is not required for these activities of NEMO. This result is consistent with previous results that have shown that mutation or deletion of Cys54 only marginally affects TNFα-induced short-term activation of NF-κB DNA binding (38, 52). It has been shown previously that TNFα-induced NF-κB signaling requires not only binding to IKKβ, but also proper folding and function of the CC2-LZ region of NEMO that mediates interactions with di-ubiquitin (33). Therefore, our results suggest that the cysteine mutations present in the 5xAla and 7xAla constructs are fully compatible with proper folding and functionality of NEMO. Our results, and those of others (56), suggest that mutating multiple cysteine residues may present a generalized way to increase yield and solubility of recombinant proteins. In this context we note that among the seven Cys residues we mutated herein, only Cys167 is conserved in the Drosophila NEMO homolog (Kenny). This finding suggests that deep phylogenetic comparison of Cys residue conservation is likely to be informative when attempting to develop Cys-directed mutagenesis strategies for solving solubility problems with recombinant proteins while retaining activity.
Several previous studies have reported the affinity of the C-terminal region of IKKβ for binding to NEMO (28, 30, 35), Direct binding measurements of the interaction of a 44-mer peptide encompassing IKKβ(701-745) to a truncated NEMO(2-200) construct using surface plasmon resonance (SPR) returned a KD value of 76 nM (35), while inhibition assays using NEMO(2-200), GST-NEMO(1-196), and GST-NEMO(2-200) gave IC50 values of 15–30 nM, 1–7 nM and 47 nM, respectively (30, 35). A study that used a longer IKKβ peptide encompassing residues 680-756 reported an affinity for binding to NEMO(38-196) of 3.4 nM by SPR but 1 μM by isothermal titration calorimetry (28). The homogeneous, full-length NEMO we describe here gave a binding affinity for IKKβ(701-745) of KD = 2.2 ± 0.8 nM (n = 4), suggesting that the true KD for the interaction of NEMO with IKKβ lies towards the lower end of the range defined by these previous studies. While some of these differences could simply reflect experimental variation, it is also possible that NEMO constructs containing only the N-terminal portion of the protein bind IKKβ less strongly than does full-length NEMO. It has been shown that NEMO can self-associate through the C-terminal region centered on residues 242-388 (22), and also through residues within the region 111-196 (30). While these portions of NEMO are not directly involved in binding IKKβ, interactions outside the IKKβ binding region might help stabilize the active conformation of NEMO’s N-terminal domain to enable high-affinity binding to IKKβ.
In the FA binding measurements, the maximum FA signal observed for wild-type NEMO was consistently 15–20% greater than that seen for the 5xAla or 7xAla constructs. This variation cannot be attributed to errors in determining the concentrations of the different NEMO constructs, or to the presence of a fraction of inactive material in any NEMO preparation, as such eventualities would be manifested as errors in the KD value and would not be expected to affect the maximum observed anisotropy. This is because in FA direct binding assays, unlike most binding assays, the ligand (tracer probe) is held at constant concentration, and it is the concentration of the receptor (NEMO) that is varied. Thus, the maximum signal seen at saturating NEMO concentrations depends only on the concentration of the tracer probe and the effective sizes (correlation times) of the bound complexes present in solution, representing a weighted average of the contributions of the different complexes present (40). The three NEMO constructs were compared using identical concentrations of FITC-IKKβ derived from the same stock solution. Therefore, the consistently higher maximum signal observed for wild-type NEMO in FA binding experiments (such as shown in Figure 3B) suggests the presence of a minor fraction of larger but still active species in the wild-type NEMO preparation, leading to a fraction of slower tumbling complexes with FITC-IKKβ that elevate the maximum FA signal. The gel filtration data (Figure 2C) do not show any evidence of major differences in the heterogeneity of wild-type NEMO as compared to 5xAla or 7xAla under the condition of 10 mM DTT that was used in the FA binding assay. Thus, we attribute the higher maximum FA signal observed for wild-type NEMO to the presence of a small percentage of high molecular weight NEMO aggregates in this protein preparation, which can bind FITC-IKKβ to form a small fraction of extremely slow tumbling complexes. This interpretation is in keeping with the greater propensity of the wild-type NEMO to aggregate (Figures 2A and B) and is also supported by the observation that a similar increase in maximum anisotropy at saturating NEMO was observed for 5xAla NEMO after treatment with 5% H2O2 (Figure 3D).
The oligomeric state of the active form of NEMO has been a topic of some debate in the literature. Various studies describing crosslinking experiments or other methods with endogenous NEMO or with recombinant truncated NEMO constructs have found evidence for NEMO dimers (2, 13, 14, 22, 27, 28, 30, 34), trimers (2, 22, 27, 34), and tetramers (13, 28, 57). Oligomerization is clearly important for NEMO function; it has been shown that inhibition of NEMO self-association using cell-permeable peptides encompassing the NEMO CC2 and/or LZ domains inhibits RANKL- and TNFα-dependent NF-κB signaling and function (58). In our binding experiments with full-length NEMO we saw no difference in IKKβ binding affinity when the 5xAla mutant, which contains Cys54 and Cys347 that can covalently cross-link the NEMO dimer, was tested under oxidizing versus reducing conditions. Moreover, the IKKβ binding affinity observed for the 5xAla mutant was indistinguishable from that of the 7xAla mutant and wild-type NEMO. If a significant fraction of NEMO existed as a monomer under reducing conditions, then IKKβ binding should be weakened relative to its interaction with a preformed covalent NEMO dimer, due to the equilibrium coupling of NEMO dimerization and subsequent IKKβ binding (Scheme 1). If NEMO were predominantly monomeric in the absence of ligand, then the apparent affinity for binding to IKKβ would be reduced, compared to the affinity observed for covalently dimeric NEMO, due to the free energy required to bring the two NEMO monomers together into the final complex. The observation that the affinity of FITC-IKKβ(701-745) is identical for oxidized versus reduced 5xAla NEMO and for 7xAla NEMO therefore indicates that, in these experiments, essentially all NEMO was dimeric even in the absence of inter-chain disulfide bonds. Thus, for full-length NEMO KDimer ≪ 5 nM (the approximate concentration of NEMO at the KD for binding IKKβ), implying that the protein exists as a constitutive noncovalent dimer even in the absence of inter-chain disulfide bonds at Cys54, Cys347 or any of the other five cysteine residues that were mutated in 7xAla. The notion that full-length NEMO is constitutively dimeric even in the absence of inter-chain disulfides is supported by our observation that the wild-type, 5xAla and 7xAla proteins contained the same α-helical content and thermal stability, as measured by CD, and that the thermal stability of 5xAla was not detectably affected by whether it was tested in an oxidized or reduced state.
Scheme 1.
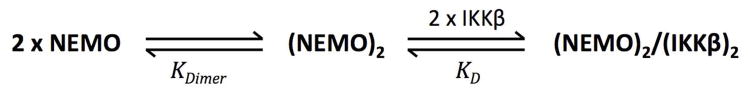
Reaction scheme showing how, in cases where inter-chain disulfide bonds are absent, the equilibrium for NEMO dimer formation is coupled to IKKβ binding.
The previous observation that NEMO fragment 44-111, which incorporates the IKKβ binding region of the protein is intrinsically disordered in the absence of ligand (30), suggesting that noncovalent interactions involving this region of NEMO are not by themselves sufficient to induce formation of a stable dimer. It therefore appears likely that noncovalent interactions involving regions of NEMO downstream of the IKKβ binding region are responsible for stabilizing the NEMO dimer, consistent with previous reports that interactions involving the CC2 and LZ domains of NEMO mediate self-association of the protein (22, 26, 34). Our results do not rule out the possibility that NEMO forms higher order oligomers at high concentrations, as proposed by Ivins et al. (13). Based on characterization of truncated NEMO constructs comprising residues 215-362, it has been suggested that NEMO dimerization is relatively weak, and that the dimer is stabilized upon di-ubiquitin binding (26). In contrast, our measurements with full-length recombinant NEMO suggest that dimerization occurs with high affinity even in the absence additional stabilization through di-ubiquitin binding, disulfide bond formation at Cys54 and/or Cys347, or other such mechanisms, and that in cells the protein exists as a constitutive noncovalent dimer.
Supplementary Material
Acknowledgments
We thank Robert Prenovitz and Natasha Gill for their preliminary work on this project, and Alexander Hoffmann (UCSD) for NEMO-deficient fibroblasts.
Funding: This research was supported by NIH grant GM094551. MH was supported by a Pre-doctoral Fellowship from the Natural Sciences & Engineering Research Council of Canada. UW, RB, KG and TE received funding from the Boston University Undergraduate Research Opportunities Program. The CD spectrometer was purchased under NSF grant CHE1126545.
Abbreviations
- CC1
the first coiled-coil domain of NEMO
- CC2
the second coiled-coil domain of NEMO
- CD
circular dichroism
- Cys
cysteine
- DTT
dithiothreitol
- FA
fluorescence anisotropy
- FITC-IKKβ
IKKβ(701-745)-derived peptide labeled with fluorescein isothiocyanate
- IκB
inhibitor of kappa-B
- IKK
inhibitor of kappa-B kinase
- LZ
leucine zipper
- NEMO
NF-κB Essential Modulator
- NF-κB
nuclear factor κB
- RANKL
RANK ligand
- rpm
revolutions per minute
- TNFα
tumor necrosis factor alpha
- TCEP
tris(2-carboxyethyl)phosphine
Footnotes
NOTES: The authors declare no competing financial interests.
Supporting Information Available: Table showing conservation of Cys residues from human NEMO across mammalian orthologs, additional results substantiating the covalent oligomeric state of wild-type, 5xAla, and 7xAla NEMO constructs after treatment under different redox conditions, thermal melting data for 5xAla NEMO under oxidizing and reducing conditions, and additional experimental details pertaining to data analysis using DynaFit. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 2.Rothwarf DM, Zandi E, Natoli G, Karin M. IKKγ is an essential regulatory subunit of the IκB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- 3.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 4.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israël A. Complementation cloning of NEMO, a component of the IκB kinase complex essential for NF-κB activation. Cell. 1998;93:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 5.Karin M, Delhase M. The IκB kinase (IKK) and NF-κB: key elements of proinflammatory signalling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 6.Shih VFS, Tsui R, Caldwell A, Hoffmann A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2010;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makris C, Roberts JL, Karin M. The carboxyl-terminal region of IκB kinase γ (IKK γ) is required for full IKK activation. Mol Cell Biol. 2002;22:6573–6581. doi: 10.1128/MCB.22.18.6573-6581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israël A, Rajewsky K, Pasparakis M. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol Cell. 2000;5:981–992. doi: 10.1016/s1097-2765(00)80263-4. [DOI] [PubMed] [Google Scholar]
- 9.Schröfelbauer B, Polley S, Behar M, Ghosh G, Hoffmann A. NEMO ensures signaling specificity of the pleiotropic IKKβ by directing its kinase activity toward IκBα. Mol Cell. 2012;47:111–121. doi: 10.1016/j.molcel.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kensche T, Tokunaga F, Ikeda F, Goto E, Iwai K, Dikic I. Analysis of NF-κB essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-κB. J Biol Chem. 2012;287:13626–12634. doi: 10.1074/jbc.M112.347195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Hadian K, Griesbach RA, Dornauer S, Wanger TM, Nagel D, Metlitzky M, Beisker W, Schmidt-Supprian M, Krappmann D. NF-κB essential modulator (NEMO) interaction with linear and Lys-63 ubiquitin chains contributes to NF-κB activation. J Biol Chem. 2011;286:26107–26117. doi: 10.1074/jbc.M111.233163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivins FJ, Montgomery MG, Smith SJM, Morris-Davies AC, Taylor IA, Rittinger K. NEMO oligomerization and its ubiquitin-binding properties. Biochem J. 2009;421:243–251. doi: 10.1042/BJ20090427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo YC, Lin SC, Rospigliosi CC, Conze DB, Wu CJ, Ashwell JD, Eliezer D, Wu H. Structural basis for recognition of diubiquitins by NEMO. Mol Cell. 2009;33:602–615. doi: 10.1016/j.molcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 16.May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 17.Tang ED. A role for NF-κB essential modifier/IκB kinase-γ (NEMO/IKKγ) ubiquitination in the activation of the IκB kinase complex by tumor necrosis factor-α. J Biol Chem. 2003;278:37297–37305. doi: 10.1074/jbc.M303389200. [DOI] [PubMed] [Google Scholar]
- 18.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 19.Cordier F, Vinolo E, Véron M, Delepierre M, Agou F. Solution structure of NEMO zinc finger and impact of an anhidrotic ectodermal dysplasia with immunodeficiency-related point mutation. J Mol Biol. 2008;377:1419–1432. doi: 10.1016/j.jmb.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 20.Sebban H, Yamaoka S, Courtois G. Posttranslational modifications of NEMO and its partners in NF-κB signaling. Trends Cell Biol. 2006;16:569–577. doi: 10.1016/j.tcb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell. 2003;115:565–576. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 22.Agou F, Ye F, Goffinont S, Courtois G, Yamaoka S, Israël A, Véron M. NEMO trimerizes through its coiled-coil C-terminal domain. J Biol Chem. 2002;277:17464–17475. doi: 10.1074/jbc.M201964200. [DOI] [PubMed] [Google Scholar]
- 23.Bloor S, Ryzhakov G, Wagner S, Butler PJG, Smith DL, Krumbach R, Dikic I, Randow F. Signal processing by its coil zipper domain activates IKKγ. Proc Natl Acad Sci USA. 2008;105:1279–1284. doi: 10.1073/pnas.0706552105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drew D, Shimada E, Huynh K, Bergqvist S, Talwar R, Karin M, Ghosh G. Inhibitor κB kinase β binding by inhibitor κB kinase γ. Biochemistry. 2007;46:12482–12490. doi: 10.1021/bi701137a. [DOI] [PubMed] [Google Scholar]
- 25.Gotoh Y, Nagata H, Kase H, Shimonishi M, Ido M. A homogeneous time-resolved fluorescence-based high-throughput screening system for discovery of inhibitors of IKKβ–NEMO interaction. Anal Biochem. 2010;405:19–27. doi: 10.1016/j.ab.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 26.Grubisha O, Kaminska M, Duquerroy S, Fontan E, Cordier F, Haouz A, Raynal B, Chiaravalli J, Delepierre M, Israël A, Véron M, Agou F. DARPin-assisted crystallography of the CC2-LZ domain of NEMO reveals a coupling between dimerization and ubiquitin binding. J Mol Biol. 2010;395:89–104. doi: 10.1016/j.jmb.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Huang GJ, Zhang ZQ, Jin DY. Stimulation of IKK-γ oligomerization by the human T-cell leukemia virus oncoprotein Tax. FEBS Lett. 2002;531:494–498. doi: 10.1016/s0014-5793(02)03590-1. [DOI] [PubMed] [Google Scholar]
- 28.Lo YC, Maddineni U, Chung JY, Rich RL, Myszka DG, Wu H. High-affinity interaction between IKKβ and NEMO. Biochemistry. 2008;47:3109–3116. doi: 10.1021/bi702312c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.May MJ. Characterization of the IκB-kinase NEMO binding domain. J Biol Chem. 2002;277:45992–46000. doi: 10.1074/jbc.M206494200. [DOI] [PubMed] [Google Scholar]
- 30.Rushe M, Silvian L, Bixler S, Chen LL, Cheung A, Bowes S, Cuervo H, Berkowitz S, Zheng T, Guckian K, Pellegrini M, Lugovskoy A. Structure of a NEMO/IKK-associating domain reveals architecture of the interaction site. Structure. 2008;16:798–808. doi: 10.1016/j.str.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 31.Strnad J, McDonnell PA, Riexinger DJ, Mapelli C, Cheng L, Gray H, Ryseck RP, Burke JR. NEMO binding domain of IKK-2 encompasses amino acids 735–745. J Mol Recognit. 2006;19:227–233. doi: 10.1002/jmr.766. [DOI] [PubMed] [Google Scholar]
- 32.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 33.Yoshikawa A, Sato Y, Yamashita M, Mimura H, Yamagata A, Fukai S. Crystal structure of the NEMO ubiquitin-binding domain in complex with Lys 63-linked di-ubiquitin. FEBS Lett. 2009;583:3317–3322. doi: 10.1016/j.febslet.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 34.Agou F, Traincard F, Vinolo E, Courtois G, Yamaoka S, Israël A, Véron M. The trimerization domain of NEMO is composed of the interacting C-terminal CC2 and LZ coiled-coil subdomains. J Biol Chem. 2004;279:27861–27869. doi: 10.1074/jbc.M314278200. [DOI] [PubMed] [Google Scholar]
- 35.Baima ET, Guzova JA, Mathialagan S, Nagiec EE, Hardy MM, Song LR, Bonar SL, Weinberg RA, Selness SR, Woodard SS, Chrencik J, Hood WF, Schindler JF, Kishore N, Mbalaviele G. Novel insights into the cellular mechanisms of the anti-inflammatory effects of NF-κB essential modulator binding domain peptides. J Biol Chem. 2010;285:13498–13506. doi: 10.1074/jbc.M109.099895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golden MS, Cote SM, Sayeg M, Zerbe BS, Villar EA, Beglov D, Sazinsky SL, Georgiadis RM, Vajda S, Kozakov D, Whitty A. Comprehensive experimental and computational analysis of binding energy hot spots at the NF-κB essential modulator/IKKβ protein-protein interface. J Am Chem Soc. 2013;135:6242–6256. doi: 10.1021/ja400914z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bagnéris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, Waksman G, Barrett T. Crystal structure of a vFlip-IKKγ complex: insights into viral activation of the IKK signalosome. Mol Cell. 2008;30:620–631. doi: 10.1016/j.molcel.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 38.Herscovitch M, Comb W, Ennis T, Coleman K, Yong S, Armstead B, Kalaitzidis D, Chandani S, Gilmore TD. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem Biophys Res Commun. 2008;367:103–108. doi: 10.1016/j.bbrc.2007.12.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thermo Scientific. Tech Tip #31: Calculate dye:protein (F/P) ratios. [Google Scholar]
- 40.Lakowicz JR. Principles of fluorescence spectroscopy. Springer; New York: 2006. [Google Scholar]
- 41.Roehrl MHA, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 42.Day ES, Cachero TG, Qian F, Sun Y, Wen D, Pelletier M, Hsu YM, Whitty A. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry. 2005;44:1919–1931. doi: 10.1021/bi048227k. [DOI] [PubMed] [Google Scholar]
- 43.Kuzmič P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 44.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2007;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Unneberg P, Merelo JJ, Chacón P, Morán F. SOMCD: method for evaluating protein secondary structure from UV circular dichroism spectra. Proteins Struct Funct Bioinforma. 2001;42:460–470. doi: 10.1002/1097-0134(20010301)42:4<460::aid-prot50>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 46.Liang MC, Bardhan S, Pace EA, Rosman D, Beutler JA, Porco JA, Jr, Gilmore TD. Inhibition of transcription factor NF-κB signaling proteins IKKβ and p65 through specific cysteine residues by epoxyquinone A monomer: Correlation with its anti-cancer cell growth activity. Biochem Pharmacol. 2006;71:634–645. doi: 10.1016/j.bcp.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 47.Gapuzan MER, Yufit PV, Gilmore TD. Immortalized embryonic mouse fibroblasts lacking the RelA subunit of transcription factor NF-κB have a malignantly transformed phenotype. Oncogene. 2002;21:2484–2492. doi: 10.1038/sj.onc.1205333. [DOI] [PubMed] [Google Scholar]
- 48.GE Healthcare. Purifying Challenging Proteins: Principles and Methods. General Electric Company; Uppsala, Sweden: 2007. [Google Scholar]
- 49.Knight CG. Active-site titration of peptidases. Methods Enzymol. 1995;248:85–101. doi: 10.1016/0076-6879(95)48008-0. [DOI] [PubMed] [Google Scholar]
- 50.Day ES, Capili AD, Borysenko CW, Zafari M, Whitty A. Determining the affinity and stoichiometry of interactions between unmodified proteins in solution using biacore. Anal Biochem. 2013;440:96–107. doi: 10.1016/j.ab.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piehler J, Brecht A, Giersch T, Hock B, Gauglitz G. Assessment of affinity constants by rapid solid phase detection of equilibrium binding in a flow system. J Immunol Methods. 1997;201:189–206. doi: 10.1016/s0022-1759(96)00222-0. [DOI] [PubMed] [Google Scholar]
- 52.Marienfeld RB, Palkowitsch L, Ghosh S. Dimerization of the IκB kinase-binding domain of NEMO is required for tumor necrosis factor alpha-induced NF-κB activity. Mol Cell Biol. 2006;26:9209–9219. doi: 10.1128/MCB.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poyet JL, Srinivasula SM, Lin JH, Fernandes-Alnemri T, Yamaoka S, Tsichlis PN, Alnemri ES. Activation of the IκB kinases by RIP via IKKγ/NEMO-mediated oligomerization. J Biol Chem. 2000;275:37966–37977. doi: 10.1074/jbc.M006643200. [DOI] [PubMed] [Google Scholar]
- 54.Fontan E, Traincard F, Levy SG, Yamaoka S, Véron M, Agou F. NEMO oligomerization in the dynamic assembly of the IκB kinase core complex. FEBS J. 2007;274:2540–2551. doi: 10.1111/j.1742-4658.2007.05788.x. [DOI] [PubMed] [Google Scholar]
- 55.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc. 2007;1:2527–2535. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kotaria R, Mayor JA, Walters DE, Kaplan RS. Oligomeric state of wild-type and cysteine-less yeast mitochondrial citrate transport proteins. J Bioenerg Biomembr. 1999;31:543–549. doi: 10.1023/a:1005460810527. [DOI] [PubMed] [Google Scholar]
- 57.Tegethoff S, Behlke J, Scheidereit C. Tetrameric oligomerization of IκB kinase (IKKγ) is obligatory for IKK complex activity and NF-κB activation. Mol Cell Biol. 2003;23:2029–2041. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Darwech I, Otero J, Alhawagri M, Dai S, Abu-Amer Y. Impediment of NEMO oligomerization inhibits osteoclastogenesis and osteolysis. J Cell Biochem. 2009;108:1337–1345. doi: 10.1002/jcb.22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.The UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2012;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.