

Abstract
Lyme neuroborreliosis, caused by the spirochete Borrelia burgdorferi, affects both peripheral and central nervous systems. We assessed a causal role for inflammation in Lyme neuroborreliosis pathogenesis by evaluating the induced inflammatory changes in the central nervous system, spinal nerves, and dorsal root ganglia (DRG) of rhesus macaques that were inoculated intrathecally with live B. burgdorferi and either treated with dexamethasone or meloxicam (anti-inflammatory drugs) or left untreated. ELISA of cerebrospinal fluid showed significantly elevated levels of IL-6, IL-8, chemokine ligand 2, and CXCL13 and pleocytosis in all infected animals, except dexamethasone-treated animals. Cerebrospinal fluid and central nervous system tissues of infected animals were culture positive for B. burgdorferi regardless of treatment. B. burgdorferi antigen was detected in the DRG and dorsal roots by immunofluorescence staining and confocal microscopy. Histopathology revealed leptomeningitis, vasculitis, and focal inflammation in the central nervous system; necrotizing focal myelitis in the cervical spinal cord; radiculitis; neuritis and demyelination in the spinal roots; and inflammation with neurodegeneration in the DRG that was concomitant with significant neuronal and satellite glial cell apoptosis. These changes were absent in the dexamethasone-treated animals. Electromyography revealed persistent abnormalities in F-wave chronodispersion in nerve roots of a few infected animals; which were absent in dexamethasone-treated animals. These results suggest that inflammation has a causal role in the pathogenesis of acute Lyme neuroborreliosis.
Lyme disease is caused by infection with the spirochete Borrelia burgdorferi (Bb). The spirochetes enter the host's skin via the bite of infected Ixodes scapularis ticks, causing an inflammatory response that may result in the appearance of a slowly radiating erythematous rash called erythema migrans, followed commonly, after spirochetal dissemination, by early flu-like symptoms, including headaches, fever, fatigue, malaise, and diffuse aches and pains.1 The disseminating spirochetes show distinct organotropisms, and manifestations of infection can include arthritis, carditis, and neurologic deficits.2,3
Nervous system involvement in Lyme disease, termed Lyme neuroborreliosis (LNB), is manifest in approximately 15% of Lyme disease patients and may affect both the central (CNS) and peripheral nervous systems (PNS). CNS involvement may result in symptoms such as headache, fatigue, memory loss, learning disability, or depression. LNB of the PNS may result in facial nerve palsy, limb pain, sensory loss, and/or muscle weakness.4–6
Clinical findings of patients with LNB typically show the neurologic triad of meningitis, cranial neuritis, and radiculoneuritis,1,7 commonly described as meningoradiculitis (also known as Garin-Bujadoux-Bannwarth syndrome). Lyme meningitis presents mostly as leptomeningitis, characterized by lymphocytic pleocytosis in the cerebrospinal fluid (CSF).8 LNB patients may experience encephalopathy, encephalitis, and encephalomyelitis concomitant with white matter inflammation in the brain and spinal cord.9–11
Neurogenic pain along the back, radiating into the legs and foot, accompanied with weakness, numbness, and tingling in the legs, described as radiculitis or radiculoneuritis, is the most common starting symptom in patients with peripheral LNB.12,13 Motor deficits are also common, and pain and motor deficits are classically dermatomal or localized to the limb closest to the tick bite, suggesting a pathology that involves sensory neurons that arise from dorsal root ganglia (DRG) in that area of the spinal cord.14 Other mononeuropathies and plexopathies that result in pain, loss of motor control, and sensory deficits also occur, with patients exhibiting electrophysiologic abnormalities indicative of widespread axonal damage.12–16 A few case reports also suggest an association with demyelinating neuropathies whereby nerve conduction studies (NCSs) showed conduction slowing and abnormal temporal dispersion, consistent with demyelinating neuropathy.17
Importantly, pathologic examinations of CNS lesions from cases of human LNB have revealed lymphocyte and plasma cell infiltration in the leptomeninges and perivascular infiltrates of immune cells adjacent to white matter lesions in the brain and transverse myelitis lesions in the spinal cord,18–25 whereas lesions from patients with PNS Lyme disease have shown inflammation in the nerve roots and DRG and patchy multifocal axonal loss accompanied with epineural perivascular inflammatory infiltrates or perineuritis.12,26,27
The rhesus macaque has proved to be an accurate model of human nervous system Lyme disease.28–31 In one study, almost all of the experimental animals demonstrated perivascular inflammatory infiltrates, multifocal axonal changes, and NCS results that were consistent with mononeuropathy multiplex.32 Sensory ganglia of rhesus macaques that were infected with Bb showed various degrees of necrosis, and peripheral nerve specimens showed multifocal axonal degeneration and regeneration and occasional perivascular inflammatory cellular infiltrates in which macrophages showed positive immunostaining with a monoclonal antibody against a 7.5-kDa lipoprotein of Bb.32 Infection in nerve roots, DRG, and involvement of the spinal cord was also observed in the rhesus monkey model of LNB.33–35
Previously, we reported that rhesus macaques that were inoculated with live Bb into the cisterna magna showed increased levels of IL-6, IL-8, chemokine ligand 2 (CCL2), and CXCL13 in the CSF within 1 week after inoculation, accompanied by a monocytic/lymphocytic pleocytosis.35 In addition, we observed elevated levels of neuronal and satellite glial cell apoptosis in the DRG of infected rhesus macaques, compared with uninfected controls. Importantly, the acute neurologic manifestations observed histopathologically as leptomeningitis and radiculitis were concomitant with the inflammatory response mounted by the Lyme disease spirochete.35 Our aim was to evaluate whether inflammation as induced by the Lyme disease spirochete has a causal role in mediating the pathogenesis of acute LNB. We hypothesized that Bb induces the production of inflammatory mediators in glial and neuronal cells and that this response has a role in potentiating glial and neuronal apoptosis. We addressed this hypothesis by evaluating the inflammatory changes induced in the CNS, spinal nerves, and DRG of rhesus macaques that were inoculated with live Bb into the cisterna magna and were either left untreated or were given the anti-inflammatory drug dexamethasone (Dex), a steroid that inhibits the expression of several immune mediators,36 or meloxicam (Mel), a nonsteroidal anti-inflammatory drug that inhibits cyclooxygenase-2.37 Rhesus macaques were studied for either 8 or 14 weeks. In accordance with our hypothesis we found that the effective suppression of inflammation by Dex treatment resulted in inhibition of glial and neuronal damage, suggesting that inflammation has a causal role in the pathogenesis of LNB. Here, we report the results of these studies.
Materials and Methods
Spirochetal Inoculum
Bb strain B31 clone 5A19 spirochetes, passage one, isolated from an ear biopsy of a previously infected mouse were grown in Barbour-Stoenner-Kelly–H medium supplemented with 6% rabbit serum and antibiotics (rifampicin at 45.4 μg/mL, phosphomycin at 193 μg/mL, and amphotericin at 0.25 μg/mL; Sigma-Aldrich, St. Louis, MO) to late logarithmic phase under microaerophilic conditions. The inoculum that contained a suspension of 1 × 108 spirochetes/mL in RPMI 1640 medium (Invitrogen, Grand Island, NY) was prepared as described earlier.35
Animals, Drug Treatments, and Intrathecal Inoculation
Fourteen 3- to 7-year-old male rhesus macaques (Macaca mulatta) of Chinese origin were used in this study. Twelve rhesus macaques were anesthetized and inoculated intrathecally into the cisterna magna with 1 × 108 live spirochetes suspended in 1 mL of RPMI 1640 medium, whereas two rhesus macaques were left uninfected but received 1 mL of RPMI 1640 medium after removing an equivalent volume of CSF, after an inoculation protocol that was approved by the Institutional Animal Care and Use Committee of the Tulane National Primate Research Center. Of the twelve Bb-inoculated rhesus macaques, four were left untreated, four were treated with Dex, and four were treated with Mel. Half of the rhesus macaques in each group of four were studied for a period of 8 weeks after inoculation and the other half for 14 weeks. One of the uninfected controls was also studied for 8 weeks and the other for 14 weeks.
Drug treatments were started 1 week before inoculation and consisted of Dex (PAR Pharmaceuticals, Spring Valley, NY), 4-mg tablets, given at a dose of 2 mg/kg once a day for 1 week and subsequently at 1 mg/kg once a day for the remainder of the study, and Mel (Metacam; Boehringer Ingelheim, St. Joseph, MO), 1.5 mg/mL, given at a dose of 0.18 mg/kg on day 1 (loading dose) and subsequently at 0.09 mg/kg once a day for the remainder of the study. These doses are consistent with standard veterinary regimens for the chosen drugs in nonhuman primates (Nonhuman Primate Formulary, http://www.primatevets.org/Content/files/Public/education/Nonhuman%20Primate%20Formulary.xls, last accessed December 22, 2014).38 The drugs (liquid or crushed tablets) were placed in a piece of fruit (bananas or apples but not citrus fruits) for daily administration. The Bb-infected untreated rhesus macaques and the uninfected controls received the same fruit devoid of medication.
Evaluation of CSF Pleocytosis, Monitoring of CNS Inflammation, and Bb Culture of CSF and CNS Tissues
The enumeration of cells in the CSF for evaluation of pleocytosis and preparation of CSF cell pellets for Bb culture was performed as previously described.35 To monitor CNS inflammation, 0.5 to 1.0 mL of CSF and serum (5 mL of blood) were collected at base line (day 0) before inoculation, subsequently after inoculation on a weekly basis for 4 weeks, and then once every 2 weeks until the end of the study for each animal. Rhesus macaques were euthanized as per a procedure consistent with the recommendations of the American Veterinary Medical Association's Panel on Euthanasia.
Tissues from various regions of the brain and spinal cord that were collected at necropsy and CSF cell pellets that were prepared from samples drawn at the various time points described in the paragraph above were incubated in 10 mL of Barbour-Stoenner-Kelly–H medium supplemented with rabbit serum and antibiotics for culture of spirochetes under microaerophilic conditions, as previously described.35
Quantification of Immune Mediators in CSF and Serum and Detection of Anti-VlsE (C6) Antibodies in Serum
The concentrations of cytokines and chemokines present in CSF and serum were quantified with the MILLIPLEX MAP Non-Human Primate Cytokine Magnetic Bead Panel, Premixed 23 Plex, PCYTMG-40 K-PX23 Cytokine-Chemokine Array kit (Millipore, Billerica, MA) according to the manufacturer's instructions. The analytes detected by this panel are as follows: granulocyte CSF, granulocyte-macrophage CSF, interferon-γ, IL-10, IL-12/23 (p40), IL-13, IL-15, IL-17, IL-18, IL-1ra, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, CCL2, CCL3, CCL4, transforming growth factor (TGF)-α, tumor necrosis factor, vascular endothelial growth factor, and sCD40L. The multiplex plate was read with a Bio-Plex 200 Suspension Array Luminex System (Bio-Rad, Hercules, CA). CXCL13 concentration was measured with a sandwich ELISA (R&D Systems, Minneapolis, MN). The cutoff line for positive sandwich ELISA values was set as the mean value for CXCL13 of all of the preinfection and control CSF specimens plus three times the SD of that mean.
The levels of serum antibody to the VlsE (C6) peptide were quantified with ELISA by using a procedure described previously.39 The mean value of all of the preimmune serum samples plus three times the SD of that mean was used to set the cutoff line for a positive C6 ELISA.
Histopathologic Evaluation, FJC Staining for Neurodegeneration, and Luxol-Fast Blue Staining for Demyelination
Tissues that were collected from various regions of the brain, spinal cord, and DRG at necropsy and fixed in formalin or Z-fixative (Anatech, Battle Creek, MI) were sectioned at 5-μm thickness and processed for hematoxylin and eosin staining for routine histopathologic evaluation.40 Tissues fixed in Z-fixative were also used for evaluation of neurodegeneration. This was performed in tissue sections cut at a thickness of 5 μm, using the Fluoro-Jade C (FJC) ready-to-dilute staining kit (Biosensis, Thebarton, SA, Australia) as per the manufacturer's instructions. FJC is able to identify degenerating neurons.41,42 Its application was followed by immunofluorescence staining with the nuclear stain TOPRO3 (Invitrogen). The slides were then air-dried and coverslipped with a nonaqueous, low-fluorescence, styrene-based mounting medium, DPX (Sigma-Aldrich). Formalin-fixed tissues cut at 5-μm thickness were used to detect demyelination after hematoxylin and eosin staining, in addition to the Luxol Fast Blue–periodic acid-Schiff–hematoxylin combination method.43,44
Determination of Phenotypes of Producer Cells, Intracytoplasmic Localization of Immune Mediators, and Detection of Cells in Inflammatory Lesions and Bb Antigen in DRG
To determine the phenotypes of producer cells and to detect intracytoplasmic localization of immune mediators, fresh tissues were collected from the DRG at necropsy and immediately processed for blocking of intracytoplasmic cytokines, followed by fixation in 2% paraformaldehyde, as previously described.45 They were then cryopreserved after cryosectioning into 16-μm sections. DRG tissue collected at necropsy, fixed in formalin (Z-fixative), and sectioned into 10-μm sections was used for the detection of Bb antigen and for the characterization of phenotypes of cells in inflammatory lesions.
Formalin-fixed tissues were subjected to antigen retrieval (Vector Laboratories, Burlingame, CA) before immunofluorescence staining. Sections were subjected to immunofluorescence staining by using combinations of primary antibodies listed in the next paragraph and corresponding secondary antibodies diluted in 10% normal goat serum, as previously described.46 Relevant isotype controls at the concentration of the corresponding primary monoclonal antibodies (Sigma-Aldrich) and universal rabbit immunoglobulin-negative controls for rabbit polyclonal antibodies (Dako Cytomation, Carpinteria, CA) were also included.
The anti-immune mediator primary antibodies used included anti-human IL-6, mouse IgG2a (ProSpec, Ness Ziona, Israel) at 1:1000; anti-human CCL2, rabbit polyclonal IgG clone ab7814 (Abcam, Cambridge, MA) at 1:50; or anti-human IL-8, polyclonal rabbit IgG (Research Diagnostics Inc., Flanders, NJ) at 10 μg/mL The primary antibodies against various phenotypic markers of cells used were anti-human 2,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), clone 11-5B mouse IgG1 (Millipore) at 10 μg/mL; anti-human S-100 (Sigma-Aldrich) at 1:500; anti-human neuronal nuclear protein (NeuN), MAB 377 clone A60, mouse IgG1 (Millipore) at 1:10; anti-human CD3 polyclonal rabbit IgG (Biocare, Concord, CA) at 1:200; anti-human CD20 clone L29, mouse IgG2a (Dako Cytomation) at 1:200; and anti-human CD68 clone KP1, mouse IgG1 at 1:50 (Dako Cytomation). Bb was stained with a rabbit polyclonal antibody against whole Borrelia (Accurate Chemicals, Westbury, NY) at 1:200.
After completion of immunofluorescence staining, slides were mounted in antiquenching agent (Sigma-Aldrich) and stored at 4°C until viewed on a confocal microscope (see Confocal Microscopy). A total of three cryosections per block of DRG tissue were evaluated per rhesus macaques for the detection of intracytoplasmic immune mediators, phenotypes of producer cells and phenotypic markers of cells in inflammatory lesions and for the detection of Bb antigen from formalin-fixed tissues, respectively.
Qualitative and Quantitative Assessment of Glial and Neuronal Apoptosis in the DRG
Glial and neuronal apoptosis by the in situ terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay was assessed by immunofluorescence staining in tissues collected from the DRG. Tissues were directly fixed in 2% paraformaldehyde and cryopreserved and frozen as previously described.46 Sections were stained for any one of the following cell markers: NeuN, S-100, or CNPase by incubation with the appropriate primary antibodies at concentrations described in the section above, followed by secondary antibodies conjugated with Alexa Flour 568 (Invitrogen). Sections were re-fixed in 2% paraformaldehyde for 15 minutes and then subjected to the TUNEL-ApopTagPlus fluorescein in situ apoptosis assay (Millipore) as per the manufacturer's instructions. Apoptosis was also evaluated by immunofluorescence staining for activated caspase-3 (AC-3), rabbit polyclonal to AC-3, clone antibody 13,847 (Abcam) at a concentration of 5 μg/mL diluted in normal goat serum followed by secondary antibody, anti-rabbit IgG conjugated to Alexa Fluor 488 (Invitrogen), in combination with antibodies to the neuronal marker NeuN and the myelinating-cell marker CNPase. Slides were finally stained with nuclear stain TOPRO3 (Invitrogen) at 1:1000 in normal goat serum for 15 minutes. Slides were washed and mounted as described above and stored at 4°C in the dark until viewed. The percentage of apoptotic cells from 10 fields were counted in each of the sections (>500 cells in all cases). The total number of NeuN- or S-100–positive cells, respectively, in each section was ascertained, followed by the percentage of cells that colocalized with the TUNEL signal for each cell marker. Similarly, the percentage of NeuN staining sensory neurons that expressed AC-3 in the DRG of all rhesus macaques was quantified. All counts were made by viewing slides under a fixed magnification of ×40 for evaluation of apoptosis by TUNEL assay for neurons and satellite glial cells and at ×20 for evaluation of neurons that express AC-3 in the DRG of all rhesus macaques, using a confocal microscope (see the next section).
Confocal Microscopy
Confocal microscopy was performed with a Leica TCS SP2 confocal microscope (Leica Microsystems, Exton, PA) as previously described.46 Photoshop CS3 (Adobe Systems Inc., San Jose, CA) was used for image processing.
NCSs and Evaluation of F-Wave Chronodispersion
NCSs were performed on rhesus macaques anesthetized with ketamine hydrochloride (10 mg/kg intramuscularly), supplemented with isofluorane by inhalation as needed. Sterile subdermal Nicolet electrodes (CareFusion, San Diego, CA) were used as recording electrodes, and cathodal surface stimulation was used for nerve excitation. For motor NCSs, recording electrodes were placed over the appropriate muscle in the hand or foot, and the nerve was stimulated at distal and proximal sites. Median, ulnar, and tibial motor responses were recorded, and conduction velocity was calculated. F waves were elicited at rest by antidromic supramaximal stimulation over the distal nerve and recording from the same muscles as in the motor NCSs.47,48 A minimum of 30 F waves were recorded, and the minimal latency, maximal latency, and the difference between the minimal and maximal latencies (chronodispersion) were measured. For median and ulnar sensory NCSs, recording electrodes were placed over the nerve at the wrist, and sensory nerve fibers were stimulated orthodromically with digital ring electrodes. All procedures were done on a Viking Select electromyographic system (Cardinal Health Systems, Dublin, OH). Limb temperature was maintained at higher than 34°C with the use of a heating lamp.
Statistical Analysis
The statistical significance of the data for pleocytosis and immune mediators in CSF and serum was calculated with the two-way analysis of variance, followed by the Bonferroni post tests, and that of the apoptosis data were evaluated by Kruskal-Wallis one-way analysis of variance nonparametric analysis, followed by the Dunn's multiple comparison test by using Prism software version 5 (GraphPad Software, Inc., La Jolla, CA).
Results
Establishment of Persistent CNS Infection as a Result of Intrathecal Inoculation with Live Bb
Culture of cell pellets from CSF collected at various time points after inoculation yielded viable spirochetes at ≥1 time points for all of the rhesus macaques that were inoculated with Bb regardless of whether they were left untreated (Bb alone), treated with Dex (Bb+Dex), or treated with Mel (Bb+Mel), as shown in Table 1. The occipital lobe of animal GB09 (Bb+Dex 8 weeks), cervical and lumbar spinal cord of animal HH83 (Bb+Mel 8 weeks), brainstem of GJ23 (Bb+Dex 14 weeks), cervical and thoracic spinal cord of GK54 (Bb+Mel 14 weeks), and the sacral spinal cord of HT72 (Bb alone) yielded positive cultures for viable spirochetes when cultured after necropsy. Cultures of CSF pellets and necropsy tissues of control rhesus macaques were negative throughout.
Table 1.
CSF Cell Pellet Culture Results for Viable Bb from CSF Collected from Rhesus Macaques Inoculated Intrathecally with Live Bb and Left Untreated or Treated in Parallel with Either Dex or Mel
| Rhesus macaques and treatments | Week 0 | Week 1 | Week 2 | Week 3 | Week 4 | Week 6 | Week 8 | Week 10 | Week 12 | Week 14 | At necropsy |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GN22 (Bb alone) | − | − | + | − | + | − | − | NA | NA | NA | − |
| GB09 (Bb+Dex) | − | + | − | + | − | + | − | NA | NA | NA | − |
| GP33 (Bb+Mel) | − | + | + | − | − | − | − | NA | NA | NA | − |
| GN21 (Bb alone) | − | − | + | − | − | + | + | NA | NA | NA | + |
| IG55 (Bb+Dex) | − | − | + | − | − | − | − | NA | NA | NA | − |
| HH83 (Bb+Mel) | − | + | − | − | − | + | − | NA | NA | NA | + |
| IK20 (control) | − | − | − | − | − | − | − | NA | NA | NA | − |
| GC59 (Bb alone) | − | + | − | − | + | − | − | − | − | + | + |
| GJ23 (Bb+Dex) | − | + | + | − | − | − | − | − | − | − | + |
| GK54 (Bb+Mel) | − | + | + | + | − | − | − | − | − | + | + |
| HT72 (Bb alone) | − | + | + | − | − | − | − | − | − | − | − |
| GM23 (Bb+Dex) | − | + | − | + | − | − | − | − | − | − | − |
| GN49 (Bb+Mel) | − | + | − | − | + | + | − | − | − | − | − |
| HT73 (control) | − | − | − | − | − | − | − | − | − | − | − |
Bb, Borrelia burgdorferi; CSF, cerebral spinal fluid; Dex, dexamethasone; Mel, meloxicam; NA, not applicable.
Dexamethasone Treatment Results in More Significant Reduction in CSF Pleocytosis as Induced by Intrathecal Inoculation of Live Bb Compared with Meloxicam
Intrathecal inoculation of live Bb into the cisterna magna of rhesus macaques resulted in CSF pleocytosis as evidenced by increased numbers of white blood cells per microliter of CSF, as early as 1 week after inoculation. The pleocytosis was primarily lymphocytic and monocytic in nature (not shown), as reported earlier.35 Dexamethasone-treated rhesus macaques showed significant reduction in pleocytosis, compared with that present in infected rhesus macaques that were left untreated, whereas infected rhesus macaques that were treated with Mel showed similar albeit slightly lower levels of pleocytosis to that of the infected untreated rhesus macaques. Specimens from uninfected controls were essentially free of cells. Similar patterns of pleocytosis were seen in both treatment groups (8 weeks and 14 weeks) (Figure 1).
Figure 1.

Dex significantly reduces CSF pleocytosis elicited by Bb. WBCs per microliter of CSF recorded over time in rhesus macaques inoculated intrathecally with live Bb alone, inoculated with Bb and treated with Dex, inoculated with Bb and treated with Mel, and uninfected controls were followed for 8 weeks (A) and 14 weeks (B) after inoculation. Data are expressed as means ± SD. n = 2 rhesus macaques. ∗∗∗P < 0.001 for the comparison of Bb+Dex with Bb alone, determined by two-way analysis of variance, Bonferroni post tests. Bb, Borrelia burgdorferi; CSF, cerebrospinal fluid; Dex, dexamethasone; Mel, meloxicam; WBC, white blood cell.
Effect of Treatment with the Anti-Inflammatory Drugs Dex and Mel on the Levels of Immune Mediators in the CSF and Serum as Induced by Intrathecal Inoculation of Live Bb
Several of the tested immune mediators were up-regulated in the CSF of the rhesus macaques that were inoculated intrathecally with Bb. These were IL-6, IL-8, CCL2, and CXCL13. The concentrations of IL-6 and IL-8 peaked by week 1 after inoculation. Levels of CCL2 and CXCL13 peaked somewhat later, between weeks 1 and 2 after inoculation for CCL2 and between weeks 1 and 3 after inoculation for CXCL13. Peak values for the untreated Bb-inoculated rhesus macaques also differed among mediators, with low levels for IL-6 (20 to 25 pg/mL) and IL-8 (70 pg/mL) and higher levels for CCL2 (700 to 800 pg/mL) and CXCL13 (1500 to 2000 pg/mL). Interestingly, peak mediator concentrations were significantly diminished when the rhesus macaques were treated with Dex but not with Mel (P < 0.05 two-way analysis of variance, Bonferroni post tests). The only exception was CXCL13, whose concentration also diminished significantly in Mel-treated rhesus macaques. The concentration of mediators in the CSF of control rhesus macaques was undetectable expect in the cases in which there was constitutive production, namely, CCL2 and IL-8. In addition, one of the controls produced low levels of constitutive IL-6. All of these patterns were similar in both treatment groups (8 weeks and 14 weeks) (Figure 2).
Figure 2.

Effect of Dex and Mel on levels of CSF immune mediators as induced by Bb. Levels of CSF IL-6 (A and B), IL-8 (C and D), CCL2 (E and F), and CXCL-13 (G and H) in rhesus macaques inoculated intrathecally with live Bb alone, inoculated with Bb and treated with Dex, inoculated with Bb and treated with Mel, and uninfected controls were followed for 8 weeks (A, C, E, and G) and 14 weeks (B, D, F, and H) after inoculation. Data are expressed as means ± SD. n = 2 rhesus macaques. ∗P < 0.05 for the comparison of Bb+Dex with Bb alone, determined by two-way analysis of variance, Bonferroni post-tests. Bb, Borrelia burgdorferi; CCL2, chemokine ligand 2; CSF, cerebrospinal fluid; Dex, dexamethasone; Mel, meloxicam.
As with CSF, the inflammatory mediators CCL2, CXCL13, and IL-8 were also elevated in the serum, but their concentrations peaked later (around 4 weeks after inoculation), except in animal HT72 (Bb alone 14 weeks) in which anti-VlsE (C6) antibody levels (described below) were undetectable throughout the duration of the study. IL-6, in contrast, was undetectable in the serum of all rhesus macaques in the study at all times. Additional mediators that were elevated in serum were IL-12, CD40L, IL-18, IL-15, and TGF-α, also reaching peak values at approximately 4 weeks after inoculation. The concentrations of all but one of the mediators that were elevated in infected/untreated rhesus macaques were reduced to baseline values or below by treatment with Dex. The exception was TGF-α, whose concentration was enhanced by Dex above the value that was observed in infected/untreated rhesus macaques. Treatment with Mel significantly diminished the concentrations of CXCL13 and CD40L but left that of the other mediators unchanged for the values observed in infected/untreated rhesus macaques (data not shown).
Evidence of Anti-VlsE (C6) Antibody in Serum
We evaluated the appearance of anti-VlsE (C6) antibodies in the serum to assess if the infection had become systemic. All rhesus macaques that were infected with Bb and left untreated except HT72 (Bb alone 14 weeks) and all infected rhesus macaques that were treated with Mel showed the presence of detectable anti-VlsE (C6) antibodies from week 3 after inoculation, but no detectable levels of anti-VlsE (C6) antibodies were found in the uninfected controls and rhesus macaques that were infected but treated with Dex, except IG55 (Bb+Dex 8 weeks; data not shown).
Differential Histopathologic Evidence of Inflammation in the Brain and Spinal Cord Regions in Infected Untreated versus Treated Rhesus Macaques
Histopathologic evaluation after hematoxylin and eosin staining40 for inflammation in tissue from rhesus macaques at necropsy of all rhesus macaques that received intrathecal inoculation of only live Bb and those that were inoculated and treated with Mel revealed lesions typical of lymphocytic meningitis in the brain and spinal cord and lymphocytic neuritis and ganglionitis in random sections of the spinal cord. No lymphocytic inflammatory lesions were seen in the brain and spinal cord of rhesus macaques that were inoculated with live Bb and treated with Dex or in the un-inoculated controls. Two rhesus macaques (one in the Bb alone group and one in the Bb+Dex group) had segmental lesions either in the brainstem or proximal cervical cord characterized by focal malacia, nerve fiber degeneration, and accumulation of enlarged phagocytic cells that consisted of distended granular cytoplasm.
Lymphocytic meningitis was observed in the dura mater in the brain and leptomeningitis was present in the spinal cord of all of the infected rhesus macaques except those that were treated with Dex. Leptomeningitis in the cervical spinal cord showed large perivascular infiltrates of mononuclear cells around blood vessels in the subarachnoid space, involving both the pia mater and arachnoid (Figure 3A). Cranial nerve neuritis showed immune cell infiltrates that consisted of small lymphocytes and a few larger histiocytic cells under the epineurium or connective tissue surrounding the nerve (Figure 3B). Vasculitis was present in the brainstem of all of the infected rhesus macaques except those treated with Dex, with lymphocytic infiltrates in the vessel walls (Figure 3C). Necrotizing myelitis and degeneration was observed in the cervical spinal cord of two rhesus macaques (Figure 3D). Severe degeneration and necrotizing myelitis near the injection site were histologically different from the lymphocytic inflammatory lesions that could be found all the way along the spinal cord to the sacral region of all of the infected rhesus macaques except those treated with Dex. A representative image of the absence of inflammatory lesions in the leptomeninges of the cervical spinal cord of Bb+Dex rhesus macaques is shown in Figure 3E.
Figure 3.

Inflammatory lesions in the brain and spinal cord in Bb-inoculated rhesus macaques. Representative images of inflammatory lesions were observed by H&E histopathologic evaluation of brain and spinal cord at necropsy of rhesus macaques that received an intrathecal inoculation of live Bb alone and those inoculated with Bb and treated with Mel show leptomeningitis in the spinal cord with Bb alone at 8 weeks (A), cranial nerve neuritis with Bb alone at 8 weeks (B), vasculitis in the brainstem with Bb+Mel at 8 weeks (C), focal degeneration and necrotizing myelitis in the cervical spinal cord with Bb alone at 14 weeks (D), and absence of inflammation in the leptomeninges of the cervical spinal cord with Bb+Dex at 8 weeks (E). Scale bar = 50 μm. Bb, Borrelia burgdorferi; Dex, dexamethasone; H&E, hematoxylin and eosin; Mel, meloxicam.
Evidence of Radiculitis in All Infected Rhesus Macaques but Those Treated with Dexamethasone
Radiculitis or inflammation in the dorsal (sensory) and ventral (motor) spinal roots affected nerve roots on both sides of the spinal cord that spanned the entire cervical region and occasionally in the thoracic region, in addition to inflammation and neuritis in the DRG in all of the rhesus macaques that were inoculated with Bb and left untreated and those rhesus macaques inoculated with Bb and treated with Mel. Lesions of radiculitis or DRG inflammation and DRG neuritis were absent in the rhesus macaques inoculated with live Bb and treated with Dex, as well as in the uninoculated controls (Figure 4). Dorsal root radiculitis in the cervical spinal cord, with lymphoid cell infiltration, and linear clear spaces around nerve fibers that are indicative of nerve fiber swelling and possible demyelination were observed (Figure 4A). Radiculitis was observed in the ventral spinal root with lymphoid infiltrates (Figure 4B). Inflammation was present in the cervical DRG with dying neuronal cell bodies and accumulation of infiltrating lymphoid mononuclear cells (Figure 4C). The presence of neurodegeneration in sensory neurons and areas corresponding to the dorsal roots that traverse the DRG (green) in an adjacent section was confirmed by FJC staining (see Evidence of Neurodegeneration). Figure 4D shows a representative image of DRG neuritis, with inflammation between the nerve fibers in a nerve root that is immediately adjacent to the DRG of the C8 cervical region. A representative image of radiculitis in the dorsal root of the cervical spinal cord at C7 of infected rhesus macaques treated with Mel at 8 weeks is shown in Figure 4E. The absence of inflammatory cell infiltrates in the DRG and dorsal root of the C8 cervical region of infected rhesus macaques treated with Dex is shown in Figure 4F.
Figure 4.

Radiculitis in Bb-inoculated rhesus macaques. Inflammatory lesions observed by H&E histopathologic evaluation of spinal nerve roots and DRG at necropsy of animals that received an intrathecal inoculation of live Bb alone and animals inoculated with Bb and treated with Mel, showing radiculitis in the dorsal root of the cervical spinal cord at C7 with Bb alone at 8 weeks (A), in the ventral spinal root (B), DRG inflammation of the C6 cervical region with Bb alone at 14 weeks (C), DRG neuritis as seen in the nerve adjacent to the C8 cervical DRG with Bb alone at 8 weeks (D), radiculitis in the dorsal root of the cervical spinal cord at C7 with Bb+Mel at 8 weeks (E), and the absence of inflammatory lesions in the DRG and dorsal root of the C8 cervical region with Bb+Dex at 8 weeks (F). Scale bar = 50 μm. Bb, Borrelia burgdorferi; Dex, dexamethasone; DRG, dorsal root ganglia; H&E, hematoxylin and eosin; Mel, meloxicam.
Detection of Bb Antigen and Characterization of Cell Phenotypes in Inflammatory Lesions in the DRG
Immunofluorescence staining and confocal microscopic evaluation of DRG tissues collected at necropsy revealed the presence of similar amounts of Bb antigen in the DRG of all of the infected rhesus macaques, including those treated with Dex. Bb antigens were present after immunofluorescence staining by using a polyclonal rabbit anti-Bb antibody, followed by the corresponding secondary antibody tagged with Alexa Fluor 488 in the DRG (Figure 5A). Antigen was localized in the spaces between the neurons. These cells appear red due to immunofluorescence staining with NeuN, a specific neuronal marker.49 Antigen was also present in the region that corresponded to the dorsal root traversing the DRG. Nuclei of all cells appear blue due to staining with TOPRO3. Spirochetal antigen was undetectable in DRG tissues from uninfected controls (Figure 5B). No green fluorescence staining was observed when universal rabbit IgG isotype control was used instead of the anti-Bb antibody, followed by the relevant secondary antibody tagged to Alexa Fluor 488 (Figure 5C).
Figure 5.

Bb antigen and inflammatory cells in the DRG of Bb-inoculated rhesus macaques. A: Confocal micrographs show the presence of Bb antigen in the DRG after immunofluorescence staining with a Borrelia-specific antibody (Bb). B: Confocal micrographs shown in the same staining applied to DRG of an uninfected control rhesus macaque. C: No fluorescence signal is observed when a DRG section from a Bb-infected rhesus macaque was stained with the relevant isotype control, followed by the corresponding secondary antibody tagged to Alexa Fluor 488. Nuclei of all cells appear blue due to staining with nuclear stain TOPRO3. Neurons appear red due to immunofluorescence staining with NeuN stain, followed by corresponding secondary antibody tagged to Alexa Fluor 568. D: A confocal microscopic image of a lesion in the dorsal root of a Bb-infected rhesus macaque shows the presence of abundant T lymphocytes that express CD3 (green), macrophages that express CD68 (red), and B lymphocytes that express CD20 (blue) after immunofluorescence staining. Unstained tissue appears gray due to differential interference contrast imaging. Bb, Borrelia burgdorferi; DIC, differential interference contrast; DRG, dorsal root ganglia; NeuN, neuronal nuclear protein.
Phenotypic characterization of cells in an inflammatory lesion from the dorsal roots of rhesus macaques that were infected with live Bb alone and those infected with Bb and treated with Mel, showed the presence of abundant lymphocytes and monocytes. Figure 5D depicts a representative image of a lesion in the dorsal root after immunofluorescence staining and confocal microscopic imaging that show abundant CD3-expressing T cells,50 CD68-expressing macrophages,51 and B cells that express CD2052
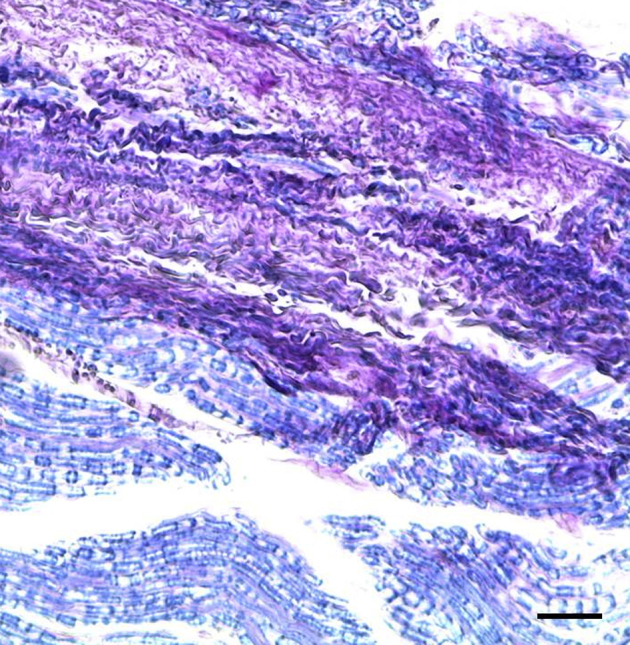
Demyelination in Spinal Nerve Roots
Demyelination was evident in the spinal nerve roots of all of the infected rhesus macaques except those treated with Dex and the uninfected controls (Figure 6).40 There was loss of the normal eosinophilic myelin coat in a bundle of nerve fibers in a cervical dorsal root. Residual Schwann cell nuclei appeared more pronounced because of loss of myelin.
Figure 6.

Demyelination in nerve roots of Bb-inoculated rhesus macaques. Demyelination in the cervical dorsal root at C6 with Bb+Mel at 14 weeks made evident by H&E stain, showing loss of normal eosinophilic myelin coat in a nerve bundle that appears gray in contrast to normally myelinated nerves that appear pink. Scale bar = 50 μm. Bb, Borrelia burgdorferi; H&E, hematoxylin and eosin; Mel, meloxicam.
Demyelination was visualized in a dorsal root after staining with Luxol-Fast Blue/periodic acid-Schiff combination stain43,44 (Supplemental Figure S1).
Qualitative and Quantitative Assessment of Glial and Neuronal Apoptosis in the DRG
We had previously observed neuronal and satellite cell apoptosis in DRG of rhesus macaques that were inoculated intrathecally with live Bb.35 We therefore evaluated the levels of apoptosis of these cells and Schwann cells in the DRG of rhesus macaques in this study, to ascertain if inflammation had a possible causal role in mediating apoptosis, as induced by Bb. The evaluation was done both qualitatively and quantitatively by using the in situ TUNEL assay53 and by immunofluorescence staining for AC-3,54 under the confocal microscope. Neuronal apoptosis in the DRG (Bb alone 14 weeks) was observed (Figure 7A). Neurons were labeled with antibody to NeuN, followed by the corresponding secondary antibody tagged to Alexa Fluor 568. Apoptotic neurons that expressed AC-3 showed colocalization with NeuN. AC-3 was also present in the area occupied by satellite glial cells around the neurons in the DRG and in the Schwann cells stained with the myelin marker CNPase55 in the dorsal roots within the DRG (not shown). The percentage of neuronal cells that showed AC-3 in the DRG for all of the rhesus macaques was calculated (Figure 7B). Dexamethasone treatment significantly reduced the percentage of neuronal cells that expressed AC-3 after 8 weeks of treatment (6.7% ± 0.5%; P < 0.05, Kruskal-Wallis test, one-way analysis of variance, Dunn's multiple comparison test) and after 14 weeks of treatment (9.3% ± 0.5%; P < 0.001) compared with infected rhesus macaques that were left untreated for 8 weeks (15.84% ± 0.89%) and 14 weeks (21.80% ± 1.86%). Meloxicam treatment for both 8 weeks (21.54% ± 0.82%) and 14 weeks (26.72% ± 1.16%) showed levels of neuronal apoptosis that were similar or slightly higher than those induced by Bb infection alone. Basal levels of neuronal apoptosis were found in the control rhesus macaques after 8 weeks (3.9% ± 0.13%) and 14 weeks (5.13% ± 0.53%).
Figure 7.

Glial and neuronal apoptosis in the DRG. A: A confocal image of DRG neuron apoptosis as visualized by immunofluorescence staining for AC-3 (green). Neurons appear red due to immunofluorescence staining for NeuN (Bb alone at 14 weeks). Apoptotic neurons that express AC-3 appear orange due to colocalization of the red (NeuN) and green (AC-3) signals. Nuclei of all cells appear blue due to staining with nuclear stain TOPRO3. B: Neuronal apoptosis as evaluated quantitatively by the AC-3 immunofluorescence assay in all of the rhesus macaques in the study (inoculated intrathecally with live Bb alone, inoculated with Bb and treated with Dex, inoculated with Bb and treated with Mel, and uninfected controls for both 8 weeks and 14 weeks after inoculation). C: A confocal image of DRG neuronal apoptosis as visualized by the TUNEL assay (Bb alone at 14 weeks). Neurons appear red due to immunofluorescence staining for NeuN, whereas TUNEL signal showing apoptotic nuclei appears green. Nuclei of all cells appear blue due to staining with nuclear stain TOPRO3. D: A representative confocal image showing abundant TUNEL+ nuclei in the dorsal region (Bb alone at 14 weeks). Neurons appear red due to immunofluorescence staining for NeuN, and nuclei of all cells appear blue due to staining with nuclear stain TOPRO3. E: A quantitative representation of neuronal apoptosis as evaluated by the TUNEL assay in all of the rhesus macaques in the study. F: A confocal image of satellite glial cells (red) after immunofluorescence staining for S-100 and TUNEL+ nuclei in green. G: A quantitative representation of satellite glial cell apoptosis as evaluated by the TUNEL assay in all of the rhesus macaques in the study. Data are expressed as means ± SEM of animals in each treatment group. n = 2 rhesus macaques. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 determined by Kruskal-Wallis test, one-way analysis of variance, Dunn's multiple comparison test. AC-3, active caspase-3; Bb, Borrelia burgdorferi; Dex, dexamethasone; DRG, dorsal root ganglia; Mel, meloxicam; NeuN, neuronal nuclear protein; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
DRG cell apoptosis was evaluated qualitatively by the TUNEL assay (Figure 7C). The positive TUNEL signal was evident in the nuclei of satellite glial cells that surround the sensory neurons. Abundant apoptotic cells in the area corresponding to the dorsal root (possibly Schwann cells) that traverses the DRG were observed (Figure 7D). The percentage of neurons undergoing apoptosis in the DRG for all of the rhesus macaques in the study was quantified by the TUNEL assay (Figure 7E). Intrathecal inoculation of live Bb resulted in a fraction of apoptotic cells of 8.4% ± 0.4% after 8 weeks and 9.5% ± 0.6% after 14 weeks after inoculation, compared with basal levels of 2.2% ± 0.3% (8 weeks) and 2.7% ± 0.2% (14 weeks) after inoculation. Dexamethasone treatment significantly reduced neuronal apoptosis as visualized by the TUNEL assay both at 8 weeks (1.46% ± 0.3%) and 14 weeks (2.6% ± 0.13%) after inoculation, whereas Mel treatment resulted in levels that were similar or slightly higher than those seen in infected rhesus macaques that were left untreated, reaching 8.01% ± 0.5% at 8 weeks and 11.88% ± 0.4% at 14 weeks after inoculation.
We used the marker S-100 to stain glial cells in the DRG and evaluated the percentage of satellite glial cells undergoing apoptosis by the TUNEL assay by counting the S-100–positive cells immediately surrounding the sensory neurons, as reported earlier35 (Figure 7F). Neuronal apoptosis was evaluated quantitatively by the TUNEL assay in satellite glial cells in all of the rhesus macaques in the study (Figure 7G). A basal level of 5% apoptosis was found in the uninfected controls both at 8 and 14 weeks after inoculation. Intrathecal inoculation of live Bb resulted in 15.05% ± 0.6% of satellite glial cell apoptosis after 8 weeks and 15.05% ± 0.7% of satellite glial cell apoptosis after 14 weeks after inoculation. Dexamethasone treatment was effective in significantly reducing neuronal apoptosis to 7.1% ± 1.2% (8 weeks) and 3.7% ± 0.2% in the Bb-infected rhesus macaques that were treated with Dex for 14 weeks after inoculation. The percentage of TUNEL-positive satellite glial cells in the infected rhesus macaques that were treated with Mel was 16.63% ± 0.44% (8 weeks) but showed only 9.12% ± 1.3% apoptosis in infected rhesus macaques that were treated with Mel for 14 weeks after inoculation, suggesting that the drug was effective in protecting against satellite glial cell apoptosis with prolonged treatment. In summary, Dex treatment was effective in protecting both satellite glial cell and neuronal apoptosis induced by Bb, as evaluated by the AC-3 and the TUNEL assays, whereas Mel treatment was only effective in protecting against satellite glial cell apoptosis after prolonged treatment after inoculation of 14 weeks.
Evidence of Neurodegeneration
We confirmed the presence of dying neurons that we found in the DRG by staining with FJC sections of tissues from the DRG and dorsal roots of all of the rhesus macaques. This anionic reagent has an affinity for degenerating neurons, which are supposed to express a basic or positive charge.41,42 The FJC stain is known to selectively label degenerating neurons in addition to certain non-neuronal elements such as the meninges and blood vessels, as was evident in the capsular area of the DRG41 (Figure 8A). The degenerating neuronal axons in a dorsal root entering the DRG in addition to degenerating neurons in the DRG of an infected rhesus macaque that was also treated with Mel (Bb+Mel 14 weeks) is shown in Figure 8B. Degenerating neurons were not detected in the DRG and axons of nerve roots from rhesus macaques that were inoculated with live Bb but treated with Dex (not shown). The uninfected control rhesus macaques showed an absence of neurodegeneration after FJC staining (Figure 8C). Neurodegeneration in a section of DRG (Bb alone 14 weeks) from an adjacent section to that shown in Figure 4C, showing the presence of neurodegeneration in sensory neurons and areas that correspond to the dorsal roots that traverse the DRG as confirmed by FJC staining, is shown in Figure 8D.
Figure 8.

Neurodegeneration in inflamed DRG. Confocal microscope image of degenerating neurons (green) as stained by FJC in the DRG (Bb alone at 14 weeks) showing degenerating neuronal axons in a dorsal root, adjacent to the DRG are also seen to stain green with FJC. Nuclei of all cells appear blue due to staining with nuclear stain TOPRO3 (A), neurodegeneration in the DRG and dorsal root entering the DRG with Bb+Mel at 14 weeks (B), absence of neurodegeneration in an uninfected control rhesus macaque at 8 weeks (C), and presence of degenerating neurons in the DRG and areas corresponding to the dorsal roots that traverse the DRG in a section parallel to that showing neurodegeneration as seen with Bb alone at 14 weeks in Figure 4C (D). Bb, Borrelia burgdorferi; DRG, dorsal root ganglia; FJC, Fluoro-Jade C; Mel, meloxicam.
CCL2 in the Dorsal Root Ganglia of Bb-Inoculated Rhesus Macaques
CCL2 in sensory neurons and in the region that corresponds to satellite glial cells in a DRG (Bb alone 14 weeks) (Supplemental Figure S2A), satellite glial cells that surround the sensory neurons (Supplemental Figure S2B), and Schwann cells (Supplemental Figure S2C) were observed.
Electrophysiologic Evidence of Abnormal F Waves/Chronodispersion in Nerve Roots of Rhesus Macaques Inoculated Intrathecally with Live Bb
We had previously observed perivascular inflammatory infiltrates and multifocal axonal changes that were consistent with mononeuropathy multiplex in rhesus macaques infected with Bb.32 Moreover, an association between demyelinating neuropathy and abnormal temporal dispersion and nerve conduction slowing was described in a few case reports of human LNB.17 We therefore performed NCSs in motor nerves (median, ulnar, and tibial) on the right side and sensory nerves (median and ulnar) bilaterally to explore the C8/T1 nerve roots in the upper limbs and S1 in the lower limb. Most marked abnormalities were found in F-wave latencies recorded 2 weeks after inoculation, in rhesus macaques infected with Bb alone (GN22 Bb alone 8 weeks, HT72 Bb alone 14 weeks) that remained abnormal until the end of the study. Some of the rhesus macaques infected with Bb in the presence of Mel (GK54, GN49) showed more marked increased F-wave latencies and chronodispersion than that seen in rhesus macaques infected with Bb alone by week 2 after inoculation and persisted until 14 weeks after inoculation, the end of the study. Only one of the four rhesus macaques that were infected and treated with Dex (GM23 Bb+Dex 14 weeks) showed a marginal increase in F-wave latencies by 2 weeks after inoculation that subsequently returned to normal by 8 weeks after inoculation. Abnormalities in F-wave latencies and chronodispersion in infected rhesus macaques, when observed, were most likely a result of pathologic change in motor nerve roots because more distal motor and sensory NCSs remained normal. No electrophysiologic abnormalities were recorded in any of the uninfected controls. A summary of the results of the F-wave studies for all of the rhesus macaques in the study is shown in Table 2.
Table 2.
F-Wave Latencies Recorded for Motor Nerves (Median, Ulnar, and Tibial) in Rhesus Macaques Inoculated Intrathecally with Live Bb and Left Untreated or Treated with Either Dex or Mel
| Rhesus macaques and treatment | F-wave latencies (msec) |
|---|---|
| GN22 (Bb alone 8 weeks) | Abnormal F waves in all nerves recorded from week 2 PI until end of study Median F waves = 14.8–20.2 (UL 14.7)∗ Ulnar F waves = 15.1–22.5 (UL 14.8)∗ Tibial F waves = 21.2–30.1 (UL 20.7)∗ |
| GB09 (Bb+Dex 8 weeks) | Normal |
| GP33 (Bb+Mel 8 weeks) | Normal |
| GN21 (Bb alone 8 weeks) | Normal |
| IG55 (Bb+Dex) | Normal |
| HH83 (Bb+Mel 8 weeks) | Normal |
| IK20 (control 8 weeks) | Normal |
| GC59 (Bb alone 14 weeks) | Normal |
| GJ23 (Bb+Dex 14 weeks) | Abnormal F waves in tibial nerve from week 2 PI but returned to normal by 8 weeks PI Tibial F waves = 17.2–25.1 at week 2; 16.5–19.6 at week 8 (UL 20.7)∗ |
| GK54 (Bb+Mel 14 weeks) | Abnormal F waves in ulnar and tibial nerves recorded from week 2 PI until end of study Ulnar F waves = 16.3–24.8 (UL 14.8)∗ Tibial F waves = 22.2–33.3 (UL 20.7)∗ |
| HT72 (Bb alone 14 weeks) | Abnormal F waves in median and ulnar nerves recorded from week 2 PI until end of study Median F waves = 15.6–22.7 (UL 14.7)∗ Ulnar F waves = 16.3–23.7 (UL 14.8)∗ |
| GM23 (Bb+Dex 14 weeks) | Normal |
| GN49 (Bb+Mel 14 weeks) | Abnormal F waves in ulnar and tibial nerves recorded from week 2 PI until end of study Ulnar F waves = 15.3–19.6 (UL 14.8)∗ Tibial F waves = 23.4–31.6 (UL 20.7)∗ |
| HT73 (control 14 weeks) | Normal |
Bb, Borrelia burgdorferi; Dex, dexamethasone; Mel, meloxicam; PI, after inoculation UL, upper limit.
UL of normal in controls was based on means + 2 SD. Unless noted, F-wave latencies were below the UL of normal.
Discussion
Here, we addressed the hypothesis that inflammation is a causal factor in LNB pathogenesis. To this end we made use of intrathecal inoculation of live Bb in rhesus macaques to facilitate the interaction of spirochetes with the CNS, a model we had previously shown to reproduce the signs of acute neuroborreliosis.35 Dex and Mel, two well-known anti-inflammatory drugs, were used to evaluate the pathogenetic effects of Bb-induced inflammation in this model. We found that Dex treatment significantly reduced both pleocytosis and the levels of CSF and serum immune mediators that were induced by Bb. Moreover, this drug inhibited the formation of inflammatory lesions in the brain and spinal cord, focal neurodegeneration, and demyelination in the cervical spinal cord, dorsal and ventral nerve roots, and DRG. In contrast, all of these signs were evident in the infected rhesus macaques that were left untreated or that were treated with Mel. Importantly, Dex was able to significantly diminish the levels of neuronal and satellite glial cell apoptosis that were induced by Bb in the DRG. Further, persistent abnormalities in F-wave chronodispersion in nerve roots, as evaluated by electromyographic studies, were found only in Bb-infected rhesus macaques that were left untreated and in those treated with the nonsteroidal anti-inflammatory drug Mel, but not in rhesus macaques treated with Dex. Thus, in accordance with our hypothesis, the effective suppression of inflammation by Dex treatment resulted in inhibition of glial and neuronal damage. These are data to suggest that inflammation has a causal role in the pathogenesis of LNB.
Intrathecal inoculation of live Bb in the cisterna magna resulted in the establishment of a CNS infection, as evidenced by recovery of live spirochetes from cultures of CSF cell pellets that were collected at one or more time points after inoculation from all of the infected rhesus macaques and from some CNS tissues of some of the animals, regardless of whether the animals were left untreated or were treated with Dex or Mel. The occurrence of pleocytosis and of increased levels of proinflammatory mediators in the CSF of Bb-infected rhesus macaques, concomitantly with the ability to culture live spirochetes from the CSF and CNS tissues and to identify Bb antigen, including in the DRG, indicates that the former phenomena occurred as a response to the presence of Bb and/or Bb antigens in the CNS and PNS (DRG). Dex significantly inhibited Bb-induced CSF pleocytosis. This effect was probably because of the parallel marked reduction in the CSF concentration of chemokines that was observed in Dex-treated rhesus macaques. Chemokines such as IL-8 and CCL2 are known to mediate influx of immune cells in the CNS compartment during bacterial meningitis.56 IL-8 is a neutrophil and T-cell attractant,57 and CCL2 attracts monocytes and T cells58 and modulates monocyte immune functions.59 Dex and Mel were both able to reduce significantly the levels of the B-cell chemokine CXCL13. This mediator is the main determinant of B-cell recruitment into the CSF during neuroinflammation.60
We previously reported that CCL2 and CXCL13 were produced by spinal cord glia of rhesus macaques that were given intrathecal inoculations with live Bb.35 In addition, we showed that Dex could inhibit production of proinflammatory mediators (IL-6, IL-8, and CCL2) secreted by both oligodendrocyte and DRG cell cultures.61,62 Hence, it is conceivable that Dex was effective in modulating the levels of immune mediators elicited by glial cells in response to Bb and the subsequent influx of immune cells from the periphery into the CNS.
The anti-inflammatory effect of Dex was also evident from the significantly reduced levels of the immune mediators detected in the serum, compared with the serum levels in infected untreated rhesus macaques. The concentration of TGF-α, in contrast, was significantly elevated in the presence of Dex. This cytokine, an epidermal growth factor receptor ligand, plays a role in the autocrine stimulation of cancer cells in addition to a myriad other functions.63 It is not, however, a proinflammatory cytokine. The observed effect, therefore, does not contradict the general anti-inflammatory ability of Dex in this experimental context. The levels of anti-VlsE (C6) antibodies in the serum of Bb-infected rhesus macaques that were treated with Dex were undetectable or low which may be because of the ability of Dex to significantly affect the levels of some serum immunoglobulins.64
Mel, like Dex, was able to significantly decrease the serum concentrations of CD40L and CXCL13. The proinflammatory mediator CD40L was implicated in altering blood-brain barrier integrity in HIV-associated neurocognitive disorders.65 Serum levels of CD40L in infected untreated rhesus macaques were as high as 20,000 pg/mL. Only Dex was effective in significantly reducing the serum levels of IL-18 and CCL2, compared with the levels observed in infected rhesus macaques that were left untreated.
An important finding of this study is that Dex treatment was effective in curbing inflammation and neurodegeneration in the DRG and demyelination in the spinal nerve roots. In addition, the drug significantly reduced the levels of Bb-induced neuronal and glial cell apoptosis in the DRG. This result, obtained in vivo, reproduces a previous result from our laboratory, whereby Dex significantly reduced apoptosis of sensory neurons in primary cultures of rhesus DRG cells, as induced by the addition of live Bb.62
Cytokine signaling and apoptosis play a main role in the regulation of neuroinflammatory responses.66–69 The lesions of inflammation and neurodegeneration that we have observed in the DRG and dorsal roots could contribute to radiculitis and consequent neurogenic pain. The latter is the most common presenting symptom in patients with LNB.12,13 Sensory neurons of the DRG play a key role in the sensation of pain,70 and the immune mediators IL-6 and CCL2 that we found in the DRG, along with IL-8 that was elevated in the CSF and serum of infected rhesus macaques that were not treated with Dex, are known to increase the sensitivity of sensory neurons to pain. They play a main role in modulating the pain response.69–73 CCL2 is involved in the signaling and up-regulation of several genes and proteins that participate in the signal transduction of the pain response both in the DRG and in the spinal cord.74–76 Disruption of CCL2 signaling was shown to block the development of neuropathic pain.77 IL-6 and IL-8 can also modulate apoptotic signaling pathways in neurons.78,79
Our electromyographic evaluations revealed that rhesus macaques that were infected and treated with Dex showed minimum abnormalities in F-wave chronodispersion. These changes shortly recovered back to normal compared with rhesus macaques that were infected with Bb and left untreated or treated with Mel. The latter rhesus macaques showed higher abnormal F-wave chronodispersions, and these persisted throughout the study period. F-wave chronodispersion refers to the difference of maximal and minimal latencies in a series of F waves and is highly sensitive for diagnosing demyelinating neuropathy, whereas persistence of F-wave chronodispersion is a measure of antidromic excitability of a particular motor neuron pool and is decreased in axonal neuropathy.48 Persistent abnormal F-wave chronodispersions that were observed in this study were localized to the nerve roots when present, suggesting that they could be a consequence of damage to axons or demyelination as caused by Bb. An association between changes in the latency of F-wave frequency, abnormal temporal dispersion, nerve conduction slowing, and the presence of demyelinating neuropathy or axonal damage was documented in several inflammatory demyelinating peripheral neuropathic disorders, including Guillain-Barré syndrome, and LNB.17,32,47
Importantly, we found necrotizing myelitis and degeneration in the spinal cord, neurodegeneration in the DRG, and demyelination in the nerve roots only when lymphocytic inflammatory lesions were also observed in both the CNS and PNS. Ongoing cytokine activation in the nervous system could contribute to the persistent symptoms of fatigue, pain, and cognitive dysfunction that patients sometimes experience despite having been treated for Lyme disease.
Our results support the hypothesis that Bb induces inflammatory mediators in glial and neuronal cells and that this inflammatory context precipitates glial and neuronal apoptosis and an array of neurologic changes typical of LNB. Consistent with this notion we found that the anti-inflammatory drug Dex significantly reduced pleocytosis, inflammatory mediators in the CSF, and neuronal and glial cell apoptosis in the DRG, in addition to preventing the appearance of inflammatory lesions, neurodegeneration, and demyelination. In contrast, the nonsteroidal anti-inflammatory drug Mel displayed a limited anti-inflammatory activity that was evident chiefly in the reduction of the expression levels of some cytokines and chemokines. Although the mechanism of action of Mel is limited to the inhibition of cyclooxygenase-2,80 a key enzyme in the process of synthesis of prostaglandins (which are mediators of inflammation), Dex interacts with numerous signaling cascades that in turn regulate both inflammation and apoptosis.81 These include the p38 mitogen-activated protein kinase and c-Jun N-terminal kinase pathways.82–85 It is possible that these signaling pathways play a role in mediating both inflammation and apoptosis as a result of Bb infection in vivo. A thorough analysis of the differences in the mechanisms of action of both drugs may provide a blueprint for the development of new treatments for LNB.
Acknowledgments
We thank Robin Rodriguez for help with formatting the figures and Mary Barnes and Melissa Pattison for technical help with the Multiplex assays.
G.R. and M.T.P. conceived and designed the experiments; G.R., P.J.D., J.D.E, L.S.-G., L.A.D.-M, D.S.M., and M.B.J. performed the research; G.R., P.J.D., J.D.E, and M.T.P. analyzed data; and G.R. and M.T.P. wrote the manuscript.
Footnotes
Supported by the NIH National Institute of Neurologic Disorders and Stroke grant NS048952 (M.T.P.) and NIH National Center for Research Resources and the Office of Research Infrastructure Programs grant P51OD011104/P51RR000164.
Disclosures: None declared.
Supplemental Data
Supplemental Figure S1.
Demyelination, as visualized in the C6 dorsal root, after staining with Luxol-Fast Blue/periodic acid-Schiff combination stain. Demyelination appears pink because of staining with periodic acid-Schiff, whereas normal nerves with intact myelin appear blue because of the Luxol-Fast Blue stain.
Supplemental Figure S2.

CCL2 in neurons (A), satellite glial cells (B), and Schwann cells (C) in dorsal root ganglia of B. burgdorferi-inoculated rhesus macaques. The nuclei of all cells appear blue because of staining with TOPRO3. CCL2, chemokine ligand 2.
References
- 1.Steere A.C. Lyme disease. N Engl J Med. 2001;345:115–125. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- 2.Bacon R.M., Kugeler K.J., Mead P.S., Centers for Disease Control and Prevention (CDC) Surveillance for Lyme disease–United States, 1992-2006. MMWR Surveill Summ. 2008;57:1–9. [PubMed] [Google Scholar]
- 3.Stanek G., Wormser G.P., Gray J., Strle F. Lyme borreliosis. Lancet. 2012;379:461–473. doi: 10.1016/S0140-6736(11)60103-7. [DOI] [PubMed] [Google Scholar]
- 4.Fallon B.A., Levin E.S., Schweitzer P.J., Hardesty D. Inflammation and central nervous system Lyme disease. Neurobiol Dis. 2010;37:534–541. doi: 10.1016/j.nbd.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 5.Bremell D., Hagberg L. Clinical characteristics and cerebrospinal fluid parameters in patients with peripheral facial palsy caused by Lyme neuroborreliosis compared with facial palsy of unknown origin (Bell's palsy) BMC Infect Dis. 2011;11:215–220. doi: 10.1186/1471-2334-11-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halperin J.J. Nervous system Lyme disease. Handb Clin Neurol. 2014;121:1473–1483. doi: 10.1016/B978-0-7020-4088-7.00099-7. [DOI] [PubMed] [Google Scholar]
- 7.Reik L., Steere A.C., Bartenhagen N.H., Shope R.E., Malawista S.E. Neurological abnormalities of Lyme disease. Medicine (Baltimore) 1979;58:281–294. doi: 10.1097/00005792-197907000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Elamin M., Monaghan T., Mulllins G., Ali E., Corbett-Feeney G., O’Connell S., Counihan T.J. The clinical spectrum of Lyme neuroborreliosis. Ir Med J. 2010;103:46–49. [PubMed] [Google Scholar]
- 9.Ackermann R., Hörstrup P., Schmidt R. Tick-borne meningopolyneuritis (Garin-Bujadoux, Bannwarth) Yale J Biol Med. 1984;57:485–490. [PMC free article] [PubMed] [Google Scholar]
- 10.Lebech A.M., Hansen K. Detection of Borrelia burgdorferi DNA in urine samples and cerebrospinal fluid samples from patients with early and late Lyme neuroborreliosis by polymerase chain reaction. J Clin Microbiol. 1992;30:1646–1653. doi: 10.1128/jcm.30.7.1646-1653.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brinar V.V., Habek M. Rare infections mimicking MS. Clin Neurol Neurosurg. 2010;112:625–628. doi: 10.1016/j.clineuro.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 12.Vallat J.M., Hugon J., Lubeau M., Leboutet M.J., Dumas M., Desproges-Gotteron R. Tick-bite meningoradiculoneuritis: clinical, electrophysiologic, and histologic findings in 10 cases. Neurology. 1987;37:749–753. doi: 10.1212/wnl.37.5.749. [DOI] [PubMed] [Google Scholar]
- 13.Dotevall L., Eliasson T., Hagberg L., Mannheimer C. Pain as presenting symptom in Lyme neuroborreliosis. Eur J Pain. 2003;7:235–239. doi: 10.1016/S1090-3801(02)00121-0. [DOI] [PubMed] [Google Scholar]
- 14.Rupprecht T.A., Koedel U., Fingerle V., Pfister H.W. The pathogenesis of lyme neuroborreliosis: from infection to inflammation. Mol Med. 2008;14:205–212. doi: 10.2119/2007-00091.Rupprecht. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Halperin J.J., Little B.W., Coyle P.K., Dattwyler R.J. Lyme disease: cause of a treatable peripheral neuropathy. Neurology. 1987;37:1700–1706. doi: 10.1212/wnl.37.11.1700. [DOI] [PubMed] [Google Scholar]
- 16.Halperin J.J., BJ Luft, Volkman D.J., Dattwyler R.J. Lyme neuroborreliosis. Peripheral nervous system manifestations. Brain. 1990;113(Pt 4):1207–1221. doi: 10.1093/brain/113.4.1207. [DOI] [PubMed] [Google Scholar]
- 17.Muley S.A., Parry G.J. Antibiotic responsive demyelinating neuropathy related to Lyme disease. Neurology. 2009;72:1786–1787. doi: 10.1212/WNL.0b013e3181a60a89. [DOI] [PubMed] [Google Scholar]
- 18.Kohler J. Lyme borreliosis: a case of transverse myelitis with syrinx cavity. Neurology. 1989;39:1553–1554. doi: 10.1212/wnl.39.11.1553-a. [DOI] [PubMed] [Google Scholar]
- 19.Benach J.L. Borrelia burgdorferi in the central nervous system. JAMA. 1992;268:872–873. doi: 10.1001/jama.268.7.872c. [DOI] [PubMed] [Google Scholar]
- 20.Oksi J., Kalimo H., Marttila R.J., Marjamäki M., Sonninen P., Nikoskelainen J., Viljanen M.K. Inflammatory brain changes in Lyme borreliosis. A report on three patients and review of literature. Brain. 1996;119(Pt 6):2143–2154. doi: 10.1093/brain/119.6.2143. [DOI] [PubMed] [Google Scholar]
- 21.Duray P.H., Steere A.C. Clinical pathologic correlations of Lyme disease by stage. Ann N Y Acad Sci. 1988;539:65–79. doi: 10.1111/j.1749-6632.1988.tb31839.x. [DOI] [PubMed] [Google Scholar]
- 22.Durovska J., Bazovska S., Pancak J., Zaborska M., Derdakova M., Traubner P. Infection with B. burgdorferi s.l., and the CNS demyelinating disease. A case report. Neuro Endocrinol Lett. 2011;32:411–414. [PubMed] [Google Scholar]
- 23.Bigi S., Aebi C., Nauer C., Bigler S., Steinlin M. Acute transverse myelitis in Lyme neuroborreliosis. Infection. 2010;38:413–416. doi: 10.1007/s15010-010-0028-x. [DOI] [PubMed] [Google Scholar]
- 24.Meurs L., Labeye D., Declercq I., Piéret F., Gille M. Acute transverse myelitis as a main manifestation of early stage II neuroborreliosis in two patients. Eur Neurol. 2004;52:186–188. doi: 10.1159/000081864. [DOI] [PubMed] [Google Scholar]
- 25.Koc F., Bozdemir H., Pekoz T., Aksu H.S., Ozcan S., Kurdak H. Lyme disease presenting as subacute transverse myelitis. Acta Neurol Belg. 2009;109:326–329. [PubMed] [Google Scholar]
- 26.Blanc F., Ballonzoli, Marcel C., de Martino S., Jaulhac B., de Seze J. Lyme optic neuritis. J Neurol Sci. 2010;295:117–119. doi: 10.1016/j.jns.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Logigian E.L. Peripheral nervous system Lyme borreliosis. Semin Neurol. 1997;17:25–30. doi: 10.1055/s-2008-1040909. [DOI] [PubMed] [Google Scholar]
- 28.Philipp M.T., Aydintug M.K., Bohm R.P., Jr., Cogswell F.B., Dennis V.A., Lanners H.N., Lowrie R.C., Jr., Roberts E.D., Conway M.D., Karacorlu M., Peyman G.A., Gubler D.J., Johnson B.J., Piesman J., Gu Y. Early and early disseminated phases of Lyme disease in the rhesus monkey: a model for infection in humans. Infect Immun. 1993;61:3047–3059. doi: 10.1128/iai.61.7.3047-3059.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts E.D., Bohm R.P., Jr., Cogswell F.B., Lanners H.N., Lowrie R.C., Jr., Povinelli L., Piesman J., Philipp M.T. Chronic lyme disease in the rhesus monkey. Lab Invest. 1995;72:146–160. [PubMed] [Google Scholar]
- 30.Roberts E.D., Bohm R.P., Jr., Lowrie R.C., Jr., Habicht G., Katona L., Piesman J., Philipp M.T. Pathogenesis of Lyme neuroborreliosis in the rhesus monkey: the early disseminated and chronic phases of disease in the peripheral nervous system. J Infect Dis. 1998;178:722–732. doi: 10.1086/515357. [DOI] [PubMed] [Google Scholar]
- 31.Pachner A.R., Delaney E., O’Neil T., Major E. Inoculation of nonhuman primates with the N40 strain of Borrelia burgdorferi leads to a model of Lyme neuroborreliosis faithful to the human disease. Neurology. 1995;45:165–172. doi: 10.1212/wnl.45.1.165. [DOI] [PubMed] [Google Scholar]
- 32.England J.D., Bohm R.P., Roberts E.D., Philipp M.T. Mononeuropathy multiplex in rhesus monkeys with chronic Lyme disease. Ann Neurol. 1997;41:375–384. doi: 10.1002/ana.410410313. [DOI] [PubMed] [Google Scholar]
- 33.Cadavid D., O’Neill T., Schaefer H., Pachner A.R. Localization of Borrelia burgdorferi in the nervous system and other organs in a nonhuman primate model of lyme disease. Lab Invest. 2000;80:1043–1054. doi: 10.1038/labinvest.3780109. [DOI] [PubMed] [Google Scholar]
- 34.Bai Y., Narayan K., Dail D., Sondey M., Hodzic E., Barthold S.W., Pachner A.R., Cadavid D. Spinal cord involvement in the nonhuman primate model of Lyme disease. Lab Invest. 2004;84:160–172. doi: 10.1038/labinvest.3700024. [DOI] [PubMed] [Google Scholar]
- 35.Ramesh G., Borda J.T., Gill A., Ribka E.P., Morici L.A., Mottram P., Jacobs M.B., Didier P.J., Philipp M.T. Possible role of glial cells in the onset and progression of Lyme neuroborreliosis. J Neuroinflammation. 2009;6:23–38. doi: 10.1186/1742-2094-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Payne D.N., Adcock I.M. Molecular mechanisms of corticosteroid actions. Paediatr Respir Rev. 2001;2:145–150. doi: 10.1053/prrv.2000.0122. [DOI] [PubMed] [Google Scholar]
- 37.Furst D.E. Meloxicam: selective COX-2 inhibition in clinical practice. Semin Arthritis Rheum. 1997;26(6 Suppl 1):21–27. doi: 10.1016/s0049-0172(97)80049-2. [DOI] [PubMed] [Google Scholar]
- 38.Dufour J.P., Phillippi-Falkenstein K., Bohm R.P., Veazey R.S., Carnal J. Excision of femoral head and neck for treatment of coxofemoral degenerative joint disease in a rhesus macaque (Macaca mulatta) Comp Med. 2012;62:539–542. [PMC free article] [PubMed] [Google Scholar]
- 39.Liang F.T., Philipp M.T. Analysis of antibody response to invariable regions of VlsE, the variable surface antigen of Borrelia burgdorferi. Infect Immun. 1999;67:6702–6706. doi: 10.1128/iai.67.12.6702-6706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyle H.M. An improved tissue technique with hematoxylin-eosin stain. Am J Med Technol. 1947;13:178–181. [PubMed] [Google Scholar]
- 41.Schmued L.C., Albertson C., Slikker W., Jr. Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 42.Schmued L.C., Stowers C.C., Scallet A.C., Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- 43.Margolis G., Pickett J.P. New applications for the Luxol fast blue myelin stain. Lab Invest. 1956;5:459–474. [PubMed] [Google Scholar]
- 44.Goto N. Discriminative staining methods for the nervous system: luxol fast blue–periodic acid-Schiff–hematoxylin triple stain and subsidiary staining methods. Stain Technol. 1987;62:305–315. doi: 10.3109/10520298709108015. [DOI] [PubMed] [Google Scholar]
- 45.Ramesh G., Alvarez X., Borda J.T., Aye P.P., Lackner A.A., Sestak K. Visualizing cytokine-secreting cells in situ in the rhesus macaque model of chronic gut inflammation. Clin Diagn Lab Immunol. 2005;12:192–197. doi: 10.1128/CDLI.12.1.192-197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramesh G., Borda J.T., Dufour J., Kaushal D., Ramamoorthy R., Lackner A.A., Philipp M.T. Interaction of the Lyme disease spirochete Borrelia burgdorferi with brain parenchyma elicits inflammatory mediators from glial cells as well as glial and neuronal apoptosis. Am J Pathol. 2008;173:1415–1427. doi: 10.2353/ajpath.2008.080483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Panayiotopoulos C.P. F chrondispersion: a new electrophysiologic method. Muscle Nerve. 1979;2:68–72. doi: 10.1002/mus.880020110. [DOI] [PubMed] [Google Scholar]
- 48.Mesrati F., Vecchierini M.F. F-waves: neurophysiology and clinical value. Neurophysiologie Clinique. 2004;34:217–243. doi: 10.1016/j.neucli.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Mullen R.J., Buck C.R., Smith A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 50.Tsoukas C.D., Landgraf B., Bentin J., Lamberti J.F., Carson D.A., Vaughan J.H. Structural and functional characteristics of the CD3 (T3) molecular complex on human thymocytes. J Immunol. 1987;138:3885–3890. [PubMed] [Google Scholar]
- 51.Pulford K.A., Rigney E.M., Micklem K.J., Jones M., Stross W.P., Gatter K.C., Mason D.Y. KP1: a new monoclonal antibody that detects a monocyte/macrophage associated antigen in routinely processed tissue sections. J Clin Pathol. 1989;42:414–421. doi: 10.1136/jcp.42.4.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Norton A.J., Isaacson P.G. Monoclonal antibody L26: an antibody that is reactive with normal and neoplastic B lymphocytes in routinely fixed and paraffin wax embedded tissues. J Clin Pathol. 1987;40:1405–1412. doi: 10.1136/jcp.40.12.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gavrieli Y., Sherman Y., Ben-Sasson S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grutter M.G. Caspases: key players in programmed cell death. Curr Opin Struct Biol. 2000;10:649–655. doi: 10.1016/s0959-440x(00)00146-9. [DOI] [PubMed] [Google Scholar]
- 55.Toma J.S., McPhail L.T., Ramer M.S. Differential RIP antigen (CNPase) expression in peripheral ensheathing glia. Brain Res. 2007;1137:1–10. doi: 10.1016/j.brainres.2006.12.053. [DOI] [PubMed] [Google Scholar]
- 56.Lahrtz F., Piali L., Spanaus K.S., Seebach J., Fontana A. Chemokines and chemotaxis of leukocytes in infectious meningitis. J Neuroimmunol. 1998;85:33–43. doi: 10.1016/s0165-5728(97)00267-1. [DOI] [PubMed] [Google Scholar]
- 57.Spanaus K.S., Nadal D., Pfister H.W., Seebach J., Widmer U., Frei K., Gloor S., Fontana A. C-X-C and C-C chemokines are expressed in the cerebrospinal fluid in bacterial meningitis and mediate chemotactic activity on peripheral blood-derived polymorphonuclear and mononuclear cells in vitro. J Immunol. 1997;158:1956–1964. [PubMed] [Google Scholar]
- 58.Carr M.W., Roth S.J., Luther E., Rose S.S., Springer T.A. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994;91:3652–3656. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Y., Beller D.I., Frendl G., Graves D.T. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J Immunol. 1992;148:2423–2428. [PubMed] [Google Scholar]
- 60.Kowarik M.C., Cepok S., Sellner J., Grummel V., Weber M.S., Korn T., Berthele A., Hemmer B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation. 2012;9:93–103. doi: 10.1186/1742-2094-9-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramesh G., Benge S., Pahar B., Philipp M.T. A possible role for inflammation in mediating apoptosis of oligodendrocytes as induced by the Lyme disease spirochete Borrelia burgdorferi. J Neuroinflammation. 2012;9:72–87. doi: 10.1186/1742-2094-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramesh G., Santana-Gould L., Inglis F.M., England J.D., Philipp M.T. The Lyme disease spirochete Borrelia burgdorferi induces inflammation and apoptosis in cells from dorsal root ganglia. J Neuroinflammation. 2013;10:88–101. doi: 10.1186/1742-2094-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh B., Coffey R.J. From wavy hair to naked proteins: the role of transforming growth factor alpha in health and disease. Semin Cell Dev Biol. 2014;28:12–21. doi: 10.1016/j.semcdb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Settipane G.A., Pudupakkam R.K., McGowan J.H. Corticosteroid effect on immunoglobulins. J Allergy Clin Immunol. 1978;62:162–166. doi: 10.1016/0091-6749(78)90101-x. [DOI] [PubMed] [Google Scholar]
- 65.Davidson D.C., Hirschman M.P., Sun A., Singh M.V., Kasischke K., Maggirwar S.B. Excess soluble CD40L contributes to blood brain barrier permeability in vivo: implications for HIV-associated neurocognitive disorders. PLoS One. 2012;7:e51793. doi: 10.1371/journal.pone.0051793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rothwell N.J., Strijbos P.J. Cytokines in neurodegeneration and repair. Int J Dev Neurosci. 1995;13:179–185. doi: 10.1016/0736-5748(95)00018-c. [DOI] [PubMed] [Google Scholar]
- 67.Raivich G., Jones L.L., Werner A., Blüthmann H., Doetschmann T., Kreutzberg G.W. Molecular signals for glial activation: pro- and anti-inflammatory cytokines in the injured brain. Acta Neurochir Suppl. 1999;73:21–30. doi: 10.1007/978-3-7091-6391-7_4. [DOI] [PubMed] [Google Scholar]
- 68.Minghetti L. Role of inflammation in neurodegenerative diseases. Curr Opin Neurol. 2005;18:315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 69.Ramesh G., MacLean A.G., Philipp M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013;2013:480739. doi: 10.1155/2013/480739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guillot X., Semerano L., Decker P., Falgarone G., Boissier M.C. Pain and immunity. Joint Bone Spine. 2012;79:228–236. doi: 10.1016/j.jbspin.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 71.Austin P.J., Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 72.Wang X.M., Hamza M., Wu T.X., Dionne R.A. Upregulation of IL-6, IL-8 and CCL2 gene expression after acute inflammation: correlation to clinical pain. Pain. 2009;142:275–283. doi: 10.1016/j.pain.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eliav E., Benoliel R., Herzberg U., Kalladka M., Tal M. The role of IL-6 and IL-1 beta in painful perineural inflammatory neuritis. Brain Behav Immun. 2009;23:474–484. doi: 10.1016/j.bbi.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 74.Miller R.J., Jung H., Bhangoo S.K., White F.A. Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol. 2009;194:417–449. doi: 10.1007/978-3-540-79090-7_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.White F.A., Jung H., Miller R.J. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jung H., Toth P.T., White F.A., Miller R.J. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abbadie C., Lindia J.A., Cumiskey A.M., Peterson L.B., Mudgett J.S., Bayne E.K., DeMartino J.A., MacIntyre D.E., Forrest M.J. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Murata Y., Rydevik B., Nannmark U., Larsson K., Takahashi K., Kato Y., Olmarker K. Local application of interleukin-6 to dorsal root ganglion induces tumor necrosis factor-α in the dorsal root ganglion and results in apoptosis of the dorsal root ganglion cells. Spine (Phila Pa 1976) 2011;36:926–932. doi: 10.1097/BRS.0b013e3181e7f4a9. [DOI] [PubMed] [Google Scholar]
- 79.Thirumangalakudi L., Yin L., Rao H.V., Grammas P. IL-8 induces expression of matrix metalloproteinases, cell cycle and pro-apoptotic proteins, and cell death in cultured neurons. J Alzheimers Dis. 2007;11:305–311. doi: 10.3233/jad-2007-11307. [DOI] [PubMed] [Google Scholar]
- 80.Hawkey C.J. COX-1 and COX-2 inhibitors. Best Pract Res Clin Gastroenterol. 2001;15:801–820. doi: 10.1053/bega.2001.0236. [DOI] [PubMed] [Google Scholar]
- 81.Parthsarathy G., Philipp M.T. The MEK/ERK pathway is the primary conduit for Borrelia burgdorferi-induced inflammation and P53-mediated apoptosis in oligodendrocytes. Apoptosis. 2014;19:76–89. doi: 10.1007/s10495-013-0913-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou Y., Ling E.A., Dheen S.T. Dexamethasone suppresses monocyte chemoattractant protein-1 production via mitogen activated protein kinase phosphatase-1 dependent inhibition of Jun N-terminal kinase and p38 mitogen-activated protein kinase in activated rat microglia. J Neurochem. 2007;102:667–678. doi: 10.1111/j.1471-4159.2007.04535.x. [DOI] [PubMed] [Google Scholar]
- 83.Bruin K.F., Hommes D.W., Jansen J., Tytgat G.N., Wouter ten Cate J., van Deventer S.J. Modulation of cytokine release from human monocytes by drugs used in the therapy of inflammatory bowel diseases. Eur J Gastroenterol Hepatol. 1995;7:791–795. [PubMed] [Google Scholar]
- 84.Abraham S.M., Lawrence T., Kleiman A., Warden P., Medghalchi M., Tuckermann J., Saklatvala J., Clark A.R. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. 2006;203:1883–1889. doi: 10.1084/jem.20060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jang B.C., Lim K.J., Suh M.K., Park J.G., Suh S.I. Dexamethasone suppresses interleukin-1beta-induced human beta-defensin 2 mRNA expression: involvement of p38 MAPK, JNK, MKP-1, and NF-kappaB transcriptional factor in A549 cells. FEMS Immunol Med Microbiol. 2007;51:171–184. doi: 10.1111/j.1574-695X.2007.00293.x. [DOI] [PubMed] [Google Scholar]