

Sickle cell disease (SCD) is one of the most common genetic disorders worldwide and is associated with episodes of acute pain and progressive multi-organ damage.1 The most common cause of SCD is homozygosity for the hemoglobin sickle (Hb S) mutation, with a minority of cases due to compound heterozygosity for Hb S and other alleles including β-thalassemia. Loss-of-function point mutations of the β-globin gene that abolish (β0) or reduce (β+) production of normal β-chains are the most common causes of β-thalassemia, with large deletions or rearrangements accounting for a small minority of β-thalassemia alleles.2,3 While patients with Hb S/β0-thalassemia generally have severe SCD, the residual production of normal β-chains in Hb S/β+-thalassemia patients is associated with lower Hb S concentration in erythrocytes and a less severe disease.4 Here, we report the unusual case of an infant who had a newborn screening profile fully consistent with sickle trait, yet was diagnosed later in childhood with typical Hb S/β+-thalassemia. Molecular testing demonstrated that the child was a compound heterozygote for the Hb S mutation and a partial deletion of the β-globin Locus Control Region (βLCR). The deletion removed two of the five DNase I hypersensitivity (HS) regions, providing valuable insight into the roles of individual HS regions in globin gene switching and expression.
The proband is now a 6-year old boy who was born to healthy non-consanguineous parents of Caribbean descent. The pregnancy and delivery were unremarkable with no evidence of perinatal anemia or jaundice. Newborn screening was negative for SCD, with the Hb profile being fully consistent with sickle trait (Hb F 79.1%, Hb A 6.0%, Hb S 4.0%, Hb Bart’s 9.1%) (Figure 1A). The patient enjoyed normal health until five years of age, when he was diagnosed with SCD during an admission for unexplained abdominal pain and enlarged spleen (splenic sequestration). Microcytosis, sickle erythrocytes, Howell-Jolly bodies and target cells were observed on the peripheral blood smear. Hemoglobin analysis using high performance liquid chromatography (HPLC) was suggestive of Hb S/β+-thalassemia (Hb A 19.4%, Hb S 72.7%, and Hb A2 2.6%) (Figure 1B). Since diagnosis, the patient has been admitted for one vasoocclusive event and had a tonsillectomy for obstructive sleep apnea.
Figure 1.

(A) BioRad HPLC traces of the proband’s hemolysate from newborn screening and (B) at five years of age. Positions of hemoglobins F, A, A2, and S are indicated above the corresponding peaks. (C) MLPA analysis showing the relative probe signals across the β-globin gene cluster (SALSA MLPA probemix P-102-B2 HBB, MRC-Holland, Amsterdam, The Netherlands). The MLPA probes that define the deletion are indicated. (D) Sequence of the deletion junction fragment compared to the normal 5′ and 3′ sequences. The first and last nucleotides of the deleted region are underlined. The deletion junction fragment was amplified as a ~0.6 kb fragment using a pair of flanking primers (forward 5′-ACTTT CAGTC CGGTC CTCA CAGT-3′, NG_000007.3 positions 8111–8133; reverse 5′-GTGGT TTCTA GTCCC TTCAC CATC TTGT-3′, NG_000007.3 positions 135523–13550), which was then sequenced using the reverse primer.
Nucleotide sequence analysis of the β-gobin gene revealed that the proband was heterozygous for the Hb S mutation (HBB:c.20A>T) with no other mutations of the β-globin gene. Deletion-specific gap-PCR demonstrated that he was also heterozygous for the rightward 3.7 kb single α-globin gene deletion (−α3.7/αα). Sequence analysis of the intact α-globin genes failed to detect any point mutations. As this genotype could not explain the reduced expression of Hb A and the SCD phenotype in the proband, we investigated the possibility of compound heterozygosity for Hb S and β+-thalassemia due to a deletion involving the βLCR. The multiplex ligation-dependent probe amplification (MLPA) and junction sequence analysis demonstrated the presence of a βLCR deletion that spans 4,860 bp and encompasses the HS3 and HS4 core regions (Figure 1C and D). The deletion has been named the Caribbean βLCR deletion (HGVS nomenclature NG_000007.3:g.8510_13369del).
DNA analysis of the parents established that the proband inherited the Hb S allele from his mother and the βLCR deletion and 3.7 kb α-thalassemia deletion from his father. The mother was also found to carry the Hb Stanleyville-II missense mutation of the α1-globin gene (HBA1:c.237C>A), which was not inherited by the proband. Hb Stanleyville-II is not associated with abnormal phenotypes in simple carriers or in combination with α- or β-thalassemia.3 Hb Stanleyville-II has the curious property of inhibiting the formation of Hb sickle fibers in vitro, and thereby may ameliorate the clinical phenotype of SCD.5 The βLCR is located approximately 5.7 kb to 21.2 kb upstream of the ε-globin gene (HBE1), and consists of five DNase I hypersensitivity sites designated HS1 through HS5. The βLCR plays a key role in high-level, tissue-specific expression of the HBB gene.6 Studies in transgenic mice have suggested that individual HS core regions may have unequal contributions to βLCR function, with HS2 having a dominant activity.7 Recently, in a mouse model, it has been demonstrated that the HS core regions differ in their degree of activity and act in an additive manner.9 Deletion of murine HS2 was associated with the greatest reduction in HBB gene expression, while deletions of HS3 or HS4 resulted in lesser reductions.8
In humans, 25 naturally occurring deletions have been reported that remove all or part of the βLCR.2,9–13 Most of these deletions include at least three HS regions and are larger than the identified deletion in our case. Typically, these deletions result in loss of expression for all of the β-like genes and a phenotype resembling (εγδβ)0-thalassemia. In heterozygous patients, neonatal hemolytic anemia is common due to impaired γ-chain synthesis required for Hb F. Once the γ→β switch has occurred during the first six months of infancy, the phenotype resolves to one of thalassemia trait with normal Hb A2. While the phenotype is well established for carriers of large deletions that remove all or most of the HS regions, the contribution of individual HS regions to β-globin gene expression in human has not been conclusively clarified. Six previously reported deletions involve the βLCR but leave intact all of the β-like globin genes (Figure 2). The phenotypes associated with these deletions are of particular interest for delineating the importance of individual HS core regions. For example, complete deletions of βLCR or the Hispanic deletion that removes HS2, HS3 and HS4 are associated with the phenotype of (εγδβ)0-thalassemia whereas the Italian deletion of HS1 and the Toledo deletion of HS3 are not associated with significant hematologic changes in carriers.9,10,13 These observations have raised the suggestion that HS2 may be the only hypersensitive site that has substantial regulatory effect on adult β-globin gene expression.
Figure 2.

Map of the β-globin gene cluster showing the locations of 8 full or partial β-LCR deletions that leave all of the β-like genes intact. The scale is relative to the GRCh38 version of chromosome 11 (NC_000011.10). Details regarding the previously unreported 12.4 kb Mediterranean deletion are provided in the Online Supplementary Appendix.
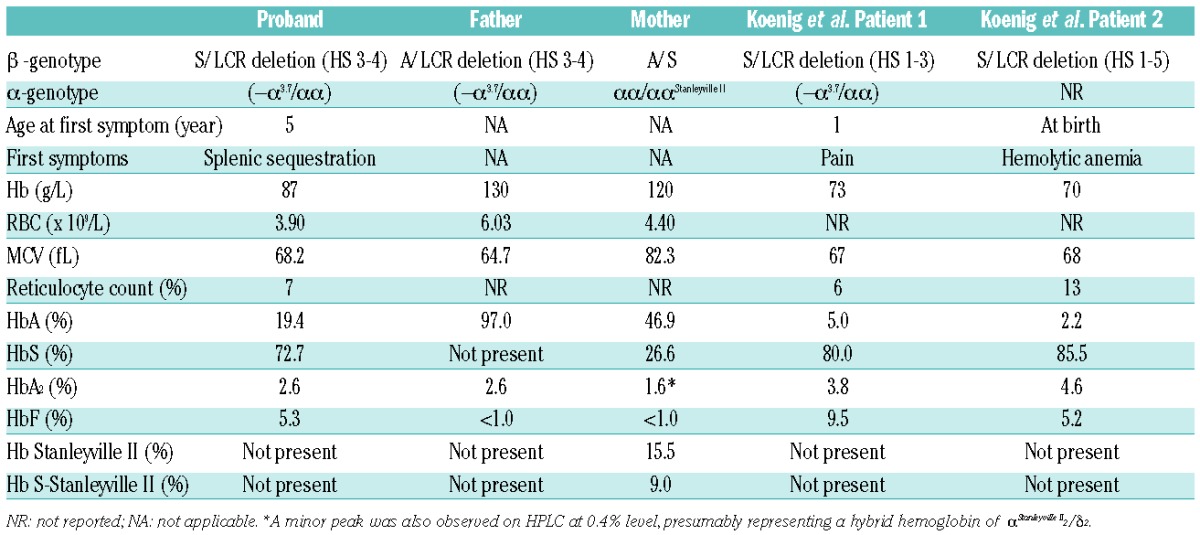
The deletion described in our patient is the smallest reported βLCR deletion that is associated with significant clinical or hematologic phenotype. Our patient had relatively mild SCD with no clinically significant hemolytic anemia at birth. This is in contrast to the more severe Hb S/β-thalassemia phenotypes reported by Keonig and colleagues in patients with more extensive LCR deletions (Table 1 and Figure 2).12 The presence of 19.4% Hb A in our patient indicates that deletions involving only HS3 and HS4 are associated with a significant reduction but not abrogation of β-globin gene expression, consistent with the murine model of independent and additive contributions of individual HS core regions.8 The level of residual β-globin gene expression associated with the deletion can be inferred from the level of Hb A present in the hemolysate of the proband, which is similar to that observed with Hb S/β+-thalassemia patients due to common promoter mutations such as −29 A→G or −88 C→T.4 Overall, the hematologic and clinical phenotype is mild and would be classified as type III Hb S/β-thalassemia.4
Table 1.
Hematologic profile and molecular genetic results for the family and the two patients reported by Koenig et al.12
It is of interest that the proband was born on Ontario, Canada, shortly after universal newborn screening (NBS) for SCD was introduced in 2006. NBS was done using blood spots and HPLC analysis, and the results were fully consistent with the child having sickle cell trait. The NBS Hb profile was Hbs F/A/S/Bart’s, with the proportion of Hb A being greater than Hb S (6.0% vs. 4.0%). This is remarkable because NBS can readily distinguish between Hb S trait and Hb S/β+-thalassemia based on the relative proportion of Hb A and Hb S. Hb S trait is characterized by a relative excess of Hb A relative to Hb S, whereas the ratio of Hb S:Hb A generally is more than 2.0 in Hb S/β+-thalassemia.14 Given that there was no record of perinatal anemia, and the ratio of Hb S:Hb A was less than 1.0 (consistent with Hb S trait as opposed to Hb S/β+-thalassemia), it is possible that 4.9 kb deletion of HS3 and HS4 did not have an appreciable impact on globin gene expression until after the γ→β globin gene switch. Similarly, carriers of the 12.4 kb Mediterranean deletion (removing HS3, HS4 and HS5) have a thalassemic phenotype without history of perinatal anemia (Online Supplementary Appendix). In comparison, complete deletions of the βLCR are associated with perinatal anemia and phenotype resembling (εγδβ)0-thalassemia, suggesting that the role and importance of individual HS regions may differ depending on the stage of development; an observation that has been previously demonstrated in experimental models15 but has yet to be observed in humans.
Overall, the present report indicates that a deletion of βLCR HS3 and HS4 may not have an appreciable impact on γ-globin gene expression during fetal development, but is clearly associated with reduced β-globin gene expression following the γ→β switch. Moreover, it is also apparent that the deletion of HS3 and HS4 results in less severe reduction of β-globin gene expression relative to deletions of the entire βLCR. Ultimately, the delineation of the roles of the individual HS regions in globin gene switching and β-like gene expression through development will depend on the identification and characterization of other naturally occurring βLCR deletions. Lastly, it is notable that adult carriers of such deletions have the phenotype of (γδβ)-thalassemia (microcytic, hypochromic anemia with normal levels of Hb A2) which could be mistaken for α-thalassemia trait. This has implications for accurate carrier screening and genetic counseling.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. [DOI] [PubMed] [Google Scholar]
- 2.Thein SL, Wood WG. The molecular basis of β thalassemia, δβ thalassemia, and hereditary persistence of fetal hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of hemoglobin. Genetics, pathophysiology, and clinical management, 2nd ed. New York: Cambridge University Press; 2009. p. 323–356. [Google Scholar]
- 3.Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42 Database issue: D1063–1069 (http://globin.cse.psu.edu/hbvar/menu.html). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serjeant GR, Serjeant BE, Fraser RA, et al. Hb S-β-thalassemia: Molecular, hematological and clinical comparisons. Hemoglobin. 2011;35(1):1–12. [DOI] [PubMed] [Google Scholar]
- 5.Burchall G, Maxwell E. Haemoglobin Stanleyville II modifies sickle cell disease phenotype. Pathology. 2010;42(3):310–312. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Peterson KR, Fang X, Stamatoyannopoulos G. Locus control regions. Blood. 2002;100(9):3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bungert J, Tanimoto K, Patel S, Liu Q, Fear M, Engel JD. Hypersensitive site 2 specifies a unique function within the human beta-globin locus control region to stimulate globin gene transcription. Mol Cell Biol. 1999;19(4):3062–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bender MA, Ragoczy T, Lee J, et al. The hypersensitive sites of the murine β-globin locus control region act independently to affect nuclear localization and transcriptional elongation. Blood. 2012;119(16):3820–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Driscoll MC, Dobkin CS, Alter BP. Gamma delta beta-thalassemia due to a de novo mutation deleting the 5′ β-globin gene activation-region hypersensitive sites. Proc Natl Acad Sci USA. 1989; 86(19):7470–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulozik AE, Bail S, Bellan-Koch A, Bartram CR, Kohne E, Kleihauer E. The proximal element of the β globin locus control region is not functionally required in vivo. J Clin Invest. 1991;87(6):2142–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joly P, Lacan P, Garcia C, Meley R, Pondarré C, Francine A. A novel deletion/insertion caused by a replication error in the β-globin gene locus control region. Hemoglobin. 2011;35(4):316–322. [DOI] [PubMed] [Google Scholar]
- 12.Koenig SC, Becirevic E, Hellberg MS, et al. Sickle cell disease caused by heterozygosity for Hb S and novel LCR deletion: Report of two patients. Am J Hematol. 2009;84(9):603–606. [DOI] [PubMed] [Google Scholar]
- 13.Nieto JM, Villegas A, De La Fuente-Gonzalo F, González FA, Ropero P. Heterozygosity for a deletion of hypersensitive site 3 in the human locus control region has an unexpected minor effect on red cell phenotype. J Hum Genet. 2014;59(10):585–587. [DOI] [PubMed] [Google Scholar]
- 14.Eastman JW, Wong R, Liao CL, Morales DR. Automated HPLC screening for sickle cell anemia and other hemoglobinopathies. Clin Chem. 1996;42(5):704–710. [PubMed] [Google Scholar]
- 15.Peterson KR, Clegg CH, Navas PA, Norton EJ, Kimbrough TG, Stamatoyannopoulos G. Effect of deletion of 5′HS3 or 5′HS2 of the human β-globin locus control region on the developmental regulation of globin gene expression in β-globin locus yeast artificial chromosome transgenic mice. Proc Natl Acad Sci USA. 1996;93(13):6605–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]