

Abstract
Serum lipoproteins (LP) are increasingly being recognized as dual purpose molecules that contribute to both cholesterol homeostasis and host innate defense. In fact, very low LP levels are associated with increased risk of bacterial infection in critically ill patients. In this respect, we reported that apolipoprotein B100 (apoB100), the 4536 amino acid structural protein of very low density lipoprotein (VLDL) produced by the liver, limits Staphylococcus aureus pathogenesis. S. aureus uses quorum-sensing (QS) via the accessory gene regulator (agr) operon and an autoinducing peptide (AIP) to coordinate expression of over 200 virulence genes. ApoB100 prevents agr activation by binding and sequestering secreted AIP. Importantly, human serum LP are produced not only by the liver, but are also produced by enterocytes, in the form of chylomicrons, during uptake of dietary lipids. In contrast to apoB100 in VLDL, human enterocytes use apoB48, the N-terminal 2152 amino acids (48%) of apoB100, as the structural component of chylomicrons. Interestingly, enteral feeding of critically ill patients has been associated with decreased risk of infectious complications, suggesting chylomicrons could contribute to host innate defense in critically ill patients when serum LP production by the liver is limited during the acute phase response. Therefore, we hypothesized that apoB48 would be sufficient to antagonize S. aureus QS. As expected, isolated apoB48-LP bound immobilized AIP and antagonized agr-signaling. ApoB48- and apoB100-LP inhibited agr activation with IC50s of 3.5 and 2.3 nM, respectively, demonstrating a conserved AIP binding site. Importantly, apoB48-LP antagonized QS, limited morbidity and promoted bacterial clearance in a mouse model of S. aureus infection. This work demonstrates that both naturally occurring forms of apolipoprotein B can antagonize S. aureus QS, and may suggest a previously unrecognized role for chylomicrons and enterocytes in host innate defense against S. aureus QS-mediated pathogenesis.
Introduction
Serum lipoproteins have historically had two primary functions: 1) to transport cholesterol and other insoluble lipids from their source to peripheral tissues to be used for cell membrane assembly, steroid production and fuel (termed forward cholesterol transport), and 2) to transport excess cholesterol from the tissues to the liver for clearance (reviewed in [1]). In recent years, however, serum lipoproteins have become increasingly recognized for their contribution to host innate defense [2–4]. Extremely low serum lipoprotein levels (hypolipoproteinemia) are associated with increased risk of bacterial infection in critically ill patients such as those experiencing trauma [4–7]. Reduced serum lipoprotein levels in these patients is in part due to the acute phase response (APR) and decreased lipoprotein released from the liver (reviewed in [4, 6]). However, in addition to very low-density lipoprotein (VLDL) and high density lipoprotein (HDL) produced by the liver, human serum lipoproteins include chylomicrons produced by intestinal enterocytes during the uptake of dietary lipids. Therefore, the absence of chylomicron production by the gut in patients unable to receive oral feeding may also contribute to hypolipoproteinemia and an increased risk of infection. Although results are varied, some studies have shown that enteral feeding of critically ill patients, which preserves the contribution of the gut to nutritional processing, is associated with reduced risk of infectious complications compared to parenteral (intravenous) feeding [8, 9]. This suggests that, beyond their role in lipid transport, chylomicrons produced by enterocytes may also contribute to limiting the pathogenesis of bacterial infection. Although VLDL, LDL (produced by lipase reduction of VLDL), HDL and their components are being increasingly recognized as host innate effectors, much less is known about the contribution of chylomicrons and their components to protection against bacterial pathogenesis.
Lipoprotein particles are able to transport water insoluble lipids to peripheral tissues largely due to the inclusion of apolipoproteins. The formation of chylomicrons and the other forward cholesterol transport molecules, VLDL and LDL, requires the structural protein, apolipoprotein B (apoB). We have shown that apoB100, the 4536 amino acid protein essential for VLDL, and thus LDL, formation (Fig 1), limits pathogenesis caused by the Gram positive pathogen Staphylococcus aureus by disrupting virulence factor expression [10, 11]. S. aureus is both a commensal and an opportunistic pathogen which causes a broad range of diseases from skin and soft tissue infections (SSTI) to life threatening conditions in critically ill patients including pneumonia, endocarditis, osteomyelitis and bacteremia. The ability of S. aureus to cause disease in such varied host niches is facilitated by the use of two-component systems, including the accessory gene regulator (agr) quorum-sensing (QS) operon, to globally coordinate gene expression [12–22]. The agr operon coordinates a density-dependent switch from a colonizing to an invasive phenotype through the up-regulated expression of over 200 virulence factors such as proteases, lipases and toxins [23–25]. Activation of the agr system is mediated by binding of a secreted autoinducing peptide (AIP) to its cognate receptor, AgrC, on the bacterial surface. S. aureus isolates consist of four possible agr alleles, and AIP from each of the four alleles (AIP1-AIP4) differs in amino acid sequence and length, ranging from seven to nine amino acids. However, all AIPs share a common five-membered thiolactone ring which provides general structural similarity [12, 26]. We have shown that apoB100 binds to and sequesters S. aureus AIPs and therefore antagonizes agr-mediated virulence [10, 11]. In contrast to apoB100 in VLDL and LDL, chylomicrons contain a truncated form of apoB, apoB48, which is only produced in humans by intestinal enterocytes. The production of apoB48, the N-terminal 2152 amino acids (48%) of apoB100 (Fig 1), results from RNA editing by an enterocyte enzyme which introduces a premature stop codon in the coding region for apoB100 [27, 28]. Importantly, the association between enteral feeding and reduced risk of infectious complications compared to parenteral feeding in critically ill patients [8, 9] suggests a potentially unrecognized role for apoB48 in host innate defense. However, whether apoB48 is sufficient to antagonize S. aureus agr-signaling and pathogenesis has not been addressed.
Fig 1. Depiction of apolipoprotein B.
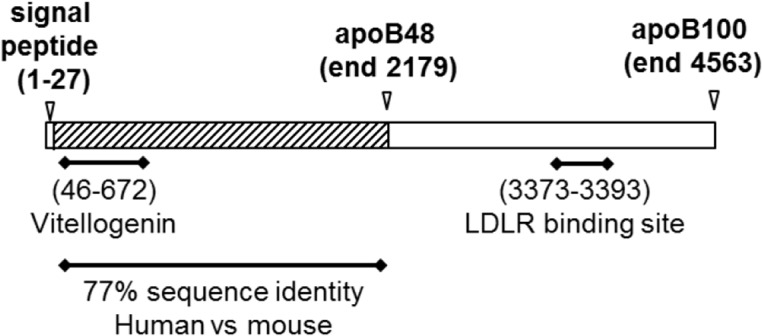
Schematic of human apoB100 (UniProt P04114) indicating the region aligning with vitellogenin, for which a crystal structure is available [57], the C-terminus of apoB48 and the low density lipoprotein receptor (LDLR) recognition site which facilitates uptake and clearance of apoB100 by the LDLR.
To test the hypothesis that apoB48 is a host innate effector against S. aureus pathogenesis, we isolated apoB48-containing lipoprotein particles and assessed their ability to disrupt agr-activation. Here we report that apoB48 directly binds AIP and antagonizes S. aureus QS in vitro. IC50s for antagonism of agr-signaling are equivalent between apoB48 and apoB100, providing evidence that the AIP binding site is conserved between these apolipoproteins. Finally, exogenous apoB48 antagonized QS and inhibited morbidity in vivo in a mouse model of S. aureus infection. Therefore, this work provides proof that the AIP binding site is located in the N-terminus of apoB, demonstrates that apoB48 is sufficient to antagonize S. aureus QS, and suggests a previously unrecognized role for chylomicrons and intestinal enterocytes in human innate defense against bacterial pathogenesis.
Results
ApoB48 is sufficient to antagonize S. aureus agr-signaling in vitro
We previously reported that apoB100, and not its associated lipids or other exchangeable apoproteins (apoA-I, apoC-I or apoE), binds and sequesters S. aureus AIP and antagonizes QS-dependent virulence [11]. To determine whether apoB48 was sufficient to inhibit S. aureus QS, we utilized serum from ApoE -/- mice as a ready source of apoB48-containing lipoprotein particles (apoB48-LP). Lipoproteins containing apoB100 are largely cleared by the LDL receptor (LDLR) which binds to the LDLR-binding sequence in the C-terminus of apoB100 (Fig 1). In contrast, clearance of apoB48 is mediated by apoE, an exchangeable apolipoprotein which facilitates uptake of chylomicron remnants via the LDLR and low-density lipoprotein related protein (LRP) [29–31]. Therefore, ApoE -/- mice amass circulatory apoB48-LP relative to wild-type mice. In order to determine whether apoB48-LP are capable of antagonizing QS, we first asked whether serum from ApoE -/- mice would antagonize S. aureus agr signaling in vitro. Because AIP binding to AgrC leads to transcriptional activation of the agr P3 promoter, we assessed agr signaling using an agr type I epidemic clone of community acquired-methicillin resistant S. aureus (CA-MRSA), USA300 LAC, with the agr::P3 promoter driving yfp expression [32]. As expected, at equivalent concentrations, serum (0.3%) from ApoE -/- mice showed increased inhibition of agr-signaling compared to serum from wild-type mice (Fig 2A). Since ApoE -/- serum is enriched in apoB48-LP compared to serum from wild-type controls, these results suggest that the increased apoB48-LP provided enhanced control of S. aureus agr-signaling in this system.
Fig 2. ApoB48-LP binds AIP and antagonizes agr-signaling.

(A) S. aureus agr-I strain AH1677 (LAC) (2 x 107 CFUs ml-1) was cultured for 2 hrs with 10 nM exogenous AIP1 and serum from apoE -/- or wild type (C57BL/6) mice (0.3%). agr::P3 promoter activation was measured by flow cytometry as mean channel fluorescence (MCF) and the MCF of the no serum control was normalized to 100%. (B) S. aureus was cultured as in (A) along with different concentrations of apoB48-LP or 50 nM human LDL (apoB100). agr::P3 promoter activation was measured by flow cytometry and the MCF of the no apoB control was normalized to 100%. Results shown are means ± SEM from three independent experiments performed in triplicate. (C-D) qRT-PCR analysis of (C) RNAIII and (D) hla expression relative to 16S rRNA under conditions described above. Results are means ± SEM from three independent experiments. (E) (Left) SDS-PAGE analysis of supernatants from 5 h cultures of LAC grown alone or with 50 nM AIP ± 50 nM apoB48-LP. Arrowhead indicates migration of Hla. (Right) Relative Hla concentration was determined by Western blot followed by quantification of band intensity compared to the + AIP control. Data are mean ± SEM of three experiments performed in triplicate. (F) Hla expression in supernatants grown as for (E), assessed via the rabbit red blood cell lysis assay. HA50 is the bacterial supernatant dilution factor required for lysis of 50% of the RBCs. Data are the mean ± SEM of triplicate experiments performed in duplicate. (G) Surface plasmon resonance (SPR) analysis of apoB48-LP binding to immobilized biotinylated AIP1. Binding was measured in resonance units (RU). (H) Anti-apoB antibody at 5-, 10- and 30-fold molar excess, but not IgG control, blocks apoB48-LP binding to AIP1. Results are the mean ± SEM of N = 3 to 5. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p≤0.0001.
To confirm that apoB48-LP were responsible for the inhibition of agr-signaling by serum from ApoE-/- mice, we used size exclusion chromatography to purify apoB48-LP from the serum. ApoB48-LP eluted in the VLDL fraction which was highly enriched in serum from ApoE -/- mice compared to wild-type C57BL/6 mice (Panel A in S1 Fig). SDS/PAGE followed by Coomassie staining of the eluted fraction showed a single band at high molecular weight (227 kDa), consistent with the predicted molecular weight of apoB48 (~240 kDa), and Western blot analysis confirmed this band was apoB (Panel B in S1 Fig). No protein band was detected corresponding to the molecular weight of apoB100 (509 kDa). Additionally, dynamic light scattering analysis revealed a unimodal peak with a Z-average diameter of 72 nm, indicative of a homogeneous sample (Panel C in S1 Fig).
If apoB48-LP contributed to QS inhibition by serum from ApoE -/- mice, then we would expect strong inhibition of agr-signaling using purified apoB48-LP. As expected, apoB48-LP inhibited agr::P3 promoter activation in a dose-dependent manner (Fig 2B). Importantly, mouse apoB48 (UniProt E9Q414), which shares 77% sequence identity with the N-terminal 48% of human apoB100 (UniProt P04114), showed equivalent inhibition of agr-signaling compared to human LDL (apoB100-LP). Furthermore, apoB48-LP mediated QS inhibition was specific to agr-signaling, and not due to an effect on bacterial viability or suppression of bacterial growth (Panels D,E in S1 Fig). As would be predicted from the agr::P3 promoter activation assays, apoB48-LP antagonized AIP-induced expression of the P3-driven agr effector molecule, RNAIII, as well as agr-mediated expression of the gene encoding alpha-hemolysin, Hla, a key S. aureus virulence factor involved in invasive infection [33–39] (Fig 2C and 2D). Furthermore, apoB48-LP antagonized AIP-induced expression of Hla protein, as measured by both Western blot and rabbit RBC lysis functional assays (Fig 2E and 2F).
We previously demonstrated that the mechanism of apoB100-mediated inhibition of agr-signaling included direct binding to immobilized AIP [10, 11]. Based on our findings above, we predicted that apoB48-LP would likewise bind to AIP. To address this, we used surface plasmon resonance (SPR) to measure binding of apoB48-LP to immobilized AIP1. As expected, apoB48-LP bound immobilized, active (cyclized) AIP1 in a dose dependent manner, but did not bind the linear, biologically inactive form of the peptide (Fig 2G). In addition, the presence of antibody specific to apoB inhibited binding of immobilized AIP by apoB48-LP as previously shown for apoB100-LP [10] (Fig 2H). Furthermore, apoB48-LP bound immobilized AIP2, AIP3 and AIP4 in a dose dependent manner (Panels A-C in S2 Fig), and antagonized agr::P3 promoter activation by agr-II, agr-III and agr-IV clinical isolates (33) (Panels D-F in S2 Fig). These data demonstrate that a binding site for each of the four S. aureus AIPs is located within apoB48 and that apoB48-LP are sufficient to antagonize agr-signaling by isolates from each of the four agr alleles.
ApoB48 and apoB100 are equally efficient inhibitors of agr-signaling
The location of the AIP binding site within the N-terminus of apoB suggests that apoB48-LP would be an equally efficient antagonist of agr-signaling compared to apoB100-LP. To address this, we tested a range of equimolar concentrations of apoB48 and apoB100 for antagonism of agr-signaling driven by a fixed amount of exogenous AIP1. Inhibition dose-response curves for both apoB48-LP and LDL were saturable and therefore exhibited the characteristic response expected from a specific ligand-receptor interacting pair. Calculated IC50 values for apoB48- and apoB100-LP were essentially identical (3.5 and 2.3 nM, respectively (Fig 3)) and below the reported EC50 for AIP1-mediated agr activation (28 nM) [40]. These data demonstrate that a high-affinity binding site for AIP is located in the N-terminus of apoB and is conformationally conserved in apoB48.
Fig 3. ApoB48- and apoB100-LP demonstrate equivalent inhibition of agr.
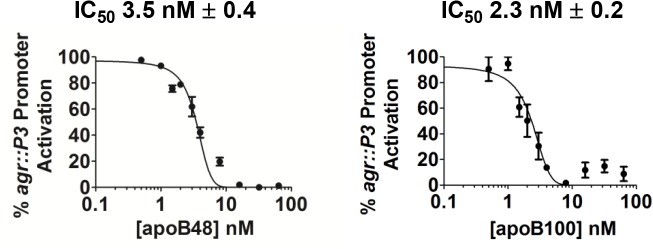
AH1677 (2x107 CFUs ml-1) was cultured with 50 nM AIP and increasing concentrations of mouse apoB48-LP or human LDL (apoB100). Data from three independent experiments are shown as normalized MCF versus the log of LP concentration.
ApoB48 antagonizes S. aureus quorum sensing and pathogenesis in vivo
Using a mouse air-pouch model of S. aureus skin and soft tissue infection (SSTI) (3, 4), we previously showed that apoB100-LP, LDL and oxLDL extravasate to the site of infection where they protect against S. aureus agr-dependent virulence [10]. Together with our in vitro data demonstrating that the AIP binding site is located within the N-terminal 48% of apoB100, we predicted that exogenous apoB48-LP would be sufficient to provide protection against agr-mediated pathogenesis in vivo. To demonstrate the potential protective role of apoB48 in critically ill patients, we utilized a hypolipidemic mouse model with high susceptibility to S. aureus infection. Specifically, we used mice lacking the phagocyte NADPH oxidase, Nox2, which were also treated with 4-aminopyrazole-(3,4-D)pyrimidine (4APP) to pharmacologically reduce serum lipoprotein levels [41, 42]. We chose Nox2 -/- mice because chronic granulomatous disease patients lack Nox2 and are highly susceptible to S. aureus infection [43]. Using the air-pouch model of SSTI, we infected the mice by injecting USA300 LAC directly into the pouch along with either apoB48-LP or vehicle control. At twenty-four hours post-infection, apoB48-treated mice showed significantly reduced weight loss and a lower clinical score, both general measures of morbidity, compared to vehicle controls (Fig 4A and 4B). As expected, bacteria recovered in pouch lavage from apoB48-treated mice showed significantly reduced agr::P3 promoter activation compared to vehicle treated mice, demonstrating a significant reduction in agr-signaling (Fig 4C). Furthermore, apoB48-treated mice had decreased bacterial burden at the site of infection compared to controls (Fig 4D). Importantly, this was not due to a negative effect on S. aureus growth caused by the addition of apoB48-LP to serum from 4APP-treated Nox2 -/- mice (Fig 4E). Having previously shown that reduction of serum lipoproteins negatively effects host clearance of agr+ but not agr- S. aureus [10, 11], and that pharmacological disruption of agr-signaling leads to enhanced immune cell-mediated bacterial clearance [44, 45], these data suggest that the reduced bacterial burden in apoB48 treated mice resulted from enhanced host mediated clearance following agr-inhibition. Together, these data demonstrate that apoB48 is sufficient to inhibit agr-signaling in vivo and to provide host protection against S. aureus agr-mediated pathogenesis.
Fig 4. apoB48-LP antagonize agr-signaling and inhibit morbidity in an air-pouch model of S. aureus SSTI.

AH1677 (3x107 CFUs) and apoB48-LP (100 nM) or vehicle control, were injected into dorsal air-pouches of 4APP-treated, Nox2 -/- mice. At 4 hrs post-infection, the mice were given a second dose of apoB48-LP or vehicle control. Twenty-four hrs post-infection, the following were determined: (A) percent weight loss; (B) clinical morbidity score (see Materials and Methods for details); (C) agr::P3 promoter activation in pouch lavage; and (D) bacterial burden in the pouch lavage and spleen. Results are the mean ± SEM of N = 7 mice/group from two independent experiments (A,B) and N = 3 mice/group (C,D). (E) Growth curves of USA300 isolate LAC grown in broth or broth with 10% serum from 4APP-treated, Nox2 -/- mice ± 100 nM apoB48-LP. Data shown are mean ± SEM from at least 2 independent experiments performed in duplicate. ns, not significant; **, p<0.01; ****, p≤0.0001.
Discussion
Whereas serum lipoproteins are essential for lipid transport and homeostasis, pathologically elevated levels of LP are major contributors to cardiovascular disease and atherosclerosis [1, 46]. This deleterious role of LP is in stark contrast to their emerging role in host innate defense and the association between very low LP levels and increased susceptibility to the pathogenesis of infection [3–6, 47]. Here we extend our understanding of the contributions of LP to host innate defense by describing a role for apoB48 in limiting S. aureus agr-regulated pathogenesis. ApoB48 binds S. aureus AIP and antagonizes agr-signaling in vitro and in vivo, and because the S. aureus agr system coordinates expression of more than 200 virulence genes [25], this antagonism could provide broad protection against S. aureus infections facilitated by agr. Importantly, IC50 values for apoB48- and apoB100-mediated antagonism of agr-I-signaling were similar and lower than the reported EC50 for AIP1 activation of AgrC [40], suggesting that both forms of apoB can compete with AgrC for AIP binding. Because apoB48 in humans is solely produced by enterocytes during the uptake and packaging of dietary lipids into chylomicrons, our work may suggest a previously unrecognized role for chylomicrons and intestinal enterocytes in limiting the pathogenesis of S. aureus infections in critically ill patients.
Although this is the first report describing a potential role for apoB48 and chylomicrons in innate defense against S. aureus QS, others have demonstrated roles for LP in host innate defense against this Gram positive pathogen, including LP binding to lipotechoic acid (LTA) as well as to certain staphylococcal toxins [48–50]. However, a broader role for LP in limiting bacterial pathogenesis lies in their ability to protect against potentially lethal endotoxic shock. LP, including chylomicrons, can bind lipopolysaccharide (LPS) from Gram negative pathogens, thus limiting LPS-induced production of pro-inflammatory cytokines and the resulting pathogenesis [51]. In this respect, we propose a model (Fig 5) whereby in a healthy subject, LP from the liver and chylomicrons from the gut are present in the vasculature from which they can act locally or extravasate to sites of peripheral infection or injury to contribute to host innate defense. In contrast, critically ill patients may have significant reductions in liver lipoprotein release during the APR. Therefore, although chylomicrons have a limited half-life in circulation [52], even a transient increase in serum lipoproteins could provide much needed defense against bacterial pathogenesis. If such patients are unable to receive nutrition perorally or via the gastrointestinal tract, needed for the formation of chylomicrons, they may have heightened susceptibility to bacterial pathogenesis. Moreover, all critically ill patients may have decreased intestinal motility resulting from anesthesia or opiate analgesia which could also impact a patient’s ability to form chylomicrons and thus their innate defense status [53, 54]. Although the full extent of the contribution of apoB and LP to host innate defense against bacterial pathogens is unknown, these studies suggest that the absence of dietary chylomicrons in critically ill patients could have previously unrecognized implications for their susceptibility to bacterial pathogenesis.
Fig 5. Model of lipoprotein access to sites of infection.
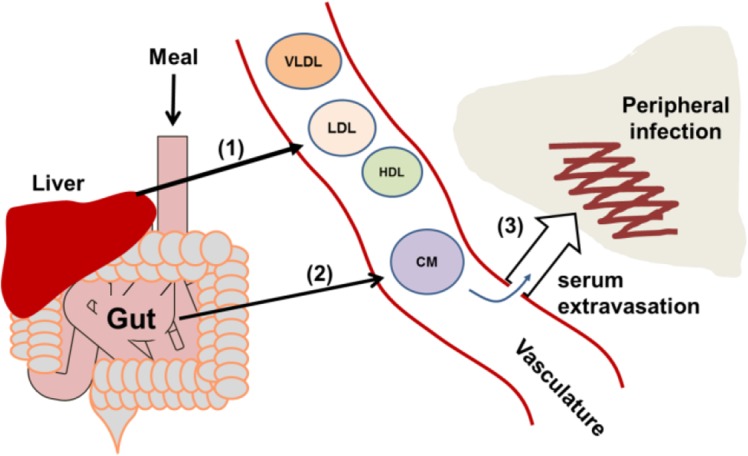
(1) In a healthy subject, the liver releases HDL and VLDL, the latter of which is reduced to LDL by lipase activity. (2) Following oral feeding, enterocytes package dietary lipids and apoB48 into chylomicrons (CM). (3) Lipoproteins are available for host innate defense in the circulation or upon serum extravasation to sites of peripheral infection or inflammation. In a critically ill patient, LP release from the liver is limited as part of the APR and oral feeding may not be possible. The resulting reductions in serum LP levels may negatively impact both peripheral and systemic host innate defense against bacterial pathogens.
While here we describe a previously unrecognized role for apoB48, the importance of the N-terminus of apoB in host innate defense is not surprising considering the evolutionary structural and functional conservation with lipid transport molecules from other species, molecules which also contribute to innate defense [55]. A prime example of this is vitellogenin (Vg) which is produced in the liver of egg laying animals and cleaved to produce lipovitellin (LV) [56, 57]. Vg is a multivalent pattern recognition receptor (PRR) capable of binding LPS, peptidoglycan and LTA through distinct recognition sites [58], and thus is able to bind to Escherichia coli and S. aureus [56, 58, 59]. Vg/LV share greater than 30% sequence similarity with the N-terminus (17%) (Fig 1) of apoB and the crystal structure of LV is often used for structural modeling of apoB [57, 60–62]. Similarly, lipophorins, which are the insect version of lipoproteins, have two non-exchangeable proteins, apolipoprotein I and II (ApoLp-I, ApoLp-II) with homology to apoB [55, 63]. Recently, ApoLp from silkworm hemolymph was shown to bind to S. aureus cell surface LTA and inhibit the SaeRS two-component regulatory system [64, 65]. These findings in both vertebrate and non-vertebrate organisms suggest that LP and apoB-like proteins have an evolutionary role as dual purpose molecules in both lipid transport and host innate defense against bacterial pathogens.
It is important to note that this work does not provide evidence that a high-fat diet (HFD) and pathologically elevated chylomicron levels are beneficial. First, the detrimental effects of a high fat diet on overall health, including systemic inflammation, are well documented (reviewed in [66]). Second, animal studies have shown that mice chronically fed a HFD have decreased survival following intravenous S. aureus challenge compared to mice on a low-fat diet (LFD) [67]. Here, decreased survival was associated with an altered cytokine profile and decreased granulocyte phagocytic function relative to mice on the LFD. Rather, in critically ill patients with already reduced serum apoB100 levels, this work suggests that the lack of dietary chylomicrons could exacerbate their susceptibility to bacterial pathogenesis, particularly under conditions in which agr-signaling contributes to disease.
Materials and Methods
Reagents
Synthetic AIPs were purchased from Biopetide Co., Inc. (San Diego, CA). Human LDL was purchased from Biomedical Technologies, Inc., (Stoughton MA). Superose 6 10/300 prepacked columns were obtained from GE Healthcare Life Sciences. Other reagents were obtained as follows: rabbit anti-mouse apoB polyclonal antibody (LS-C20729, LifeSpan Biosciences); alkaline phosphatase conjugated anti-rabbit IgG (Sigma-Aldrich); nitro-blue tetrazolium and 5-bromo-4-chloro-3′-indolyphosphate (NBT/BCIP, Pierce), Modified Lowry Protein Assay Kit (Thermo Scientific); 3–8% Tris-Acetate protein gel (NuPAGE, Novex; Life Technologies); 4-aminopyrazole-(3,4-D)pyrimidine (4APP), (Sigma-Aldrich).
Bacterial strains and culture
The agr::P3-yfp strains used in this study were generously provided by Dr. Alex Horswill (University of Iowa): AH1677 (agr-I isolate LAC), AH430 (agr-II isolate 502a), AH1747 (agr-III isolate MW2), and AH1872 (agr-IV isolate MN TG) [32]. Bacteria were grown in Trypticase Soy Broth (TSB) and early exponential phase bacteria were prepared as previously described [68]. CFU were determined by plating on TSA with 5% sheep blood (Becton, Dickinson and Company).
ApoB48-lipoprotein particle purification
Serum from 12 week old male ApoE -/- mice on a C57BL/6 background was purchased from The Jackson Laboratory (strain B6.129P2-Apoetm1Unc/J). Serum was centrifuged to remove particulates and ≤ 600 μl aliquots were fractioned by gel filtration chromatography. Briefly, two Superose 6 10/300 prepacked columns were connected in series and equilibrated in PBS, pH 7.4 [69, 70]. One milliliter fractions were collected using an ÄKTA FPLC system with a flow rate of 0.3 ml min-1. The leading protein peak, identified by the absorbance at 280 nm, is the VLDL/apoB48-containing fraction. This fraction was pooled and concentrated.
Protein purity was assessed using SDS/PAGE (3–8% Tris-Acetate gel) and the protein identity was confirmed by Western blot using anti-mouse apoB polyclonal antibody and alkaline phosphatase conjugated anti-rabbit IgG. A FluorChem R system with AlphaView SA software was used for imaging (ProteinSimple, Santa Clara, CA). Protein concentrations of apoB48-LP and human LDL were determined using the modified Lowry Protein Assay. The hydrodynamic radius and particle size distribution of the purified fraction was determined by dynamic light scattering photon correlation spectroscopy (DLS) using a Malvern Zetasizer Nano-ZS at 25°C.
agr::P3-yfp promoter activation assays
Early exponential phase, non-fluorescing bacteria (2x107 ml-1) were incubated in TSB along with the indicated concentration of AIP, lipoprotein or vehicle control. Following 2 hours (unless otherwise noted) incubation with shaking at 37°C, bacteria were washed in PBS with 0.1% Triton X-100 by centrifugation at 1820 x g for 4 min at 4°C. Bacteria were then sonicated, cultured for CFU where indicated, and fixed for 5 minutes at room temperature in 10% buffered formalin phosphate. agr::P3 promoter activation was measured using the mean channel fluorescence (MCF) of induced YFP expression using an Accuri C6 (BD Accuri Cytometers, Ann Arbor, MI). Goat anti-human LDL and goat IgG in PBS were used in experiments for antibody reversal of apoB-mediated agr inhibition.
IC50 values were determined as follows. Fluorescence values were normalized by setting the highest mean value per replicate to 100%, and the lowest mean value per replicate to zero. IC50 values were calculated by fitting a normalized response–variable hill slope function to the data , where h is the Hill slope (GraphPad Prism 5.04). The positive feedback loop in the agr regulon necessitates the use of a variable Hill slope for these calculations.
Growth curves
Growth curves were generated using the CLSI microbroth dilution method, with minor modifications. Briefly, LAC was diluted in Mueller Hinton II broth (MH; BD and Co., Sparks, MD) to 1 x 106 CFU ml-1, confirmed by serial dilution plating on blood agar. Sera or lipoproteins were diluted in MH and mixed 1:1 in a 96-well plate to attain 5 x 105 CFU ml-1 and the indicated concentration of serum or lipoprotein. Plates were incubated in an Infinite 200 plate reader (Tecan, Mannedorf, CH) at 37°C and maximum orbital shaking, for 18 h with OD600 measured every 15 min.
qRT-PCR analysis
Cultures for quantitative RT-PCR were prepared as described above for agr::P3-yfp promoter activation assays. Bacterial RNA was isolated and purified using the Qiagen RNAprotect Bacteria Reagent and RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA generation and qPCR analysis was carried out as previously described (3). RNAIII and hla were quantified relative to 16S RNA. Each experiment was performed in duplicate and samples assayed in triplicate using previously described primers and probes [10].
Alpha-hemolysin (Hla) assays
LAC was cultured in 5 ml TSB for 5 h with DMSO, 50 nM AIP, or 50 nM AIP with 50 nM apoB48-LP. Bacteria were pelleted by centrifugation and supernatants were filtered through a 0.2 μm HT Tuffryn membrane (Pall, Port Washington, NY). Supernatants were diluted to equivalent protein concentrations based on A280 (Nanodrop 1000; Thermo Fisher Scientific Inc., Grand Island, NY). Briefly, supernatant (~9 μg protein) was separated by SDS-PAGE on a Bolt 4–12% BisTris gel (Thermo Fisher Scientific Inc.) and either stained with SYPRO Ruby (Lonza, Rockland, ME) according to manufacturer’s directions, or transferred to a polyvinylidene fluoride membrane. Membranes were blocked for 2 h at 22°C with TBST (20 mM Tris [pH 7.5], 150 mM NaCl, 0.1% Tween 20) and 5% nonfat dry milk. Membranes were probed with anti-Hla antibody (ab15948; Abcam, Cambridge, MA) at a 1:1,000 dilution, washed with TBST, and Hla detected with alkaline phosphatase secondary antibody (ab97127; Abcam). Bands were developed with nitroblue tetrazolium / bromo-chloro-indolyl-phosphate (NBT/BCIP) for 15 min at 22°C. Band relative intensities compared to the + AIP control were measured using ImageJ software [71].
The rabbit red blood cell lysis assay was performed as previously described [72], with minor modifications. Serial two-fold dilutions of filter supernatants were incubated at 37°C for 1 h in a 4% solution of rabbit red blood cells (rRBCs). Lysis was assessed spectrophotometrically at OD650. Data were analyzed by non-linear regression fit to a four-parameter logistic curve and represented as the HA50. The HA50 is 1/dilution number required for 50% rRBC lysis compared to a 0.1% Triton X-100 control.
AIP binding analysis
A Biacore X100 surface plasmon resonance instrument (Biacore Life Sciences, GE Healthcare) was used to assess lipoprotein binding to N-terminally biotinylated AIPs immobilized on a streptavidin chip as previously described [10, 11]. Binding stability is reported as the resonance units (RU) from the binding of the lipoprotein to the immobilized AIP ligand minus the RU from the lipoprotein binding to the unmodified reference surface. Polyclonal rabbit anti–mouse VLDL antibody and rabbit IgG in PBS were used in experiments for antibody reversal of lipoprotein binding to AIP.
Mouse infection model
Animal studies were conducted in strict accordance with recommendations in the Guide for the Care and Use of Laboratory Animals, the Animal Welfare Act and U.S. federal law. The protocol was approved by the University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee (IACUC) (approval number 14-101174-HSC). All infections were performed under anesthesia (inhaled isoflurane) and all efforts were made to minimize suffering. Mice were euthanized by carbon dioxide inhalation. Male, 8–12 week old Nox2 -/- mice on the C57BL/6 background [43] were purchased from Jackson Laboratory (Bar Harbor, ME). Subcutaneous air pouches were created on the backs of the mice as previously described [10, 11, 68, 73–77]. At 48, 24 and 0 hours before infection, the mice were injected i.p. with 4-aminopyrazole-(3,4-D)pyrimidine (4APP) (500 μg) or buffer control. Subcutaneous air pouches were infected with 3x107 CFU of AH1677. Twenty-four hours post infection, weight loss was measured and morbidity scored according to the following clinical scale: appearance: 0–4; natural behavior: 0–3; hydration status (skin pinch test): 0–3; provoked behavior: 0–4. The overall clinical score is the sum of the scores in the 4 categories with a maximum of 14. Pouch lavage and spleen CFU were determined by plating on blood agar. In addition, agr::P3 promoter activation of bacteria from the pouch lavage was measured as MCF as described above.
Statistical analyses
Data are shown as mean ± SEM. Statistical analysis of all in vitro data was performed with the two-tailed Student’s t-test. In vivo data were analyzed by the Mann-Whitney U test for non-parametric data.
Supporting Information
(A) Fractionation of serum from ApoE -/- or wild-type mice by size exclusion chromatography. Major lipoprotein containing peaks are indicated. ApoB48-containing LP were eluted in the VLDL fraction. (B) SDS-PAGE and Western blot of ApoE -/- serum and the pooled VLDL fraction containing highly purified apoB48-LP. (C) Dynamic light scattering (DLS) analysis of purified apoB48-LP showing a unimodal size distribution peak. (D) Bacterial count of AH1677 grown in the presence or absence of 50 nM apoB48-LP from experiment shown in Fig 2D. (E) Growth curves of USA300 isolate LAC grown in broth, or broth with either 100 nM or 0.1 nM purified apoB48-LP added. Data shown are mean ± SEM from at least 2 independent experiments performed in duplicate.
(TIF)
SPR analysis of apoB48-LP binding to immobilized (A) AIP2, (B) AIP3 and (C) AIP4. (D-F) agr::P3 promoter activation assay with the indicated strain at 2x107 CFUs ml-1, 50 nM of the appropriate exogenous AIP and 50 nM apoB48-LP: (D) AH430 (agr-II, 5 hrs); (E) AH1747 (agr-III, 4.5 hrs) and (F) AH1872 (agr-IV, 2 hrs). Results are the mean ± SEM from triplicate experiments. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p≤0.0001.
(TIF)
Acknowledgments
We thank Dr. Richard S. Larson and Dr. Scott W. Burchiel for use of critical equipment. We thank Seth Daly for expert technical assistant and contributions to preparation of this manuscript, and Moriah Castleman and Brett Manifold-Wheeler for critical reading of the manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by National Institutes of Health grant AI091917 to PRH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Daniels TF, Killinger KM, Michal JJ Jr RWW, Jiang Z. Lipoproteins, cholesterol homeostasis and cardiac health. Int J Biol Sci. 2009;5(5):474–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han R. Plasma lipoproteins are important components of the immune system. Microbiol Immunol. 2010;54(4):246–53. 10.1111/j.1348-0421.2010.00203.x PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 3. Feingold KR, Grunfeld C. Lipids: a key player in the battle between the host and microorganisms. J Lipid Res. 2012;53(12):2487–9. 10.1194/jlr.E033407 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45(7):1169–96. 10.1194/jlr.R300019-JLR200 PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 5. Femling JK, West SD, Hauswald EK, Gresham HD, Hall PR. Nosocomial infections after severe trauma are associated with lower apolipoproteins B and AII. The Journal of Trauma and Acute Care Surgery. 2013;74(4):1067–73. 10.1097/TA.0b013e3182826be0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vyroubal P, Chiarla C, Giovannini I, Hyspler R, Ticha A, Hrnciarikova D, et al. Hypocholesterolemia in clinically serious conditions–review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008;152(2):181–9. [DOI] [PubMed] [Google Scholar]
- 7. Leardi S, Altilia F, Delmonaco S, Cianca G, Pietroletti R, Simi M. Blood levels of cholesterol and postoperative septic complications. Ann Ital Chir. 2000;71(2):233–7. [PubMed] [Google Scholar]
- 8. Kattelmann KK, Hise M, Russell M, Charney P, Stokes M, Compher C. Preliminary evidence for a medical nutrition therapy protocol: enteral feedings for critically ill patients. J Am Diet Assoc. 2006;106(8):1226–41. 10.1016/j.jada.2006.05.320 PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 9. Heyland DK, Dhaliwal R, Drover JW, Gramlich L, Dodek P, Canadian Critical Care Clinical Practice Guidelines C. Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients. JPEN J Parenter Enteral Nutr. 2003;27(5):355–73. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 10. Hall PR, Elmore BO, Spang CH, Alexander SM, Manifold-Wheeler BC, Castleman MJ, et al. Nox2 Modification of LDL Is Essential for Optimal Apolipoprotein B-mediated Control of agr Type III Staphylococcus aureus Quorum-sensing. PLoS Path. 2013;9(2). ARTN e1003166 10.1371/journal.ppat.1003166 PubMed PMID: WOS:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peterson MM, Mack JL, Hall PR, Alsup AA, Alexander SM, Sully EK, et al. Apolipoprotein B Is an innate barrier against invasive Staphylococcus aureus infection. Cell Host Microbe. 2008;4(6):555–66. 10.1016/j.chom.2008.10.001 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Novick RP, Geisinger E. Quorum sensing in staphylococci. Annu Rev Genet. 2008;42:541–64. 10.1146/annurev.genet.42.110807.091640 PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 13. Malachowa N, Whitney AR, Kobayashi SD, Sturdevant DE, Kennedy AD, Braughton KR, et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One. 2011;6(4):e18617 10.1371/journal.pone.0018617 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palazzolo-Ballance AM, Reniere ML, Braughton KR, Sturdevant DE, Otto M, Kreiswirth BN, et al. Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus . J Immunol. 2008;180(1):500–9. [DOI] [PubMed] [Google Scholar]
- 15. McAdow M, DeDent AC, Emolo C, Cheng AG, Kreiswirth BN, Missiakas DM, et al. Coagulases as determinants of protective immune responses against Staphylococcus aureus . Infect Immun. 2012;80(10):3389–98. 10.1128/IAI.00562-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. 2010;6(8):e1001036 10.1371/journal.ppat.1001036 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui J, et al. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe. 2007;1(1934–6069; 2):109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith EJ, Visai L, Kerrigan SW, Speziale P, Foster TJ. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus . Infect Immun. 2011;79(9):3801–9. 10.1128/IAI.05075-11 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheung AL, Bayer AS, Zhang G, Gresham H, Xiong YQ. Regulation of virulence determinants in vitro and in vivo in Staphylococcus aureus . FEMS Immunol Med Microbiol. 2004;40(0928–8244; 1):1–9. [DOI] [PubMed] [Google Scholar]
- 20. Yarwood JM, McCormick JK, Paustian ML, Kapur V, Schlievert PM. Repression of the Staphylococcus aureus accessory gene regulator in serum and in vivo. J Bacteriol. 2002;184(0021–9193; 4):1095–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koprivnjak T, Weidenmaier C, Peschel A, Weiss JP. Wall teichoic acid deficiency in Staphylococcus aureus confers selective resistance to mammalian group IIA phospholipase A(2) and human beta-defensin 3. Infect Immun. 2008;76(5):2169–76. 10.1128/IAI.01705-07 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun. 2010;2(6):546–59. 10.1159/000319855 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. George EA, Muir TW. Molecular mechanisms of agr quorum sensing in virulent staphylococci. Chembiochem. 2007;8(1439–4227; 8):847–55. [DOI] [PubMed] [Google Scholar]
- 24. Diep BA, Otto M. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 2008;16(8):361–9. 10.1016/j.tim.2008.05.002 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheung GY, Wang R, Khan BA, Sturdevant DE, Otto M. Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect Immun. 2011;79(5):1927–35. 10.1128/IAI.00046-11 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. Peptide signaling in the staphylococci. Chem Rev. 2011;111(1):117–51. 10.1021/cr100370n PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakamuta M, Chang BH, Zsigmond E, Kobayashi K, Lei H, Ishida BY, et al. Complete phenotypic characterization of apobec-1 knockout mice with a wild-type genetic background and a human apolipoprotein B transgenic background, and restoration of apolipoprotein B mRNA editing by somatic gene transfer of Apobec-1. J Biol Chem. 1996;271(42):25981–8. [DOI] [PubMed] [Google Scholar]
- 28. Tennyson GE, Sabatos CA, Higuchi K, Meglin N, Jr HBB. Expression of apolipoprotein B mRNAs encoding higher- and lower-molecular weight isoproteins in rat liver and intestine. Proc Natl Acad Sci U S A. 1989;86(2):500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: tests of the hypothesis in "knockout" mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci U S A. 1994;91(10):4431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bergeron N, Kotite L, Verges M, Blanche P, Hamilton RL, Krauss RM, et al. Lamellar lipoproteins uniquely contribute to hyperlipidemia in mice doubly deficient in apolipoprotein E and hepatic lipase. Proc Natl Acad Sci U S A. 1998;95(26):15647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71(2):343–53. [DOI] [PubMed] [Google Scholar]
- 32. Malone CL, Boles BR, Lauderdale KJ, Thoendel M, Kavanaugh JS, Horswill AR. Fluorescent reporters for Staphylococcus aureus . J Microbiol Methods. 2009;77(3):251–60. 10.1016/j.mimet.2009.02.011 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, et al. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J Infect Dis. 2011;204(6):937–41. 10.1093/infdis/jir441 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, et al. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis. 2010;202(7):1050–8. 10.1086/656043 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ragle BE, Wardenburg JB. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect Immun. 2009;77(7):2712–8. 10.1128/IAI.00115-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17(10):1310–4. 10.1038/nm.2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inoshima N, Wang Y, Bubeck Wardenburg J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J Invest Dermatol. 2012;132(5):1513–6. 10.1038/jid.2011.462 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilke GA, Wardenburg JB. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A. 2010;107(30):13473–8. 10.1073/pnas.1001815107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sampedro GR, DeDent AC, Becker RE, Berube BJ, Gebhardt MJ, Cao H, et al. Targeting Staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J Infect Dis. 2014;210(7):1012–8. 10.1093/infdis/jiu223 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lyon GJ, Wright JS, Muir TW, Novick RP. Key determinants of receptor activation in the agr autoinducing peptides of Staphylococcus aureus . Biochemistry (John Wiley & Sons). 2002;41(0006–2960; 31):10095–104. [DOI] [PubMed] [Google Scholar]
- 41. Mounkes LC, Zhong W, Silva HVd, Handumrongkul C, Desai B, Tse E, et al. Evaluation of the role of lipoprotein metabolism genes in systemic cationic liposome-mediated gene transfer in vivo. Hum Gene Ther. 2001;12(1043–0342; 16):1939–54. [DOI] [PubMed] [Google Scholar]
- 42. Shiff TS, Roheim PS, Eder HA. Effects of high sucrose diets and 4-aminopyrazolopyrimidine on serum lipids and lipoproteins in the rat. J Lipid Res. 1971;12(5):596–603. [PubMed] [Google Scholar]
- 43. Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9(1061–4036; 2):202–9. [DOI] [PubMed] [Google Scholar]
- 44. Daly SM, Elmore BO, Kavanaugh JS, Triplett KD, Figueroa M, Raja HA, et al. omega-Hydroxyemodin Limits Staphylococcus aureus Quorum Sensing-Mediated Pathogenesis and Inflammation. Antimicrob Agents Chemother. 2015;59(4):2223–35. 10.1128/AAC.04564-14 PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, et al. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 2014;10(6):e1004174 10.1371/journal.ppat.1004174 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Collaboration TERF. Major Lipids, Apolipoproteins and Risk of Vascular Disease. JAMA. 2009;302:1993–2000. 10.1001/jama.2009.1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han C, Lin Y, Shan G, Zhang Z, Sun X, Wang Z, et al. Plasma concentration of malaria parasite-derived macrophage migration inhibitory factor in uncomplicated malaria patients correlates with parasitemia and disease severity. Clin Vaccine Immunol. 2010;17(10):1524–32. 10.1128/CVI.00149-10 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhakdi S, Tranum-Jensen J, Utermann G, Fussle R. Binding and partial inactivation of Staphylococcus aureus alpha-toxin by human plasma low density lipoprotein. J Biol Chem. 1983;258(9):5899–904. [PubMed] [Google Scholar]
- 49. Sigel S, Bunk S, Meergans T, Doninger B, Stich K, Stulnig T, et al. Apolipoprotein B100 is a suppressor of Staphylococcus aureus-induced innate immune responses in humans and mice. Eur J Immunol. 2012;42(11):2983–9. 10.1002/eji.201242564 [DOI] [PubMed] [Google Scholar]
- 50. Surewaard BG, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA. Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog. 2012;8(3):e1002606 10.1371/journal.ppat.1002606 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harris HW, Grunfeld C, Feingold KR, Rapp JH. Human very low density lipoproteins and chylomicrons can protect against endotoxin-induced death in mice. J Clin Invest. 1990;86(3):696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Park Y, Damron BD, Miles JM, Harris WS. Measurement of human chylomicron triglyceride clearance with a labeled commercial lipid emulsion. Lipids. 2001;36(2):115–20. [DOI] [PubMed] [Google Scholar]
- 53.Leslie JB, Viscusi ER, Jr JVP, Panchal SJ. Anesthetic Routines: The Anesthesiologist's Role in GI Recovery and Postoperative Ileus. Adv Prev Med. 2011;2011. [DOI] [PMC free article] [PubMed]
- 54. Bueno L, Fioramonti J. Action of opiates on gastrointestinal function. Baillieres Clin Gastroenterol. 1988;2(1):123–39. [DOI] [PubMed] [Google Scholar]
- 55. Horst DJVd Roosendaal SD, Rodenburg KW. Circulatory lipid transport: lipoprotein assembly and function from an evolutionary perspective. Mol Cell Biochem. 2009;326(1–2):105–19. 10.1007/s11010-009-0068-7 [DOI] [PubMed] [Google Scholar]
- 56. Zhang S, Wang S, Li H, Li L. Vitellogenin, a multivalent sensor and an antimicrobial effector. The International Journal of Biochemistry & Cell Biology. 2011;43(3):303–5. [DOI] [PubMed] [Google Scholar]
- 57. Thompson JR, Banaszak LJ. Lipid-protein interactions in lipovitellin. Biochemistry. 2002;41(30):9398–409. [DOI] [PubMed] [Google Scholar]
- 58. Li Z, Zhang S, Liu Q. Vitellogenin functions as a multivalent pattern recognition receptor with an opsonic activity. PLoS One. 2008;3(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tong Z, Li L, Pawar R, Zhang S. Vitellogenin is an acute phase protein with bacterial-binding and inhibiting activities. Immunobiology. 2010;215(11):898–902. 10.1016/j.imbio.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 60. Mann CJ, Anderson TA, Read J, Chester SA, Harrison GB, Kochl S, et al. The structure of vitellogenin provides a molecular model for the assembly and secretion of atherogenic lipoproteins. J Mol Biol. 1999;285(1):391–408. [DOI] [PubMed] [Google Scholar]
- 61. Segrest JP, Jones MK, Dashti N. N-terminal domain of apolipoprotein B has structural homology to lipovitellin and microsomal triglyceride transfer protein: a "lipid pocket" model for self-assembly of apob-containing lipoprotein particles. J Lipid Res. 1999;40(8):1401–16. [PubMed] [Google Scholar]
- 62. Raag R, Appelt K, Xuong NH, Banaszak L. Structure of the lamprey yolk lipid-protein complex lipovitellin-phosvitin at 2.8 A resolution. J Mol Biol. 1988;200(3):553–69. [DOI] [PubMed] [Google Scholar]
- 63. Smolenaars MM, Kasperaitis MA, Richardson PE, Rodenburg KW, Horst DJVd. Biosynthesis and secretion of insect lipoprotein: involvement of furin in cleavage of the apoB homolog, apolipophorin-II/I. J Lipid Res. 2005;46(3):412–21. [DOI] [PubMed] [Google Scholar]
- 64. Hanada Y, Sekimizu K, Kaito C. Silkworm apolipophorin protein inhibits Staphylococcus aureus virulence. J Biol Chem. 2011;286(45):39360–9. 10.1074/jbc.M111.278416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Omae Y, Hanada Y, Sekimizu K, Kaito C. Silkworm apolipophorin protein inhibits hemolysin gene expression of Staphylococcus aureus via binding to cell surface lipoteichoic acids. J Biol Chem. 2013;288(35):25542–50. 10.1074/jbc.M113.495051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Huang EY, Devkota S, Moscoso D, Chang EB, Leone VA. The role of diet in triggering human inflammatory disorders in the modern age. Microbes and Infection. 2013;15(12):765–74. 10.1016/j.micinf.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 67. Strandberg L, Verdrengh M, Enge M, Andersson N, Amu S, Onnheim K, et al. Mice chronically fed high-fat diet have increased mortality and disturbed immune response in sepsis. PLoS One. 2009;4(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rothfork JM, Dessus-Babus S, Wamel WJV, Cheung AL, Gresham HD. Fibrinogen depletion attenuates Staphyloccocus aureus infection by preventing density-dependent virulence gene up-regulation. J Immunol. 2003;171(0022–1767; 10):5389–95. [DOI] [PubMed] [Google Scholar]
- 69. Gerdes LU, Gerdes C, Klausen IC, Faergeman O. Generation of analytic plasma lipoprotein profiles using two prepacked superose 6B columns. Clin Chim Acta. 1992;205(1–2):1–9. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 70. Garber DW, Kulkarni KR, Anantharamaiah GM. A sensitive and convenient method for lipoprotein profile analysis of individual mouse plasma samples. J Lipid Res. 2000;41(6):1020–6. PubMed PMID: . [PubMed] [Google Scholar]
- 71. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bernheimer AW. Assay of hemolytic toxins. Methods Enzymol. 1988;165(0076–6879):213–7. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 73. Rothfork JM, Timmins GS, Harris MN, Chen X, Lusis AJ, Otto M, et al. Inactivation of a bacterial virulence pheromone by phagocyte-derived oxidants: new role for the NADPH oxidase in host defense. Proc Natl Acad Sci U S A. 2004;101(38):13867–72. 10.1073/pnas.0402996101 PubMed PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yoshikawa Y, Morikawa K, Nonaka M, Torii I. Effect of arbekacin on a methicillin-resistant Staphylococcus aureus-induced biofilm in a rat model. J Infect Chemother. 2004;10(1341–321; 5):268–73. [DOI] [PubMed] [Google Scholar]
- 75. Sin YM, Wong MK. Effect of sodium aurothiomalate on carrageenan induced inflammation of the air pouch in mice. Ann Rheum Dis. 1992;51(1):112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Brauner E, Gorbach S, Davey P. Comparative study of clindamycin, imipenem, oxacillin and vancomycin in the infected granuloma pouch model. J Antimicrob Chemother. 1989;23(6):891–8. [DOI] [PubMed] [Google Scholar]
- 77. Yasuda H, Ajiki Y, Koga T, Kawada H, Yokota T. Interaction between biofilms formed by Pseudomonas aeruginosa and clarithromycin. Antimicrob Agents Chemother. 1993;37(9):1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Fractionation of serum from ApoE -/- or wild-type mice by size exclusion chromatography. Major lipoprotein containing peaks are indicated. ApoB48-containing LP were eluted in the VLDL fraction. (B) SDS-PAGE and Western blot of ApoE -/- serum and the pooled VLDL fraction containing highly purified apoB48-LP. (C) Dynamic light scattering (DLS) analysis of purified apoB48-LP showing a unimodal size distribution peak. (D) Bacterial count of AH1677 grown in the presence or absence of 50 nM apoB48-LP from experiment shown in Fig 2D. (E) Growth curves of USA300 isolate LAC grown in broth, or broth with either 100 nM or 0.1 nM purified apoB48-LP added. Data shown are mean ± SEM from at least 2 independent experiments performed in duplicate.
(TIF)
SPR analysis of apoB48-LP binding to immobilized (A) AIP2, (B) AIP3 and (C) AIP4. (D-F) agr::P3 promoter activation assay with the indicated strain at 2x107 CFUs ml-1, 50 nM of the appropriate exogenous AIP and 50 nM apoB48-LP: (D) AH430 (agr-II, 5 hrs); (E) AH1747 (agr-III, 4.5 hrs) and (F) AH1872 (agr-IV, 2 hrs). Results are the mean ± SEM from triplicate experiments. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p≤0.0001.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.