

Abstract
Serum glucocorticoid kinase 1 (SGK1) has been shown to be protective in models of Parkinson's disease, but the details by which it confers benefit is unknown. The current study was designed to investigate the details by which SGK1 confers neuroprotection. To do this we employed a cellular neurodegeneration model to investigate c-Jun N-terminal kinase (JNK) signaling and endoplasmic reticulum (ER) stress induced by 6-hydroxydopamine. SGK1-expressing adenovirus was created and used to overexpress SGK1 in SH-SY5Y cells, and dexamethasone was used to increase endogenous expression of SGK1. Oxidative stress, mitochondrial dysfunction, and cell death were monitored to test the protective effect of SGK1. To investigate the effect of SGK1 overexpression in vivo, SGK1-expressing adenovirus was injected into the striatum of mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, and protection of dopaminergic neurons was quantitatively assessed by tyrosine hydroxylase immunohistochemistry. SGK1 overexpression was found to decrease reactive oxygen species generation, alleviate mitochondrial dysfunction, and rescue cell death in vitro and in vivo by inactivating mitogen-activated protein kinase kinase 4 (MKK4), JNK, and glycogen synthase kinase 3β (GSK3β) and thereby decreasing ER and oxidative stress. These results suggest that therapeutic strategies for activation of SGK1 may have the potential to be neuroprotective by deactivating the JNK and GSK3β pathways.
INTRODUCTION
Serum- and glucocorticoid-inducible kinase 1 (SGK1) belongs to the AGC family of kinases and has been shown to have various cellular functions, including the promotion of cell survival (1–3). SGK1 is activated by insulin and growth factors via phosphoinositide 3-kinase (PI3K), 3-phosphoinositide-dependent kinase 1 (PDK1), and mammalian target of rapamycin complex 2 (mTORC2) (4, 5). SGK1 shares its functions and some substrates with another kinase from the AGC family, protein kinase B (PKB/Akt). Akt, like SGK1, has been shown to mediate cell survival through various signaling cascades and gets activated by a wide range of extracellular stimuli (6). SGK1 lacks the pleckstrin homology (PH) domain that tethers Akt to the plasma membrane, making SGK1 more accessible to cytosolic and nuclear sites and thereby providing it with cellular functions and substrates that do not overlap those of Akt (1, 6). SGK1 plays a protective role in oxidative stress conditions as small interfering RNA (siRNA) knockdown of SGK1 has shown an increase in oxidative stress-induced cell death in HEK293 cells (7). Oxidative stress is a hallmark of neurodegenerative disorders, such as Parkinson's disease (PD), Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD) (8). In a study published in 2005 by Schoenebeck et al., upregulation of SGK1 was seen in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxin model and in a transgenic model of ALS (SOD1-G93A), and protection from cell death was observed for animals treated with dexamethasone (Dex), which is known to upregulate SGK1 expression, prior to treatment with the neurotoxin (1). In another study, analysis of cortical tissue from patients with severe Alzheimer's disease (AD) showed an increase not only of SGK1 activity but also of its substrates, N-myc downstream-regulated gene 1 (NDRG-1) and forkhead box 3a protein (FoxO3a) (9–12). SGK1 shares the latter substrate with Akt. Two recent studies have shown a neuroprotective role for SGK1 in a 6-hydroxydopamine (6-OHDA) neurotoxin mouse model and in an ischemia reperfusion rat model (13, 14). These findings underscore the importance of SGK1 in neurodegeneration, but the details of signaling molecules that contribute to neuroprotection are not well defined.
The c-Jun N-terminal kinases (JNK) are mitogen-activated protein (MAP) kinases responsive to physiological and environmental stress. JNK activation has been observed in various neurodegenerative disorders where the JNK signaling cascade has been shown to cause neuronal cell death (15–19). Importantly, postmortem studies, along with MPTP and 6-OHDA animal models of neurodegeneration, showed an important role for JNK in the disease pathogenesis (15, 16, 19). There is very little literature which links JNK and SGK1. In 2007, Kim et al. utilized HEK293 cells to show by Western analysis that SGK1-mediated phosphorylation of mitogen-activated protein kinase kinase 4 (MKK4) on serine 80 results in abrogation of MKK4 binding to JNK and thereby inhibits the JNK signaling cascade (20). In 2011, Xu et al. utilized primary cerebellar granular neurons (CGNs) from compound JNK-deficient mice to identify JNK as a negative regulator of FoxO-dependent autophagy in neurons (21). FoxO activation in neurons leads to the expression of proapoptotic BH3-only protein (Bim). Bim gets phosphorylated by JNK, which leads to its dissociation from prosurvival protein Mcl-1, leading to apoptosis (21). SGK1, in parallel with Akt, has also been shown to negatively regulate the activation and proapoptotic function of FoxO proteins (12). Another cellular event where SGK1 and JNK pathways converge involves an important cellular kinase, glycogen synthase kinase 3β (GSK3β). SGK1 has been shown to phosphorylate and inhibit activity of GSK3β in mouse dendritic cells (22). In a separate proapoptotic pathway, JNK phosphorylates Mcl-1 and primes it for phosphorylation by GSK3β, which ultimately leads to the proteosomal degradation of Mcl-1 (23). Therefore, these studies suggest that cross talk between JNK and SGK1/Akt signaling cascades would define cellular fate under stress conditions. These intriguing observations led us to hypothesize that SGK1 activation may indeed exert its neuroprotective effects via impacting the JNK pathway. To test this hypothesis we established 6-OHDA cell culture models and studied the impact of overexpression of SGK1 in an MPTP animal model of neurodegeneration to establish the link between SGK1 and JNK and to understand the mechanism by which SGK1 may exert its neuroprotective effect. The main findings from our study showed that SGK1 overexpression rescued neurotoxin-induced mitochondrial dysfunction and subsequent cell death. It did this by regulating the expression of endoplasmic reticulum (ER) and mitochondrial stress marker proteins and by inactivating JNK and GSK3β.
MATERIALS AND METHODS
Kinase-Glo phosphorylation assay.
A recombinant substrate, inactive MKK4 (Millipore), was diluted to various concentrations in JNK activity buffer (25 mM HEPES, pH 7.4, 10 mM MgCl2, 2 mM dithiothreitol [DTT], 1 mg/ml bovine serum albumin [BSA], and 1 μM ATP). The reaction was initiated with the addition of 0.75 μM active SGK1 (GeneTex). The reaction mixture was incubated at 30°C for 60 min. The reaction was stopped by the addition of 50 mM EDTA. The reaction product was combined with the Kinase-Glo reagent (Promega) at a 1:1 ratio and then incubated at room temperature (RT) for 10 min. Luminescence was monitored on a Spectromax M5e plate reader (Molecular Devices) with an integration of 500 ms. ATP quantitation was determined based on values interpolated onto an ATP standard curve. Data are reported as percent SGK1 activity based on uninhibited, active SGK1/substrate phosphorylation.
Cell culture and treatments.
SH-SY5Y cells (ATCC) were grown under normal cell culture conditions (37°C and 5% CO2) in Dulbecco's modified Eagle's medium (DMEM)–F-12 medium (Invitrogen) supplemented with 10% fetal bovine serum and penicillin-streptomycin. To ensure that the cells were actively growing, only cells at ∼80% confluence and between passages 5 and 15 were used in our experiments. Cells were treated with 35 μM 6-OHDA solubilized in dimethyl sulfoxide (DMSO) for 5 h for the different analyses. Cells were treated with 10 μM dexamethasone for 24 h prior to neurotoxin treatment to induce SGK1 expression. JNK inhibitor, SR-3306, and Akt inhibitor, MK-2206 (Selleck Chemicals), were added to cells 1 h before the treatment with the neurotoxin.
Adenovirus-mediated SGK1 expression.
Adenovirus-mediated overexpression of SGK1 was achieved using adenovirus particles purchased from Vector Biolabs. The viral backbone was adenoviral type 5 (dE1/dE3) with a synapsin promoter and yellow fluorescent protein (YFP) tag to visualize the cells/tissue after the adenoviral infection. SH-SY5Y cells were infected at a multiplicity of infection (MOI) of 5.
Mitochondrial superoxide production.
Mitochondrial superoxide generation was monitored by MitoSOX red (Invitrogen) fluorescence. SH-SY5Y cells (50,000 cells/well) were seeded into a black-wall, clear-bottom 96-well plate. After treatments, cells were stained with 2.5 μM MitoSOX red for 25 min under growth conditions. The cells were washed twice in Hanks' buffered salt solution (HBSS) and placed in prewarmed HBSS for imaging. MitoSOX red fluorescence was detected by exciting the fluorophore at 510 nm and monitoring the emission at 580 nm on a SpectraMax M5e plate reader (Molecular Devices). Mitochondrial superoxide was normalized to cell abundance by staining the cells with Hoechst 33342 (excitation, 350 nm; emission, 450 nm). Rotenone was used as a positive control for superoxide generation. Cells were treated with 100 nM rotenone for 24 h to generate a quality signal.
Mitochondrial membrane potential.
Mitochondrial membrane potential was monitored using a JC-1 dye kit (Cayman Chemical). Cells were seeded in a 96-well black-wall, clear-bottom plate as described in MitoSOX staining above. The cells were then stained with 5 mM JC-1 for 20 min as indicated in the manufacturer's protocol. The cells were washed three times in HBSS and covered in prewarmed HBSS for fluorescence measurements. The green JC-1 species were detected by excitation at 460 nm and monitoring emission at 488 nm. JC-1 staining was normalized to Hoechst 33342 staining.
Cell viability.
Cell viability of SH-SY5Y cells was monitored by an MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (Cayman Chemical). Cells (50,000 cells/well) were seeded in a 96-well plate (clear bottom) and treated as described in the text. At the culmination of each treatment, the cells were treated with the MTT reagent. Absorbance was monitored in a SpectraMax M5e plate reader (Molecular Devices).
Cell lysis and Western blot analysis.
SH-SY5Y (1 × 105) cells were plated on a six-well plate. To acquire protein for Western blot analysis after an experiment, cells were washed twice in phosphate-buffered saline (PBS). Cell lysis buffer (Cell Signaling Technology) supplemented with protease inhibitors (1 mm phenylmethylsulfonyl fluoride [PMSF] and 1× Halt protease inhibitor cocktail [Thermo Scientific]) and 1× phosphatase inhibitors (Halt phosphatase inhibitor cocktail; Thermo Scientific) was added to the cells. Cells were incubated in lysis buffer at 4°C for 5 min with gentle rocking. Cells were scraped from the culture surface and transferred to a microcentrifuge tube. Following a 2-min incubation on ice, the cells were disrupted by brief sonication. The cell lysate was centrifuged at 14,000 × g for 15 min to remove cellular debris. The concentration of protein in the supernatant was quantified by bicinchoninic acid (BCA) analysis using the Pierce kit protocol. For Western blot analysis, proteins (25 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes for 1.5 h. Membranes were incubated with blocking buffer (SuperBlock blocking buffer; Thermo Scientific) for 1 h at room temperature or overnight at 4°C. After 2 h of incubation with a secondary antibody linked to horseradish peroxidase (HRP), membranes were visualized using an ECL detection system (Thermo Scientific). The antibodies used for Western blot analysis were the following (all from Cell Signaling Technology): phospho-JNK (catalog number 9255), phospho-c-Jun (3270), Hsp70 (4876), MKK4 (3346), phospho-MKK4 (Ser257/Thr261) (9156), phospho-MKK4 (Ser80) (9155), phospho-MKK7 (4171), phospho-Akt (4060), SGK1 (12103), phospho-SGK1 (5599), BiP (3183) α-tubulin (2125), SOD1 (4266), secondary anti-rabbit IgG, HRP linked (7074), and secondary anti-mouse IgG, HRP linked (7076). The primary antibodies were used at a dilution of 1:1,000, according to the manufacturer's instructions. The secondary antibody was used at a dilution of 1:2,000.
Replicates and statistics.
For each Western blot analysis, three biological replicates were acquired. For cell viability assays, mitochondrial superoxide detection, and mitochondrial membrane potential, three sample replicates were measured for the three biological replicates obtained for each treatment. Finally, all statistical analysis was performed using a Student t test of significance. A P value of 0.05 was used to determine significance.
Reverse transcription-PCR (RT-PCR) analysis of SH-SY5Y cells.
Total RNA was isolated from cells using an RNeasy minikit (Qiagen). One microgram of RNA was reverse transcribed using an Omniscript reverse transcription kit (Qiagen) using the reverse primer 5′-ACGAACTACCCCGACCGTAACCGCCA.
Adenovirus injections in mice.
Eleven-week-old male C57BL/6J mice (Jackson Laboratories) weighing in the range of 25 to 30 g were used. Mice were acclimated for 1 week prior to initiation of study. Mice were anesthetized via an intraperitoneal injection of ketamine and xylazine and placed into a stereotaxic frame (Stoelting) with the head positioned flat. Unilateral injections of adenovirus-expressing SGK1 (AdV-SGK1) were made into the right striatum at 1.5 mm, 2.0 mm, and 2.5 mm mediolateral from the bregma, with two depths at 3.6 mm and 3.0 mm dorsoventral below the dura, for a total of six points. A volume of 1 μl of virus at a concentration of 1 × 1010 virus particles (VP) or a corresponding amount of saline alone was injected with a 30-gauge needle and an automatic pump (Stoelting) at each coordinate at a rate of 0.5 μl/min for a total of 6 μl (n = 17 for both saline and SGK1 injections). The needle was left in place for 3 min following each injection before being withdrawn to prevent backflow. Gene expression was allowed to proceed for 21 days until the mice were challenged with an acute MPTP lesion-inducing regimen. All animal experiments were approved by the Scripps Florida IACUC.
Immunohistochemistry.
Animals were sacrificed by an overdose of ketamine and xylazine, followed by cardiac perfusion with 0.9% saline and then with 4% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4. The brains were removed and further postfixed in 4% paraformaldehyde at 4°C for 1 day, followed by cryoprotection in 30% sucrose for 3 to 4 days. Brains were embedded and frozen in optimal cutting temperature compound and stored frozen at −80°C until sectioning. Symmetrical 40-μm-thick sections were cut on a cryostat (CM3050S; Leica) between approximately −2.46 and −4.36 mm relative to the bregma for approximately 48 sections. Every other section (∼21 to 24 sections) was processed for immunohistochemistry. Free-floating sections were pretreated in 0.3% H2O2 in Tris-buffered saline (TBS) for 15 min and in blocking solution (4% bovine serum albumin [BSA] in TBS containing 0.1% Triton X-100) for 1 h at room temperature. In between steps, sections were washed three times for 15 min in TBS with 0.1% Triton X-100. For proper identification of the substantia nigra pars compacta (SNpc), all sections were incubated overnight at 4°C with polyclonal rabbit anti-tyrosine hydroxylase (TH; 1:500) (ab112; Abcam) in 4% BSA in TBS containing 0.1% Triton X-100. Sections were then washed with TBS containing 0.1% Triton X-100 and incubated with anti-rabbit secondary antibody (1:500; Alexa Fluor 488) in 4% BSA in TBS containing 0.1% Triton X-100 for 2 h at room temperature in the dark. In between steps, sections were washed three times for 15 min in TBS. Sections were mounted on Superfrost Plus slides, and a drop of Fluoroshield mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI; 1:4) was applied to each section. Slides were coverslipped, sealed with nail polish, and stored at 4°C.
Detection of SGK1, TH, and phospho-c-Jun (p-c-Jun).
SGK1 expression in the SNpc was recognized via a yellow fluorescent tag (Ad-Syn-hSGK1_v2-YFP; Vector Biolabs) in one section (bregma −3.0 mm) from each group used for immunohistochemical staining of TH neurons for stereological counting. A YFP filter (U-MF2; Olympus) was used to detect SGK1 expression at the same exposure time for each group. A Texas Red filter was used for detection of TH in the same section used to detect SGK1 in a similar manner.
From a separate set of mice unilaterally injected with SGK1-expressing adenovirus, 20-μm sections from the SNpc (bregma −3.0 mm) from each group were subjected to double immunofluorescence staining for p-c-Jun and TH. Free-floating sections were pretreated in 0.1% H2O2 in Tris-buffered saline (TBS) for 5 min and in blocking solution (4% bovine serum albumin [BSA] in TBS containing 0.1% Triton X-100) for 1 h at room temperature. In between steps, sections were washed three times for 15 min in TBS with 0.1% Triton X-100. Sections were incubated overnight at 4°C with polyclonal sheep anti-tyrosine hydroxylase (1:500) (ab113; Abcam) and rabbit anti-p-c-Jun (Ser73) (1:100) (9164; Cell Signaling Technology) in 2% BSA in TBS containing 0.1% Triton X-100. Sections were then washed with TBS containing 0.1% Triton X-100 and incubated with anti-sheep and anti-rabbit secondary antibodies (1:500) (A11015 and A11012, respectively; Life Technologies) in 2% BSA in TBS containing 0.1% Triton X-100 for 2 h at room temperature in the dark. Sections were washed three times for 15 min in TBS and then mounted on Superfrost Plus slides for imaging.
MPTP challenge and stereological counting of TH+ dopaminergic cells.
The acute MPTP lesion-inducing regimen, immunostaining of TH, stereological evaluation of TH-positive (TH+) cells in the SNpc, and safety precautions as outlined by Hunot et al. and Jackson-Lewis and Przedborski were followed (16, 24). Mice were injected intraperitoneally (i.p.) four times at 2-h intervals over 1 day with 18 mg/kg MPTP-HCl (Sigma) dissolved in 0.9% saline or a corresponding volume of saline alone. Mice were sacrificed 7 days after the MPTP administration and evaluated for counting of SNpc dopaminergic neurons (n = 4 for vehicle group for MPTP [saline plus saline], n = 4 for vehicle group for SGK1 [SGK1 plus saline], n = 13 for MPTP group [saline plus MPTP], and n = 13 for SGK1 group [SGK1 plus MPTP]). Beginning at bregma −2.70 mm, brains were sectioned at 40 μm for 48 sections, with every other section discarded up to bregma −4.04 mm. Of the 28 sections, 12 sections between bregma −2.80 and −3.80 mm were counted. Unbiased stereological counting by a blinded investigator of TH-positive cells in the SNpc was quantitated (standard error of the mean [SEM]) using the Stereo Investigator (version 8.0) software program (MicroBrightField). Statistical analysis of the TH counting data was performed utilizing a Mann-Whitney U test.
RESULTS
SGK1 is upregulated in response to neurotoxic stress, and its overexpression rescues 6-OHDA-induced mitochondrial dysfunction and cell death in SH-SY5Y cells.
In order to assess whether the expression profile of SGK1 was altered in response to induced neurodegeneration, SH-SY5Y neuroblastoma cells were treated with 35 μM 6-OHDA for various amounts of time to induce neurotoxic stress and death. Treatment with 6-OHDA showed increases in SGK1 mRNA levels over time (Fig. 1A). Western immunoblot analysis of SH-SY5Y cells treated with 6-OHDA showed an increase in endogenous SGK1 protein expression with time (peaking at 24 h), and a corresponding increase in levels of phospho-SGK1 (Ser80) was also observed (Fig. 1B). These observations corroborate previous studies in in vivo MPTP and transgenic ALS mouse models as well as in postmortem brain sections from AD and PD patients, where increased SGK1 expression at both mRNA and protein levels has been observed (1, 2, 25).
FIG 1.

SGK1 is upregulated in response to neurotoxic stress, and its overexpression induced by dexamethasone rescues 6-OHDA-mediated cell death and mitochondrial dysfunction in SH-SY5Y cells. (A) SGK1 mRNA expression profile showed an increase in expression on treatment with 35 μM 6-OHDA over time. (B) Western immunoblot analysis showed that endogenous SGK1 expression is upregulated and that SGK1 is phosphorylated in response to cellular stress, such as that induced by 6-OHDA. (C, D, and E) SGK1 expression was induced by adding 10 μM dexamethasone (Dex) to SH-SY5Y cells 24 h prior to 6-OHDA treatment. 6-OHDA (35 μM) was used to induce mitochondrial dysfunction and cell death. Mitochondrial superoxide production (C), membrane depolarization (D), and cell death (E) were monitored in SH-SY5Y cells, and protection by SGK1 was compared to protection from a selective JNK inhibitor. (F) An allosteric Akt inhibitor, MK-2206, was used. DMSO was used as a small-molecule control. Data presented in this figure represent measurements from three biological replicates with three samples per replicate. Statistical significance (P < 0.05) was determined using Student's t test. Differences between values for 6-OHDA-treated and mock- or Dex-treated cells are shown by a single asterisk. Differences between values for 6-OHDA and Dex plus 6-OHDA treatment groups or between the 6-OHDA and Dex plus Akt inhibitor plus 6-OHDA treatment groups are shown by a double asterisk. MW, molecular weight (in thousands).
We next investigated the role of SGK1 in mitochondrial dysfunction by measuring generation of mitochondrial reactive oxygen species (ROS) and changes in the mitochondrial membrane potential caused by 6-OHDA injury to the cells. Cell death was also measured. Endogenous SGK1 expression was induced in SH-SY5Y cells by treatment with 10 μM dexamethasone, which has been shown to upregulate transcription and expression of SGK1 (26). Treatment with dexamethasone showed protection from neurotoxin-induced mitochondrial dysfunction and cell death (Fig. 1C, D, and E) at a magnitude similar to that of the protection seen from the selective JNK inhibitor SR-3306. Since SGK1 shares various cell-regulatory roles with Akt, an Akt allosteric inhibitor, MK-2206, was added to the cells overexpressing SGK1 and prior to the addition of the neurotoxin to determine whether the protective effects of SGK1 were mainly due to its overexpression. The data showed that neuroprotection was a result of dexamethasone-induced SGK1 overexpression in these cells and that the absence of Akt activity did not significantly increase cell death, therefore suggesting that SGK1 compensated for Akt activity in cells undergoing stress (Fig. 1E and F).
To further elucidate the role of SGK1 in neuroprotection, type 5 (dE1/E3) adenovirus was prepared to induce SGK1 expression (AdV-SGK1) in SH-SY5Y cells. Immunofluorescence images from 6-OHDA-treated SH-SY5Y cells showed that adenovirus-induced SGK1 overexpression in these cells was able to reduce neurotoxin-mediated mitochondrial ROS compared to the level in PBS-6-OHDA-treated cells (Fig. 2A) in a manner similar to the reduction seen with JNK inhibition by SR-3306. A similar reduction in mitochondrial ROS by the JNK inhibitor SR-3306, which was used as a positive control in our studies (Fig. 2A), was reported by our lab previously (15, 19). Quantitative analysis of MitoSOX red and JC-1 staining revealed that mitochondrial superoxide levels and membrane depolarization were raised by treatment with 35 μM 6-OHDA and alleviated in cells treated with the SGK1-expressing adenovirus prior to their treatment with the neurotoxin (Fig. 2B and C). A similar effect was seen for the viability of these cells (Fig. 2D). Use of the Akt inhibitor MK-2206 produced the same results as the AdV-SGK1 treatment (Fig. 2D), indicating Akt activity compensation by AdV-SGK1-induced SGK1 expression. These studies show an important role of SGK1 in rescuing neuronal cells from mitochondrial dysfunction and cell death induced by a neurotoxin. Thus, cells overexpressing SGK1, induced by either the adenovirus or dexamethasone, showed a 2-fold decrease in mitochondrial ROS production and membrane depolarization and were rescued from 6-OHDA-induced cell death.
FIG 2.

Adenovirus-mediated overexpression of SGK1 rescued 6-OHDA-induced mitochondrial superoxide production, membrane depolarization, and cell death. SH-SY5Y cells were infected with AdV-SGK1 at an MOI of 5 and were incubated at 37°C and 5% CO2 for 48 h for protein expression. Cells were then treated with 35 μM 6-OHDA for 5 h. Mock green fluorescent protein-infected cells were used as cellular background controls, while DMSO was used as a small-molecule control. Mitochondrial superoxide production was monitored by MitoSOX red staining for both live-cell imaging (A) and a quantitative measure (B). JC-1 staining was used to measure mitochondrial membrane depolarization (C), and cell death was measured by using an MTT cell viability assay (D). Data presented represent measurements from three biological replicates with three samples per replicate. Statistical significance (P < 0.05) was determined using Student's t test. Differences between values for 6-OHDA-treated and mock- or AdV-SGK1-treated cells are shown by a single asterisk, and differences between values for 6-OHDA and AdV-SGK1 plus 6-OHDA treatment groups or between 6-OHDA and AdV-SGK1 plus Akt inhibitor plus 6-OHDA treatment groups are shown by a double asterisk.
Overexpression of SGK1 in SH-SY5Y cells regulates 6-OHDA-induced changes in cellular stress marker proteins.
Mitochondrial dysfunction, ER stress triggered by the unfolded protein response, and subsequent cell death are preceded by expression and activation of important regulatory proteins such as heat shock protein 70 (Hsp70), the ER stress marker protein binding immunoglobulin protein (BiP), the mitochondrial stress marker superoxide dismutase 1 (SOD1), and various others (27–32). ER and cellular stress marker proteins such as BiP and Hsp70 have been shown to have elevated expression levels in response to toxic stress in various neurodegenerative disorders, and many reports have detailed the relationship between JNK and Hsp70 (28, 29, 33–35). Mitochondrial protein SOD1 has been implicated in ALS, and its activity is critical against oxidative stress, a hallmark of various neurodegenerative diseases (36). To understand the role of SGK1 in cellular stress and investigate the relationship between SGK1 and JNK, we measured many of these stress markers in our cell system. Western blot analysis of SH-SY5Y cells treated with 35 μM 6-OHDA for 5 h showed an increase in the expression of BiP (Fig. 3A) and Hsp70 (Fig. 3B) and a decrease in SOD1 (Fig. 3C) protein levels, as expected, under cellular stress conditions. Expression of BiP and Hsp70 was rescued in cells overexpressing SGK1 induced by either Dex or AdV-SGK1 (Fig. 3A and B). Inhibition of JNK activity by SR-3306 in these cells also rescued the expression of these ER stress marker proteins and restored SOD1 levels. These data suggest that SGK1 rescues neurotoxin-induced cellular stress by directly or indirectly regulating the expression of these proteins that have been implicated in various neurodegenerative disorders.
FIG 3.

Overexpression of SGK1 rescues 6-OHDA-induced expression of cellular stress marker proteins in SH-SY5Y cells. SH-SY5Y cells were infected with AdV-SGK1 or treated with 10 μM dexamethasone (Dex) to induce SGK1 expression, and cell lysates were examined for BiP (A), Hsp70 (B), and SOD1 (C) expression, as indicated. α-Tubulin was used as a loading control. BiP, Hsp70, and SOD1 expression was also monitored in 6-OHDA-treated cells treated with 500 nM JNK inhibitor SR-3306. Adenovirus expressing green fluorescent protein was used as a control for AdV-SGK1 infection. Data presented here are representative Western blots of three biological replicates.
AdV-SGK1-induced expression in SH-SY5Y cells inhibits its downstream substrates, FoxO3a and NDRG-1.
The mechanism by which SGK1 confers neuroprotection is not well known, and it could potentially be via various cell signaling pathways. Two well-recognized substrates of SGK1 are NDRG-1 and FoxO3a (6, 9). The former is exclusively a substrate of SGK1, whereas the latter also gets phosphorylated by Akt. SGK1 phosphorylation of NDRG-1 has been shown to prime it for phosphorylation by GSK3β, but there is not sufficient information available about the biological consequences of NDRG-1 phosphorylation by SGK1 (10, 11, 22). To study this in vitro we infected SH-SY5Y cells with AdV-SGK1 and monitored phosphorylation levels of NDRG-1 and FoxO3a. The results shown in Fig. 4 indicate that there was a marked increase in expression of phospho-NDRG-1 and phospho-FoxO3a in SH-SY5Y cells treated with AdV-SGK1. These higher expression levels coincided with the neuroprotective effects of SGK1 in these cells as described in the previous paragraphs (Fig. 1 to 3). As noted previously, overexpression of SGK1 compensated for the absence of Akt activity, and therefore phospho-FoxO3a levels were not changed in the presence of an Akt inhibitor (data not shown). These studies substantiate that one of the ways by which SGK1 exerts its prosurvival effects is by phosphorylating and inhibiting these substrates, which are key proteins for regulating apoptotic events in neurons.
FIG 4.
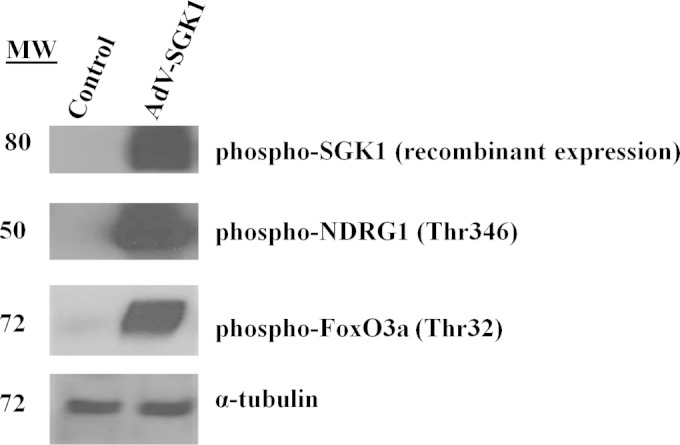
Western immunoblot analysis for phosphorylation of the SGK1 substrates, NDRG-1 and FoxO3a, in cells overexpressing SGK1. SH-SY5Y cells were infected with AdV-SGK1 and incubated for 48 h for protein expression as described in Materials and Methods. α-Tubulin is shown as a loading control. Data presented are representative of Western blots from three biological replicates. MW, molecular weight (in thousands).
SGK1 rescues the degradation of cell survival protein Mcl-1 and its phosphorylation by inactivating GSK3β in SH-SY5Y cells.
Because GSK3β and Mcl-1 are regulated by SGK1, we investigated the protective effects of SGK1 overexpression on 6-OHDA-induced phosphorylation of GSK3β and Mcl-1 (22). Figure 5A presents the relative levels of active phospho-GSK3β (Ser 390) in SH-SY5Y cells that were untreated or treated with 35 μM 6-OHDA. 6-OHDA treatment increased the relative levels of active phospho-GSK3β by 5-fold, and overexpression of SGK1 reduced these levels 2-fold (Fig. 5A). Conversely, the relative levels of Ser9 phospho-GSK3β (representative of an inhibitory phosphorylation caused by SGK1) were very low in 35 μM 6-OHDA-treated cells and increased to almost untreated control cell levels in SGK1-infected cells even though they had been treated with 35 μM 6-OHDA (Fig. 5B). In a similar manner to inhibitory GSK3β (Ser9), Mcl-1 expression was reduced relative to the level of the control in the presence of 35 μM 6-OHDA, and the levels were increased when SGK1 was overexpressed (Fig. 5C). Similarly, there was a corresponding increase in phosphorylated Mcl-1 levels (Fig. 5D) in the presence of 35 μM 6-OHDA, and the levels were increased when SGK1 was overexpressed (Fig. 5D). Higher levels of GSK3β phosphorylated at Ser9 (Fig. 5B) in these samples indicated that SGK1 inhibited the activation of GSK3β, leading to reduced phospho-Mcl-1 levels. These data show a link between the neuroprotective role of SGK1 and the way by which it inhibits the activation of GSK3β, thereby restoring Mcl-1 levels in the cell.
FIG 5.

SGK1 rescued the degradation of cell survival protein Mcl-1 and its phosphorylation by inactivating GSK3β in SH-SY5Y cells. The following ratios were determined under different cell treatment conditions, as indicated: GSK3β (Ser390) phosphorylation/α-tubulin levels (A), phospho-GSK3β (Ser9)/α-tubulin levels (B), Mcl-1/α-tubulin levels (C), and phospho-Mcl-1/α-tubulin levels (D). Cells were either untreated, untreated and infected with AdV-SGK1, treated with PBS and 35 μM 6-OHDA, or treated with 35 μM 6-OHDA and infected with AdV-SGK1.
SGK1 phosphorylated MKK4 at serine 80 and prevented activation of MKK4.
MKK4 has been shown to activate JNK signaling, and it also has been shown that MKK4 is phosphorylated at serine 80 by SGK1 and is subsequently inactivated (20, 37, 38). To confirm that MKK4 is indeed a substrate for SGK1, a Kinase-Glo assay was performed with increasing concentrations of recombinant inactive MKK4 and active SGK1 in the presence of 1 μM ATP (Fig. 6A). Western blot analysis using these reaction mixtures confirmed MKK4 phosphorylation at serine 80 by activated SGK1 (Fig. 6B). These in vitro data were corroborated by cell-based experiments looking at inhibition of MKK4 activity by SGK1 phosphorylation of Ser80 in MKK4. SH-SY5Y cells were treated with 6-OHDA to activate MKK4 and, consequently, the JNK signaling pathway. Cells treated with 6-OHDA showed 12-fold higher levels of active MKK4 than control cells, and overexpression of SGK1 reduced the level of active MKK4 to background levels (Fig. 6C). Cells that overexpressed SGK1 showed increased levels of phospho-MKK4 (Ser80) (Fig. 6D); 6-OHDA treatment reduced these levels back to around baseline, whereas overexpression of SGK1, even in the presence of 6-OHDA, restored the phospho-MKK4 (Ser80) levels indicative of full inactivation (Fig. 6D). Activation of MKK7 was also seen for cells stressed with 6-OHDA, but those levels were reduced in cells overexpressing SGK1 and treated with 6-OHDA, as expected (Fig. 6E). Activation of MKK4 and MKK7 coincided with high phospho-JNK levels in cells treated with the neurotoxin (Fig. 6F). As a consequence of MKK4 and MKK7 inactivation in cells overexpressing SGK1, low levels of phospho-JNK were seen in these cells (Fig. 6F). Lowering of phospho-MKK7 levels in cells that overexpressed SGK1 could be due to the alleviation of overall stress in the cells, leading to cell survival and reduced activation of MKK7, instead of to direct inhibition by SGK1. These findings support the observed inhibition of Mcl-1 degradation, as shown in Fig. 5A, and also demonstrate the contribution of SGK1 in disabling MKK4-mediated JNK activation in neurons.
FIG 6.

Effect of SGK1 expression on the phosphorylation state of MKK4, MKK7, and JNK in the presence and absence of 6-OHDA in vitro. (A) A Kinase-Glo assay was performed using 0.75 μM active SGK1 protein and different concentrations of inactive MKK4 (0 to 10 μM) in the presence of 1 μM ATP. MKK7 protein was used as a negative control in this assay and, as expected, showed no phosphorylation in the presence of active SGK1 and ATP. (B) Western immunoblot analysis for phosphorylation of MKK4 on serine 80 by active SGK1 in the presence of 1 μM and 10 μM ATP. (C to F) Western immunoblot analysis was performed using SH-SY5Y cells untreated and treated with 35 μM 6-OHDA in the presence and absence of AdV-SGK1 to determine the following ratios: phospho-MKK4 (Ser257/Thr261)/α-tubulin (C), phospho-MKK4 (Ser80)/α-tubulin (D), phospho-MKK7 (Ser271/Thr275)/α-tubulin (E), and phospho-JNK/α-tubulin (F). Expression levels of MKK4, MKK7, and JNK did not change significantly between different treatments (data not shown).
SGK1 overexpression protects MPTP-induced TH cell death in substantia nigra in C57BL/6J mice by inhibiting MKK4-mediated JNK activation.
To test whether the protective effects of SGK1 seen in vitro could be manifested in vivo, we assessed if SGK1 could block dopaminergic cell loss in the substantia nigra pars compacta (SNpc) of mice with MPTP-induced lesions. To do this, we stereotactically injected 1 × 1010 AdV-SGK1 particles into the right striatum, and gene expression was allowed to proceed for 21 days, following which mice were challenged with an acute MPTP lesion-inducing regimen as described in Materials and Methods. Figure 7 presents the YFP-SGK1 expression in the SNpc in the region of −3.0 bregma 28 days after injection into the striatum for both the saline treatment and the MPTP-treated mouse. A high level of YFP-SGK1 expression was seen for both treatment groups, and this expression coincided with the TH immunoreactivity in the SNpc, as seen in the merged panels. Figure 8A shows immunohistochemical staining of TH neurons in the SNpc for one representative mouse for each of the three groups. Figure 8B, panel i, presents the unbiased stereological count for the number of TH+ cells in the ipsilateral SNpc for the three following treatment groups: saline/saline, saline/AdV-SGK1, saline/MPTP, and AdV-SGK1/MPTP. As expected, the MPTP treatment reduced the number of TH+ dopaminergic neurons by 50% compared with the number in saline-treated or AdV-SGK1-injected animals (P < 0.05), and overexpression of SGK1 restored those levels by ∼40% (P < 0.05). The data in Fig. 8B, panel ii, show that the total area measured in the SNpc following the MPTP treatment was the same for all groups. The increased TH+ cell count in AdV-SGK1/MPTP group compared to that of the saline/MPTP group indicated that neurons overexpressing SGK1 were protected from MPTP-induced lesions (Fig. 8A and Bi). To test if the JNK pathway was activated by MPTP and subsequently protected by SGK1 overexpression, we monitored various components of the pathway in vivo by Western analysis. Western immunoblot analysis of the SNpc tissue samples showed increased levels of phospho-MKK4 (Ser257/Thr261), phospho-MKK7 (Ser272/Thr275), phospho-JNK, and phospho-c-Jun in the MPTP-treated group (Fig. 8Ci). This increase was rescued in groups treated with AdV-SGK1 prior to the MPTP treatment. The control group showed lower levels of these active phosphorylated proteins. Phospho-MKK4 (Ser78) (serine 80 in humans) levels, however, were low in the MPTP-treated group compared to levels in the AdV-SGK1/MPTP-treated group. To corroborate the Western immunoblot findings, we also looked at p-c-Jun levels by immunohistochemical analysis in the three treatment groups (Fig. 9). The data show that p-c-Jun levels were significantly increased in the SNpc and colocalized with TH neurons after MPTP treatment and that overexpression of SGK1 modestly reduced these levels (Fig. 9) in a similar manner to that seen in the Western analysis. These results indicate that SGK1 inhibited MKK4 by phosphorylating it on serine 78 (serine 80 in humans) and consequently inhibited JNK activity in vivo and protected dopaminergic neurons from MPTP-induced death.
FIG 7.

YFP-SGK1 Overexpression in the SNpc after 28 days. AdV-YFP-SGK1 was injected into the striatum of mice treated with saline or treated with MPTP, and 28 days later YFP-SGK1 expression was assessed. TH immunohistochemistry is shown for a representative saline-treated mouse and for an MPTP-treated mouse in which AdV-SGK1 was overexpressed for 21 days prior to MPTP treatment. A total of 1 × 1010 viral particles of AdV-SGK1 were injected into the striatum 21 days prior to an acute treatment regimen of 18 mg/kg MPTP (1 dose every 2 h for 4 doses).
FIG 8.

SGK1 overexpression protected MPTP-induced dopaminergic neuronal cell death in the substantia nigra in C57BL/6J mice by inhibiting MKK4-mediated JNK activation. (A) TH immunohistochemistry for a representative saline-treated mouse, an MPTP-treated mouse, and an MPTP-treated mouse in which AdV-SGK1 was overexpressed for 21 days prior to MPTP treatment. A total of 1 × 1010 viral particles of AdV-SGK1 were injected into the striatum 21 days prior to an acute treatment regimen of 18 mg/kg MPTP (1 dose every 2 h for 4 doses). (B) Stereological quantification of TH-positive neurons in different treatment groups (i) and the measured volume of midbrain section used for the TH neuron analysis (ii). ISI, ipsilateral; IP, intraperitoneal. *, P < 0.01 between either saline plus saline or AdV-SGK1 plus saline and saline plus MPTP; **, P < 0.01 between saline plus MPTP and Adv-SGK1 plus MPTP. (C) Western immunoblot analysis of phospho-SGK1, phospho-MKK4 (Ser257/Thr261), phosphor-MKK4 (Ser78), phospho-JNK, and phospho-c-Jun in midbrains treated with saline, saline/MPTP, and MPTP/AdV-SGK1 was performed (i). Expression of these proteins was quantitated and normalized to that of α-tubulin for the respective treatments (ii).
FIG 9.

Phospho-c-Jun (p-c-Jun) immunohistochemical analysis in the substantia nigra in C57BL/6J mice. p-c-Jun immunohistochemistry (red) is shown for a representative saline-treated mouse, an MPTP-treated mouse, and an MPTP-treated mouse in which AdV-SGK1 was overexpressed for 21 days prior to MPTP treatment. TH immunohistochemistry (green) is shown for a representative saline-treated mouse, an MPTP-treated mouse, and an MPTP-treated mouse in which AdV-SGK1 was overexpressed for 21 days prior to MPTP treatment. A total of 1 × 1010 viral particles of AdV-SGK1 were injected into the striatum 21 days prior to an acute treatment regimen of 18 mg/kg MPTP (1 dose every 2 h for 4 doses).
DISCUSSION
The role of SGK1 in neurobiology has not been studied extensively, and therefore very little is known regarding the details by which it confers neuroprotection. The JNK signaling pathway, on the other hand, has been well investigated in neurodegeneration and underscores the importance of JNK in the brain (15, 16, 18, 39, 40). The work presented here for the first time links the neuroprotective molecular components of SGK1 with the JNK signaling cascade in a PD neurotoxin cell and animal model.
Various studies have shown upregulation of SGK1 as a cell survival response in transgenic models of PD and ALS (1–3, 25). In line with previously published data, in our study we, too, observed an increase in SGK1 expression at both mRNA and protein levels and a corresponding increase in phosphorylated and activated levels of SGK1 in response to neurotoxic stress induced by 6-OHDA in SH-SY5Y cells, which enabled us to utilize this neurotoxin cellular model to study SGK1 biology in neurodegeneration. Neuronal apoptosis has been shown to be accompanied and preceded by mitochondrial dysfunction (41), and our work here for the first time shows that induction of SGK1 expression had a direct effect on mitochondrial ROS production and mitochondrial membrane depolarization alleviation and protected cells from neurotoxin-induced death. Since SGK1 shares some its functions and substrates with Akt (6), it is not very clear in much of the previously published data whether the neuroprotective effects seen are due solely to SGK1 expression or to a combination of effects. Our studies showed that SGK1 overexpression likely compensated for the absence of Akt and therefore conferred protection in neuronal cells primed for apoptosis. Phosphorylation of SGK1 substrates NDRG-1 and FoxO3a, one of which is shared with Akt, was shown to be augmented in our SGK1 overexpression cell model. These increases could either directly or indirectly lead to cell survival by regulating the transcription of key anti- and proapoptotic proteins and signaling pathways.
ER and mitochondrial stress are hallmarks of neuronal apoptosis, which is tightly regulated by various proteins responsible for cell survival and apoptosis. Proteins such as BiP, Hsp70, and SOD1, which were investigated in our study, are well-known indicators of ER and mitochondrial stress (28, 29, 32, 36), and their expression is critical for the progression of neuronal cell death. The restoration of expression levels of BiP and Hsp70 to normal levels and the increases in SOD1 levels in our cellular neurodegeneration model when SGK1 expression was high signified an essential role for SGK1 in relieving mitochondrial and ER stress by regulating the expression of these key cellular dysfunction marker proteins. These changes in expression levels for stress marker proteins could be either a direct or an indirect effect of SGK1 overexpression and will require further detailed study to determine whether SGK1 induces protection by directly interacting with these molecules or through its substrates, NDRG-1 and FoxO3a.
Signaling cascades upstream of SGK1 are well studied, but insufficient information is available about downstream events in neurodegeneration. In order to delineate the neuroprotective effect of SGK1 in our neurotoxin models, we investigated opposing signaling pathways that mediate cell survival and apoptosis involving SGK1 and JNK. Expression of the anti-apoptotic protein Mcl-1 has been shown to decrease under cellular stress as it gets phosphorylated by JNK and subsequently by GSK3β, priming it for degradation (23). Our findings supported these observations and showed that SGK1 overexpression in SH-SY5Y cells rescued the degradation of the prosurvival protein Mcl-1, which was accompanied by a marked decrease in the activated levels of its kinases GSK3β and JNK. Rescue of Mcl-1 protein degradation and inactivation of GSK3β by SGK1 overexpression indicate a cross talk between the cell survival and apoptotic pathways that are engaged in these neurons. Stressed cells are directed toward the prosurvival pathway when there is more SGK1 to inactivate GSK3β and JNK, consequently restoring Mcl-1 levels and cell viability.
The data from our study suggest a role for SGK1 in protecting neuroblastoma cells from neurotoxic cell death by alleviating mitochondrial dysfunction and by regulating the expression of ER and mitochondrial stress marker proteins. SGK1 does so by suppressing well-known proapoptotic signaling cascades. JNK activation by MKK4 during cellular stress is well known (38). Our biochemical and cell-based experiments corroborated previous studies that show phosphorylation of MKK4 by SGK1 (20). Our results showed that SGK1 overexpression in 6-OHDA-treated or untreated SH-SY5Y cells triggered significant increases in phosphorylation of MKK4 at serine 80 and inhibited its activity in these cells, as was shown by lower phospho-JNK levels. The higher levels of inhibited phospho-MKK4 (Ser80) which were found in cells overexpressing SGK1 and treated with the neurotoxin indicated that SGK1 inhibited MKK4 activity, which in turn inhibited activation of the JNK signaling cascade. SGK1-mediated JNK inactivation also explains the rescue of Mcl-1 degradation in cells treated with SGK1 adenovirus prior to stress. These experiments demonstrated a more direct role of SGK1 in JNK inactivation, which also supports the observation of augmented Mcl-1 levels in stressed cells with increased SGK1 activity and rescue of cellular stress on the whole. Chen et al. showed in a 6-OHDA PD mouse model that overexpressing SGK1 in the mesencephalic dopamine neurons protected neurons from 6-OHDA-induced apoptosis (13). We attempted to test this observation in an MPTP mouse model of neurodegeneration. Our in vivo findings corroborated the 6-OHDA finding from Chen et al. as well as our cell-based findings, where we found that by inducing the expression of SGK1 using the adenoviral vector transfer, the dopaminergic neurons could be protected from MPTP-induced lesions. This observation was accompanied by an increased expression of phospho-MKK4 (Ser78) (Ser80 in humans) and, as a result, reduced JNK activity, as seen by reduced levels of phospho-JNK and its nuclear product, phospho-c-Jun.
Through a series of in vitro and in vivo studies, we have begun to elucidate the molecular contributors to cytoprotection by SGK1. Our data suggest the following proposed contributions to SGK1 signaling and cell survival. First, overexpression of SGK1 caused inactivating phosphorylation on GSK3β and MKK4, as well as phosphorylation of FoxO3a and NDRG-1. The inactivation of MKK4 subsequently caused inactivation of JNK. The inactivation of GSK3β along with the lack of JNK activation caused Mcl-1 not to be phosphorylated, preventing it from being degraded and thus promoting cell survival. In addition, the phosphorylation on FoxO3a and NDRG-1 also served to be neuroprotective, thereby promoting cell survival (Fig. 10). The data also suggest that even though the levels of endogenous SGK1 increase in the cell under stress, the increase is not sufficient to promote cell survival in our neurodegeneration model. On the other hand, cell survival mechanisms dominate when exogenous SGK1 is added to the neuronal cells. However, careful observations must be made of the differences in cellular signaling outcomes for different pathologies affecting tissues involving SGK1 activation or overexpression.
FIG 10.

Molecular mechanism for SGK1 signaling and cell survival.
Collectively, our findings help to further define the underlying pathogenic mechanisms in neurodegeneration models where complex I deficiencies and mitochondrial dysfunction may be the underlying sources for dopaminergic neuronal death. This work elucidated the interplay of various apoptotic and cell survival mechanisms involving SGK1 and JNK under neurotoxin-induced cellular stress conditions, and an upregulation of survival pathways in these cells significantly reduced apoptosis in neurons. Additional studies need to be undertaken to further dissect the role of SGK1 in JNK and other cell death signaling cascades so that this molecular understanding of protein interactions and modulations can be exploited in developing small-molecule SGK1 activators that would promote cell survival in neurodegeneration.
ACKNOWLEDGMENTS
This work was supported by Department of Defense grant W81XWH-12-1-0431, NIH grant U01-NS057153, NIH grant GM103825, the Michael J. Fox Foundation/23andMe, the Saul and Theresa Esman Foundation, and a gift from the McCubbin Family.
REFERENCES
- 1.Schoenebeck B, Bader V, Zhu XR, Schmitz B, Lübbert H, Stichel CC. 2005. Sgk1, a cell survival response in neurodegenerative diseases. Mol Cell Neurosci 30:249–264. doi: 10.1016/j.mcn.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Stichel CC, Schoenebeck B, Foguet M, Siebertz B, Bader V, Zhu XR, Lübbert H. 2005. sgk1, a member of an RNA cluster associated with cell death in a model of Parkinson's disease. Eur J Neurosci 21:301–316. doi: 10.1111/j.1460-9568.2005.03859.x. [DOI] [PubMed] [Google Scholar]
- 3.Lang F, Strutz-Seebohm N, Seebohm G, Lang UE. 2010. Significance of SGK1 in the regulation of neuronal function. J Physiol 588:3349–3354. doi: 10.1113/jphysiol.2010.190926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lang F, Artunc F, Vallon V. 2009. The physiological impact of the serum and glucocorticoid-inducible kinase SGK1. Curr Opin Nephrol Hypertens 18:439–448. doi: 10.1097/MNH.0b013e32832f125e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang F, Shumilina E. 2013. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J 27:3–12. doi: 10.1096/fj.12-218230. [DOI] [PubMed] [Google Scholar]
- 6.Pearce LR, Komander D, Alessi DR. 2010. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 7.Nasir O, Wang K, Föller M, Gu S, Bhandaru M, Ackermann TF, Boini KM, Mack A, Klingel K, Amato R, Perrotti N, Kuhl D, Behrens J, Stournaras C, Lang F. 2009. Relative resistance of SGK1 knockout mice against chemical carcinogenesis. IUBMB Life 61:768–776. doi: 10.1002/iub.209. [DOI] [PubMed] [Google Scholar]
- 8.Radi E, Formichi P, Battisti C, Federico A. 2014. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis 42:S125–S152. doi: 10.3233/JAD-132738. [DOI] [PubMed] [Google Scholar]
- 9.Sahin P, McCaig C, Jeevahan J, Murray JT, Hainsworth AH. 2013. The cell survival kinase SGK1 and its targets FOXO3a and NDRG1 in aged human brain. Neuropathol Appl Neurobiol 39:623–633. doi: 10.1111/nan.12023. [DOI] [PubMed] [Google Scholar]
- 10.Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, Lang F, Wulff P, Kuhl D, Cohen P. 2004. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem J 384:477–488. doi: 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.García-Martínez JM, Alessi DR. 2008. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J 416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 12.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. 2001. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Tagliaferro P, Kareva T, Yarygina O, Kholodilov N, Burke RE. 2012. Neurotrophic effects of serum- and glucocorticoid-inducible kinase on adult murine mesencephalic dopamine neurons. J Neurosci 32:11299–11308. doi: 10.1523/JNEUROSCI.5910-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, Qian C, Li S. 2014. Protective effect of SGK1 in rat hippocampal neurons subjected to ischemia reperfusion. Cell Physiol Biochem 34:299–312. doi: 10.1159/000363000. [DOI] [PubMed] [Google Scholar]
- 15.Crocker CE, Khan S, Cameron MD, Robertson HA, Robertson GS, LoGrasso P. 2011. JNK inhibition protects dopamine neurons and provides behavioral improvement in a rat 6-hydroxydopamine model of Parkinson's disease. ACS Chem Neurosci 2:207–212. doi: 10.1021/cn1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA. 2004. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A 101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L, Kunwar AJ, Walton JC, Nagahara AH, Lu KP, Nelson RJ, Tuszynski MH, Huang K. 2012. JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75:824–837. doi: 10.1016/j.neuron.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. 2001. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A 98:10433–10438. doi: 10.1073/pnas.181182298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chambers JW, Pachori A, Howard S, Ganno M, Hansen D, Kamenecka T, Song X, Duckett D, Chen W, Ling YY, Cherry L, Cameron MD, Lin L, Ruiz CH, Lograsso P. 2011. Small molecule c-jun-N-terminal kinase (JNK) inhibitors protect dopaminergic neurons in a model of Parkinson's disease. ACS Chem Neurosci 2:198–206. doi: 10.1021/cn100109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MJ, Chae JS, Kim KJ, Hwang SG, Yoon KW, Kim EK, Yun HJ, Cho J-H, Kim J, Kim B-W, Kim H-C, Kang SS, Lang F, Cho S-G, Choi E-J. 2007. Negative regulation of SEK1 signaling by serum- and glucocorticoid-inducible protein kinase 1. EMBO J 26:3075–3085. doi: 10.1038/sj.emboj.7601755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu P, Das M, Reilly J, Davis RJ. 2011. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 25:310–322. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russo A, Schmid E, Nurbaeva MK, Yang W, Yan J, Bhandaru M, Faggio C, Shumilina E, Lang F. 2013. PKB/SGK-dependent GSK3-phosphorylation in the regulation of LPS-induced Ca2+ increase in mouse dendritic cells. Biochem Biophys Res Commun 437:336–341. doi: 10.1016/j.bbrc.2013.06.075. [DOI] [PubMed] [Google Scholar]
- 23.Morel C, Carlson SM, White FM, Davis RJ. 2009. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol 29:3845–3852. doi: 10.1128/MCB.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson-Lewis V, Przedborski S. 2007. Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc 2:141–151. doi: 10.1038/nprot.2006.342. [DOI] [PubMed] [Google Scholar]
- 25.Arteaga MF, Coric T, Straub C, Canessa CM. 2008. A brain-specific SGK1 splice isoform regulates expression of ASIC1 in neurons. Proc Natl Acad Sci U S A 105:4459–4464. doi: 10.1073/pnas.0800958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webster MK, Goya L, Firestone GL. 1993. Immediate-early transcriptional regulation and rapid mRNA turnover of a putative serine/threonine protein kinase. J Biol Chem 268:11482–11485. [PubMed] [Google Scholar]
- 27.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. 2001. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol 21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorbatyuk MS, Gorbatyuk OS. 2013. The molecular chaperone GRP78/BiP as a therapeutic target for neurodegenerative disorders: a mini revies. J Genet Syndr Gene Ther 4:128. doi: 10.4172/2157-7412.1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. 2001. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet 10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 30.Jana NR, Tanaka M, Wang Gh, Nukina N. 2000. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet 9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 31.Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, Derbeneva O, Khusainova R, Khidiyatova I, Magzhanov R, Wallace DC. 2008. A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson's disease. Ann N Y Acad Sci 1147:1–20. doi: 10.1196/annals.1427.001. [DOI] [PubMed] [Google Scholar]
- 32.Milani P, Gagliardi S, Cova E, Cereda C. 2011. SOD1 transcriptional and posttranscriptional regulation and its potential implications in ALS. Neurol Res Int 2011:458427. doi: 10.1155/2011/458427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shukla AK, Pragya P, Chaouhan HS, Tiwari AK, Patel DK, Abdin MZ, Chowdhuri DK. 2014. Heat shock protein-70 (Hsp-70) suppresses paraquat-induced neurodegeneration by inhibiting JNK and caspase-3 activation in Drosophila model of Parkinson's disease. PLoS One 9:e98886. doi: 10.1371/journal.pone.0098886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong Z, Wolfer DP, Lipp H-P, Büeler H. 2005. Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther 11:80–88. doi: 10.1016/j.ymthe.2004.09.007.. [DOI] [PubMed] [Google Scholar]
- 35.Auluck PK, Chan HYE, Trojanowski JQ, Lee VMY, Bonini NM. 2002. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 36.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 37.Lisnock J, Griffin P, Calaycay J, Frantz B, Parsons J, O'Keefe SJ, LoGrasso P. 2000. Activation of JNK3α1 requires both MKK4 and MKK7: kinetic characterization of in vitro phosphorylated JNK3α1. Biochemistry 39:3141–3148. doi: 10.1021/bi992410+. [DOI] [PubMed] [Google Scholar]
- 38.Weston CR, Davis RJ. 2002. The JNK signal transduction pathway. Curr Opin Genet Dev 12:14–21. doi: 10.1016/S0959-437X(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 39.Chambers JW, Pachori A, Howard S, Iqbal S, LoGrasso PV. 2013. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J Biol Chem 288:4000–4011. doi: 10.1074/jbc.M112.406777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ries V, Silva RM, Oo TF, Cheng H-C, Rzhetskaya M, Kholodilov N, Flavell RA, Kuan C-Y, Rakic P, Burke RE. 2008. JNK2 and JNK3 combined are essential for apoptosis in dopamine neurons of the substantia nigra, but are not required for axon degeneration. J Neurochem 107:1578–1588. doi: 10.1111/j.1471-4159.2008.05713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schüssel K, Müller WE. 2003. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol 66:1627–1634. doi: 10.1016/S0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]