

Abstract
Candida albicans is an important human fungal pathogen in both immunocompetent and immunocompromised individuals. C. albicans regulation has been studied in many contexts, including morphological transitions, mating competence, biofilm formation, stress resistance, and cell wall synthesis. Analysis of kinase- and phosphatase-deficient mutants has made it clear that protein phosphorylation plays an important role in the regulation of these pathways. In this study, to further our understanding of phosphorylation in C. albicans regulation, we performed a deep analysis of the phosphoproteome in C. albicans. We identified 19,590 unique peptides that corresponded to 15,906 unique phosphosites on 2,896 proteins. The ratios of serine, threonine, and tyrosine phosphosites were 80.01%, 18.11%, and 1.81%, respectively. The majority of proteins (2,111) contained at least two detected phosphorylation sites. Consistent with findings in other fungi, cytoskeletal proteins were among the most highly phosphorylated proteins, and there were differences in Gene Ontology (GO) terms for proteins with serine and threonine versus tyrosine phosphorylation sites. This large-scale analysis identified phosphosites in protein components of Mediator, an important transcriptional coregulatory protein complex. A targeted analysis of the phosphosites in Mediator complex proteins confirmed the large-scale studies, and further in vitro assays identified a subset of these phosphorylations that were catalyzed by Cdk8 (Ssn3), a kinase within the Mediator complex. These data represent the deepest single analysis of a fungal phosphoproteome and lay the groundwork for future analyses of the C. albicans phosphoproteome and specific phosphoproteins.
INTRODUCTION
Candida species are both human commensals and the most common human fungal pathogens. Candidiasis includes both superficial and invasive fungal infections (1–3). Candida albicans is the most common cause of invasive candidiasis worldwide (4), and this disease is associated with very high mortality rates, as high as 35% in some populations (5).
In the context of the host, C. albicans is exposed to numerous physical and chemical signals that can trigger a variety of responses, including morphological transitions between yeast and filamentous forms, switching between different cell types, the formation of multicellular structures, including biofilms, and altered cell wall states (6–9). The ability of C. albicans to respond to different stimuli contributes to its virulence (3). Hyphal growth and the coordinated expression of hypha-specific genes are important virulence traits, since they participate in surface-associated growth and escape after phagocytosis (10, 11). Many kinases and phosphatases of biological importance have been described (12–15). Hyphal morphology in C. albicans is initiated and maintained by the Ras1-Cyr1 (adenylate cyclase)-protein kinase A (PKA) pathway, in which the PKA catalytic subunits Tpk1 and Tpk2 positively regulate hyphal growth (16–18). Mitogen-activated protein (MAP) kinase pathways also control many important processes, including filamentation, mating, biofilm formation, and stress resistance, through phosphorylation/dephosphorylation cycles (19).
Advances in mass spectrometry (MS)-based proteomics technologies and phosphopeptide enrichment methods have greatly enhanced our ability to identify and characterize phosphorylation sites (P sites) in the proteome of any organism with a sequenced genome (20–22). In many fungi, phosphoproteome characterization is at an early stage. The best-characterized fungal phosphoproteome is that in Saccharomyces cerevisiae. Using 12 publicly available data sets, Amoutzias et al. (21) identified 2,781 phosphorylated proteins with 9,783 unique P sites, with an average of four P sites per phosphoprotein. In the fungus Fusarium graminearum, 2,902 putative phosphopeptides on 1,496 different proteins were identified (23). Phosphoproteomics data sets for the fungi Cryptococcus neoformans (24), Aspergillus nidulans (25), Alternaria brassicicola (26), Botrytis cinerea (26), Neurospora crassa (27), and Schizosaccharomyces pombe (28) have also been published.
Here we report the first large-scale analysis of the phosphoproteome in C. albicans in cells grown under hypha-inducing conditions. We observed 19,590 unique P sites on 2,896 proteins. Consistent with phosphoproteomic analyses in S. cerevisiae, there was an enrichment of cytoskeleton proteins among those that were highly phosphorylated. P sites within the central regulatory Ras1-Cyr1-PKA signaling pathway in C. albicans were also found. Differences in the types of proteins, categorized by Gene Ontology (GO) term analysis, with serine and threonine P sites versus tyrosine P sites were identified. P sites within the global transcriptional coregulatory Mediator complex were found both in the large-scale data set and in a targeted follow-up study. Cdk8 is the sole kinase component of Mediator, and subsequent in vitro assays identified P sites within Mediator that were phosphorylated by Cdk8 in vitro at sites consistent with the previously described Cdk8 target motif (phospho-S/T-P) (29–33). This data set gives a first in-depth analysis of the C. albicans phosphoproteome and is the largest fungal phosphopeptide data set at this time. These data should serve as an important resource for researchers studying pathways that are regulated by phosphorylation.
MATERIALS AND METHODS
Preparation of C. albicans lysates.
A single colony of C. albicans strain SC5314 (34) was grown overnight in 50 ml of yeast extract-peptone-dextrose (YPD) medium. From that overnight culture, four flasks with 1 liter of YPD containing 5 mM N-acetylglucosamine (GlcNAc) were each inoculated to an optical density at 600 nm (OD600) of 0.05 and then incubated at 37°C at 150 rpm for 4 to 5 h. The cells were collected by centrifugation in 250-ml centrifugation cups for 5 min at 6,000 rpm. Cell pellets were washed once in 20 ml of phosphate-buffered saline and then pelleted in 50-ml Falcon tubes (5,000 rpm for 10 min). The cell pellets were frozen at −80°C for at least 1 h. The frozen cell pellets were lyophilized overnight in a freeze dryer (Labconco), and dry weights were measured. The dried cell mass was ground to a fine powder using a mortar and pestle in the presence of liquid nitrogen. The ground cells were transferred to fresh 50-ml Falcon tubes and stored at −80°C.
Enrichment and analysis of C. albicans phosphopeptides and mass spectrometric analysis.
Lyophilized C. albicans powder was homogenized in ice-cold lysis buffer containing 8 M urea, 25 mM Tris-HCl (pH 8.1), 150 mM NaCl, phosphatase inhibitors (2.5 mM beta-glycerophosphate, 1 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium molybdate, 1 mM sodium tartrate), and protease inhibitors (1 mini-complete EDTA-free tablet per 10 ml of lysis buffer; Roche Life Sciences) by means of sonication. The lysate was clarified by centrifugation, reduced, alkylated, and trypsin digested (35). After overnight digestion with trypsin at 37°C, the digest was acidified with trifluoroacetic acid (TFA), and peptides were desalted and lyophilized (22). Two rounds of phosphopeptide enrichment using titanium dioxide microspheres were performed, and phosphopeptides were separated by strong cation exchange chromatography (SCX) (22). After separation, the SCX fractions were dried, desalted, combined, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). LC-MS/MS analysis was performed on a Q-Exactive Plus mass spectrometer (Thermo Scientific) equipped with an Easy-nLC 1000 (Thermo Scientific). Peptides were redissolved in 5% acetonitrile (ACN)–1% formic acid and loaded onto a trap column (ReproSil, C18 AQ, 5 μm, 200-Å pore [Dr. Maisch, Ammerbuch, Germany]) and eluted across a fritless analytical resolving column (30-cm length, 100-μm inner diameter, ReproSil, C18 AQ, 3 μm, 200-Å pore) pulled in-house with a 90-min gradient of 4 to 30% LC-MS buffer B (LC-MS buffer A includes 0.0625% formic acid and 3% ACN; LC-MS buffer B includes 0.0625% formic acid and 95% ACN) at 400 nl/min. Raw data were searched using COMET in high-resolution mode (36) against a target decoy (reversed) (37) version of the C. albicans proteome sequence database with a precursor mass tolerance of ±1 Da and requiring tryptic peptides with up to two miscleavages, carbamidomethylcysteine as fixed modification, and oxidized methionine and phosphorylated serine, threonine, and tyrosine as variable modifications. The C. albicans proteome sequence database was generated by combining the UniProt C. albicans database (downloaded February 2013; 18,226 total [forward and reverse] proteins) with proteins unique to the Candida Genome Database (CGD) (at least one amino acid difference between the sequence in the CGD and any sequence in the UniProt C. albicans database [downloaded February 2013]). The resulting peptide spectral matches were filtered to a <1% false-discovery rate (FDR) based on reverse-hit counting, mass measurement accuracy (MMA) within ±3.5 ppm, a delta-XCorr (dCn) of more than 0.12, and appropriate XCorr values for +2- and +3-charge state peptides (37). Probability of phosphorylation site localization was determined by PhosphoRS (38). Peptide motif analysis was performed based on reference 39.
GO term analysis.
The Gene Ontology (GO) Slim annotation was used for GO term analysis. The gene association file, created 15 December 2014, was downloaded from the CGD website (http://www.candidagenome.org), and only annotations assigned to C. albicans (taxon 5476) were used. In total, 6,314 unique genes had at least one associated GO term and served as the background distribution of observed gene ontologies for the C. albicans genome in our study. GO enrichment analysis of the total phosphorylation (P-all), phospho-S/T (P-S/T), and phospho-Y (P-Y) were evaluated independently using the gene ontologies Biological Process, Cellular Component, and Molecular Function. GO::TermFinder (40) as implemented via The Generic GO Term Finder (http://go.princeton.edu/cgi-bin/GOTermFinder) was used to test for ontology enrichment among phosphorylated peptides. To determine significance of enrichment of terms, a Bonferroni corrected P value cutoff of 0.05 was used. GO::TermFinder calculates a P value using the hypergeometric distribution comparing the number of terms found in a set (phosphorylated genes) to the overall background of the genome (40). No evidence codes were excluded from the analysis. The CGD Gene Ontology Slim Mapper available from the CGD website and an R script (GOstats.R, bioconductor) in which a GSEAGOHyperGParams function is used for calculating a P value were also used to determine significant GO term enrichment, and similar term enrichments were found. Heat maps of the categories were generated by calculating nonparametric ranking of percent values for each GO term using the “heatmap.2” function in the “gplots” package (41) in R (R Foundation for Statistical Computing, Vienna, Austria).
Strain construction and Mediator complex purification.
Wild-type C. albicans Mediator complex was purified from cTTR03 through 6×His-3×Flag-tagged Med8 (42). To allow for the affinity purification of Mediator complex from mutant strains, one or both copies of MED8 were fused to a sequence encoding a C-terminal 6×His-3×Flag tag. In addition, Med8 was tagged in backgrounds lacking different Mediator components, including AZC52 (med12Δ/Δ [42]), AZC63(med9Δ/Δ [42]), cTTR01 (med15Δ/Δ [42]), and SN913 (ssn3Δ/Δ [43]) using PCR products amplified by primer pair LM025/LM014 (44) from the plasmids pFA6a-6×His-3×Flag-HIS1 and pFA6a-6×His-3×Flag-ARG4. These two template plasmids were generated by replacing the SAT1 marker on pFA6a-6×His-3×Flag-SAT1 (44) between the AscI and PmeI sites with HIS1 and ARG4 markers subcloned from pFA6a-TAP-HIS1 and pFA6a-TAP-ARG4 (45), respectively. Similarly, both copies of SSN3 and SSN8 were 6×His-3×Flag tagged in BWP17 (46) by using primer pairs SSN3tag_sense/SSN3tag_anti and SSN8tag_sense/SSN8tag_anti (47) to generate yLM256 and yLM255, respectively.
Each species of Mediator complex used in this study was isolated from cultures grown at 30°C in YPD without addition of any filamentous-growth-inducing agents. The Mediator complex was purified from the high-salt elution fraction of the heparin column by Flag affinity purification through the 6×His-3×Flag tag placed on the C terminus of the Med8 (cTTR03) or Ssn3 (yLM256) subunit as previously described (44). Free CDK8 module, containing only Ssn3/Cdk8, Ssn8/CycC, Ssn5/Med12, and Ssn2/Med13, was purified from the heparin flowthrough fraction of yLM255 lysate by Flag purification. The Flag purification method was identical to that described above except that the salt concentration of the heparin flowthrough was adjusted from 150 mM potassium acetate (KOAc) to 300 mM KOAc before application to the Flag-agarose. All strains used in this study are presented in Table S1 in the supplemental material.
Analysis of Cdk8-dependent phosphorylation of Mediator.
For the analysis of C. albicans mediator Cdk8 kinase assay, trichloroacetic acid (TCA)-precipitated, reduced, and alkylated proteins were trypsin digested and dried. Reductive dimethyl labeling was performed essentially as described previously (48, 49). Dried peptides were resuspended in 100 μl of 100 mM triethyl ammonium bicarbonate (TEAB) by vortexing. For “light” labeling, 0.7 μl of 37% formaldehyde was added to the sample, followed by vortexing and addition of 4 μl of 0.6 M cyanoborohydride. For “heavy” labeling, 1 μl of 30% formaldehyde-13C d2 was added to the sample, followed by vortexing and addition of 4 μl of 0.6 M cyanoborodeuteride. Samples were incubated at room temperature for 1 h. Afterwards, reactions were quenched by the addition of 10 μl of 2% NH3 solution, followed by vortexing and then incubation for 5 min at room temperature. Reactions were further quenched by the addition of 16 μl of 5% formic acid, followed by vortexing and 5 min of incubation at room temperature. Heavy and light samples were mixed, acidified with 6 μl of 20% TFA, desalted, dried, and analyzed by LC-MS/MS. LC-MS/MS analysis was performed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific) equipped with an Easy-nLC 1000 (Thermo Scientific). Peptides were redissolved in 5% ACN–1% formic acid and loaded onto a trap column (ReproSil, C18 AQ, 5 μm, 200-Å pore [Dr. Maisch, Ammerbuch, Germany]) and eluted across a fritless analytical resolving column (30-cm length, 100-μm inner diameter, ReproSil, C18 AQ, 3 μm 200-Å pore) pulled in-house with a 50-min gradient of 4 to 30% LC-MS buffer B (LC-MS buffer A includes 0.0625% formic acid and 3% ACN; LC-MS buffer B includes 0.0625% formic acid and 95% ACN) at 400 nl/min. Raw data were searched using COMET in high-resolution mode (36) against a target decoy (reversed) (37) version of the C. albicans proteome sequence database with a precursor mass tolerance of ±1 Da and requiring tryptic peptides with up to two miscleavages, carbamidomethylcysteine, and dimethylation at peptide amino termini and lysines, as fixed modifications. Oxidized methionine and phosphorylated serine, threonine, and tyrosine, isotopically heavy labeled dimethylation at peptide amino termini, and lysines were searched as variable modifications. The resulting peptide spectral matches were filtered to a <1% FDR, based on reverse hit. Probability of phosphorylation site localization was determined by PhosphoRS (38). Quantification of LC-MS/MS spectra was performed using MassChroQ (50).
Proteomics data accession number.
The proteomics data have been deposited to the ProteomeXchange Consortium (51) via the PRIDE partner repository with the data set identifier PXD001844 and 10.6019/PXD001844 and also into the Candida Genome Database (http://www.candidagenome.org).
RESULTS AND DISCUSSION
Description of the C. albicans SC5314 phosphoproteome.
We aimed to perform a deep qualitative characterization of the C. albicans phosphoproteome, and we chose to do so using C. albicans hyphae. Total proteins were extracted from growing C. albicans hyphae cultured in YPD at 37°C with 5 mM GlcNAc. Extracted proteins were trypsin digested, and phosphopeptides were enriched using titanium dioxide (TiO2). The resultant fractions were analyzed by LC-MS/MS (analysis scheme shown in Fig. 1). Eighty-six percent of the unique peptides (19,590 out of 22,783) identified contained a phosphorylation site (P site), indicating strong enrichment for phosphorylated peptides; this level of enrichment is similar to what has been reported in other studies (22). We found that 66.3% of identified phosphopeptides had a PhosphoRS score of 0.99 or higher, which corresponds to a false-localization rate (FLR) of approximately 1% (38, 52) The 19,590 phosphopeptides had an average length of 18.5 amino acids (aa), with the range being between 7 and 48 amino acids (see Table S2 in the supplemental material). We found that 11,348 (57.9%) peptides had one P site, 6,964 peptides (35.5%) had two P sites, 1,210 peptides (6.2%) had 3 P sites, and 68 peptides (0.3%) had 4 P sites (Fig. 2A).
FIG 1.
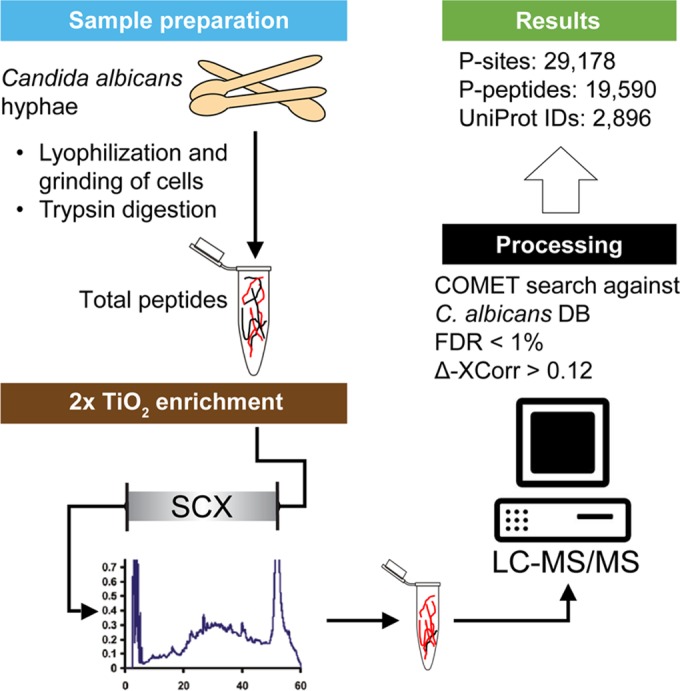
Overview of the analytical workflow. C. albicans cells, grown as hyphae, were harvested, lyophilized, and ground to a fine powder. After trypsin digestion, peptides were enriched by two rounds of TiO2 treatment, separated by a strong cation exchange (SCX) chromatography, and analyzed by LC-MS/MS. For peptide identification, spectra were searched against a custom C. albicans database using COMET. Probability of phosphorylation site localization was determined by PhosphoRS. We identified a total of 29,718 P sites (15,905 unique) on 19,590 unique peptides that can be clustered to 2,896 proteins.
FIG 2.
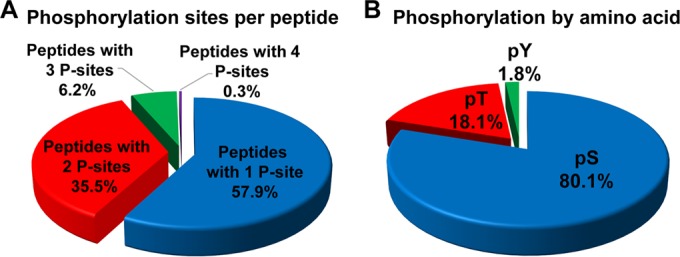
Phosphorylation sites per peptide and distribution of phosphorylated residues. (A) Percentage of singly, doubly, triply, and quadruply phosphorylated peptides; (B) distribution of unique phosphosites by modified amino acids: serine (pS), threonine (pT), and tyrosine (pY) sites.
The 19,690 phosphopeptides mapped to 15,905 unique P sites (see Table S2). We identified 12,736 (80.1%) serine, 2,881 (18.1%) threonine, and 288 (1.8%) tyrosine phosphorylation sites (Fig. 2B). This distribution is similar to what has been observed in other eukaryotic organisms, including C. neoformans (S, 81.7%; T, 17%; and Y, 1.3%) (24), Tetrahymena thermophila (S, 80.5%; T, 15.7%; and Y, 3.8%) (53), and humans (S, 85.3%; T, 12.9%; and Y, 1.8%) (54). Phosphorylation can also occur on aspartate, lysine, arginine, and histidine residues; however, those phosphorylations are comparatively difficult to detect under our experimental conditions due to their labile character under acidic conditions (55, 56). While our analysis approach was not designed to look for phosphohistidines, C. albicans does have three histidine kinases, Nik1, Sln1, and Chk1, which can autophosphorylate in response to specific cues in order to initiate a phosphorelay that results in a change in transcriptional regulation (57, 58). We detected 6 P sites (3 serine and 3 threonine) on Nik1 and 25 P sites (22 serine and 3 threonine) on Sln1; Chk1 peptides were not detected (see Table S3 in the supplemental material).
Identification of phosphorylated proteins.
To assign protein identities to the peptide spectra, we assembled a custom database by merging the Swiss-Prot/TrEMBL C. albicans database (59) and the Candida Genome Database (CGD) (60). Of the 19,590 detected phosphopeptides, 13,825 matched a single identifier (ID) in our database; 5,765 phosphopeptides matched with more than one ID. Peptides with two IDs generally reflected UniProt IDs for the same protein referring to the two alleles, since C. albicans is a diploid organism. For the peptides assigned three or more IDs, often there was a previous CGD ID that could be converted into one of the two UniProt IDs. We identified 54 phosphopeptides with 6 or more IDs. Interestingly, those peptides can be clustered into 3 groups. The first group consists of 42 phosphopeptides with the same amino acid sequence, varying only in phosphorylation pattern, within proteins belonging to a set of fungus-specific putative Rho GTPase-activating proteins (see Table S2). The second group consists of 11 phosphopeptides that matched with 7 open reading frames (ORFs) within a family of FGR6-related (fungal growth regulator) genes in the RB2 repeat sequence of the genome (61). The last group contains a phosphopeptide found within 13 of the 14 Tlo protein orthologs of S. cerevisiae Med2 (ScMed2), a component of Mediator complex (discussed further below); only Tlo protein Tlo4 does not contain this peptide sequence. To simplify the downstream analyses, CGD ORF numbers were replaced with UniProt IDs when available, and the first ID that was assigned to a cluster as the protein identifier was used. When nonoverlapping peptides with the same UniProt ID were clustered, we identified 2,896 unique UniProt IDs with the caveat that in some cases it is not clear if the peptide identified belongs to one or more than one distinct proteins.
Analysis of proteins with tyrosine phosphorylations.
While C. albicans has numerous serine/threonine kinases (14), only a single putative tyrosine kinase has been described (62). In addition, some serine/threonine kinases are known to also phosphorylate tyrosine residues, like members of the MEK/Ste7 and Wee1/Mik1 families (63, 64). We detected 253 proteins with unique UniProt identifiers (8.7% of all phosphoproteins) with tyrosine P sites. The majority of these proteins contained only one detected phosphotyrosine, only 23 proteins were found to have two phosphorylated tyrosine residues, and 6 proteins had more than three detected tyrosine P sites. The majority of proteins with phosphotyrosines also had detected phosphorylations on serines and threonines. Only 16 proteins were found to have only phosphorylated tyrosines (see Table S3 in the supplemental material). For example, the only two P sites on ribosomal protein L15 were Y6 and Y81 (see Table S3). Ddr48 was found to have the most phosphotyrosines, with seven, and Gin4 was second, with four. Gin4, a serine/threonine protein kinase that plays a role in C. albicans hypha formation and septin organization (15), was the most phosphorylated protein, with 74 unique P sites (57 serine, 13 threonine, and 4 tyrosine residues) (see Table S3). In S. cerevisiae, Gin4 interacts physically with Swe1, a kinase with the ability to phosphorylate S, T, and Y residues. Swe1 inhibits the activity of Cdc28 (Cdk1) by phosphorylation of tyrosine residue 19 in S. cerevisiae and Y18 in C. albicans, and this phosphorylation controls a morphogenesis checkpoint that delays mitosis and prevents a switch from polarized growth to isometric growth (15, 65, 66). We detected phospho-Y18 in C. albicans Cdc28, along with Cdc28 P sites at S5 and T166, suggesting that this phosphorylation pattern also exists in C. albicans hyphae (see Table S2).
GO term analysis of phosphoproteins.
To determine if any classes of proteins were enriched among the detected phosphoproteins, the percentage of proteins assigned to Gene Ontology (GO) term categories for all annotated proteins was compared to the percentages for proteins with either phospho-S/T or phospho-Y residues. The distribution of C. albicans genome-wide GO terms includes 6,314 reported ORFs with at least one GO term annotation, and this number was used to calculate the percentage of proteins within each GO category (Fig. 3, Genome). There were 2,577 proteins detected as phosphoproteins (Fig. 3, P-all). We also analyzed the GO term distribution for the 2,368 proteins found to have serine and/or threonine phosphorylations (Fig. 3, P-S/T) and the 235 proteins with tyrosine phosphorylation sites (Fig. 3, P-Y). Only GO terms that had significant enrichment of proteins were used for further analyses, and the significance cutoff for shared GO terms was a P value of <0.05 with a Bonferroni correction. The enrichment of proteins within a subset of GO Slim assignments within the general categories Biological Process, Molecular Function, and Cellular Component, are presented in Fig. 3; the percentage of proteins in the GO term category set is provided in Table S4 in the supplemental material. For each GO term where significance was met in all three phosphoprotein data sets, P-all equals the sum of P-S/T and P-Y minus any overlap between these two data sets. A gray box is in place for cases where a GO term did not make the P value cutoff of 0.05 (Fig. 3).
FIG 3.

GO enrichment analysis of the C. albicans phosphoproteome. Heat maps indicate the rank abundance of proteins within selected significant GO Slim categories (P value cutoff = 0.05) within the Biological Process (A), Molecular Function (B), and Cellular Component (C) gene ontologies. For each set of proteins (Genome, P-all, P-S/T, and P-Y), the percentages of proteins associated with each GO term were used to rank the distribution of proteins across the GO terms analyzed. See Table S4 in the supplemental material for the data used in these analyses.
Comparison of the rank-based distribution of the GO assignments of the total phosphorylation data set to the whole predicted proteome revealed a similar distribution of terms, showing no detectable bias in what classes of proteins were phosphorylated (Fig. 3). As the vast majority of P sites were on serine or threonine residues, it was not surprising that P-S/T GO term distribution was similar to that for the genome and the complete phosphoprotein data set. In contrast, the small set of proteins with detected tyrosine phosphorylations differed in their GO Slim term distribution (Fig. 3). For example, the P-Y proteins had a larger fraction of proteins with Biological Process classification of “response to extracellular stimuli,” “signal transduction,” and “regulation of cellular process,” while there was no significant enrichment in the GO term “metabolic process,” which contained the largest percentage of proteins encoded in the genome-, P-all, and P-S/T data sets (Fig. 3A). In the Molecular Function ontology, tyrosine-phosphorylated proteins were present in only 3 of the 8 selected GO terms. Interestingly, there was a larger fraction of P-Y proteins in three Molecular Function categories (DNA binding, kinase, and signal transducer activity) than in the other data sets; only P-Y phosphoproteins were enriched for the “signal transducer activity” GO term category (Fig. 3B). Analyses of proteins with the GO terms in the Cellular Component ontology revealed that tyrosine phosphoproteins were predominantly in the cytoplasm and the nucleus and were generally absent from membranes or membrane-bound organelles. In contrast, S/T phosphoproteins were not specifically enriched in or excluded from either membrane-associated categories or the cytoplasm (Fig. 3C). Analysis of all phosphoproteins (P-all) revealed that the most represented Cellular Component categories were “nucleus” and “cytoplasm,” and this result is comparable to what was observed in Aspergillus nidulans (25).
Phosphorylation sites per protein.
The average number of phosphorylation sites per protein is 5.5; however, the distribution of P sites across proteins is skewed. For example, 786 of 2,896 proteins had a single detected P site and 509 proteins contained two detected P sites (Fig. 4). In contrast, 19 proteins had more than 45 detected P sites (Table 1). These highly phosphorylated proteins have an average length of 1,419 aa, which is larger than the average protein length in C. albicans (480 aa) (67). Thus, the large size of the protein may account for some, but not all, of the higher-level phosphorylation. In addition to the highly phosphorylated Gin4 protein, Hsl1, a related kinase also involved in the regulation of Swe1, was also highly phosphorylated (Table 1) (15). Hsl1 had 62 P sites (52 serine and 10 threonine residues), including P sites at S782 and S1299. These Hsl1 P sites are consistent with the findings of Umeyama et al. (68), which showed that C. albicans Hsl1 is phosphorylated at S33, S782, and S1299 by Cdc28 and other kinases.
FIG 4.
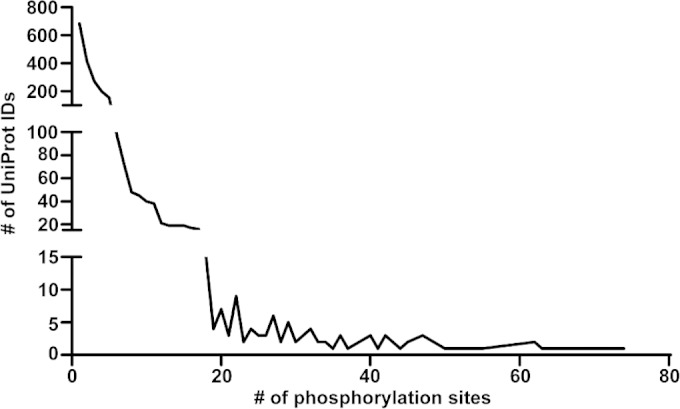
Phosphorylation sites per protein. Of the 2,896 unique proteins detected, 685 had only 1 P site, 415 had 2 P sites, and 271 had 3 P sites. More heavily phosphorylated proteins were less commonly observed. The protein with the most P sites (74 P sites) is the serine/threonine-protein kinase Gin4 (Table 1).
TABLE 1.
Phosphorylated proteins with >45 P sites
| No. of phosphorylation sites | Gene name | Length of protein (aa) | Feature |
|---|---|---|---|
| 74 | GIN4 | 1,349 | Serine/threonine protein kinase Gin4 |
| 73 | CaO19.14160 | 1,352 | Putative uncharacterized protein |
| 72 | BUD4, INT1 | 1,711 | Bud site selection protein Bud4 |
| 66 | CTA3 | 1,217 | Potential EH domains and endocytosis protein |
| 63 | SPA2 | 1,466 | Putative uncharacterized protein Spa2 |
| 62 | ZDS1 | 1,645 | Potential regulator of cell polarity |
| 62 | HSL1 | 1,462 | Serine/threonine protein kinase Hsl1 |
| 58 | FAB1 | 2,624 | Uncharacterized protein |
| 57 | BNI4 | 1,655 | Possible bud neck-localizing protein phosphatase subunit Bni4 |
| 53 | CaO19.9831 | 1,309 | Uncharacterized protein |
| 50 | CaO19.11901 | 1,073 | Potential GRAM domain protein |
| 47 | CaO19.11117 | 1,563 | Uncharacterized protein |
| 47 | BOI2 | 1,172 | Putative uncharacterized protein Boi2 |
| 47 | RGA2 | 1,176 | Putative uncharacterized protein Rga2 |
| 46 | CaO19.8830 | 1,043 | Uncharacterized protein |
| 45 | CaO19.7006 | 1,094 | Putative uncharacterized protein |
| 45 | CaO19.7738 | 896 | Uncharacterized protein |
| 45 | KIN1 | 1,212 | Likely protein kinase |
| 45 | SEC16 | 1,947 | Putative uncharacterized protein Sec16 |
Among the highly phosphorylated proteins, we observed an enrichment of proteins that are localized to the bud neck and are involved in septin ring formation, including Bud4, Bni4, and Spa2 in addition to Gin4 and Hsl1 (69, 70). These proteins have been associated with hyphal growth and establishment or maintenance of cytoskeleton polarity (71). Future studies will reveal how changes in phosphorylation state of these proteins participate in the induction and maintenance of different growth states.
Phosphorylation sites within the Ras1-Cyr1-PKA pathway.
The Ras/cyclic AMP (cAMP)/PKA signaling pathway (72), a central regulatory pathway involved in the induction and maintenance of hyphal growth, may be regulated by phosphorylation in a variety of different ways (Fig. 5). Ras1, which is active in its GTP-bound state and inactive in its GDP-bound conformation, is controlled by a Ras-GTPase-activating protein (GAP), Ira2, and the guanine nucleotide exchange factor (GEF) Cdc25. We found P sites on both Ira2 and Cdc25. Ras1 interacts activates adenylate cyclase Cyr1 (73), and the resulting increase in cAMP activates the catalytic subunits of protein kinase A (Tpk1 and -2) by causing their release from the regulatory subunit Bcy1. The activated PKA subunits likely have many targets, such as transcription factors, including Efg1 (72, 74). In S. cerevisiae, Bcy1 itself is regulated by phosphorylation, in part by PKA subunits. Bcy1 that has been phosphorylated at S145 by PKA catalytic subunits causes Bcy1 to be a more efficient inhibitor of Tpk1, Tpk2, and Tpk3 (75). An alignment of ScBcy1 and C. albicans Bcy1 (CaBcy1) shows that the two proteins are highly similar; ScBcy1 S145, found within the canonical RRxS/T PKA motif, corresponds to CaBcy1 S180 (76) (Fig. 5B). In Fig. 5B, blue arrows indicate the six phosphorylation sites that we identified for Bcy1 in our data set, including S180.
FIG 5.

Phosphorylation pattern of proteins in the Ras1-Cyr1-PKA pathway in C. albicans. (A) Schematic model and the phosphorylation states of the Ras1-Cyr1-PKA pathway in C. albicans. Ras1 activity is governed by Ira2 and Cdc25, which both contain P sites. Ras1 induces adenylate cyclase (Cyr1) to produce cAMP, which inhibits the formation of the protein kinase A complex consisting of the catalytic subunits Tpk1 and -2, and the regulatory subunit Bcy1, by directly binding to Bcy1. Phosphorylation states are depicted by color (open ovals, no phosphorylation; green ovals, <3 P sites; orange ovals, between 4 and 10 P sites; and red jagged ovals, >10 P sites). (B) An alignment of ScBcy1 and CaBcy1 shows high overall similarity. S. cerevisiae Ser145 is within an RRxS/T motif which is recognized by PKA (red box); this serine corresponds to Ser180 in C. albicans. The blue arrows indicate the Bcy1 P sites (Q9HEW1; see Table S3 in the supplemental material).
Besides Bcy1, other proteins known to be phosphorylated by PKA in S. cerevisiae were also phosphorylated within a putative PKA motif in our data set. For example, the forkhead transcription factor Fhl1 and its regulator Ifh1 (77) and the autophagy regulators Atg1 and Atg13 (78) were found. Efg1, reported as a PKA target at T206 (18, 79, 80), was found to be phosphorylated at six sites (5 serine and 1 threonine) but not at the T206 position, suggesting that PKA-mediated phosphorylation of Efg1 is dynamic (79). The amino acid sequence of Efg1 around the important threonine 206 is IRPRVTTT (the underline indicates threonine 206), which varies only by one amino acid from the PKA consensus sequence.
Cdk8-dependent and independent phosphorylation of Mediator.
The multiprotein complex Mediator is a coactivator of RNA polymerase II transcription. In other species, the Mediator complex is known to be phosphorylated by multiple kinases, and the phosphorylation states of proteins within the complex can impact gene expression (81). The Mediator complex in C. albicans is composed of at least 23 subunits plus a Med2 ortholog, which in C. albicans is encoded by 14 different paralogs (referred to as Tlo proteins), which are held in the tail module through interaction with Med3 (44). We observed P sites in 17 out of the 23 subunits with Med15, the most phosphorylated Mediator protein, with 16 distinct P sites (Table 2). A peptide that is conserved in 13 of the 14 Tlo proteins also contained a P site, indicating phosphorylation of one or more Tlo subunits.
TABLE 2.
Mediator complex P sites
| UniProt ID | Gene ID | P site(s)a as detected by: |
||
|---|---|---|---|---|
| Large-scale phosphoproteomics | In vitro Cdk8 kinase assay (ATP dependent) | In vitro Cdk8 kinase assay (ATP independent) | ||
| Q5AHH0 | Med1 | S445, S446 | S445, S446 | |
| Q59Q94 | Med3 | S117, T125, T133 | T140 | S117, S181, S183, T119, T121, T125, T132, T133 |
| Q59U73 | Med4 | S9, S13, S17, S33, S322, T36, T37, Y321 | S9, S33, T36, T37 | S9, S13 |
| Q59KC0 | Med5 | S252 | S252 | |
| Q5A2Z1 | Med6 | |||
| Q5AEN6 | Med7 | S285 | ||
| Q59TD3 | Med8 | T37 | ||
| Q5A0D0 | Med9 | S102 | T109 | |
| Q5A3K2 | Med10 | S170 | ||
| Q59S43 | Med11 | S132, S142, T140, T144, T149 | S120, S132, S142, T140 | |
| Q59MN9 | Med12 | S594, S1033, S1579, S1680, S1682, S1713, T1039 | S63 | |
| Q5AG31 | Med14 | S385, T395 | S14, S385, S678, S763, S1170 | |
| Q5A757 | Med15 | S898, S939, S948, S1035, S1043, S1046, S1084, S1097, S1110, T903, T906, T913, T914, T937, T943, T1042 | S898, S900, S1082, S1084, T906, T913, T1030 | S898, S900, S948, S1043, S1082, S1083, S1084, T977, T1030, T1042 |
| Q59PP6 | Med16 | S380 | ||
| Q5AHZ7 | Med17 | S43, S73, T70 | ||
| Q59X40 | Med18 | S207, T162 | ||
| Med19 | ||||
| C4YHG8 | Med20 | |||
| Q5ACU4 | Med21 | S79, S83 | S79 | S79, S85, S87 |
| Q5A917 | Med22 | |||
| Q59P87 | Med31 | |||
| Q5ALX5 | Ssn2 (Med13) | S399, S478, S493, S605, S615, S650, S652, S840, T476, T494, T774, T1449 | S493, T476, T774, T1449 | S348, S605, S833, S1343, S1345 |
| Q5AHK2 | Ssn3 (Cdk8) | S209 | S209 | |
| Q5A4H9 | Ssn8 | |||
| Tlo | S39 | T38 | ||
P sites detected in the large-scale phosphoproteomics data set and the ATP-dependent in vitro Cdk8 kinase assay are in bold, and P sites detected in the large-scale phosphoproteomics data set and the ATP-independent in vitro Cdk8 kinase assay are in italics.
Cdk8/Ssn3 is a kinase that is part of the CDK8 module of Mediator, and it has been shown to modulate Mediator and transcription factor activity in ways that impact hyphal growth, switching, metabolism, and stress resistance (42, 43, 47, 82). To directly determine if Cdk8 catalyzes the phosphorylation of Mediator subunits, we performed in vitro kinase assays using purified Mediator or mixtures of Mediator subcomplexes. First, we found that when the native complex, including the Cdk8 module, was incubated with [32P]ATP, we detected phosphorylations on proteins with mobilities that most likely corresponded to those of Cdk8, Med8, Med9, and Med15. In addition, we detected phosphorylation of protein(s) in a band whose molecular mass corresponded to either Med12 or Med13, as well as another band (Mp48) whose molecular mass corresponded to either Med4, Med7, or Med18 (Fig. 6A). Subsequent phosphoproteomics, discussed below, indicate that the bands that could not be unambiguously assigned were most likely Med13 and Med4, respectively. All [32P]ATP-dependent phosphorylations were absent when Mediator was purified from a med12Δ/Δ strain, in which the Cdk8-kinase containing module is no longer associated with the core Mediator complex, or from a cdk8Δ/Δ strain lacking the Cdk8 kinase itself (Fig. 6A). This shows that the likelihood of contaminating kinases contributing to phosphorylation in these assays is low. The identified phosphorylation events could be rescued by providing purified Cdk8 module in trans to purified Mediator complex from either med12Δ/Δ or cdk8Δ/Δ (Fig. 6A). Additional experiments were performed to verify the identities of Med8, Med9, Med15, and Cdk8 as targets. In vitro assays performed using Mediator purified from mutants lacking either Med15 or Med9 showed that the respective bands corresponding to those phosphorylated proteins were missing in these samples (Fig. 6B, lanes 1 and 2). Furthermore, when the kinase assay was performed with Mediator from a strain bearing either Cdk8-HF or Med8-HF (6×His-3×FLAG), we detected the expected mass shifts in the corresponding bands (Fig. 6B, lanes 3 and 4). All proteins other than Med8 that showed in vitro phosphorylation were also detected as phosphorylated proteins in our large phosphoproteomics data set (Fig. 7). The absence of Med8 from the large-scale data set is not surprising, since the tryptic peptide generated form the N terminus of Med8, which contains the most likely Cdk8 P sites, is above the molecular mass typically detected by this methodology. To determine if Cdk8 can phosphorylate a Tlo protein family member and Med3, we performed an [32P]ATP kinase assay in which purified Mediator was incubated with a complex of recombinant Tloα12 bound to 6×His-tagged Med3 (44). The phosphorylation pattern showed that the recombinant proteins were detected in a dose-dependent manner at the expected sizes (Fig. 6C). Phosphorylated bands for the native Tlo proteins associated with Mediator were not observed, likely because any one of the 14 different Tlo paralogs, with varying molecular masses, were not sufficiently abundant.
FIG 6.

Western blot analysis of phosphorylation of Mediator subunits by Cdk8. (A) Autoradiographic images of in vitro kinase assay data are shown for reaction mixtures containing [32P]ATP and the Mediator complex purified from C. albicans via the Med8-HF tag (“HF” refers to a 6×His-3×Flag tag) (lane 1), Mediator purified from a Δmed12 strain (which lacks the Cdk8 module) without and with purified Cdk8 module (lanes 2 and 3, respectively), and Mediator purified from a cdk8Δ/Δ strain (Δcdk8) without and with purified Cdk8 module (lanes 4 and 5, respectively). Cdk8 module alone shows autophosphorylation within the module (lane 6). (B) Similar experiments were performed with med15Δ/Δ (Δmed15) and med9Δ/Δ (Δmed9) to confirm the identity of those bands by showing their absence in the appropriate strain. In vitro kinase assays with strains expressing either Cdk8-HF or Med8-HF also demonstrated mass shifts that confirmed protein identities. (C) Purified Mediator with Cdk8 as the sole kinase was used in an experiment to demonstrate that Cdk8 phosphorylates the Tlo protein family and Med3. Spiked-in recombinant Tloα12 and 6×His-tagged Med3 from C. albicans could be detected in a dose-dependent manner at the expected sizes.
FIG 7.

Summary of phosphorylation analyses of Mediator complex. The overall structure of the Mediator complex is shown. Subunits phosphorylated in vitro by the Cdk8 kinase are indicated by a bold outline. Subunits with P sites identified in the large-scale phosphoproteomics analysis are shown in color. Subunits labeled in bold letters have been identified in a phosphoproteomics analysis of in vitro CDK8 activity using purified Mediator. P sites in purified Mediator that are enriched after incubation with ATP are indicated in parentheses.
To further define the P sites that were attributable to Cdk8 activity and compare these sites to those detected in the large-scale phosphoproteomics effort, we performed an in vitro analysis using purified Mediator. To quantitatively determination which Mediator P sites could be a consequence of Cdk8 activity, we analyzed purified Mediator, in which Cdk8 is the only protein with kinase activity, after incubation with and without ATP. Differential methyl labeling of the samples incubated with and without ATP prior to sample mixing allowed for direct quantitative analysis of newly phosphorylated sites versus those present on purified Mediator prior to ATP addition. The results confirmed and refined our previous assignments of phosphorylated subunits (Fig. 6) and identified the specific sites phosphorylated by Cdk8 in purified Mediator (Table 2). Consistent with the data shown in Fig. 6, Cdk8-dependent P sites were found in subunits Med3, Med8, Med9, Cdk8, and Med15 (Table 2). Of Med4, Med7, and Med18, which could not be distinguished in the [32P]ATP in vitro assays (Fig. 6A), Med4 was the only one that was found to be phosphorylated; 4 of the 5 Med4 P sites were heavily enriched among the ATP-dependent P sites. Ssn2/Med13, but not Med12, had also many sites enriched in the ATP-dependent P sites. This analysis also revealed ATP-independent P sites already present in the purified Mediator, which could have resulted from the in vivo activity of Cdk8 or other kinases (Table 2).
Comparison of the ATP-dependent P sites identified in the in vitro Cdk8 kinase assay to those identified in a phosphoproteomics analysis of S. cerevisiae Mediator (81) revealed some general similarities but no single P site that was clearly highly conserved. For instance, both our data and the S. cerevisiae work showed high phosphorylation of the C terminus of Med15, which is thought to be associated with suppression of stress-induced transcription (81). Our work advances the S. cerevisiae study, since those authors were unable to identify P sites in the Cdk8 module due the absence of the module in their purified Mediator sample. We found an autophosphorylation site in Ssn3/Cdk8 as well as several ATP (Cdk8)-dependent and independent P sites in Ssn2/Med13 that potentially play regulatory roles. The results also showed that the large-scale phosphoproteomics analysis of whole-cell extracts was reasonably adept at picking up both Cdk8-dependent and independent P sites in Mediator. In the large-scale phosphoproteomics data set, we identified 68 P sites on 18 of the 24 Mediator subunits plus the Tlo protein family. The in vitro Cdk8 kinase analyses found 20 ATP-dependent P sites, with 12 that were also present in the large-scale experiment (Fig. 7; Table 2). It is interesting that a majority of the Mediator P sites detected in the large-scale proteomics were not detected in the purified sample; however, the discrepancy could be explained by the different growth conditions that were used to perform the two experiments, and it is well known that phosphorylation levels of the Mediator change in response to external stimuli (81). This finding also suggests that the large-scale methodology may be highly valuable in identifying highly labile P sites that do not survive purification of the complex.
Conclusions.
To our knowledge, this report is the first comprehensive phosphoproteomics study of hyphae in the human fungal pathogen C. albicans. In this data set, we found phosphorylation targets that have been previously identified experimentally both in either C. albicans and in S. cerevisiae, such as those in the highly conserved Ras1-Cyr1-PKA pathway and in the Mediator complex. It will be interesting to see if the level of conservation of phosphorylation sites across other species in different pathways as more studies characterizing the phosphoproteome are published. These phosphoproteomics data can be used as a resource for future targeted and large-scale phosphoprotein analyses in this fungus or as a reference for other fungi.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by National Institutes of Health (NIH) grant R01GM108492 (D.A.H. and J.E.S.), the Hitchcock Foundation (L.C.M.), American Cancer Society research grant IRG-82-003-30 (A.N.K.), and the Cystic Fibrosis Research Development Program (STANTO07R0) (S.D.W.). The Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific) was acquired with support from the NIH (S10-OD016212).
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
We thank Allia Lindsay for her analysis of Cdk8 phosphorylation sites in preliminary studies.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00011-15.
REFERENCES
- 1.Trofa D, Gacser A, Nosanchuk JD. 2008. Candida parapsilosis, an emerging fungal pathogen. Clin Microbiol Rev 21:606–625. doi: 10.1128/CMR.00013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 3.Calderone RA, Fonzi WA. 2001. Virulence factors of Candida albicans. Trends Microbiol 9:327–335. doi: 10.1016/S0966-842X(01)02094-7. [DOI] [PubMed] [Google Scholar]
- 4.Calderone RA, Clancy CJ. 2012. Candida and candidiasis. ASM Press, Washington, DC. [Google Scholar]
- 5.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naglik JR, Richardson JP, Moyes DL. 2014. Candida albicans pathogenicity and epithelial immunity. PLoS Pathog 10:e1004257. doi: 10.1371/journal.ppat.1004257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soll DR. 2014. The role of phenotypic switching in the basic biology and pathogenesis of Candida albicans. J Oral Microbiol 6:22993 http://dx.doi.org/10.3402/jom.v6.22993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonhomme J, d'Enfert C. 2013. Candida albicans biofilms: building a heterogeneous, drug-tolerant environment. Curr Opin Microbiol 16:398–403. doi: 10.1016/j.mib.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Hall RA, Gow NA. 2013. Mannosylation in Candida albicans: role in cell wall function and immune recognition. Mol Microbiol 90:1147–1161. doi: 10.1111/mmi.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saville SP, Lazzell AL, Monteagudo C, Lopez-Ribot JL. 2003. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot Cell 2:1053–1060. doi: 10.1128/EC.2.5.1053-1060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumamoto CA, Vinces MD. 2005. Alternative Candida albicans lifestyles: growth on surfaces. Annu Rev Microbiol 59:113–133. doi: 10.1146/annurev.micro.59.030804.121034. [DOI] [PubMed] [Google Scholar]
- 12.Monge RA, Roman E, Nombela C, Pla J. 2006. The MAP kinase signal transduction network in Candida albicans. Microbiology 152:905–912. doi: 10.1099/mic.0.28616-0. [DOI] [PubMed] [Google Scholar]
- 13.Bruckmann A, Kunkel W, Hartl A, Wetzker R, Eck R. 2000. A phosphatidylinositol 3-kinase of Candida albicans influences adhesion, filamentous growth and virulence. Microbiology 146(Part 11):2755–2764. [DOI] [PubMed] [Google Scholar]
- 14.Blankenship JR, Fanning S, Hamaker JJ, Mitchell AP. 2010. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog 6:e1000752. doi: 10.1371/journal.ppat.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wightman R, Bates S, Amornrrattanapan P, Sudbery P. 2004. In Candida albicans, the Nim1 kinases Gin4 and Hsl1 negatively regulate pseudohypha formation and Gin4 also controls septin organization. J Cell Biol 164:581–591. doi: 10.1083/jcb.200307176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindsay AK, Deveau A, Piispanen AE, Hogan DA. 2012. Farnesol and cyclic AMP signaling effects on the hypha-to-yeast transition in Candida albicans. Eukaryot Cell 11:1219–1225. doi: 10.1128/EC.00144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bockmühl DP, Krishnamurthy S, Gerads M, Sonneborn A, Ernst JF. 2001. Distinct and redundant roles of the two protein kinase A isoforms Tpk1p and Tpk2p in morphogenesis and growth of Candida albicans. Mol Microbiol 42:1243–1257. doi: 10.1046/j.1365-2958.2001.02688.x. [DOI] [PubMed] [Google Scholar]
- 18.Sonneborn A, Bockmuhl DP, Gerads M, Kurpanek K, Sanglard D, Ernst JF. 2000. Protein kinase A encoded by TPK2 regulates dimorphism of Candida albicans. Mol Microbiol 35:386–396. doi: 10.1046/j.1365-2958.2000.01705.x. [DOI] [PubMed] [Google Scholar]
- 19.Berg JM TJ, Stryer L. 2002. Biochemistry, 5th ed W H Freeman, New York, NY. [Google Scholar]
- 20.Yachie N, Saito R, Sugiyama N, Tomita M, Ishihama Y. 2011. Integrative features of the yeast phosphoproteome and protein-protein interaction map. PLoS Comput Biol 7:e1001064. doi: 10.1371/journal.pcbi.1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amoutzias GD, He Y, Lilley KS, Van de Peer Y, Oliver SG. 2012. Evaluation and properties of the budding yeast phosphoproteome. Mol Cell Proteomics 11:M111.009555. doi: 10.1074/mcp.M111.009555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kettenbach AN, Gerber SA. 2011. Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal Chem 83:7635–7644. doi: 10.1021/ac201894j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rampitsch C, Tinker NA, Subramaniam R, Barkow-Oesterreicher S, Laczko E. 2012. Phosphoproteome profile of Fusarium graminearum grown in vitro under nonlimiting conditions. Proteomics 12:1002–1005. doi: 10.1002/pmic.201100065. [DOI] [PubMed] [Google Scholar]
- 24.Selvan LD, Renuse S, Kaviyil JE, Sharma J, Pinto SM, Yelamanchi SD, Puttamallesh VN, Ravikumar R, Pandey A, Prasad TS, Harsha HC. 2014. Phosphoproteome of Cryptococcus neoformans. J Proteomics 97:287–295. doi: 10.1016/j.jprot.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 25.Ramsubramaniam N, Harris SD, Marten MR. 2014. The phosphoproteome of Aspergillus nidulans reveals functional association with cellular processes involved in morphology and secretion. Proteomics 14:2454–2459. doi: 10.1002/pmic.201400063. [DOI] [PubMed] [Google Scholar]
- 26.Davanture M, Dumur J, Bataille-Simoneau N, Campion C, Valot B, Zivy M, Simoneau P, Fillinger S. 2014. Phosphoproteome profiles of the phytopathogenic fungi Alternaria brassicicola and Botrytis cinerea during exponential growth in axenic cultures. Proteomics 14:1639–1645. doi: 10.1002/pmic.201300541. [DOI] [PubMed] [Google Scholar]
- 27.Xiong Y, Coradetti ST, Li X, Gritsenko MA, Clauss T, Petyuk V, Camp D, Smith R, Cate JH, Yang F, Glass NL. 2014. The proteome and phosphoproteome of Neurospora crassa in response to cellulose, sucrose and carbon starvation. Fungal Genet Biol 72:21–33. doi: 10.1016/j.fgb.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson-Grady JT, Villen J, Gygi SP. 2008. Phosphoproteome analysis of fission yeast. J Proteome Res 7:1088–1097. doi: 10.1021/pr7006335. [DOI] [PubMed] [Google Scholar]
- 29.Raithatha S, Su TC, Lourenco P, Goto S, Sadowski I. 2012. Cdk8 regulates stability of the transcription factor Phd1 to control pseudohyphal differentiation of Saccharomyces cerevisiae. Mol Cell Biol 32:664–674. doi: 10.1128/MCB.05420-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chi Y, Huddleston MJ, Zhang X, Young RA, Annan RS, Carr SA, Deshaies RJ. 2001. Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev 15:1078–1092. doi: 10.1101/gad.867501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hallberg M, Polozkov GV, Hu GZ, Beve J, Gustafsson CM, Ronne H, Bjorklund S. 2004. Site-specific Srb10-dependent phosphorylation of the yeast Mediator subunit Med2 regulates gene expression from the 2-μm plasmid. Proc Natl Acad Sci U S A 101:3370–3375. doi: 10.1073/pnas.0400221101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirst M, Kobor MS, Kuriakose N, Greenblatt J, Sadowski I. 1999. GAL4 is regulated by the RNA polymerase II holoenzyme-associated cyclin-dependent protein kinase SRB10/CDK8. Mol Cell 3:673–678. doi: 10.1016/S1097-2765(00)80360-3. [DOI] [PubMed] [Google Scholar]
- 33.Nelson C, Goto S, Lund K, Hung W, Sadowski I. 2003. Srb10/Cdk8 regulates yeast filamentous growth by phosphorylating the transcription factor Ste12. Nature 421:187–190. doi: 10.1038/nature01243. [DOI] [PubMed] [Google Scholar]
- 34.Gillum AM, Tsay EY, Kirsch DR. 1984. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol Gen Genet 198:179–182. doi: 10.1007/BF00328721. [DOI] [PubMed] [Google Scholar]
- 35.Villén J, Beausoleil SA, Gerber SA, Gygi SP. 2007. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A 104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eng JK, Jahan TA, Hoopmann MR. 2013. Comet: an open-source MS/MS sequence database search tool. Proteomics 13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 37.Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 38.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. 2011. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 39.Wang T, Kettenbach AN, Gerber SA, Bailey-Kellogg C. 2012. MMFPh: a maximal motif finder for phosphoproteomics datasets. Bioinformatics 28:1562–1570. doi: 10.1093/bioinformatics/bts195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. 2004. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, Maechler M, Magnusson A, Moeller S, Schwartz M, Venables B. 2015. Various R programming tools for plotting data, 2.16.0. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 42.Zhang A, Liu Z, Myers LC. 2013. Differential regulation of white-opaque switching by individual subunits of Candida albicans mediator. Eukaryot Cell 12:1293–1304. doi: 10.1128/EC.00137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C, Noble SM. 2012. Post-transcriptional regulation of the Sef1 transcription factor controls the virulence of Candida albicans in its mammalian host. PLoS Pathog 8:e1002956. doi: 10.1371/journal.ppat.1002956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang A, Petrov KO, Hyun ER, Liu Z, Gerber SA, Myers LC. 2012. The Tlo proteins are stoichiometric components of Candida albicans mediator anchored via the Med3 subunit. Eukaryot Cell 11:874–884. doi: 10.1128/EC.00095-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lavoie H, Sellam A, Askew C, Nantel A, Whiteway M. 2008. A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans. BMC Genomics 9:578. doi: 10.1186/1471-2164-9-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson RB, Davis D, Mitchell AP. 1999. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol 181:1868–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindsay AK, Morales DK, Liu Z, Grahl N, Zhang A, Willger SD, Myers LC, Hogan DA. 2014. Analysis of Candida albicans mutants defective in the Cdk8 module of mediator reveal links between metabolism and biofilm formation. PLoS Genet 10:e1004567. doi: 10.1371/journal.pgen.1004567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. 2009. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 49.Kettenbach AN, Sano H, Keller SR, Lienhard GE, Gerber SA. 2015. SPECHT—single-stage phosphopeptide enrichment and stable-isotope chemical tagging: quantitative phosphoproteomics of insulin action in muscle. J Proteomics 114:48–60. doi: 10.1016/j.jprot.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valot B, Langella O, Nano E, Zivy M. 2011. MassChroQ: a versatile tool for mass spectrometry quantification. Proteomics 11:3572–3577. doi: 10.1002/pmic.201100120. [DOI] [PubMed] [Google Scholar]
- 51.Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. 2014. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marx H, Lemeer S, Schliep JE, Matheron L, Mohammed S, Cox J, Mann M, Heck AJ, Kuster B. 2013. A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics. Nat Biotechnol 31:557–564. doi: 10.1038/nbt.2585. [DOI] [PubMed] [Google Scholar]
- 53.Tian M, Chen X, Xiong Q, Xiong J, Xiao C, Ge F, Yang F, Miao W. 2014. Phosphoproteomic analysis of protein phosphorylation networks in Tetrahymena thermophila, a model single-celled organism. Mol Cell Proteomics 13:503–519. doi: 10.1074/mcp.M112.026575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song C, Ye M, Liu Z, Cheng H, Jiang X, Han G, Songyang Z, Tan Y, Wang H, Ren J, Xue Y, Zou H. 2012. Systematic analysis of protein phosphorylation networks from phosphoproteomic data. Mol Cell Proteomics 11:1070–1083. doi: 10.1074/mcp.M111.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cieśla J, Fraczyk T, Rode W. 2011. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim Pol 58:137–148. [PubMed] [Google Scholar]
- 56.Hohenester UM, Ludwig K, Krieglstein J, Konig S. 2010. Stepchild phosphohistidine: acid-labile phosphorylation becomes accessible by functional proteomics. Anal Bioanal Chem 397:3209–3212. doi: 10.1007/s00216-009-3372-x. [DOI] [PubMed] [Google Scholar]
- 57.Kruppa M, Jabra-Rizk MA, Meiller TF, Calderone R. 2004. The histidine kinases of Candida albicans: regulation of cell wall mannan biosynthesis. FEMS Yeast Res 4:409–416. doi: 10.1016/S1567-1356(03)00201-0. [DOI] [PubMed] [Google Scholar]
- 58.Li D, Agrellos OA, Calderone R. 2010. Histidine kinases keep fungi safe and vigorous. Curr Opin Microbiol 13:424–430. doi: 10.1016/j.mib.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 59.The UniProt Consortium. 2014. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42:D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inglis DO, Arnaud MB, Binkley J, Shah P, Skrzypek MS, Wymore F, Binkley G, Miyasato SR, Simison M, Sherlock G. 2012. The Candida genome database incorporates multiple Candida species: multispecies search and analysis tools with curated gene and protein information for Candida albicans and Candida glabrata. Nucleic Acids Res 40:D667–D674. doi: 10.1093/nar/gkr945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uhl MA, Biery M, Craig N, Johnson AD. 2003. Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J 22:2668–2678. doi: 10.1093/emboj/cdg256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Z, Jin Q, Xu JR, Liu H. 2014. Identification of a fungi-specific lineage of protein kinases closely related to tyrosine kinases. PLoS One 9:e89813. doi: 10.1371/journal.pone.0089813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fattaey A, Booher RN. 1997. Myt1: a Wee1-type kinase that phosphorylates Cdc2 on residue Thr14. Prog Cell Cycle Res 3:233–240. [DOI] [PubMed] [Google Scholar]
- 64.Dhanasekaran N, Premkumar Reddy E. 1998. Signaling by dual specificity kinases. Oncogene 17:1447–1455. doi: 10.1038/sj.onc.1202251. [DOI] [PubMed] [Google Scholar]
- 65.Sia RA, Herald HA, Lew DJ. 1996. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell 7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harvey SL, Kellogg DR. 2003. Conservation of mechanisms controlling entry into mitosis: budding yeast wee1 delays entry into mitosis and is required for cell size control. Curr Biol 13:264–275. doi: 10.1016/S0960-9822(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 67.Braun BR, van Het Hoog M, d'Enfert C, Martchenko M, Dungan J, Kuo A, Inglis DO, Uhl MA, Hogues H, Berriman M, Lorenz M, Levitin A, Oberholzer U, Bachewich C, Harcus D, Marcil A, Dignard D, Iouk T, Zito R, Frangeul L, Tekaia F, Rutherford K, Wang E, Munro CA, Bates S, Gow NA, Hoyer LL, Kohler G, Morschhauser J, Newport G, Znaidi S, Raymond M, Turcotte B, Sherlock G, Costanzo M, Ihmels J, Berman J, Sanglard D, Agabian N, Mitchell AP, Johnson AD, Whiteway M, Nantel A. 2005. A human-curated annotation of the Candida albicans genome. PLoS Genet 1:36–57. doi: 10.1371/journal.pgen.0010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Umeyama T, Kaneko A, Nagai Y, Hanaoka N, Tanabe K, Takano Y, Niimi M, Uehara Y. 2005. Candida albicans protein kinase CaHsl1p regulates cell elongation and virulence. Mol Microbiol 55:381–395. doi: 10.1111/j.1365-2958.2004.04405.x. [DOI] [PubMed] [Google Scholar]
- 69.Pruyne D, Bretscher A. 2000. Polarization of cell growth in yeast. I. Establishment and maintenance of polarity states. J Cell Sci 113(Part 3):365–375. [DOI] [PubMed] [Google Scholar]
- 70.Gale CA, Bendel CM, McClellan M, Hauser M, Becker JM, Berman J, Hostetter MK. 1998. Linkage of adhesion, filamentous growth, and virulence in Candida albicans to a single gene, INT1. Science 279:1355–1358. doi: 10.1126/science.279.5355.1355. [DOI] [PubMed] [Google Scholar]
- 71.Zheng XD, Wang YM, Wang Y. 2003. CaSPA2 is important for polarity establishment and maintenance in Candida albicans. Mol Microbiol 49:1391–1405. doi: 10.1046/j.1365-2958.2003.03646.x. [DOI] [PubMed] [Google Scholar]
- 72.Hogan DA, Sundstrom P. 2009. The Ras/cAMP/PKA signaling pathway and virulence in Candida albicans. Future Microbiol 4:1263–1270. doi: 10.2217/fmb.09.106. [DOI] [PubMed] [Google Scholar]
- 73.Wang Y. 2013. Fungal adenylyl cyclase acts as a signal sensor and integrator and plays a central role in interaction with bacteria. PLoS Pathog 9:e1003612. doi: 10.1371/journal.ppat.1003612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inglis DO, Sherlock G. 2013. Ras signaling gets fine-tuned: regulation of multiple pathogenic traits of Candida albicans. Eukaryot Cell 12:1316–1325. doi: 10.1128/EC.00094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Budhwar R, Lu A, Hirsch JP. 2010. Nutrient control of yeast PKA activity involves opposing effects on phosphorylation of the Bcy1 regulatory subunit. Mol Biol Cell 21:3749–3758. doi: 10.1091/mbc.E10-05-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zelada A, Castilla R, Passeron S, Giasson L, Cantore ML. 2002. Interactions between regulatory and catalytic subunits of the Candida albicans cAMP-dependent protein kinase are modulated by autophosphorylation of the regulatory subunit. Biochim Biophys Acta 1542:73–81. doi: 10.1016/S0167-4889(01)00168-9. [DOI] [PubMed] [Google Scholar]
- 77.Martin DE, Soulard A, Hall MN. 2004. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119:969–979. doi: 10.1016/j.cell.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 78.Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. 2005. An evolutionary proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A 102:13933–13938. doi: 10.1073/pnas.0501046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bockmühl DP, Ernst JF. 2001. A potential phosphorylation site for an A-type kinase in the Efg1 regulator protein contributes to hyphal morphogenesis of Candida albicans. Genetics 157:1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lassak T, Schneider E, Bussmann M, Kurtz D, Manak JR, Srikantha T, Soll DR, Ernst JF. 2011. Target specificity of the Candida albicans Efg1 regulator. Mol Microbiol 82:602–618. doi: 10.1111/j.1365-2958.2011.07837.x. [DOI] [PubMed] [Google Scholar]
- 81.Miller C, Matic I, Maier KC, Schwalb B, Roether S, Strasser K, Tresch A, Mann M, Cramer P. 2012. Mediator phosphorylation prevents stress response transcription during non-stress conditions. J Biol Chem 287:44017–44026. doi: 10.1074/jbc.M112.430140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uwamahoro N, Qu Y, Jelicic B, Lo TL, Beaurepaire C, Bantun F, Quenault T, Boag PR, Ramm G, Callaghan J, Beilharz TH, Nantel A, Peleg AY, Traven A. 2012. The functions of Mediator in Candida albicans support a role in shaping species-specific gene expression. PLoS Genet 8:e1002613. doi: 10.1371/journal.pgen.1002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.