

Abstract
Quinones and other oxygenated polycyclic aromatic hydrocarbons (oxy-PAHs) are toxic and/or genotoxic compounds observed to be cocontaminants at PAH-contaminated sites, but their formation and fate in contaminated environmental systems have not been well studied. Anthracene-9,10-dione (anthraquinone) has been found in most PAH-contaminated soils and sediments that have been analyzed for oxy-PAHs. However, little is known about the biodegradation of oxy-PAHs, and no bacterial isolates have been described that are capable of growing on or degrading anthraquinone. PAH-degrading Mycobacterium spp. are the only organisms that have been investigated to date for metabolism of a PAH quinone, 4,5-pyrenequinone. We utilized DNA-based stable-isotope probing (SIP) with [U-13C]anthraquinone to identify bacteria associated with anthraquinone degradation in PAH-contaminated soil from a former manufactured-gas plant site both before and after treatment in a laboratory-scale bioreactor. SIP with [U-13C]anthracene was also performed to assess whether bacteria capable of growing on anthracene are the same as those identified to grow on anthraquinone. Organisms closely related to Sphingomonas were the most predominant among the organisms associated with anthraquinone degradation in bioreactor-treated soil, while organisms in the genus Phenylobacterium comprised the majority of anthraquinone degraders in the untreated soil. Bacteria associated with anthracene degradation differed from those responsible for anthraquinone degradation. These results suggest that Sphingomonas and Phenylobacterium species are associated with anthraquinone degradation and that anthracene-degrading organisms may not possess mechanisms to grow on anthraquinone.
INTRODUCTION
Oxygenated polycyclic aromatic hydrocarbons (oxy-PAHs) such as quinones are cocontaminants in PAH-contaminated soils and sediments (1–3). They are of concern because they have been identified to be toxic and/or genotoxic, either as pure compounds (2, 4–6) or by association with genotoxic fractions of fractionated extracts from contaminated soils and sediments (7–9). In addition, because they are more polar than the parent PAHs, oxy-PAHs can exhibit greater mobility within a contaminated environmental system (2, 10). Little is known about the fate of oxy-PAHs in contaminated systems, in part because relatively few studies have attempted to identify these compounds and the analytical methods have not yet been standardized (11, 12).
It is not possible to assess the source(s) or fate of oxy-PAHs in contaminated systems from observation of their presence alone, although it has been suggested that the ratio of an oxy-PAH to the parent PAH can be diagnostic of the source (3, 13). Oxy-PAHs can be present in the same source as the PAHs (2) (such as coal tars), formed in the atmosphere by heterogeneous reactions on particles containing PAHs (14) that can reach soil or sediment by deposition, or produced by chemical or photochemical oxidation of PAHs in situ (2). Oxy-PAHs can also result from microbial transformation of the parent PAHs in situ (10, 15–19) or as a result of biological treatment of contaminated soil (15, 17, 20). A number of bacterial isolates have been observed to produce oxy-PAHs as extracellular products during aerobic metabolism of PAHs (21–26). However, little is known about the bacteria that can degrade oxy-PAHs in the environment or the mechanisms of degradation.
Anthracene-9,10-dione (anthraquinone) is among the most commonly found oxy-PAHs in soil and sediment samples in which oxy-PAHs have been analyzed. It has been found in contaminated soils at former manufactured-gas plant (MGP) sites (9, 20, 27, 28) and creosote-contaminated sites (1, 9, 27), in contaminated surface water sediments (7, 13), and in groundwater at several sites contaminated with tar (29). It has been observed to be produced as a result of microbial activity in soil spiked with anthracene (10) or with PAH mixtures (18), whereas net anthraquinone removal has been observed during active biological treatment of field-contaminated soils (20, 28).
In this study, we used DNA-based stable-isotope probing (SIP) with uniformly 13C-labeled anthraquinone to identify anthraquinone-degrading bacteria in contaminated soil from a former MGP site, both before and after treatment of the soil in an aerobic, slurry-phase bioreactor. To assess whether the anthraquinone degraders can also grow on anthracene, we conducted SIP with [U-13C]anthracene in parallel for the untreated soil and also compared the anthraquinone degraders identified in this study to the anthracene degraders in bioreactor-treated soil recently identified by SIP with anthracene (30).
MATERIALS AND METHODS
Soil.
PAH-contaminated soil was obtained from a former manufactured-gas plant located in Salisbury, NC, and processed as described elsewhere (31). Briefly, the soil was air dried, sieved through a 10-mm wire screen, blended, and sieved again through no. 6 mesh before being stored in the dark at 4°C until use. The processed soil (64% sand, 30% silt, 6% clay [pH 7.6]) was treated in a bench-scale, semicontinuous, aerobic, slurry-phase bioreactor (32); 20% of the treated soil slurry was replaced every 7 days with untreated, processed soil in a buffer containing 5 mM potassium phosphate (pH 7.5) supplemented with 2.5 mM NH4NO3 (“reactor buffer”). The untreated, processed soil is also referred to below as “feed soil,” and the material removed from the bioreactor is referred to as “treated soil.” The sample of treated soil for SIP incubations was obtained from the slurry removed from the bioreactor at the end of a 7-day cycle, before new feed soil was added to the bioreactor.
Chemicals.
Unlabeled anthracene was obtained from Eastman Kodak (Rochester, NY). Anthracene-9,10-dione of 99.2% purity was purchased from Sigma-Aldrich (St. Louis, MO). All solvents were molecular biology or high-pressure liquid chromatography (HPLC) grade. [U13-C]anthracene was synthesized as described previously (33). [U-13C]anthracene-9,10-dione ([U13-C]anthraquinone) was prepared by oxidation of [U-13C]anthracene with CrO3. Briefly, [U-13C]anthracene (11 mg) in acetic acid (500 μl) was treated with CrO3 (15 mg), and the mixture was refluxed for 15 min. After cooling to ambient temperature, the mixture was diluted with water and the precipitate was collected and washed with hot water, 1 N NaOH, and water successively. The crude product was then recrystallized from acetic acid to afford pure [U-13C]anthracene-9,10-dione. The identity and purity of the product were verified by proton and carbon nuclear magnetic resonance (NMR) spectrometry, as well as gas chromatography and electron impact mass spectrometry (GC-EIMS) (see the supplemental material). Results are summarized as follows: 1H NMR (CDCl3, 400 MHz), 8.31 (broad doublet, J = 166 Hz, H-1, H-4, H-5, H-8), 7.80 (broad doublet, J = 166 Hz, H-2, H-3, H-6, H-7);13C NMR, 126.43 to 128.17 (multiplet, C-1, C-4, C-5, C-8), 132.74 to 134.89 (multiplet, C-2, C-3, C-6, C-7, C-8a, C-9a, C-4a, C-10a), 182.74 to 184.02 (multiplet, C-9, C-10) ppm; GC-EIMS, m/z 222 (100%, M+), 193 (90), 164 (80), 82 (90).
Stable-isotope probing.
Feed soil (approximately 1 g [dry weight]) was preincubated at room temperature by shaking at 225 rpm in the dark for 2 days in 10 ml of reactor buffer to decrease the native PAH concentrations before the SIP experiment (34). The preincubated feed soil and slurry removed from the bioreactor were centrifuged, and in each case an amount of centrifuged soil corresponding to 1 g (dry weight) was placed in each of replicate 125-ml screw-cap flasks containing 30 ml of reactor buffer. Duplicate flasks containing feed soil were amended with one of the following materials, as described by Jones et al. (34): 625 μg of unlabeled anthraquinone, 625 μg of unlabeled anthracene, 625 μg of 13C-labeled anthraquinone, or 625 μg of 13C-labeled anthracene. Duplicate flasks containing treated soil were amended with 625 μg of unlabeled anthraquinone, and a second set of duplicates was amended with 625 μg of 13C-labeled anthraquinone. For both feed soil and treated soil, duplicate acid-inhibited (phosphoric acid to pH ∼2) controls were prepared for each substrate (anthracene or anthraquinone). All incubations were conducted by shaking (225 rpm) at room temperature in the dark.
One milliliter of soil slurry (∼33 mg [dry weight]) was sampled from each duplicate flask containing unlabeled anthracene or anthraquinone at the initial condition (0 days) and after 3 days, 7 days, 15 days, and 20 days for DNA extraction. DNA from the sample collected on day 20 was used for isolation of 13C-enriched DNA and generation of clone libraries as described below. An additional 1 ml of soil slurry was collected at the same time points for analysis of residual anthracene or anthraquinone. All incubations were terminated at 20 days to be consistent with our previous study on SIP of anthracene in the feed soil (34) and based on substantial removal of anthraquinone by 20 days as determined in preliminary incubations with treated soil (data not shown).
Analysis of anthracene and anthraquinone.
Each sample removed from incubation flasks was extracted with 5 ml of dichloromethane and 5 ml of hexanes in a 30-ml glass bottle. Bottles were crimp-sealed and shaken in the dark overnight on a wrist action shaker. The contents were transferred to a sterile 50-ml conical-bottom tube and centrifuged. Supernatant was filtered (0.2-μm pore size) into a crimp-top amber vial with no headspace. Samples for anthracene analysis were diluted into acetonitrile 1:50 (vol/vol), followed by a 1:20 dilution, and stored at 4°C until analysis. Samples for anthraquinone analysis were diluted 1:10 (vol/vol) in methanol and stored at 4°C.
Extracts containing anthracene were analyzed by HPLC with fluorescence detection as described elsewhere (35). Extracts containing anthraquinone were analyzed by HPLC on the same system but with UV detection (Applied Biosystems [Ramsey, NJ] Kratos Analytical Spectroflow 757 UV absorbance detector). The column was a Supelcosil LC-PAH column (3-μm particle size, 10 cm by 4.6 mm [inside diameter]; Supelco, Bellefonte, PA), and detection was at a fixed wavelength of 254 nm; anthraquinone concentrations were quantified against external standards. Initial conditions consisted of 80% methanol and 20% water at a flow rate of 0.65 ml/min for 7 min followed by a linear increase to 100% methanol over 1 min and held at 100% methanol for 3 min. The mobile phase was then returned to the initial conditions for 9 min between injections of 10 μl. The column temperature was maintained at 30°C.
DNA isolation.
Each slurry sample from incubations with unlabeled or labeled substrates was divided into portions containing approximately 250 mg (dry weight) and centrifuged. Each 250-mg aliquot of soil was extracted using the FastDNA spin kit for soil (MP Biomedicals, Solon, OH) as previously described (34). For incubations with the 13C-labeled substrates, the DNA extracted from each 250-mg aliquot of a given replicate was combined and mixed with SYBR Safe (Invitrogen, Carlsbad, CA) (36) and unlabeled DNA from a culture of Escherichia coli K-12 grown on nutrient broth (37) in a cesium chloride solution (ρ = 1.72 g/ml) in a 6-ml polyallomer Ultracrimp tube (Kendro Laboratory Products, Newtown, CT). The tubes were crimp-sealed and ultracentrifuged in a Sorvall (Newtown, CT) RC70 ultracentrifuge using a TV-1665 rotor (Sorvall) at 175,800 × g for 40 h at 20°C. DNA bands were visualized using the Safe Imager blue light transilluminator (Invitrogen, Carlsbad, CA). Fractions of 250 μl each were collected from the bottom of each tube as described previously (37). Total DNA in each fraction was quantified using a NanoDrop 3300 fluorospectrometer (NanoDrop Products, Wilmington, DE) with the Quant-iT PicoGreen double-stranded DNA (dsDNA) assay kit (Invitrogen, Eugene, OR).
To identify fractions containing amplifiable DNA, PCR was performed with 16S rRNA gene primers 341F/517R (Table 1) (38). To assist in determining which fractions contained unlabeled DNA, PCR was performed using E. coli-specific primers (Table 1) (39). PCR was done with a Mastercycler gradient thermocycler (Eppendorf, Hauppauge, NY) using 5 Prime mastermix (5 Prime, Gaithersburg, MD) in a 25-μl reaction mixture. To identify shifts in the community DNA resulting from isotopic enrichment, denaturing gradient gel electrophoresis (DGGE) was performed on each fraction using primers 341FGC/517R (38) and a 10% acrylamide gel with a linear denaturing gradient of 30 to 60% on a DCode universal mutation detection system (Bio-Rad Laboratories, Hercules, CA). Fractions that corresponded to heavy DNA in extracts from each flask containing 13C-labeled substrate were identified based on total DNA, DGGE analysis, and results of the E. coli-specific PCR.
TABLE 1.
PCR primers used in this study
| Target group | Primer | Primer sequence | Tma (°C) | qPCR standardb | Amplicon length (bp) | Reference |
|---|---|---|---|---|---|---|
| Bacteria | 341F | CCTACGGGAGGCAGCAG | 60 | 177 | 38 | |
| 517R | ATTACCGCGGCTGCTGG | |||||
| Anthracene group 1 | AG1.1F | TTGGCAAGTCAGGGGT | 55.5 | AT1 Untreated 44 | 64 | 34 |
| AG1.1R | CAAGCGAGGCAGTTTC | (KP128835) | ||||
| Phenylobacterium | Caulo.1F | TTGGGCACTCTAGTGGGACT | 57 | AQ1 Untreated 33 | 146 | This study |
| Caulo.1R | AGGGATTAGCTCACCATCGC | (KP128832) | ||||
| Pyrene group 2 | PAH.4F | CGGCTTGTTAAGTCGGATGT | 57 | AT1 Untreated 42 | 88 | This study |
| PAH.4R | CACTTCCCTCTACCACACTCTA | (KP128833) | ||||
| Sphingomonas | Sphing.1F | GAGGAACTGCCGGTGATAAG | 60 | AQ1 Treated 20 | 104 | This study |
| Sphing.1R | CCCTCTGTACTTGCCATTGT | (KP128831) | ||||
| Variovorax | VARIO.2F | AGCTGTGCTAATACCGCATA | 55 | AT1 Untreated 43 | 67 | 34 |
| VARIO.2R | TCCATTCGCGCAAGGTCTTG | (KP128834) |
Tm, melting (or midpoint) temperature.
Representative clones, designated according to the incubation substrate (AT, anthracene; AQ, anthraquinone), replicate number, incubation inoculum (untreated or treated soil), and clone number. GenBank accession number is in parentheses.
Clone libraries.
The DNA from ultracentrifuge fractions containing heavy DNA from a given replicate incubation was pooled to generate a 16S rRNA gene clone library for that replicate. PCR was performed using 1 μl of pooled DNA as a template using primers 8F (40) and 1492R (41) and 5 Prime mastermix in a 25-μl reaction. The PCR program began with 10 min at 95°C followed by 25 cycles of 1 min at 94°C, 1 min at 50°C, and 1 min 30 s at 72°C, with a final elongation of 15 min at 72°C. Clone libraries were produced from the amplified products using the TOPO TA cloning kit for sequencing (Invitrogen, Carlsbad, CA). Clones were picked at random for sequencing, and colony PCR was performed to amplify the inserts using vector-specific primers M13F and M13R. The PCR product was sequenced with primer 8F (Eton Biosciences, Research Triangle Park, NC). Sequence analysis was performed in Sequencher (42); sequences were grouped on the basis of at least 97% similarity. Phylogenetic analysis of clone sequences was conducted in MEGA version 5 (43). Chimeras and singleton sequences were removed from the analysis (44). The major groups and their closest relatives in each library were determined from the Ribosomal Database Project (RDP) classifier (45) and BLASTN analysis of the GenBank database.
16S rRNA gene quantification.
Sequences representing the major groups found in the clone libraries were quantified by quantitative PCR (qPCR). Primers specific for Variovorax and anthracene group 1 were as previously described (34). Primers for sequences most closely related to pyrene group 2, Sphingomonas, and Caulobacteraceae were developed for this study. Primer specificity was validated in silico by conducting NCBI Primer-BLAST for each primer sequence. The specificity was tested experimentally by doing PCR and qPCR on DNA that was known to contain the target sequence and PCR on DNA obtained from a different soil sample that was not otherwise known to contain the target sequence to confirm that there was no product produced. The qPCR program comprised 15 min at 95°C followed by 45 cycles of 15 s at 95°C, 30 s at the annealing temperature, and 30 s at 72°C. Data were collected during primer extension, and the products of the reaction were analyzed by melt curve analysis between 65 and 95°C. The r2 value for each qPCR standard curve was ≥0.995, and the amplification efficiencies were close to 2.0. Primers and qPCR conditions are summarized in Table 1.
Pyrosequencing.
DNA was extracted from the bioreactor slurry in June 2012 and April 2013; the latter date was closer to the timing of the SIP experiment. The 2012 sample was extracted using the FastDNA spin kit for soil (MP Biomedicals, Solon, OH), and the 2013 bioreactor sample and feed soil were extracted using the PowerSoil DNA extraction kit. Bacterial 16S rRNA genes were amplified using the FastStart high-fidelity PCR system (Roche, Indianapolis, IN). The PCR mixture was composed of 1 μM (each) concentrations of the forward and reverse primers (16S_577F [AYTGGGYDTAAAGNG] and 16S_926R [CCGTCAATTCMTTTRAGT]), 0.2 mM deoxynucleoside triphosphates (dNTPs), and 150 ng/μl of bovine serum albumin (BSA) in a total volume of 20 μl with 1× buffer and 1 unit of enzyme per reaction. Approximately 25 ng of template DNA was added to each reaction. The PCR protocol consisted of 95°C for 2 min followed by 30 cycles of 95°C for 45 s, 55°C for 45 s, and 72°C for 1 min, with a final elongation at 72°C for 4 min. All reactions were done in triplicate. The amplified products were separated on a 1% agarose gel.
The product (1 μl) was reamplified for 5 cycles with bar-coded 16S primers (IDT DNA Technologies, Coralville, IA) and at the same temperatures as in the initial PCR protocol. The bar-coded PCR products were separated on a 1% agarose gel. Triplicate reactions were pooled during gel purification with the Qiagen gel extraction kit and DNA quantified with the Nanodrop spectrophotometer. The samples were run at the University of North Carolina at Chapel Hill Microbiome Core Facility on a Roche 454 FLX Titanium sequencer. Sequences were analyzed using the mothur software package (46). The sequence data were queried for major sequences identified by SIP using locally installed BLAST (47).
Nucleotide sequence accession numbers.
Sequences from representative clones were deposited in GenBank with accession numbers KP128831 to KP128835.
RESULTS
Incubations with unlabeled anthraquinone or anthracene were conducted in parallel with incubations containing 13C-labeled substrates to track substrate removal. After 20 days of incubation, less than 10% of the spiked anthraquinone or anthracene remained, whereas no removal was observed in acid-inhibited controls (see Fig. S4 in the supplemental material).
“Heavy” (13C-enriched) DNA recovered from incubations with 13C-labeled anthracene or anthraquinone was ultracentrifuged and separated into fractions. The fractions containing heavy DNA were identified using a combination of methods, including total DNA quantification (Fig. 1; see also Fig. S5 to S10 in the supplemental material), DGGE analysis of fractions from incubations with the 13C-labeled substrate (see Fig. S5 to S10), and identification by PCR of the fractions containing the unlabeled E. coli DNA that was used to spike the samples before ultracentrifugation (data not shown). For the incubations with 13C-labeled anthraquinone, there was distinct separation between unlabeled and 13C-enriched DNA (Fig. 1; see also Fig. S8). For the incubation of 13C-labeled anthracene with feed soil, a clear separation was not observed, but there was a significant shift of DNA to the lower fractions (see Fig. S9 and S10). DGGE analysis showed a shift in banding patterns between the unlabeled and 13C-enriched fractions in all of the samples (see Fig. S5 to S10).
FIG 1.
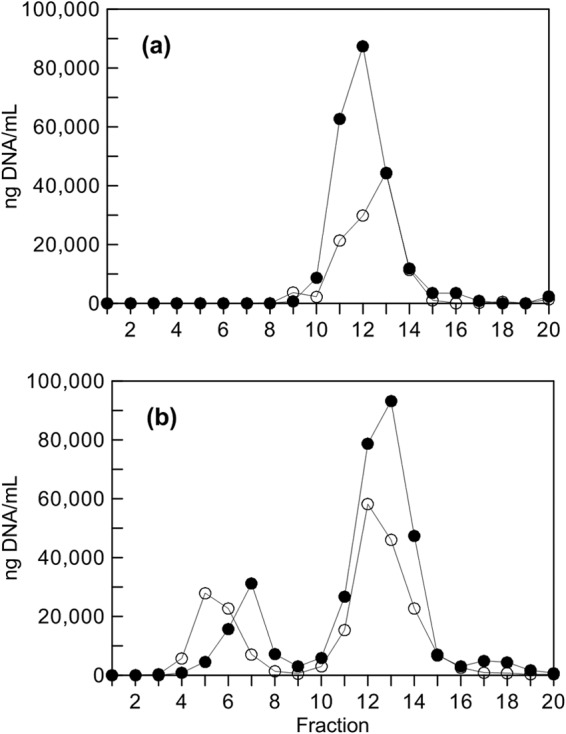
Example of DNA quantification in fractions obtained after ultracentrifugation of extracts from each duplicate incubation of feed soil with unlabeled anthraquinone (a) and [U-13C]anthraquinone (b). Fractions are numbered from the bottom of the centrifuge tube up; for both of the incubations with 13C-labeled anthraquinone, fractions 5 to 8 were selected as those containing heavy (13C-enriched) DNA. The data for the different duplicate incubations are represented by different symbols in each panel.
16S rRNA clone libraries.
Clone libraries of 16S rRNA genes were constructed from the pooled DNA from heavy fractions of each duplicate incubation with 13C-labeled anthraquinone or anthracene. Collectively, 249 total clones were retained after chimeric and singleton sequences were removed. The predominant sequences recovered from each incubation condition and their closest matches are summarized in Table 2 (the data for each of the duplicates from a given incubation are shown in Table S1 in the supplemental material). All sequences from enrichment of the feed soil with anthraquinone were classified as Phenylobacterium within the Caulobacteraceae family (100% RDP confidence threshold). Very different results were obtained from enrichment of the bioreactor-treated soil with anthraquinone, for which the vast majority (92%) of sequences were related to Sphingomonas (98% RDP confidence threshold). Sphingomonas sp. strain MM-1 (48) was the closest BLAST match to the Sphingomonas sequence found in this study, with 96% identity. Sequences related to Phenylobacterium were only 5% of the library from incubation of the treated soil with 13C-labeled anthraquinone.
TABLE 2.
Sequences recovered in clone libraries of 16S rRNA genes in 13C-enriched DNA from SIP incubations, as percentage of total sequences for each incubation conditiona
| Genus or groupb | % sequence for indicated incubation condition |
||
|---|---|---|---|
| Feed soil, [U-13C]anthracene | Feed soil, [U-13C]anthraquinone | Treated soil, [U-13C]anthraquinone | |
| Pyrene group 2 | 51.4 | ||
| Anthracene group 1c | 20.2 | ||
| Variovoraxc | 17.6 | ||
| Bradyrhizobium | 5.4 | ||
| Pigmentiphagac | 2.7 | ||
| Phenylobacterium | 100 | 4.8 | |
| Sphingomonas | 91.7 | ||
| Sphingomonadaceae | 3.6 | ||
Numbers of clones sequenced (sum of duplicate clone libraries for each condition): feed soil incubated with anthracene, 74; feed soil incubated with anthraquinone, 87; and treated soil incubated with anthraquinone, 84. Two clones containing unclassified sequences are excluded from the data for the incubation of feed soil with anthracene.
Best match from RDP Classifier or BLASTN analysis of the NCBI database.
Group or genus previously associated with growth on anthracene by SIP of feed soil (34).
Sequences related to uncultivated gammaproteobacteria collectively referred to as pyrene group 2 (PG2) were half of the sequences recovered from incubation of feed soil with 13C-labeled anthracene (Table 2); the representative PG2 sequence recovered in this study has 99% similarity to the first reported PG2 sequence (49), PYR10d11 (GenBank accession number DQ123671). Other predominant groups from incubation of feed soil with anthracene were Variovorax (18% of clones) and an uncultivated group of bacteria referred to as anthracene group 1 (20% of clones). Anthracene group 1 and Variovorax were the two most predominant sequences observed in an earlier SIP study of anthracene-degrading bacteria on a different sample of the same feed soil as evaluated in this study (34), although pyrene group 2 was not identified as an anthracene degrader in the earlier study. The anthracene group 1 sequence is most closely related to Altererythrobacter (98% RDP confidence threshold).
Quantification of bacterial sequences identified by SIP.
Once the major groups present in the SIP enrichments were identified, qPCR primers were developed to quantify the sequences associated with those groups to verify that they increased in abundance during incubation with anthracene or anthraquinone (Fig. 2). In the feed soil incubated with anthracene, a clear increase in the number of 16S rRNA genes associated with the three major groups identified in the clone libraries (Table 2) was observed during the first 3 to 7 days of incubation; the greatest increase was observed for PG2 sequences, which increased by 2 orders of magnitude within 7 days. Increases were also seen within the first 3 to 7 days for sequences associated with the major groups identified in SIP incubations with anthraquinone in treated soil (Sphingomonas) and feed soil (Phenylobacterium).
FIG 2.
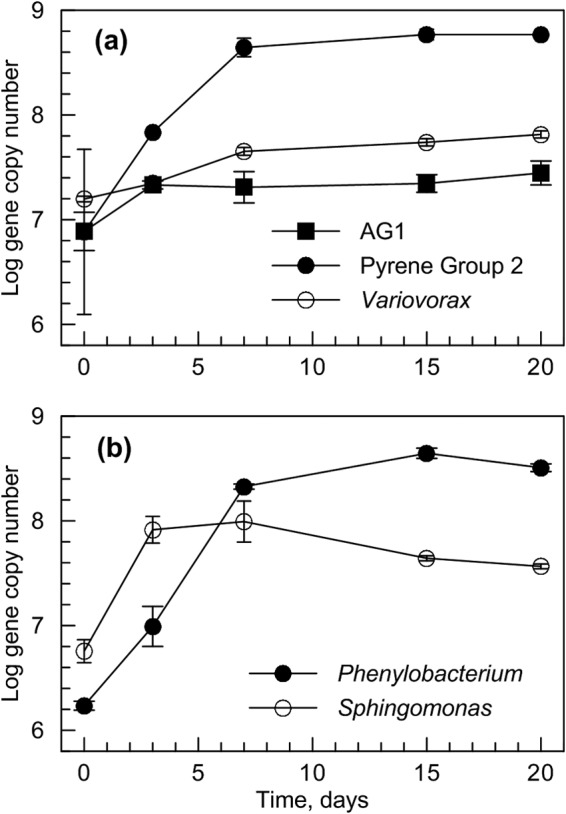
Quantification over time of major sequences identified in clone libraries of heavy DNA from incubations with [U-13C]anthracene in feed soil (a) and [U-13C]anthraquinone in feed soil (Phenylobacterium) and treated soil (Sphingomonas) (b). Error bars represent standard deviations from triplicate qPCRs; if not visible, the error bars are within the size of the symbol. AG1, anthracene group 1.
The major groups associated with degradation of anthracene and anthraquinone were also quantified in the ultracentrifuge fractions from incubations with either the 13C-labeled or unlabeled substrates to verify that they were associated with heavy DNA in the incubations with the 13C-labeled substrates (see Fig. S11). In each case there was a shift toward heavy DNA in the incubations with 13C-labeled substrate, although the shift was less pronounced for PG2. Nevertheless, PG2 sequences were 1 to 2 orders of magnitude greater in the designated heavy fractions from the incubation with [U-13C]anthracene than in the corresponding fractions from the incubations with unlabeled anthracene (see Fig. S11 in the supplemental material), and the increased abundance of PG2 organisms was clearly demonstrated over the course of the incubation with anthracene (Fig. 2).
Pyrosequencing.
Pyrosequencing was performed on two samples of bioreactor-treated soil (10 months apart) and one sample of feed soil, and the sequence data were mined for the 5 major groups identified as anthraquinone or anthracene degraders by SIP. Pyrosequencing data obtained previously from the feed and treated soils shortly after the bioreactor began operation (32) were also probed for the same 5 groups. Data from this study and the previous study are summarized in Table S1 in the supplemental material. Sequences representing Phenylobacterium are highly abundant in the most recently collected feed soil sample (17% of the library), a substantial shift from their almost complete absence in the feed soil collected 3 years earlier; we assume that this change resulted from growth during long-term storage of the feed soil at 4°C. Their high abundance in feed soil is reflected in their relatively high abundance in the bioreactor (treated soil samples obtained during this study, from 5 to 8%). Pyrene group 2 sequences in treated soil were less abundant in the present study than in the samples collected in 2010. Sequences representing Variovorax and Sphingomonas were present at low relative abundances in both the present and earlier studies.
DISCUSSION
PAH metabolism results in oxygenated products, such as quinones, that can accumulate at contaminated sites during bioremediation and are also found as cocontaminants with PAHs in the source materials (2). Because quinones and other oxy-PAHs are known to be toxic, their net formation or removal during bioremediation efforts is important to understand. To date, the only bacteria known to metabolize PAH quinones are the pyrene-degrading bacteria Mycobacterium sp. PYR100 (50) and Mycobacterium vanbaalenii PYR-1 (51), each of which produces two quinone reductases that convert PAH o-quinones to the corresponding catechols; M. vanbaalenii PYR-1 has also been observed to grow on 4,5-pyrenequinone (26). Because anthraquinone is not an o-quinone, it is possible that quinone reductases of the type found in PAH-degrading Mycobacterium spp. would not act on anthraquinone. A strain of Sphingomonas xenophaga can degrade the substituted anthraquinone bromoamine acid (52) but has not been reported to degrade unsubstituted anthraquinone.
Using DNA-based SIP, we showed that bacteria closely related to the genera Phenylobacterium and Sphingomonas assimilate carbon from anthraquinone. On the basis of these findings, combined with evidence that these sequences increased substantially in abundance during the first 3 to 5 days of incubation (Fig. 2), we conclude that the corresponding organisms utilize anthraquinone as a growth substrate. A Phenylobacterium sequence was the only sequence associated with anthraquinone degradation in the untreated feed soil for our laboratory-scale, slurry-phase bioreactor. A sequence related to Phenylobacterium was also recovered in the incubation of bioreactor-treated soil with 13C-labeled anthraquinone, although it was only 5% of the clone library (Table 2). Phenylobacterium is not a well-studied genus, with the first species reported in 1985 (53) and remaining the only reported species until the mid-2000s (54). Sequences related to Phenylobacterium have been observed previously in petroleum reservoirs (55) and polychlorinated biphenyl-contaminated soil (56).
Despite the high relative abundances of Phenylobacterium sequences in the feed soil and in the bioreactor (see Table S1 in the supplemental material), the most predominant sequence associated with anthraquinone degradation in the bioreactor-treated soil is related to Sphingomonas. In previous studies we have similarly seen that the major degraders of a PAH in the bioreactor are not the same as found from SIP of the untreated soil (30, 57). These findings suggest that SIP with an untreated soil will not necessarily predict that the identified organism(s) will be predominant degraders of the same compound under bioremediation conditions. Sphingomonas species are already well known for their involvement in the biodegradation of aromatic pollutants, including PAHs (23, 58). Our results extend the range of pollutants that can be degraded by organisms in this genus to a quinone commonly found as a cocontaminant in PAH-contaminated systems.
Identification of anthracene-degrading bacteria.
Sequences associated with uncultivated organisms in groups referred to as pyrene group 2 and anthracene group 1, as well as sequences closely related to Variovorax, were the most abundant sequences recovered in incubations of feed soil with 13C-labeled anthracene (Table 2). A significant increase in abundance was observed in all three groups by the third day of incubation, with the maximum increase occurring in 1 week (Fig. 2), indicating an immediate response of these groups to the presence of anthracene.
PG2 organisms have been associated with the degradation of pyrene, benz[a]anthracene, and fluoranthene in the same feed soil as used in this study (59), although they were not previously associated with anthracene degradation (34). Consistent with the present results, in the earlier SIP study with anthracene, both anthracene group 1 and Variovorax organisms were the predominant anthracene degraders observed in the feed soil (34). In a separate SIP study, organisms in the genus Altererythrobacter were the predominant anthracene degraders in bioreactor-treated soil (30).
In this study, neither of the major anthraquinone degraders recovered by SIP were related to the major anthracene degraders found in feed soil or bioreactor-treated soil. This finding suggests that anthracene-degrading bacteria might not possess the metabolic capability to grow on anthraquinone. Mycobacterium species are able to metabolize both 4,5-pyrenequinone and the parent compound pyrene because they possess quinone reductases that reduce the quinone to an o-dihydroxylyated intermediate that is part of the pyrene metabolic pathway. The details of anthraquinone metabolism, however, will remain unknown until more quinone-degrading bacteria can be isolated and studied. Bacteria capable of quinone metabolism are crucial in understanding the degradation of PAH metabolites, which, in turn, is valuable information for the study and management of PAH-contaminated sites.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the National Institute of Environmental Health Sciences (grant 5 P42ES005948).
We thank David Singleton for assistance with SIP and interpretation of the sequence data.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00033-15.
REFERENCES
- 1.Meyer S, Cartellieri S, Steinhart H. 1999. Simultaneous determination of PAHs, hetero-PAHs (N, S, O), and their degradation products in creosote-contaminated soils. Method development, validation, and application to hazardous waste sites. Anal Chem 71:4023–4029. [Google Scholar]
- 2.Lundstedt S, White PA, Lemieux CL, Lynes KD, Lambert IB, Öberg L, Haglund P, Tysklind M. 2007. Sources, fate, and toxic hazards of oxygenated polycyclic aromatic hydrocarbons (PAHs) at PAH-contaminated sites. Ambio 36:475–485. doi: 10.1579/0044-7447(2007)36[475:SFATHO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 3.Wilcke W, Bandowe BAM, Lueso MG, Ruppenthal M, del Valle H, Oelmann Y. 2014. Polycyclic aromatic hydrocarbons (PAHs) and their polar derivatives (oxygenated PAHs, azaarenes) in soils along a climosequence in Argentina. Sci Total Environ 473-474:317–325. doi: 10.1016/j.scitotenv.2013.12.037. [DOI] [PubMed] [Google Scholar]
- 4.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. 2000. Role of quinones in toxicology. Chem Res Toxicol 13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 5.Zielinska-Park J, Nakamura J, Swenberg JA, Aitken MD. 2004. Aldehydic DNA lesions in calf thymus DNA and HeLa S3 cells produced by bacterial quinone metabolites of fluoranthene and pyrene. Carcinogenesis 25:1727–1733. doi: 10.1093/carcin/bgh174. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, Dai J, Zhong R, She Y, Liu R, Wei H. 2011. Production of polycyclic aromatic hydrocarbon metabolites from a peroxynitrite/iron(III) porphyrin biomimetic model and their mutagenicities. Environ Toxicol Chem 30:723–729. doi: 10.1002/etc.430. [DOI] [PubMed] [Google Scholar]
- 7.Fernández P, Grifoll M, Solanas AM, Bayona JM, Albaigés J. 1992. Bioassay-directed chemical analysis of genotoxic components in coastal sediments. Environ Sci Technol 26:817–829. doi: 10.1021/es00028a024. [DOI] [Google Scholar]
- 8.Lemieux CL, Lambert IB, Lundstedt S, Tysklind M, White PA. 2008. Mutagenic hazards of complex polycyclic aromatic hydrocarbon mixtures in contaminated soil. Environ Toxicol Chem 27:978–990. doi: 10.1897/07-157.1. [DOI] [PubMed] [Google Scholar]
- 9.Park J, Ball LM, Richardson SD, Zhu H-B, Aitken MD. 2008. Oxidative mutagenicity of polar fractions from polycyclic aromatic hydrocarbon-contaminated soils. Environ Toxicol Chem 27:2207–2215. doi: 10.1897/07-572.1. [DOI] [PubMed] [Google Scholar]
- 10.Weigand H, Totsche KU, Kögel-Knabner I, Annweiler E, Richnow HH, Michaelis W. 2002. Fate of anthracene in contaminated soil: transport and biochemical transformation under unsaturated flow conditions. Eur J Soil Sci 53:71–81. doi: 10.1046/j.1365-2389.2002.00427.x. [DOI] [Google Scholar]
- 11.Bandowe BAM, Wilcke W. 2010. Analysis of polycyclic aromatic hydrocarbons and their oxygen-containing derivatives and metabolites in soils. J Environ Qual 39:1349–1358. doi: 10.2134/jeq2009.0298. [DOI] [PubMed] [Google Scholar]
- 12.Lundstedt S, Bandowe BAM, Wilcke W, Boll E, Christensen JH, Vila J, Grifoll M, Faure P, Biache C, Lorgeoux C, Larsson M, Frech Irgum K, Ivarsson P, Ricci M. 2014. First intercomparison study on the analysis of oxygenated polycyclic aromatic hydrocarbons (oxy-PAHs) and nitrogen heterocyclic polycyclic aromatic compounds (N-PACs) in contaminated soil. Trends Anal Chem 57:83–92. doi: 10.1016/j.trac.2014.01.007. [DOI] [Google Scholar]
- 13.McKinney RA, Pruell RJ, Burgess RM. 1999. Ratio of the concentration of anthraquinone to anthracene in coastal marine sediments. Chemosphere 38:2415–2430. doi: 10.1016/S0045-6535(98)00435-4. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Zhu T. 2014. Formation of nitroanthracene and anthraquinone from the heterogeneous reaction between NO2 and anthracene adsorbed on NaCl particles. Environ Sci Technol 48:8671–8678. doi: 10.1021/es501543g. [DOI] [PubMed] [Google Scholar]
- 15.Eriksson M, Faldt J, Dalhammar G, Borg-Karlson AK. 2001. Determination of hydrocarbons in old creosote contaminated soil using headspace solid phase microextraction and GC-MS. Chemosphere 44:1641–1648. doi: 10.1016/S0045-6535(00)00371-4. [DOI] [PubMed] [Google Scholar]
- 16.Meyer S, Steinhart H. 2001. Fate of PAHs and hetero-PAHs during biodegradation in a model soil/compost-system: formation of extractable metabolites. Water Air Soil Pollut 132:215–231. doi: 10.1023/A:1013252021388. [DOI] [Google Scholar]
- 17.Saponaro S, Bonomo L, Petruzzelli G, Romele L, Barbafieri M. 2002. Polycyclic aromatic hydrocarbons (PAHs) slurry phase bioremediation of a manufacturing gas plant (MGP) site aged soil. Water Air Soil Pollut 135:219–236. doi: 10.1023/A:1014716502484. [DOI] [Google Scholar]
- 18.Wischmann H, Steinhart H. 1997. The formation of PAH oxidation products in soils and soil/compost mixtures. Chemosphere 35:1681–1698. doi: 10.1016/S0045-6535(97)00249-X. [DOI] [Google Scholar]
- 19.Wilcke W, Kiesewetter M, Musa Bandowe BA. 2014. Microbial formation and degradation of oxygen-containing polycyclic aromatic hydrocarbons (OPAHs) in soil during short-term incubation. Environ Pollut 184:385–390. doi: 10.1016/j.envpol.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 20.Lundstedt S, Haglund P, Öberg L. 2003. Degradation and formation of polycyclic aromatic compounds during bioslurry treatment of an aged gasworks soil. Environ Toxicol Chem 22:1413–1420. doi: 10.1002/etc.5620220701. [DOI] [PubMed] [Google Scholar]
- 21.Stringfellow WT, Aitken MD. 1995. Competitive metabolism of naphthalene, methylnaphthalenes, and fluorene by phenanthrene-degrading pseudomonads. Appl Environ Microbiol 61:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grifoll M, Selifinov SA, Chapman PJ. 1994. Evidence for a novel pathway in the degradation of fluorene by Pseudomonas sp. strain F274. Appl Environ Microbiol 60:2438–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Story SP, Kline EL, Hughes TA, Riley MB, Hayasaka SS. 2004. Degradation of aromatic hydrocarbons by Sphingomonas paucimobilis strain EPA505. Arch Environ Contam Toxicol 47:168–176. doi: 10.1007/s00244-004-3069-2. [DOI] [PubMed] [Google Scholar]
- 24.Moody JD, Freeman JP, Doerge DR, Cerniglia CE. 2001. Degradation of phenanthrene and anthracene by cell suspensions of Mycobacterium sp. strain PYR-1. Appl Environ Microbiol 67:1476–1483. doi: 10.1128/AEM.67.4.1476-1483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kazunga C, Aitken MD, Gold A, Sangaiah R. 2001. Fluoranthene-2,3- and -1,5-diones are novel products from the bacterial transformation of fluoranthene. Environ Sci Technol 35:917–922. doi: 10.1021/es001605y. [DOI] [PubMed] [Google Scholar]
- 26.Kazunga C, Aitken MD. 2000. Products from the incomplete metabolism of pyrene by polycyclic aromatic hydrocarbon-degrading bacteria. Appl Environ Microbiol 66:1917–1922. doi: 10.1128/AEM.66.5.1917-1922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundstedt S, Haglund P, Oberg L. 2006. Simultaneous extraction and fractionation of polycyclic aromatic hydrocarbons and their oxygenated derivatives in soil using selective pressurized liquid extraction. Anal Chem 78:2993–3000. doi: 10.1021/ac052178f. [DOI] [PubMed] [Google Scholar]
- 28.Hu J, Adrion AC, Nakamura J, Shea D, Aitken MD. 2014. Bioavailability of (geno)toxic contaminants in polycyclic aromatic hydrocarbon-contaminated soil before and after biological treatment. Environ Eng Sci 31:176–182. doi: 10.1089/ees.2013.0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlanges I, Meyer D, Palm W-U, Ruck W. 2008. Identification, quantification and distribution of PAC-metabolites, heterocyclic PAC and substituted PAC in groundwater samples of tar-contaminated sites from Germany. Polycycl Aromat Compd 28:320–338. doi: 10.1080/10406630802377807. [DOI] [Google Scholar]
- 30.Dunlevy SR, Singleton DR, Aitken MD. 2013. Biostimulation reveals functional redundancy of anthracene-degrading bacteria in polycyclic aromatic hydrocarbon-contaminated soil. Environ Eng Sci 30:697–705. doi: 10.1089/ees.2013.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richardson SD, Aitken MD. 2011. Desorption and bioavailability of PAHs in contaminated soil subjected to long-term in situ biostimulation. Environ Toxicol Chem 30:2674–2681. doi: 10.1002/etc.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singleton DR, Richardson SD, Aitken MD. 2011. Pyrosequence analysis of bacterial communities in aerobic bioreactors treating polycyclic aromatic hydrocarbon-contaminated soil. Biodegradation 22:1061–1073. doi: 10.1007/s10532-011-9463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z, Sangaiah R, Gold A, Ball LM. 2011. Synthesis of uniformly 13C-labeled polycyclic aromatic hydrocarbons. Org Biomol Chem 9:5431–5435. doi: 10.1039/c0ob01107j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones MD, Singleton DR, Sun W, Aitken MD. 2011. Multiple DNA extractions coupled with stable-isotope probing of anthracene-degrading bacteria in contaminated soil. Appl Environ Microbiol 77:2984–2991. doi: 10.1128/AEM.01942-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson SD, Lebron BL, Miller CT, Aitken MD. 2011. Recovery of phenanthrene-degrading bacteria after simulated in situ persulfate oxidation in contaminated soil. Environ Sci Technol 45:719–725. doi: 10.1021/es102420r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martineau C, Whyte LG, Greer CW. 2008. Development of a SYBR safeTM technique for the sensitive detection of DNA in cesium chloride density gradients for stable isotope probing assays. J Microbiol Methods 73:199–202. doi: 10.1016/j.mimet.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 37.Singleton DR, Powell SN, Sangaiah R, Gold A, Ball LM, Aitken MD. 2005. Stable-isotope probing of bacteria capable of degrading salicylate, naphthalene or phenanthrene in a bioreactor treating contaminated soil. Appl Environ Microbiol 71:1202–1209. doi: 10.1128/AEM.71.3.1202-1209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muyzer G, De Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabat G, Rose P, Hickey WJ, Harkin JM. 2000. Selective and sensitive method for PCR amplification of Escherichia coli 16S rRNA genes in soil. Appl Environ Microbiol 66:844–849. doi: 10.1128/AEM.66.2.844-849.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards U, Rogall T, Blöcker H, Emde M, Böttger EC. 1989. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 17:7843–7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–175. In Stackebrandt E, Goodfellow M (ed), Nucleic acid sequencing techniques in bacterial systematics. John Wiley & Sons, New York, NY. [Google Scholar]
- 42.Gene Codes Corporation. 2013. Sequencher version 5.0.1 sequence analysis software. Gene Codes Corporation, Ann Arbor, MI. [Google Scholar]
- 43.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright E, Yilmaz L, Noguera D. 2012. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol 78:717–725. doi: 10.1128/AEM.06516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 48.Tabata M, Ohtsubo Y, Ohhata S, Tsuda M, Nagata Y. 2013. Complete genome sequence of the γ-hexachlorocyclohexane-degrading bacterium Sphingomonas sp. strain MM-1. Genome Announc 1(3):e00247-13. doi: 10.1128/genomeA.00247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singleton DR, Sangaiah R, Gold A, Ball LM, Aitken MD. 2006. Identification and quantification of uncultivated proteobacteria associated with pyrene degradation in a bioreactor treating PAH-contaminated soil. Environ Microbiol 8:1736–1745. doi: 10.1111/j.1462-2920.2006.01112.x. [DOI] [PubMed] [Google Scholar]
- 50.Kim YH, Engesser KH, Cerniglia CE. 2003. Two polycyclic aromatic hydrocarbon o-quinone reductases from a pyrene-degrading Mycobacterium. Arch Biochem Biophys 416:209–217. doi: 10.1016/S0003-9861(03)00297-2. [DOI] [PubMed] [Google Scholar]
- 51.Kim YH, Moody JD, Freeman JP, Brezna B, Engesser KH, Cerniglia CE. 2004. Evidence for the existence of PAH-quinone reductase and catechol-O-methyltransferase in Mycobacterium vanbaalenii. J Ind Microbiol Biotechnol 31:507–516. doi: 10.1007/s10295-004-0178-x. [DOI] [PubMed] [Google Scholar]
- 52.Qu Y, Wang J, Zhou J, Xing L. 2005. Decolorization of bromoamine acid by a newly isolated strain of Sphingomonas xenophaga QYY and its resting cells. Biochem Eng J 27:104–109. doi: 10.1016/j.bej.2005.08.005. [DOI] [Google Scholar]
- 53.Lingens F, Blecher R, Blecher H, Blobel F, Eberspächer J, Fröhner C, Görisch H, Görisch H, Layh G. 1985. Phenylobacterium immobile gen. nov., sp. nov., a Gram-negative bacterium that degrades the herbicide chloridazon. Int J Syst Bacteriol 35:26–39. doi: 10.1099/00207713-35-1-26. [DOI] [Google Scholar]
- 54.Eberspächer J, Lingens F. 2006. The genus Phenylobacterium. Prokaryotes 5:250–256. doi: 10.1007/0-387-30745-1_13. [DOI] [Google Scholar]
- 55.Lenchi N, İnceoğlu Ö, Kebbouche-Gana S, Gana ML, Llirós M, Servais P, García-Armisen T. 2013. Diversity of microbial communities in production and injection waters of Algerian oilfields revealed by 16S rRNA gene amplicon 454 pyrosequencing. PLoS One 8:e66588. doi: 10.1371/journal.pone.0066588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nogales B, Moore ERB, Llobet-Brossa E, Rossello-Mora R, Amann R, Timmis KN. 2001. Combined use of 16S ribosomal DNA and 16S rRNA to study the bacterial community of polychlorinated biphenyl-polluted soil. Appl Environ Microbiol 67:1874–1884. doi: 10.1128/AEM.67.4.1874-1884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones MD, Rodgers-Vieira EA, Hu J, Aitken MD. 2014. Association of growth substrates and bacterial genera with benzo[a]pyrene mineralization in contaminated soil. Environ Eng Sci 31:689–697. doi: 10.1089/ees.2014.0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi T, Fredrickson JK, Balkwill DL. 2001. Biodegradation of polycyclic aromatic hydrocarbons by Sphingomonas strains isolated from the terrestrial subsurface. J Ind Microbiol Biotechnol 26:283–289. doi: 10.1038/sj.jim.7000130. [DOI] [PubMed] [Google Scholar]
- 59.Jones MD, Crandell DW, Singleton DR, Aitken MD. 2011. Stable-isotope probing of the polycyclic aromatic hydrocarbon-degrading bacterial guild in a contaminated soil. Environ Microbiol 13:2623–2632. doi: 10.1111/j.1462-2920.2011.02501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.