

Abstract
Background
Renal cell carcinoma (RCC) is the most common cancer in adult kidney. The accuracy of current diagnosis and prognosis of the disease and the effectiveness of the treatment for the disease are limited by the poor understanding of the disease at the molecular level. To better understand the genetics and biology of RCC, we profiled the expression of 7,129 genes in both clear cell RCC tissue and cell lines using oligonucleotide arrays.
Methods
Total RNAs isolated from renal cell tumors, adjacent normal tissue and metastatic RCC cell lines were hybridized to affymatrix HuFL oligonucleotide arrays. Genes were categorized into different functional groups based on the description of the Gene Ontology Consortium and analyzed based on the gene expression levels. Gene expression profiles of the tissue and cell line samples were visualized and classified by singular value decomposition. Reverse transcription polymerase chain reaction was performed to confirm the expression alterations of selected genes in RCC.
Results
Selected genes were annotated based on biological processes and clustered into functional groups. The expression levels of genes in each group were also analyzed. Seventy-four commonly differentially expressed genes with more than five-fold changes in RCC tissues were identified. The expression alterations of selected genes from these seventy-four genes were further verified using reverse transcription polymerase chain reaction (RT-PCR). Detailed comparison of gene expression patterns in RCC tissue and RCC cell lines shows significant differences between the two types of samples, but many important expression patterns were preserved.
Conclusions
This is one of the initial studies that examine the functional ontology of a large number of genes in RCC. Extensive annotation, clustering and analysis of a large number of genes based on the gene functional ontology revealed many interesting gene expression patterns in RCC. Most notably, genes involved in cell adhesion were dominantly up-regulated whereas genes involved in transport were dominantly down-regulated. This study reveals significant gene expression alterations in key biological pathways and provides potential insights into understanding the molecular mechanism of renal cell carcinogenesis.
Background
Renal cell carcinoma (RCC) accounts for 3% of all malignancies with about 30,000 new cases and 12,000 deaths each year in the United States. RCC is the most common cancer in adult kidney and the most lethal cancer of the urinary system. The incidence of RCC has been increasing at a rate of 3% per year in the United States and Europe. Histopathologically, RCC is a heterogeneous disease. The five distinct types of RCC include clear cell (70–80%), papillary (15–20%), chromophobe (4–5%), collecting duct (<1%) and medullary cell (<1%) [1]. The most common RCC, clear cell RCC, is believed to originate from the proximal tubule epithelium. It is mostly sporadic, unilateral, and unifocal [2]. The main genetic alterations of clear cell RCC have been identified to be chromosome 3 alterations and Von Hippel-Landau gene mutations [2].
The diagnosis of RCC is often confirmed by imaging studies such as computed tomography and X-ray, but the possible existence of benign renal tumors can be a serious challenge to the diagnosis. Previous studies have shown that RCC is one of the most therapy-resistant cancers. It responds very poorly or not at all to chemotherapy, hormonal therapy and radiation therapy [2,3]. Even for the immunotherapy, the response rate is only 10–15% and mostly the response is partial [2]. Surgery thus remains to be the main method of treatment for RCC although it is effective only in about 70% of early-stage and localized RCC [2,4]. The prognosis of RCC is mainly based on the clinical stage and pathological grade of the disease. A review of the Cleveland Clinic Foundation's nephrectomy database with a 10-year follow-up revealed that the size and stage of tumor had the best prognostic value whereas the surgical margin width was not significant [5,6]. This suggests that patients' outcomes with surgery are primarily dependent on tumor biology. Therefore, the advances in our understanding of the genetics and biology of RCC are essential to improve the current diagnosis, treatment and prognosis of RCC.
The emergence of DNA microarray technology made it possible to investigate the expression of thousands of genes simultaneously [7-13]. The large-scale analysis of the gene expression levels can provide insights into the underlying molecular mechanism of RCC and possibly lead to the finding of molecular tumor markers that can potentially be used for more accurate diagnosis, prognosis and possibly can serve as drug targets for effective therapies. Recently, microarray gene expression profiling has been performed to identify gene expression patterns for many solid and hematological malignancies such as colon cancer, breast cancer, prostate cancer, leukemia, and lymphoma [14-20]. Molecular profiling of RCC using cDNA microarrays has also been reported [21-26]. Using a 31,500-element cDNA array, Boer et al. identified 1,738 differentially expressed genes in clear cell RCC. Three hundred and twenty-one of them were annotated for biological processes [22]. Takahashi et al. identified 109 differentially expressed genes in 29 clear cell RCC samples. Approximately 40 genes were then used in a simulation to verify the clinical outcomes of 29 patients. The accuracy of the prediction was reported to exceed that of prediction based on staging [23]. Young et al. analyzed the gene expression patterns of 7075 genes for four types of RCC including clear cell RCC and identified 189 differentially expressed genes among the four different types [24]. More recently, Higgins et al. compared the gene expression profiles of diverse histological types including clear cell, papillary and chromophobe RCC. One thousand five hundred and fifty differentially expressed genes were identified [26].
To better understand the genetics and biology of clear cell RCC, we profiled the expression of 7129 genes in two pooled RCC tissue samples, two patient-matched normal tissues and two pooled RCC cell lines using oligonucleotide arrays. The gene expression profiles were analyzed and visualized using singular value decomposition analysis. A subset of differentially expressed genes identified in this study is common to those discovered previously. Based on biological process ontology, selected genes were annotated and clustered extensively. The analysis of the expression profiles of genes in the annotated functional groups provides insights into biological pathways of RCC. Moreover, comparison of expression patterns in RCC tissue samples and RCC cell lines reveals significant differences between the two types of samples.
Methods
Tissue samples and RCC cell lines
Six clear cell RCC tissue samples (four of them were Fuhrman grade 3, one Fuhrman grade 2, one Fuhrman grade 1) along with six corresponding patient-matched normal kidney tissue samples were obtained from patients undergoing partial or radical nephrectomy at the Cleveland Clinic Foundation. Institutional review board approval and informed consent from patients were obtained and tissue samples were frozen and stored at -80°C immediately after surgery. All the RCC tumors were at the early stages of development (five at stage 1 and one at stage 2). The ages of patients were around sixty years old. Five patients were white male and one was white female. Metastatic RCC cell lines, RCC13 and RCC54 [27] were obtained from Memorial Sloan-Kettering Cancer Center.
RNA extraction and microarray experiments
For microarray experiments, six pairs of RCC tissues and patient-matched normal kidney tissues (total of twelve frozen tissue samples) were mechanically disrupted in TriZol reagent (Life Technologies, Inc.) using a PowerGen 35 tissue homogenizer (Fisher Scientific) and total RNA was immediately isolated from each tissue sample following the manufacturer's procedures (Invitrogen, Carlsbad, CA). Six RCC tissue samples were divided into two groups with three RCC tissue samples in each group. One group includes two Fuhrman grade 3 RCC tissue samples and one Fuhrman grade 1 RCC tissue sample. The other group includes two Fuhrman grade 3 RCC tissue samples and one Fuhrman grade 1 RCC tissue sample. Six corresponding patient-matched normal samples were divided into two groups accordingly. A total of four groups of tissue samples were thus generated, two RCC tissue groups and two normal kidney tissue groups. For each group, 10 μg of total RNA from each tissue sample were pooled. Four pooled total RNA samples from tissues were thus generated. Also, one pooled total RNA sample was generated by pooling 10 μg of total RNA isolated from each of the two RCC cell lines. Double-stranded cDNAs were synthesized from 10 μg of each total RNA sample using SuperScript Choice double-stranded cDNA synthesis kit from Invitrogen following the manufacturer's protocol. cDNAs were purified by phenol/chloroform extraction and ethanol precipitation. Biotin-labeled cRNAs were synthesized by an in vitro transcription reaction using the BioArray HighYield RNA Transcript Labeling Kit (Enzo Diagnostics, Farmingdale, NY). cRNAs were purified from the in vitro transcription reaction using RNeasy Mini kit (Qiagen, Valencia, CA). The fragmentation of biotin-labeled cRNAs and hybridization of the fragments to HuFL Oligonucleotide Arrays (Affymetrix, Santa Clara, CA) were performed following the manufacturer's protocol. The Oligonucleotide Arrays were washed and stained according to the Affymetrix protocol Midi-3 Euk2v3, and scanned using a Hewlett-Parkard GeneArray scanner (Hewlett-Parkard, Palo Alto, CA) with a 570 nM filter and a pixel size of 3 μM. For RT-PCR experiments, total RNAs were isolated from eight additional pairs of RCC tissues and patient-matched normal kidney tissues using the same methods described above.
Data preprocessing
Raw data were acquired using Microsuite 5.1 software of Affymetrix and normalized following a standard practice of scaling the average of all gene signal intensities to a common arbitrary value. The 7,129 genes were preprocessed to eliminate the genes whose signal intensities were not significantly different from their background levels and thus labeled as "Absent" by MicroSuite 5.1. After elimination, 3,145 genes remained for further analysis.
Functional clustering analysis
To analyze the expression profiles of genes in different biological functional groups, 1340 of the 3145 selected genes were annotated for biological process using the software GeneSpring from Silicon Genetics. The ontology is based on the description of the Gene Ontology Consortium [28]. The annotated genes were then categorized into functional groups and analyzed based on the gene expression levels.
RT-PCR
cDNA was synthesized by reverse transcription of 2.5 μg total RNA using oligo(dT)12–18 and SuperScript II RNA H-reverse transcriptase (Invitrogen). Using the same cDNA preparation as template, DNA fragments of SLC6A3, BIGH3 and vWF were amplified by 35 PCR cycles using Taq polymerase (Invitrogen). Normalization was made using α-tubulin. Primers used for the PCR amplifications are as follows:
SLC6A3, sense: 5'-CACCGTGCGTGCCACATCAATAAC-3',
antisense: 5'-CTCCCACCGAGCATTACACT-3';
BIGH3, sense: 5'-CACCCCGCACCATAATGAGATGTG-3',
antisense: 5'-GGCTGGATTGCTTGATTCAT-3';
vWF, sense: 5'-GTGACGGTGAATGGGAGACTGG-3',
antisense: 5'-GTCATTGGCTCCGTTCTCATCAC-3';
α-tubulin, sense: 5'-CTGCCATTGCCACCATCAAAACCAA-3',
antisense: 5'-ATTCAGGGAGCATGACATGCAGCAG-3'.
The PCR products were separated on 2% agrose gels, stained with ethidium bromide and visualized using the Flouro-Chem imaging system (Alpha InnoTech Cooperation).
Singular value decomposition
Singular value decomposition is a very powerful method to analyze and compare the subspaces associated with a matrix. It has been widely used in data compression and visualization [29]. Recently there have been many applications of SVD to analyze microarray gene expression data [30-34]. Following the notation of van Loan [29], the SVD of a real m-by-n (m≥n) matrix A can be written as:
A = UΣVT,
where U = [u1,u2,...,un]∈ Rmxn and V = [v1,v2,...,vn]∈ Rnxn are orthogonal matrices and Σ = diag(σ1, ... ,σn)∈ Rnxn is a diagonal matrix and σ1≥ ... ≥σr≥σr+1 = ... = σn = 0. The vectors ui and vi are the ith left and right singular vectors respectively, σi are the singular values of A, and r is called the rank of A. Based on the structure of the decomposition, the SVD expansion can be readily obtained
![]()
The magnitudes of singular values indicate how close a given matrix A is to a matrix of lower rank. In gene expression data analysis, each column of A represents the expression profile of a corresponding sample and each row represents the transcriptional response of a specific gene. The singular values indicate how well a lower dimensional linear projection of the expression data can represent the original data. The projection onto a subspace spanned by the first p left/right singular vectors can be described by
![]()
Analyzing and visualizing a resulting lower dimensional projection can provide a great insight into understanding the inherent structure of the original data. In this study, the gene expression data was projected onto a 2-D subspace spanned by the first two left singular vectors.
Results
Patterns of gene expression alterations in RCC tissue samples
To analyze the expression profiles of genes in different biological functional groups, 1340 of the 3145 selected genes were annotated for their biological processes. The gene ontology tree that describes the biological process is shown in Figure 1. The 1340 annotated genes were associated with 72 nodes within the ontology. As shown in Figure 1, there are three numerical numbers following the name of each category node. The first integer represents the number of genes associated with the category. The first percent number stands for the percentage of genes in the category that are at least two-fold up-regulated in average. The second number is the percentage of down-regulated genes. Notably, 16% of the total 1340 genes are up-regulated while only 9% of them are down-regulated. The majority (75%) of the genes are not differentially expressed. The biological process ontology includes two major categories: cell communication and signal transduction. Higher percentages of genes in signal transduction are up-regulated compared with that for cell communication. In many functional groups such as cell adhesion, cell motility, proliferation, stress response, G-protein signaling, Ca++ dependent receptor signaling, integrin receptor signaling, transduction, viral life cycle, and pathogenesis, a majority of the differentially expressed genes are up-regulated. This suggests that these gene categories are in the up-regulated pathways and likely play significant roles in carcinogenesis. On the other hand, only very few categories such as biogenesis, gamma aminobutyric acid signaling, nitric oxide mediated signal transduction, and respiration appear to be in the down-regulated pathways. More notably, significant numbers of genes in metabolism and transport are down-regulated, although some important genes in these two groups, such as manganese superoxide dismutase, are up-regulated and its over-expression at protein level is also observed [35]. These interesting expression patterns reflection on ontology suggest important functional gene regulation pathways and also reveal variations in the gene expression levels even within a functional group such as metabolism.
Figure 1.

Biological process ontology tree of 1,340 genes associated with RCC tissues. The first integer following the name of each functional group represents the number of genes associated with the group. The first percent number stands for the percentage of genes in the group that are at least two-fold up regulated in average. The second number is the percentage of down-regulated genes.
Based on expression levels, 74 differentially expressed genes with at least 5-fold change in expression level in both pooled RCC tissue samples were identified and are shown in Tables 1 and 2. Many of the gene expression patterns revealed in the tables are consistent with those in Figure 1, although more genes are down-regulated than up-regulated when at least 5-fold change was used as a selection criterion. Table 1 describes the 32 up-regulated genes in RCC. The over-expression of 19 of them was reported in the literature [22-24,26]. The expression alteration of 3 of them, dopamine transporter (SLC6A3), transforming growth factor-beta induced gene product (BIGH3) and von Willebrand factor (vWF), were verified here by RT-PCR. The results are shown in Figure 2. The transcript levels of SLC6A3 and vWF were remarkably higher in nearly all the RCC tissues than in the normal tissues. The transcript levels of BIGH3 were also significantly higher in almost all the RCC tissues examined. Table 2 describes 42 down-regulated genes, of which 29 were reported previously [22-24,26]. This group of highly differentially expressed genes may be useful as molecular tumor markers that can potentially be used for more accurate diagnosis, prognosis and possibly can serve as drug targets for effective therapies.
Table 1.
Commonly up-regulated genes in both RCC tissue samples
| Accession number | Gene name* | Avg. fold-change |
| M95167 | Dopamine transporter (SLC6A3) | 55.6 |
| HG3044-HT3742 | Fibronectin 1 1,2,4 | 44.2 |
| U29195 | Neuronal pentraxin II (NPTX2) | 38.6 |
| X51441 | Serum amyloid A (SAA) (_s_at)1 | 37.4 |
| J03474 | Serum amyloid A (SAA)1 | 34.4 |
| U90546 | Butyrophilin (BTF4) | 32.5 |
| X51441 | Serum amyloid A (SAA) (_at)1 | 29.2 |
| M77349 | Transforming growth factor-beta induced gene product (BIGH3)1,3,4 | 28.4 |
| X51956 | Enolase2 (ENO2)2,4 | 26.2 |
| U10485 | Lymphoid-restricted membrane protein (Jaw1) | 25.9 |
| D88667 | Cerebroside sulfotransferase | 21.3 |
| U17077 | BENE4 | 17.3 |
| M10321 | von Willebrand factor (vWF)1,2,3 | 15.5 |
| U45878 | Inhibitor of apoptosis protein 1 | 14.4 |
| X06256 | Fibronectin receptor alpha subunit1 | 12.8 |
| L27624 | Tissue factor pathway inhibitor-2 | 12.1 |
| M20902 | Apolipoprotein C-I (VLDL)1,2,4 | 12.0 |
| M24766 | Alpha-2 collagen type IV (COL4A2)1,4 | 11.8 |
| X90908 | I-15P (I-BABP) | 11.7 |
| M15796 | Cyclin4 | 11.5 |
| U52101 | YMP | 11.4 |
| X66839 | MaTu MN for p54/58N | 10.7 |
| M35878 | Insulin-like growth factor-binding protein-31,2,4 | 10.0 |
| U24577 | LDL-phospholipase A2 | 9.4 |
| L34155 | Laminin-related protein (LamA3) | 9.2 |
| M16591 | Hemopoietic cell protein-tyrosine kinase (HCK)4 | 9.1 |
| U60644 | HU-K44 | 8.9 |
| X91911 | RTVP-11,4 | 8.4 |
| M94250 | Retinoic acid inducible factor (MK) | 6.6 |
| X07743 | Pleckstrin (P47)4 | 6.3 |
| X51405 | Carboxypeptidase E (EC 3.4.17.10) | 6.0 |
| X02761 | Fibronectin 11,2,4 | 5.9 |
*Over-expression also reported in clear cell RCC by 1Boer et al., 2Takahashi et al., 3Young et al., and/or 4Higgins et al.
Table 2.
Commonly down-regulated genes in both RCC tissue samples
| Accession number | Gene name* | Avg. fold change |
| M10050 | Liver fatty acid binding protein (FABP1)2 | 118.8 |
| M11437 | Kininogen (KNG) (cds2)1,2,3 | 64.9 |
| X91220 | Na/Cl electroneutral thiazide-sensitive cotransporter | 51.0 |
| U02388 | Cytochrome P450 4F2 (CYP4F2)3 | 44.9 |
| M95549 | Sodium/glucose cotransporter protein1 | 43.9 |
| J05257 | Microsomal dipeptidase (MDP)1 | 42.0 |
| L13258 | Renal Na/Pi-cotransporter1 | 36.1 |
| M29874 | Cytochrome P450-IIB (hIIB1) | 26.7 |
| M11437 | Kininogen (KNG) (cds1) 1,2,3 | 23.3 |
| X53961 | Lactoferrin (LTF) | 23.0 |
| U17418 | Parathyroid hormone-related peptide receptor1 | 21.1 |
| D31628 | 4-hydroxyphenylpyruvic acid dioxygenase (HPD)1,3 | 15.6 |
| D12620 | Cytochrome P-450LTBV | 13.9 |
| M31153 | Steroid 17-alpha-hydroxylase | 13.5 |
| M13149 | Histidine-rich glycoprotein1 | 13.4 |
| HG2841-HT2969 | Albumin 3 | 12.9 |
| M34276 | Plasminogen1,2,4 | 12.5 |
| M21642 | Antithrombin III (ATIII) Utah gene4 | 11.9 |
| J03258 | Vitamin D receptor | 11.0 |
| L12760 | Phosphoenolpyruvate carboxykinase (PCK1)1,2,4 | 10.9 |
| X92720 | Phosphoenolpyruvate carboxykinase (PCK1)1,2,4 | 10.9 |
| U95090 | Chromosome 19 cosmid F19541 | 10.5 |
| J03910 | Metallothionein-IG (MT1G)2,4 | 10.3 |
| U01120 | Glucose-6-phosphatase | 9.3 |
| L47726 | Phenylalanine hydroxylase (PAH) 2,3 | 9.2 |
| L32961 | 4-aminobutyrate aminotransferase (GABAT) | 8.6 |
| M76180 | Aromatic amino acid decarboxylase | 8.6 |
| X59766 | Zn-alpha2-glycoprotein1,4 | 8.5 |
| U21931 | Fructose-1,6-biphosphatase (FBP1)1 | 8.4 |
| M15656 | Aldolase B (ALDOB)1,2 | 8.3 |
| Y07755 | S100A2, exon 1, 2 and 31 | 7.8 |
| U27333 | Alpha (1,3) fucosyltransferase (FUT6) | 7.7 |
| X63359 | UDP glucuronosyltransferase1 | 7.6 |
| X01630 | Argininosuccinate synthetase1,2,3,4 | 7.5 |
| M24902 | Prostatic acid phosphatase | 6.8 |
| HG3286-HT3463 | Crystallin, Alpha A1 | 6.7 |
| X59065 | FGF, exon 3 | 6.5 |
| D78011 | Dihydropyrimidinase1,3,4 | 6.0 |
| L07548 | Aminoacylase-1 (ACY1)1,4 | 5.9 |
| L05144 | Phosphoenolpyruvate carboxykinase (PCK1)1,2,4 | 5.7 |
| U27326 | Alpha (1,3/1,4) fucosyltransferase (FUT3) | 5.7 |
| U22961 | Similar to L-glycerol-3-phosphate:NAD oxidoreductase and albumin1 | 5.6 |
*Under-expression also reported in clear cell RCC by 1Boer et al., 2Takahashi et al., 3Young et al., and/or 4Higgins et al.
Figure 2.
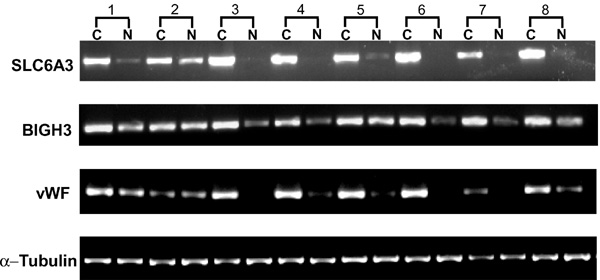
Semi-quantitative RT-PCR of SLC6A3, BIGH3 and vWF. Total RNA was extracted from 8 pairs of RCC tissue (C) and patient-matched normal kidney tissue (N). The over-expression of SLC6A3 was seen in all 8 tissue pairs and the over-expression of BIGH3 and vWF was seen in 7 of the 8 tissue pairs (sample 1, 3, 4, 5, 6, 7, 8). Amplification of DNA fragment of α-tubulin was used as quantitative control.
Patterns of gene expression alterations in RCC cell line
We also performed a microarray experiment using total RNA from RCC cell lines in parallel with those using total RNA from tissue samples. The average gene expression level of normal tissue samples was used as the normal reference. The same data preprocessing and analysis as described above was performed. The ontology tree of 75 nodes together with the statistics of the 1383 selected genes was generated (Ontology tree not shown). Table 3 compares the numbers of the differentially expressed genes in the RCC cell lines and RCC tissues. It is clear that a much higher percentage of genes in the RCC cell lines are differentially expressed than that in the RCC tissue samples. This finding suggests that the gene expression profile in RCC cell lines is significantly different from that in RCC tissue. Interestingly, two of the up-regulated genes in the RCC cell lines, myosin heavy chain 11 and calponin 1 were also reported to be among the 17 signature genes associated with metastasis in primary solid tumors of lung, breast, prostate, colorectal, uterus and ovary [20]. The over-expression of the two genes in RCC tissues was not observed. Despite the significant difference between the expression profiles of the RCC cell lines and RCC tissue samples, many important patterns were preserved. Thirty-nine out of the 42 at least 5-fold down-regulated genes in the RCC tissue samples are also found to be at least 5-fold under-expressed in the RCC cell lines while 21 of the 32 at least 5-fold up-regulated genes in the RCC tissue samples were also found to be at least 5-fold over-expressed in the RCC cell lines.
Table 3.
Numbers of differentially expressed genes in RCC tissues and RCC cell lines
| Fold change | RCC tissuea | RCC tissueb | RCC cell line |
| ≥2 | 373 | 660 | 1472 |
| ≥5 | 74 | 143 | 541 |
| ≥10 | 23 | 47 | 300 |
a) commonly up-regulated/down-regulated b) based on average expression level
Singular value decomposition analysis
To visualize and classify the gene expression profiles of the tissue and cell line samples, the expression matrix of the five pooled samples was analyzed. Based on the 3145 selected genes, the data were was decomposed using singular value decomposition (SVD). The resulting singular values {0.588, 0.176, 0.128, 0.0656, 0.0428} form a spectrum. It is clear from the magnitude of the values that the first two singular vectors account for more than 76% of the total variance in the expression data. The projections of the five expression profiles onto the first two singular vectors are displayed in Figure 3. The gene expression profiles of the two normal tissue sample clustered together. The difference between the two normal profiles reflects the variations among different patients. The gene expression profiles of the two RCC tissue samples were clustered into a distinct group. More notably, the profile of the RCC cell lines is well separated from the tissue groups, indicating that the cell line gene expression profile is very different from the profiles of either normal kidney or RCC tissue samples.
Figure 3.
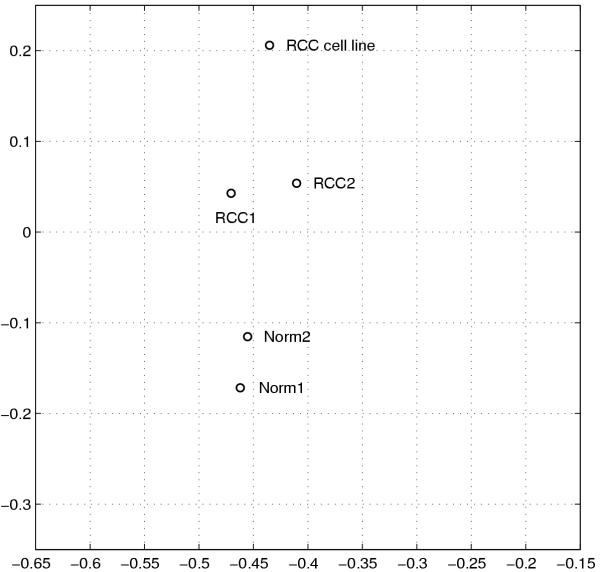
SVD projections of the five expression profiles based on 3,145 genes. The horizontal axis represents the first singular vector. The vertical axis is for the second singular vector. Normal tissue sample profiles are clustered together while RCC tissue sample profiles are grouped into a distinct group. The cell line profile is well separated from the tissue profiles.
Discussion
We analyzed the gene expression profiles of both clear cell RCC tissues and a RCC cell lines using functional clustering analysis and singular value decomposition. The expression levels of the genes in certain functional groups, such as cell adhesion and transport, were either mainly up-regulated or down-regulated, while the expression levels of many other groups such as metabolism, do not show clear patterns. Interestingly, all of the five at least 5-fold differentially expressed genes in the cell adhesion group are up-regulated in RCC (Table 1). The five genes are laminin A3, fibronectin 1, fibronectin receptor alpha subunit, vWF, and BIGH3. This greatly altered cell adhesion expression is very likely associated with carcinogenesis, tumor invasion and metastasis. Laminin A3 is the alpha 3 chain of laminin 5, which is an adhesive glycoprotein in the extracellular matrix. The laminins mediate the attachment, migration and organization of cells into tissues by interacting with other extracellular matrix components [36]. The association of laminin A3 with carcinogenesis has been reported for other types of cancers such as lung cancer [37]. Interestingly, the expression of laminin A3 was suppressed in lung cancer in contrast with its over-expression in RCC found in this study. Fibronectin 1 is another adhesive protein that binds to the external face of the plasma membrane and enables cells to interact with the extracellular matrix [38]. The cell-binding region of fibronectin 1 binds and releases integrin, a complex of proteins that span the plasma membrane. vWF is a plasma protein. It mediates platelet adhesion to the injured vessel wall and carries and protects coagulation factor VIII. At the site of vascular damage, vWF binds immediately to exposed collagens, thereby facilitating the adhesion of platelets [39]. BIGH3 encodes a secreted adhesion molecule, which is believed to be involved in tumor progression by regulating integrin receptors [40]. The over-expression of fibronectin 1, fibronectin receptor alpha subunit, vWF, and BIGH3 have all been reported previously in RCC and/or other types of cancers [22-24,26,41,42]. Like many other genes, the five genes are playing many different roles in biological processes. Laminin A3, fibronectin receptor alpha subunit, and BIGH3 are also associated with integrin receptor signaling. BIGH3 is also involved in cell growth and proliferation, while fibronectin 1 is also involved in cell motility and transduction in developmental processes. vWF plays roles in blood coagulation. The 27 remaining over-expressed genes are mainly associated with cell growth, metabolism, proliferation, and transduction in developmental processes.
Table 2 shows the genes that are at least 5-fold down-regulated in RCC tissue. These include five of the six at least 5-fold differentially expressed genes involved in transport are found in the table. They include the renal Na/Pi cotransporter, Na/Cl electro-neutral thiazide-sensitive cotransporter, sodium/glucose cotransporter, liver fatty acid binding protein (FABP1), and lactoferrin (LTF). Notably, the cotransporters are all remarkably down-regulated. The renal Na/Pi-cotransporter is localized at the apical membrane of the proximal tubular cells [43]. It is believed to play an important role in the maintenance of phosphate homeostasis in the kidney. Reabsorption of phosphate in the kidney occurs predominately in the proximal tubule. This process is mediated mainly by Na+ dependent Na/Pi cotransporter in the brush border membrane and is regulated by a variety of hormones, including insulin-like growth factor. Na/Cl electro-neutral thiazide-sensitive cotransporter is highly and specifically expressed in epithelial cells of distal convoluted tubule of the kidney [44]. It drives the movement of chloride across the membrane of epithelial cells and thus maintains to the chloride homeostasis. The sodium/glucose cotransporter is located in the early proximal convoluted tubule. It is involved in the reabsorption of D-glucose in the kidney [45]. The remarkable under-expression of the cotransporters clearly indicates that the disruption of electrolyte homeostasis maintained by ion transport systems is associated with RCC carcinogenesis. FABP1 is in a family of small, highly conserved, cytoplasmic proteins that bind long-chain fatty acids and other hydrophobic ligands. LTF is an important member of the transferrin family that plays an essential role in the transport of iron to all tissue cells. Since LTF is carried in the bloodstream, its level can be easily monitored, with the gene expression alteration of LTF having a greater potential to be used for early diagnosis of RCC.
The comparison of gene expression profiles in RCC tissues and the cell lines shows significant differences between the expression patterns of the two different types of samples. Three factors that might contribute to these differences include (1) the RCC cell lines were derived from metastatic RCC. Therefore, possibly more gene mutations were accumulated and thus more genes were differentially expressed; (2) the RCC cell lines are a pure population of cancer cells in contrast to the RCC tissue samples that contain many other different types of cells besides cancer cells. Thus the expression intensity of differentially expressed genes was magnified in the RCC cell lines; (3) the in vitro culture of the RCC cell lines may have introduced changes in the gene expression profile as compared with in vivo cancer cells in the RCC tissue samples. On the other hand, many important genes were consistently expressed in both types of samples, suggesting that some of the gene expression patterns in RCC tissues can be recognized through the study of the gene expression in RCC cell lines. In this study, all the genes associated with cell adhesion that were discussed above, laminin A3, fibronectin 1, fibronectin receptor alpha subunit, and BIGH3 except vWF were consistently up-regulated in the RCC cell lines. The expression intensity of vWF in the RCC cell lines was not significantly different from the background, thus vWF was not selected for the analysis of the cell line profile. We note that the up-regulation of vWF in RCC tissue samples has been reported [22-24]. The five genes involved in transport, renal Na/Pi cotransporter, Na/Cl electro-neutral thiazide-sensitive cotransporter, sodium/glucose cotransporter, FABP1 and LTF are all remarkably down-regulated in the RCC cell lines, which is consistent with the results from the RCC tissue samples.
Conclusions
This study has identified 74 gene expression alterations in clear cell RCC. The majority (~64%) of these alterations has been reported in RCC previously. Extensive annotation, clustering and analysis of a large number of genes based on the gene functional ontology revealed many interesting gene expression patterns in RCC. Most notably, genes involved in cell adhesion were up-regulated whereas genes involved in transport were down-regulated. The identified alterations of gene expression will likely give insight into RCC carcinogenesis and tumor progression. Our initial, detailed comparison of gene expression profiles in RCC tissue and cell lines revealed significant differences of gene expression patterns between the two types of samples.
Competing interest
None declared.
Abbreviations
RCC, cell renal cell carcinoma
RT-PCR, reverse transcription polymerase chain reaction
SVD, singular value decomposition
SLC6A3, dopamine transporter
BIGH3, transforming growth factor-beta induced gene product
VWF, von Willebrand factor
FABP1, liver fatty acid binding protein
LTF, lactoferrin
Author contributions
ST participated in the data analysis, conducted RT-PCR experiments and participated in the writing of all drafts and the final version of the manuscript. LL initiated the project, prepared tissue samples and provided gene chips and participated in the writing of drafts. ZD performed the SVD and functional clustering analysis and participated in the writing of drafts and the final version of the manuscript. PS isolated the total RNA. MS provided pathology review of all tissue samples. SD performed the microarray experiment. AN participated in the data interpretation. JH and AA helped with the use of GeneSpring software and critically reviewed the final version of the manuscript. JD directed the whole project and participated in the writing of all drafts and the final version of the manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
The work was partly supported by grants to J. A. DiDonato from the National Institute of Health, Department of Defense, and the Wilson Foundation and a start-up grant to Z-H. Duan from The University of Akron and an National Institute of Health grant (CA 82858) to A. Almasan.
Contributor Information
Louis S Liou, Email: lioul@ccf.org.
Ting Shi, Email: shiti@ccf.org.
Zhong-Hui Duan, Email: duan@uakron.edu.
Provash Sadhukhan, Email: sadhukp@ccf.org.
Sandy D Der, Email: sandy.der@utoronto.ca.
Andrew A Novick, Email: novicka@ccf.org.
John Hissong, Email: hissonj@ccf.org.
Marek Skacel, Email: skacelm@ccf.org.
Alexandru Almasan, Email: almasaa@ccf.org.
Joseph A DiDonato, Email: didonaj@ccf.org.
References
- Reuter VE, Presti JC. Contemporary approach to the classification of renal epithelial tumors. Semin Oncol. 2000;27:124–37. [PubMed] [Google Scholar]
- Walsh PC, Retik AB, Vaughan ED, Wein AJ, Kavoussi LR, Novick AC, Partin AW, Peters CA. Campbell's Urology. 8. Philadelphia, London, New York, St. Louis, Sydney, Toronto: Saunders; 2003. [Google Scholar]
- Amato RJ. Chemotherapy for renal cell carcinoma. Semin Oncol. 2000;27:177–86. [PubMed] [Google Scholar]
- Russo P. Renal cell carcinoma: presentation, staging, and surgical treatment. Semin Oncol. 2000;27:160–176. [PubMed] [Google Scholar]
- Fergany AF, Hafez KS, Novick AC. Long-term results of nephron sparing surgery for localized renal cell carcinoma: 10-year followup. J Urol. 2000;163:442–445. [PubMed] [Google Scholar]
- Castilla EA, Liou LS, Abrahams NA, Fergany A, Rybicki LA, Myles J, Novick AC. Prognostic importance of the width of the resection margin after nephron-sparing surgery for renal cell carcinoma. Urology. 2002;60:993–997. doi: 10.1016/S0090-4295(02)01983-0. [DOI] [PubMed] [Google Scholar]
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 1996;14:457–60. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–80. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci. 1996;93:10614–9. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, Woolley DE, Davis RW. Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci. 1997;94:2150–2155. doi: 10.1073/pnas.94.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, Pergamenschikov A, Lee JC, Lashkari D, Shalon D, Myers TG, Weinstein JN, Botstein D, Brown PO. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- King HC, Sinha AA. Gene expression profile analysis by DNA microarrays: promise and pitfalls. JAMA. 2001;286:2280–2288. doi: 10.1001/jama.286.18.2280. [DOI] [PubMed] [Google Scholar]
- Xu J, Stolk JA, Zhang X, Silva SJ, Houghton RL, Matsumura M, Vedvick TS, Leslie KB, Badaro R, Reed SG. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer Res. 2000;60:1677–1682. [PubMed] [Google Scholar]
- Belov L, de la Vega O, dos Remedios CG, Mulligan SP, Christopherson RI. Immunophenotyping of leukemias using a cluster of differentiation antibody microarray. Cancer Res. 2001;61:4483–4489. [PubMed] [Google Scholar]
- Brenton JD, Aparicio SA, Caldas C. Molecular profiling of breast cancer: portraits but not physiognomy. Breast Cancer Res. 2001;3:77–80. doi: 10.1186/bcr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Ross DT, Kadin ME, Brown PO, Wasik MA. Comparative genome-scale analysis of gene expression profiles in T cell lymphoma cells during malignant progression using a complementary DNA microarray. Am J Pathol. 2001;158:1231–1237. doi: 10.1016/S0002-9440(10)64073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemasa I, Higuchi H, Yamamoto H, Sekimoto M, Tomita N, Nakamori S, Matoba R, Monden M, Matsubara K. Construction of preferential cDNA microarray specialized for human colorectal carcinoma: molecular sketch of colorectal cancer. Biochem Biophys Res Commun. 2001;285:1244–1249. doi: 10.1006/bbrc.2001.5277. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet. 2002;31:141–149. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Moch H, Schraml P, Bubendorf L, Mirlacher M, Kononen J, Gasser TH, Mihatsch MJ, Kallioniemi OP, Sauter G. Identification of Potential Prognostic Parameters for Renal Cell Carcinoma by Tissue Microarray Analysis and cDNA Microarrays Screening. Verh Dtsch Ges Path. 1999;83:225–232. [PubMed] [Google Scholar]
- Boer JM, Huber WK, Sültmann H, Wilmer F, von Heydebreck A, Haas S, Korn B, Gunawan B, Vente A, Füzesi L, Vingron M, Poustka A. Identification and Classification of Differentially Expressed Genes in Renal Cell Carcinoma by Expression Profiling on a Global Human 31,500-Element cDNA Array. Genome Research. 2001;11:1861–1870. doi: 10.1101/gr.184501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Rhodes DR, Furge KA, Kanayama HO, Kagawa S, Haab BB. Gene expression profiling of clear cell renal cell carcinoma: Gene identification and prognostic classification. Proc Natl Acad Sci. 2001;98:9754–9759. doi: 10.1073/pnas.171209998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AN, Amin MB, Moreno CS, Lim SD, Cohen C, Petros JA, Marshall FF, Neish AS. Expression Profiling of Renal Epithelial Neoplasms: A Method for Tumor Classification and Discovery of Diagnostic Molecular Markers. American Journal of Pathology. 2001;158:1639–1651. doi: 10.1016/S0002-9440(10)64120-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Rubin MA. Molecular Markers for renal cell carcinoma: Impact on diagnosis and treatment. Seminars in Urologic Oncology. 2001;19:80–87. [PubMed] [Google Scholar]
- Higgins JPT, Shinghal R, Gill H, Reese JH, Terris M, Cohen RJ, Fero M, Pollack JR, Van de Rijn M, Brooks JD. Gene Expression Patterns in Renal Cell Carcinoma Assessed by Complementary DNA Microarray. Am J Pathol. 2003;162:925–932. doi: 10.1016/S0002-9440(10)63887-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert T, Bander NH, Finstad CL, Ramsawak RD, Old LJ. Establishment and characterization of human renal cancer and normal kidney cell lines. Cancer Res. 1990;50:5531–5536. [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulob GH, van Loan CF. Matrix computations. 3. Baltimore and London: The Johns Hopkins University Press; 1996. [Google Scholar]
- Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holter NS, Mitra M, Maritan A, Cieplak M, Banavar JR, Fedoroff NV. Fundamental patterns underlying gene expression profiles: Simplicity from complexity. Proc Natl Acad Sci. 2000;97:8409–8414. doi: 10.1073/pnas.150242097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holter NS, Maritan A, Cieplak M, Fedoroff NV, Banavar JR. Dynamic modeling of gene expression data. Proc Natl Acad Sci. 2001;98:1693–1698. doi: 10.1073/pnas.98.4.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MKS, Tegner J, Collins JJ. Reverse engineering gene networks using singular value decomposition and robust regression. Proc Natl Acad Sci. 2002;99:6163–6168. doi: 10.1073/pnas.092576199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall ME, Rechtsteiner A, Rocha LM. Singular Value Decomposition and Principal Component Analysis. In: Berrar DP, Dubitzky W, Granzow M, editor. In A Practical Approach to Microarray Data Analysis. Boston: Kluwer Academic Publishers; 2003. pp. 91–109. [Google Scholar]
- Shi T, Dong F, Liou LS, Duan ZH, Novick AC, DiDonato JA. Differential protein Profiling in renal-cell carcinoma. Mol Carcinog. 2004;40:47–61. doi: 10.1002/mc.20015. [DOI] [PubMed] [Google Scholar]
- Kariya Y, Tsubota Y, Hirosaki T, Mizushima H, Puzon-McLaughlin W, Takada Y, Miyazaki K. Differential regulation of cellular adhesion and migration by recombinant laminin-5 forms with partial deletion or mutation within the G3 domain of alpha3 chain. J Cell Biochem. 2003;88:506–20. doi: 10.1002/jcb.10350. [DOI] [PubMed] [Google Scholar]
- Akashi T, Ito E, Eishi Y, Koike M, Nakamura K, Burgeson RE. Reduced expression of laminin alpha 3 and alpha 5 chains in non-small cell lung cancers. Jpn J Cancer Res. 2001;92:293–301. doi: 10.1111/j.1349-7006.2001.tb01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Olden K. Fibronectins: adhesive glycoproteins of cell surface and blood. Nature. 1978;275:179–84. doi: 10.1038/275179a0. [DOI] [PubMed] [Google Scholar]
- Denis CV. Molecular and cellular biology of von Willebrand factor. Int J Hematol. 2002;75:3–8. doi: 10.1007/BF02981972. [DOI] [PubMed] [Google Scholar]
- Zhao YL, Piao CQ, Hei TK. Overexpression of Betaig-h3 gene downregulates integrin alpha5beta1 and suppresses tumorigenicity in radiation-induced tumorigenic human bronchial epithelial cells. Br J Cancer. 2002;86:1923–1928. doi: 10.1038/sj.bjc.6600304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonmez HS, Karaarslan I, Baloglu H, Kokoglu E. Tissue fibronectin levels of human prostatic cancer, as a tumor marker. Cancer Biochem Biophys. 1995;15:107–110. [PubMed] [Google Scholar]
- Suer S, Sonmez H, Karaaslan I, Baloglu H, Kokoglu E. Tissue sialic acid and fibronectin levels in human prostatic cancer. Cancer Lett. 1996;99:135–7. doi: 10.1016/0304-3835(95)04084-6. [DOI] [PubMed] [Google Scholar]
- Murer H, Hernando N, Forster I, Biber J. Regulation of Na/Pi transporter in the proximal tubule. Annu Rev Physiol. 2003;65:531–42. doi: 10.1146/annurev.physiol.65.042902.092424. [DOI] [PubMed] [Google Scholar]
- Obermuller N, Bernstein P, Velazquez H, Reilly R, Moser D, Ellison DH, Bachmann S. Expression of the thiazide-sensitive Na-Cl cotransporter in rat and human kidney. Am J Physiol. 1995;269:900–10. doi: 10.1152/ajprenal.1995.269.6.F900. [DOI] [PubMed] [Google Scholar]
- Santer R, Kinner M, Lassen CL, Schneppenheim R, Eggert P, Bald M, Brodehl J, Daschner M, Ehrich JH, Kemper M, Li Volti S, Neuhaus T, Skovby F, Swift PG, Schaub J, Klaerke D. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J Am Soc Nephrol. 2003;14:2873–82. doi: 10.1097/01.asn.0000092790.89332.d2. [DOI] [PubMed] [Google Scholar]