

The methylcytosine dioxygenase TET2 is frequently mutated in hematological disorders, including acute myeloid leukemia (AML), and has been suggested to protect CpG islands and promoters from aberrant DNA methylation. Rasmussen et al. used a novel Tet2-dependent leukemia mouse model to show that the primary effect of Tet2 loss in preleukemic hematopoietic cells is progressive and widespread DNA hypermethylation affecting up to 25% of active enhancer elements.
Keywords: leukemia, TET2, DNA methylation, enhancer
Abstract
DNA methylation is tightly regulated throughout mammalian development, and altered DNA methylation patterns are a general hallmark of cancer. The methylcytosine dioxygenase TET2 is frequently mutated in hematological disorders, including acute myeloid leukemia (AML), and has been suggested to protect CG dinucleotide (CpG) islands and promoters from aberrant DNA methylation. In this study, we present a novel Tet2-dependent leukemia mouse model that closely recapitulates gene expression profiles and hallmarks of human AML1-ETO-induced AML. Using this model, we show that the primary effect of Tet2 loss in preleukemic hematopoietic cells is progressive and widespread DNA hypermethylation affecting up to 25% of active enhancer elements. In contrast, CpG island and promoter methylation does not change in a Tet2-dependent manner but increases relative to population doublings. We confirmed this specific enhancer hypermethylation phenotype in human AML patients with TET2 mutations. Analysis of immediate gene expression changes reveals rapid deregulation of a large number of genes implicated in tumorigenesis, including many down-regulated tumor suppressor genes. Hence, we propose that TET2 prevents leukemic transformation by protecting enhancers from aberrant DNA methylation and that it is the combined silencing of several tumor suppressor genes in TET2 mutated hematopoietic cells that contributes to increased stem cell proliferation and leukemogenesis.
Methylation of DNA at CG dinucleotides (CpGs) plays an important function in the maintenance of cell identity as well as the regulation of developmental processes (Messerschmidt et al. 2014). A large number of studies have shown that patterns of DNA methylation are perturbed in human diseases such as imprinting disorders and cancer (Baylin and Jones 2011). In cancer, these studies have almost exclusively focused on CpG island methylation, and it has been shown that several tumor suppressor genes are silenced by this mechanism (Baylin and Jones 2011). Although it is well established that the CpG islands becomes increasingly methylated as a function of age (so-called “epigenetic drift”), the mechanisms leading to the changes in DNA methylation patterns are not well understood (for review, see Teschendorff et al. 2013; Issa 2014; Schoofs et al. 2014). However, the recent discovery that primary tumors carry mutations in genes involved in the regulation of DNA methylation, including DNMT3A, IDH1, IDH2, and TET2, suggests that somatic mutations in specific genes might contribute to the altered DNA methylation patterns in cancer (Shih et al. 2012).
TET2 is a member of the TET family of proteins (TET1–3) that can convert 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and promotes site-specific DNA demethylation (Pastor et al. 2013). TET2 is the only gene of the TET family that is mutated with high frequency in patients suffering from a wide variety of hematopoietic diseases (for review, see Solary et al. 2014), including malignancies such as myelodysplastic syndrome (MDS) (Delhommeau et al. 2009; Langemeijer et al. 2009; Messerschmidt et al. 2014), chronic myelomonocytic leukemia (CMML) (Kosmider et al. 2009; Baylin and Jones 2011), acute myeloid leukemia (AML) (Baylin and Jones 2011; Weissmann et al. 2012), and B- and T-cell lymphomas (Quivoron et al. 2011; Asmar et al. 2013; Teschendorff et al. 2013; Issa 2014; Schoofs et al. 2014). Genetic inactivation of Tet2 in the mouse hematopoietic system confers a competitive advantage to stem and progenitor cells and disrupts terminal differentiation, resulting in a CMML-like phenotype (Li et al. 2011; Moran-Crusio et al. 2011; Quivoron et al. 2011; Shide et al. 2012; Shih et al. 2012). Although this leads to increased susceptibility to cellular transformation, the resulting hematopoietic malignancies occur with low penetrance. Therefore, in both human patients and mouse models, the kinetics of disease development suggests that cooperating mutations are necessary to achieve full malignant transformation. In accordance, cooperation of Tet2 deficiency with KIT activation (Soucie et al. 2012; Pastor et al. 2013) and with inactivation of the Notch pathway (Lobry et al. 2013; Solary et al. 2014) was recently demonstrated. However, the mechanistic role of Tet2 loss in this process remains unknown.
Despite several reports, it is not clear how TET2 mutations affect DNA methylation patterns in the genome and contribute to hematological disorders. Initial analysis revealed global hypomethylation in TET2 mutated versus TET2 wild-type CMML patients (Ko et al. 2010). Subsequently, this observation was partly validated by an additional study that found the majority of differentially methylated promoters (43 out of 56) in CMML patients to be hypomethylated (Pérez et al. 2012). In contrast, another group found increased methylation in 129 promoters in AML patients with TET2 mutations (Figueroa et al. 2010). Finally, Yamazaki et al. (2012) found that CMML patients with TET2 mutations had global increase in DNA methylation, and since they were not able to detect increased methylation at several loci investigated, they speculated that the increase in DNA methylation most likely occurred outside of CpG islands and gene promoters. In support of this notion, two recent reports revealed a potential role of Tet proteins in the maintenance of DNA methylation on enhancer elements (Hon et al. 2014; Lu et al. 2014); however, the relevance of this observation for hematopoietic cells and tumorigenesis is not clear at present. To investigate the role of Tet2 in the regulation of DNA methylation in hematopoietic cells and how its loss can contribute to hematopoietic disorders, we generated a mouse model for Tet2-deficient AML. Using this model in combination with in vitro studies on preleukemic cells, we demonstrated that loss of Tet2 led to a genome-wide increase in DNA methylation of active enhancers over time. Several of these enhancers regulate the expression of tumor suppressor genes, and we propose that the combined silencing of these contributes to increased stem cell proliferation and tumorigenesis.
Results
Loss of Tet2 and AML1-ETO (AE) expression collaborate to induce AML
To understand the role of TET2 in the development of leukemia, we sought to develop a mouse model of human AML dependent on the loss of Tet2 activity. The combination of TET2 mutations and the t(8:21)(q22:q22) translocation has been observed in both pediatric and adult de novo AML patients (Supplemental Table S1). We therefore decided to combine Tet2 deficiency with expression of AE, the oncofusion protein emanating from the t(8;21) translocation. We first investigated the effect of disrupting Tet2 in a serial replating assay using Kit-enriched hematopoietic stem and progenitor cells (HSPCs) expressing AE or empty vector (EV). Whereas both Tet2−/−;EV and Tet2fl/fl;AE cells showed a low basal colony formation capacity, the combination of Tet2 disruption and AE expression led to a dramatic and permanent increase in colony-forming unit (CFU) numbers and colony sizes, indicating a strong synergistic effect (Fig. 1A; Supplemental Fig. S1A).
Figure 1.

Loss of Tet2 and AE expression collaborate to induce AML. (A) Serial replating assay in methylcellulose-containing medium of Tet2fl/fl or Tet2−/− Kit-enriched HSPCs transduced with either EV- or AE-expressing retrovirus. Bars represent mean value (n = 3), and error bars indicate SD. (*) P < 0.025 (two-way ANOVA); (n.s) not significant. (B) Kaplan-Meier plot showing overall recipient mouse survival upon primary (1°) or secondary (2°) transplantation. (Left) Lethally irradiated (900 Rad) recipient SJL mice transplanted with Tet2fl/fl (n = 13) or Tet2−/− (n = 12) Kit-enriched HSPCs transduced with AE-expressing retrovirus. (Right) Sublethally irradiated (650 Rad) recipient mice (n = 5) transplanted with 1 × 106 splenocytes isolated from moribund leukemic Tet2−/−;AE mice (LeuA). (****) P-value < 0.0001 (Wilcoxon test). LeuA and LeuB mark the origins of two Tet2−/−;AE leukemias from independent mice used for secondary transplantation experiments and gene expression analysis. (C) Peripheral blood parameters of wild-type (WT) mice (n = 4) or moribund mice transplanted with Tet2−/−;AE HSPCs (n = 7). (*) P < 0.01 (Student's t-test). (D) Splenomegaly observed in moribund mice transplanted with Tet2−/−;AE HSPCs, as indicated by spleen versus body weight ratio (n = 4) (left panel) or visual inspection (n = 3) (right panel). (***) P < 0.0001 (Student's t-test). (E) Representative FACS analysis plots of peripheral blood from recipient mice showing the CD45.2-positive Tet2fl/fl;AE (top panel) or Tet2−/−;AE (bottom panel) cells as well as the CD45.1-positive helper cells (wild type). Early signs of disease include the appearance of CD45-negative and GFP-positive leukemic blasts in circulation. (F) Unsupervised hierarchical clustering of mouse wild-type GMPs and leukemic GMPs (L-GMPs; LeuA and LeuB) together with human AML samples from the MILE study (Haferlach et al. 2010) (left) and the Cancer Genome Atlas (TCGA) study (The Cancer Genome Atlas Research Network 2013) (right). For clarity, each karyotypic subgroup in the MILE study is limited to 10 patients showing the highest intrasample correlation. In addition, TCGA patients with co-occurring mutations in multiple genes involved in DNA methylation (TET1, DNMT3A, DNMT3B, IDH1, and IDH2) were excluded, and only patients with TET2 mutations and/or t(8;21) translocations as well as an APL control group are shown. Samples with TET2 mutation are reported with red stars. See Supplemental Figure S1 for additional clustering analysis.
Next, we investigated AML development in vivo using mouse bone marrow transplantation assays. Lethally irradiated SJL mice were transplanted with Tet2fl/fl or Tet2−/− HSPCs transduced with AE and monitored for hematopoietic abnormalities. The majority of mice (12 out of 13) transplanted with Tet2−/−;AE HSPCs succumbed to leukemia with a median latency of 6 mo, whereas mice receiving Tet2fl/fl;AE HSPCs (12 out of 12) remained healthy over a 12-mo observation period. Of note, splenic cells from moribund leukemic mice gave rise to leukemia with accelerated onset when transplanted into sublethally irradiated secondary recipient mice (Fig. 1B). Loss of Tet2 also greatly accelerated the onset of disease when combined with a truncated version of the AE fusion protein (AML1-ETO9a) that is sufficient to induce leukemia (Supplemental Fig. S1B; Yan et al. 2006).
The diseased mice transplanted with Tet2−/−;AE HSPCs suffered from leukocytosis and severe anemia, indicated by increased numbers of white blood cells (WBCs) and a reduction of red blood cells (RBCs) and hematocrit value in the peripheral blood (Fig. 1C). In addition, the mice showed greatly increased spleen sizes (Fig. 1D) accompanied by disruption of splenic architecture and infiltration of leukemic blasts in the spleen, liver, and bone marrow (Supplemental Fig. S1C). Further analysis of peripheral blood and bone marrow using flow cytometry revealed that the GFP-positive leukemic blasts had lost expression of CD45 (Fig. 1E), a hallmark of t(8:21) AML patients (Lo et al. 2012), and displayed an immature GMP-like myeloid immunophenotype (Supplemental Fig. S1D,E).
To investigate the validity of this model, we performed gene expression analysis on sorted leukemic GMP (L-GMP) cells from secondary recipient animals transplanted with splenocytes isolated from two independent moribond mice (LeuA and LeuB) (Fig. 1B). Unsupervised hierarchical clustering with human AML samples in the MILE study (Haferlach et al. 2010) and TCGA study (The Cancer Genome Atlas Research Network 2013) using the 10% most varying gene homologs revealed that, whereas wild-type GMP cells from control mice clustered with normal human GMP cells, murine Tet2−/−;AE L-GMP cells clustered closely together with t(8:21) AML patients as well as patients carrying TET2 mutations (Fig. 1F; Supplemental Fig. S1F). Hence, disruption of Tet2 collaborates with AE in vitro and in vivo to induce a lethal and transplantable leukemia that recapitulates the hallmarks of human t(8;21) and TET2 mutated AML disease.
Deletion of Tet2 in preleukemic hematopoietic cells expressing AE leads to accelerated in vitro cell proliferation and gene expression changes associated with human AML with TET2 mutations
Development of leukemia in an in vivo mouse model is a complex process often resulting in a heterogeneous pool of leukemic cells with diverse differentiation states and self-renewal capabilities. Thus, to investigate the primary regulatory effects of Tet2 disruption in hematopoietic cells, we developed an in vitro-based cell culture system. We crossed mice harboring the inducible Rosa26-CreERT2 allele with Tet2fl/fl mice to obtain Tet2fl/fl;Rosa26+/CreERT2 (referred to as Tet2fl/fl: CreER) double-transgenic animals. HSPCs isolated from mice of this genotype were transduced with AE, grown for three rounds of serial replatings in methylcellulose, and shifted to culture in liquid medium supplemented with cytokines. This resulted in independent cell cultures characterized by a population of in vitro GMP (iGMP) cells (B220+, CD3e−, Gr1−, CD11b−, and CD16/32+) that had gained self-renewal potential but retained the capacity to differentiate to mature cells of the granulocytic lineage (Gr1+CD11b+). Treatment with 4-hydroxytamoxifen (4-OHT) for two passages resulted in complete loss of full-length Tet2 mRNA with no effect on the level of AE expression (Supplemental Fig. S2A–D). In addition, the cells showed a twofold to fourfold reduction in endogenous 5-hmC levels, indicating that the catalytic activity of Tet2 was efficiently disrupted (Fig. 2A). Due to the high efficiency of recombination, EtOH control and 4-OHT-treated cultures derived from Tet2fl/fl;AE;CreER animals are hereafter referred to as Tet2fl/fl;AE and Tet2−/−;AE, respectively. In contrast to previous observations in Tet2 knockout animals, morphological and flow cytometry analysis revealed that granulocytic differentiation was unperturbed in Tet2−/−;AE cultures (Fig. 2B; Supplemental Fig. S2E,G). However, similar to what was observed in semisolid medium, Tet2−/−;AE cells showed a pronounced and lasting acceleration of cell proliferation compared with Tet2fl/fl;AE cells (average doubling time ∼20 h vs. ∼27 h) (Fig. 2C). In part, this was due to a cell type-specific decrease of apoptotic rate in iGMP cells in the Tet2−/−;AE cultures, indicating that loss of Tet2 confers a distinct survival advantage to this cell population (Supplemental Fig. S2H).
Figure 2.

Disruption of Tet2 in preleukemic AE cell cultures leads to gene expression changes similar to those found in human AML with TET2 mutations. (A) Overview of experimental setup (top) and quantification of global 5hmC levels by dot blotting (bottom). Genomic DNA isolated from biological duplicate cultures (Rep1 and Rep2) of Tet2fl/fl;AE;CreER cells treated with EtOH (control) or 4-OHT (induction of Tet2 disruption) were assayed. To show specificity, genomic DNA from wild-type or DNMT triple-knockout embryonic stem (ES) cells (Tsumura et al. 2006) as well as synthetic oligonucleotides were included. (B) Representative FACS plots showing populations of immature iGMP cells (GFP+CD3e−B220−Gr1−Mac1−CD16/32+) as well as mature Gr1+Mac1+ myeloid cells. Both Tet2fl/fl;AE (top panel) and Tet2−/−;AE (bottom panel) cultures show sign of granulocytic differentiation. (C) Accelerated in vitro proliferation of AE cells upon disruption of Tet2. Cumulative cell numbers or average doubling time (inset) of Tet2fl/fl;AE and Tet2−/−;AE cell cultures (n = 3). Cells were counted and plated at equal cell density every second day for a period of 20 d. Symbols show mean cell number, and error bars indicate SD. (****) P < 0.0001 (nonlinear regression). (D) Heat map of the top differentially expressed genes (false discovery rate [FDR] < 0.05, fold change >2) in sorted iGMP cells isolated from triplicate Tet2fl/fl;AE and Tet2−/−;AE cultures grown for two and 10 passages, respectively. Individual expression values were normalized, and the z-scores for each gene are presented. See Supplemental Table S2 for a full list of differentially expressed genes. (E) Tet2−/−;AE cells cluster closely with TET2 mutated AML patients, indicating that gene expression changes observed in iGMP cells upon Tet2 deletion are relevant to gene signatures of TET2 mutated AML patients. Differential gene expression-based hierarchical clustering of preleukemic Tet2fl/fl;AE and Tet2−/−;AE cells together with patients from TCGA (The Cancer Genome Atlas Research Network 2013) are shown. For clarity, patients with co-occurring mutations in multiple genes involved in DNA methylation (TET1, DNMT3A, DNMT3B, IDH1, and IDH2) were excluded, and only patients with TET2 mutations and/or t(8;21) translocations as well as an APL control group are shown. See also Supplemental Figure S2 for additional clustering analysis.
To gain insight into the onset and progression of effects associated with Tet2 disruption in iGMP cells, we performed gene expression analysis at both early (passage 2) and late (passage 10) stages. Tet2 disruption resulted in significant deregulation of 225 genes, of which 47 were changed more than twofold (Fig. 2D; Supplemental Table S2). Among these, many genes with known roles in stem cell function, leukemogenesis, and cancer were identified. These include several down-regulated putative tumor suppressor genes (e.g., Mtss1, Las2, Lxn, Ctdspl, Grap2, etc.) and up-regulated putative oncogenes (e.g., Aff3, Pim2, Nepn, Notch3, Igf1r, etc.) as well as many genes of unknown significance. The deregulated expression of many of these genes was validated by quantitative RT–PCR (qRT–PCR) on independently derived cultures (Supplemental Fig. S2I). Of note, these genes were not affected in nonleukemic GMP cells with Tet2 disruption, implying a context-specific role of Tet2 in gene regulation (Supplemental Fig. S2J).
Finally, we tested whether the gene expression changes observed in the iGMP cells were similar to those occurring in human AML patients. Remarkably, clustering analyses based on differentially expressed genes (Supplemental Table S2) showed that iGMP cells, upon disruption of Tet2, cluster closely with AML patient samples carrying TET2 mutations as compared with Tet2fl/fl;AE iGMP cells, t(8;21) patients, and an APL control group (Fig. 2E). Furthermore, clustering based on sample correlation revealed that two out of three passage 10 Tet2−/−;AE samples clustered specifically together with a patient carrying a t(8:21)/TET2 double mutation, corroborating the relevance of prolonged cell passaging and Tet2 deletion to human AML with these aberrations (Supplemental Fig. S2K). Thus, the preleukemic Tet2fl/fl;AE and Tet2−/−;AE in vitro-cultured cells represent a physiologically relevant and tractable cellular system to study the primary role of Tet2 disruption on DNA methylation and gene expression in hematopoietic cells.
5hmC is specifically lost at enhancer elements
The lack of Tet2-specific antibodies for chromatin immunoprecipitation (ChIP) prompted us to investigate the specific distribution of 5hmC as a surrogate marker of Tet2 occupancy and catalytic activity. Therefore, we performed 5hmC DNA immunoprecipitation (DIP) sequencing (5hmC-DIP-seq) on biological duplicate Tet2fl/fl;AE and Tet2−/−;AE cultures. Despite globally reduced 5hmC levels, initial analysis of summarized read densities across different genomic elements previously mapped in MEL cells revealed only a modest decrease of 5hmC at DNaseI sites, with little or no change at CpG islands, promoters, gene bodies, and Ctcf sites (Supplemental Fig. S3A). Thus, to discern the effect on DNaseI sites, we mapped enhancers and promoters based on duplicate ChIP-seq (ChIP combined with deep sequencing) profiles of H3K4me1, H3K4me3, and H3K27ac from Tet2fl/fl;AE and Tet2−/−;AE cells. Heat map visualization of normalized 5hmC read densities in wild-type cells showed that out of the 8739 enhancers mapped, about one-third was enriched in 5hmC. Upon Tet2 disruption, the same set of enhancers showed a marked depletion of 5hmC, which was also evident in summarized reads over all enhancers (Fig. 3A,B). This specific depletion of 5hmC at enhancers was further confirmed by quantitative analysis of differential peaks in both replicate samples (Supplemental Fig. S3B). Finally, depletion of 5hmC was also confirmed on several enhancers using 5hmC-DIP-qPCR on independently derived cell cultures (Fig. 3C,D; Supplemental Fig. S3C). Together, these results demonstrate that 5hmC is specifically lost from enhancer elements upon Tet2 disruption in hematopoietic cells.
Figure 3.

5hmC is specifically lost at enhancers. (A) Heat map showing an overview of 5hmC-DIP-seq read density on all 8739 enhancers in duplicate cultures of Tet2fl/fl;AE and Tet2−/−;AE cells. The top panel shows 2999 enhancers enriched in 5hmC in wild-type and depleted of 5hmC upon Tet2 knockout (mean log2 fold change >0.5), whereas the bottom panel shows the remaining enhancers. Each row represents a 20-kb window centered on an enhancer and extends 10 kb upstream and 10 kb downstream. (B) Summarized 5hmC-DIP-seq read densities across all 8739 enhancers in duplicate cultures of Tet2fl/fl;AE and Tet2−/−;AE cells. See Supplemental Figure S3 for other genomic elements. (C) Representative UCSC tracks showing specific loss of a 5hmC peak in an enhancer in the Zfp128 locus. The top tracks show 5hmC-DIP-seq enrichment data from biological duplicate cultures of Tet2fl/fl;AE and Tet2−/−;AE, respectively. The bottom tracks represent histone ChIP-seq experiments performed on Tet2fl/fl;AE and Tet2−/−;AE cells showing enrichment of H3K27ac, H3K4me1, and H3K4me3, respectively. (D) 5hmC-DIP followed by qPCR in independent biological triplicate samples. Primers directed against the Zfp128 enhancer as well as an unchanging downstream peak are shown. Bars represent mean enrichment over input, and error bars indicate SD. (*) P-value < 0.05 (Student's t-test). See also Supplemental Figure S3 for validation of additional loci.
Loss of TET2 leads to preferential hypermethylation of enhancer elements
Hydroxylation of 5mC has been implicated in both active and passive demethylation processes (for review, see Pastor et al. 2013). We therefore hypothesized that the specific loss of 5hmC on enhancer elements would make these prone to increased DNA methylation. To test this, we performed enhanced reduced representation bisulfite sequencing (eRRBS) Akalin et al. 2012) on early (passage 2) and late (passage 10) stage Tet2fl/fl;AE and Tet2−/−;AE cultures. Approximately 70% of reads could be aligned to the genome, yielding information about ∼2.3 million individual CpGs, each covered by >10 reads in all samples (see Supplemental Fig. S4A for detailed information of genome coverage).
We then focused on analyzing the differences in methylation across the four conditions. We reasoned that the methylation state of a given locus could be influenced by the absence of Tet2 catalytic activity (Tet2-dependent effect) but also by prolonged passaging of cells in culture (passage-dependent and age-related changes known as epigenetic drift). Interestingly, analysis of average DNA methylation (Supplemental Fig. S4B) or DNA methylation of individual CpGs within enhancer elements (Fig. 4A,B) revealed a striking and progressive Tet2-dependent hypermethylation at both early and late passage cells. However, when comparing all genomic elements, we noted that a large fraction of the CpGs covered in the analysis did not change in any condition. To exclude these and obtain a comparable measure of methylation change, we defined variable CpGs within each genomic element [Q-value < 0.05, abs(diff) > 20%] and calculated the mean difference in methylation. In this measure, two-way changes in methylation state (equal hypermethylation and hypomethylation) would result in a value close to 0, whereas a strong one-way effect (e.g., predominant hypermethylation) would result in deviation from this baseline. First, we analyzed Tet2fl/fl;AE cells at early and late passage numbers to evaluate passage-dependent (and Tet2-independent) drift in DNA methylation levels. Interestingly, and in agreement with previous reports studying primary cell culture systems (Meissner et al. 2008; Landan et al. 2012) as well as aged mice and MDS patients (Maegawa et al. 2014), we could observe a strong passage-dependent hypermethylation of CpG sites within CpG islands and promoters (Fig. 4C; Supplemental Fig. S4B). However, strikingly, when we analyzed the Tet2-dependent effect in Tet2−/−;AE versus Tet2fl/fl;AE cells at passages 2 and 10 following Tet2 disruption, we observed a significant hypermethylation of CpG sites in enhancer elements at both early and late passage cells, with little or no effect on DNA methylation in other genomic elements (Fig. 4D). It is noteworthy that Tet2 disruption had no detectable effect on DNA methylation of either CpG islands or promoters.
Figure 4.

eRRBS reveals preferential DNA hypermethylation in enhancer elements. (A) DNA methylation scatter plot showing methylation levels of individual CpG sites in enhancer elements. Methylation levels are shown for Tet2fl/fl;AE (Y-axis) and Tet2−/−;AE (X-axis) cultures at passage 2 (left panel) or passage 10 (right panel) after Tet2 disruption. CpG sites that are changing significantly [Q-value < 0.05; abs(diff) > 20%] are marked in red. (B) Cumulative distribution plot showing the methylation levels on all covered CpGs in enhancer elements. (Inset) Tet2-deficient cells have overall increased level of DNA methylation on enhancers upon cell passaging (p10 vs. p2 Tet2−/−;AE cells), indicating progressive hypermethylation. (C) Bar chart showing passage-dependent effects on DNA methylation in Tet2fl/fl;AE cells. Bars represent mean change of DNA methylation in the indicated genomic elements between passage 10 and passage 2. (D) Bar chart showing Tet2-dependent effects on DNA methylation at passage 2 (left panel) and passage 10 (right panel). Bars represent mean change of DNA methylation in the indicated genomic elements between Tet2−/−;AE (knockout) and Tet2fl/fl;AE (wild type). In both C and D, only CpG sites that showed significantly different DNA methylation levels [Q-value < 0.05; abs(diff) > 20%] were considered. The effect size of methylation change at each element was measured with Cohen's d. (*) Cohen's d between 0.5 and 1.0; (**) Cohen's d > 1.0. See Supplemental Figure S4 for details. (E) Serial replating assay of Tet2fl/fl or Tet2−/− Kit-enriched HSPCs transduced with MLL-AF9-expressing retrovirus. The cells were replated in methylcellulose-containing medium (M3534) every 5 d for a total of four replatings. Bars represent mean numbers of CFUs (n = 3), and error bars indicate SD. (F) Cumulative growth curve of Tet2fl/fl;MLL-AF9 or Tet2−/−;MLL-AF9 cultures (n = 3) grown for 10 passages. Cells were counted and plated at equal cell density every second day for a total of 20 d. (ns.) not significant (nonlinear regression). (G) Bar chart showing mean change of DNA methylation in the indicated genomic elements between Tet2−/−;MLL-AF9 (knockout) and Tet2fl/fl;MLL-AF9 (wild-type) cells 20 passages after Tet2 disruption. Mean changes of DNA methylation and genomic elements are presented in C and D. (*) Cohen's d between 0.5 and 1.0.
Since the increased DNA methylation of enhancer elements could be a result of a selection process dependent on the AE transgene and not only a result of Tet2 deletion, we decided to test the effect of Tet2 disruption in the context of the unrelated MLL-AF9 human oncofusion protein. Importantly, loss of Tet2 in HSPCs immortalized with the MLL-AF9 oncogene does not give any selective advantage as measured by colony formation in semisolid medium (Fig. 4E) or cell growth during prolonged passaging in liquid culture (Fig. 4F). Consistent with the observation using AE-dependent preleukemic cells, loss of Tet2 in immortalized MLL-AF9 cells grown for 20 passages led to selective hypermethylation of putative enhancer elements (Fig. 4G). In summary, these results show that Tet2 disruption leads to preferential DNA hypermethylation of enhancer elements during hematopoietic transformation.
Increased DNA methylation on enhancers correlates with loss of enhancer activity and lower expression of nearby genes
Using independent data sets of 5hmC and DNA methylation patterns, we showed that Tet2 disruption predominantly affects the epigenetic state of enhancers. Hence, we asked whether the overlap of these data sets could be used to find direct and high-confidence targets of Tet2. To do so, we defined continuous regions within enhancers (at least three CpGs within 1 kb, covered by >10 reads) and calculated the mean DNA methylation difference to identify differentially methylated regions (DMRs). This yielded 2017 enhancer regions in passage 10 cells, of which 22.7% was hypermethylated, and 1.1% was hypomethylated [abs(mean difference) >10%]. As anticipated, a significant proportion of these hypermethylated enhancer DMRs were also associated with loss of 5hmC (Fig. 5A; Supplemental Fig. S5A), and we could successfully validate the 5hmC and 5mC changes using independently derived triplicate Tet2fl/fl;AE and Tet2−/−;AE cultures (Fig. 5B; Supplemental Fig. S3C). To assess the effect of DNA methylation on enhancers, we defined the local (enhancer DMR + 250-base-pair [bp] flanking region) differential H3K27ac enrichment as a surrogate marker of enhancer activity as well as the normalized expression of genes within 100 kb of each enhancer DMR in Tet2fl/fl;AE and Tet2−/−;AE cells. Interestingly, increased DNA methylation correlates with loss of H3K27ac, suggesting that enhancers lose activity upon DNA hypermethylation (Fig. 5C; Supplemental Fig. S5C). Conversely, enhancers that are lost upon Tet2 disruption (mapped in wild type but not in knockout) also show the greatest increase in DNA methylation (Supplemental Fig. S5D). Accordingly, increased DNA methylation also correlates to some degree with lower gene expression of nearby genes, albeit only for enhancer DMRs with >30% increase in methylation (Fig. 5D). Attempts to correlate loss of 5hmC to differential H3K27ac and gene expression were unsuccessful, suggesting that loss of 5hmC is not sufficient to yield a detectable effect (Supplemental Fig. S5C).
Figure 5.

Loss of 5hmC and gain of methylation lead to decreased H3K27ac and expression of neighboring genes. (A) Overview of 2107 regions in enhancers covered by eRRBS. The percentage of enhancer DMRs with an average DNA methylation increase (blue; mean difference >10%) or decrease (black; mean difference <−10%) are shown (left pie chart) as well as the fraction of hypermethylated enhancer DMRs associated with loss of 5hmC (log2 fold depletion >0.5) upon Tet2 knockout (right pie chart). See Supplemental Table S3 for full list of enhancer regions. (B) Quantitative bisulfite pyrosequencing validation of DNA methylation changes at four enhancer DMRs (Mtss1, Fam105a, Tmem123, and Las2 locus) associated with loss of 5hmC and gain of DNA methylation in Tet2−/−;AE cells. Bar graphs show methylation levels of each individual CpG within the locus in cells grown for two or 10 passages following Tet2 disruption. Bars represent mean methylation (n = 3), and error bars indicate SEM. (*) P-value < 0.05; (**) P-value < 0.01; (****) P-value > 0.0001 (two-way ANOVA). (C) Increase in DNA methylation levels at enhancer DMRs correlates with loss of H3K27ac, a surrogate marker of enhancer activity. Violin plots show the mean and lower and upper quartiles of the log2 transformed differential H3K27ac read density (knockout/wild type) for enhancer DMR + 250-bp flanking sequence. The 2107 enhancer DMRs have been divided into groups with <10% increase, 10%–30% increase, or >30% increase in DNA methylation levels upon Tet2 knockout. (*) P-value < 0.05; (****) P-value < 0.0001 (unpaired Student's t-test). (D) Increase in DNA methylation levels at enhancer DMRs correlates with decreased expression of genes within 100 kb. Violin plots show the mean and lower and upper quartiles of average z-scores (knockout/wild type) of genes within 100 kb of enhancer DMRs. Subgroups and significance levels are presented as in C. (E) Convergent gene regulation of AE and Tet2. Venn diagrams showing overlaps and corresponding P-values (Fisher's test, Yates-corrected) of genes deregulated in Tet2−/−;AE cells and genes bound by AE in AML patients (as described in Martens et al. 2012; Ptasinska et al. 2012). (Right panel) Heat map representing gene expression changes for genes differentially regulated by Tet2 knockout and bound by AE in both studies.
To better understand the determining factors of hypermethylation and gene expression changes, we searched for known transcription factor-binding motifs within hypermethylated enhancer DMRs (Supplemental Fig. S5D). Interestingly, both a known PU.1 motif (E-value 2.3 × 10−004), a key hematopoietic transcription factor, and a Runx1 motif (E-value 2.3 × 10−002) were enriched in the enhancer DMRs, suggesting that the hypermethylated enhancers are important for normal hematopoiesis and that AE (through its Runt domain) is affecting at least part of the genes deregulated upon Tet2 knockout. In support of this, 52 of the 190 orthologous genes deregulated in the preleukemic cells have previously been found to be bound by AE in t(8;21) patients (Fig. 5E; Supplemental Fig. S5E; Supplemental Table S1). Thus, these data suggest that AE and TET2 are simultaneously affecting a subset of enhancers as well as genes.
Disruption of Tet2 leads to DNA hypermethylation of enhancers in GMP cells, embryonic stem (ES) cells, and murine and human AML cells
To extend the findings in the in vitro grown preleukemic cells to primary murine AML cells, we examined enhancer methylation patterns in FACS-sorted GMP and L-GMP cells (Fig. 6A). Quantitative bisulfite pyrosequencing of three enhancers identified in the preleukemic in vitro system confirmed a strong and specific Tet2-dependent enhancer hypermethylation in in vivo isolated GMP cells (Fig. 6B). Specifically, we detected a reproducible increase in methylation at enhancers upon disruption of Tet2 alone, whereas leukemic transformation with AE exacerbated this effect (∼15% vs. ∼43% mean increase). As observed previously in the in vitro-grown cells (Fig. 4D), we also detected a slight hypomethylation in GMP cells with AE alone (∼12% mean decrease). Hence, these results suggest that the function of Tet2 is linked to enhancer methylation both during normal hematopoiesis and in malignant cells. Furthermore, we found that Tet2 knockout mouse ES cells also show a pronounced and specific enhancer hypermethylation, extending the role of Tet2 in enhancer methylation to nonhematopoietic and nonmalignant cells (Supplemental Fig. S6).
Figure 6.

Disruption of Tet2 leads to DNA hypermethylation of enhancers in murine and human AML cells. (A) Overview of the strategy for in vivo isolation of cells. GMP cells were sorted from the bone marrow of recipient mice 1 mo after transplantation of either wild-type or Tet2−/− bone marrow, AE-transduced cKit cells, or splenic cells from moribond Tet2−/−;AE leukemic mice (LeuA and LeuB) (see Fig. 1B). (B) DNA methylation levels assayed by quantitative bisulfite pyrosequencing at three enhancers (Mtss1, Tmem123, and Pde2a locus) in FACS-sorted wild-type (n = 4), Tet2−/− (n = 4), AE-transduced (n = 4), or leukemic Tet2−/−;AE (n = 8) GMP cells. Bars represent mean locus DNA methylation, and error bars show SD. (*) P-value < 0.05; (**) P-value < 0.01; (***) P-value < 0.001; (****) P-value < 0.0001 (unpaired Student's t-test). (C) Distribution of DNA methylation changes of significantly changing CpGs (P-value < 0.05 [Wilcoxon two-sample test, abs(diff) > 20%] in AML patients with TET2 mutations (left panel), t(8;21) translocation (middle panel), and DNMT3A mutations (right panel). The percentages of hypermethylated CpG sites versus all significantly changing sites are indicated on the plots. (D) Bar chart showing the distribution of the hypermethylated CpG sites in AML patients with TET2 mutation or t(8:21) translocation across various genomic elements. The number of hypermethylated CpG sites observed in each element was normalized to the number expected by random distribution. The P-value for enhancer CpGs in TET2 mutated patients is shown (Fisher's exact test, Yates correction). (DNMT3A mutated patients) Not sufficient hypermethylated sites for analysis. (E) As in C, but for probes hypomethylated in AML patients with DNMT3A mutations or t(8:21) translocation. (TET2 mutated patients) Not sufficient hypomethylated sites for analysis.
Finally, to explore whether enhancer hypermethylation could be observed in human AML patients with TET2 mutations, we took advantage of a publicly available data set (The Cancer Genome Atlas Research Network 2013) in which 194 de novo AML patients were analyzed using the Illumina Infinium 450k methylation array. Patients were filtered based on the mutational status of genes involved in regulation of DNA methylation, and the most variable probes between an AML patient control group having wild-type TET2 (n = 103) and patient groups with either a TET2 mutation (n = 9), a t(8;21) translocation (n = 6), or a DNMT3A mutation (n = 29) were extracted. Consistent with previous studies, patients with TET2 mutations showed an overall hypermethylation phenotype, with 96.6% of significantly changing CpG sites showing an increase in methylation (Fig. 6C). In contrast, patients expressing mutant DNMT3A showed pronounced hypomethylation, and patients with t(8:21) translocation had both hypermethylated and hypomethylated CpG sites (Fig. 6C). Although the Infinium array is biased to assay the methylation state of promoters and CpG islands, detailed annotation revealed 8328 probes interrogating CpGs within predicted human enhancers. Remarkably, analysis of various genomic elements revealed a pronounced enrichment of hypermethylated CpG sites in enhancers in TET2 mutated patients (approximately fivefold over background) (Fig. 6D). Conversely, this was not observed in either hypermethylated CpG sites in t(8:21) patients or hypomethylated CpG sites in t(8;21) patients and patients with mutant DNMT3A (Fig. 6D,E). Finally, we observed a depletion of hypermethylated sites at CpG islands in TET2 mutated patients, suggesting, in agreement with the in vitro system, that disruption of TET2 does not result in CpG island hypermethylation. Taken together, this analysis shows that disruption of TET2 leads to specific hypermethylation of enhancer elements in human AML cells, and this increase in hypermethylation appears to be independent of the other transforming genetic events leading to leukemia.
Discussion
By generating and using a mouse model of human AE-induced AML, we showed that loss of Tet2 in hematopoietic cells led to a specific and progressive enhancer hypermethylation phenotype associated with altered gene expression, increased rate of cell accumulation, and development of leukemia. In contrast, CpG islands and gene promoters were not affected by Tet2 disruption but experienced a slow increase of DNA methylation related to the number of cell doublings; i.e., age-related epigenetic drift. Hence, data from this and another study (Maegawa et al. 2014) suggest that normal hematopoietic stem cells will be subject to a slow accumulation of CpG island DNA methylation and that an acquired TET2 mutation will lead to a relatively rapid increase in enhancer DNA methylation conferring proliferation and survival advantage. In turn, this combined effect on the DNA methylome is likely to directly or indirectly make the cell vulnerable to additional oncogenic hits and full-blown leukemic transformation (Fig. 7).
Figure 7.
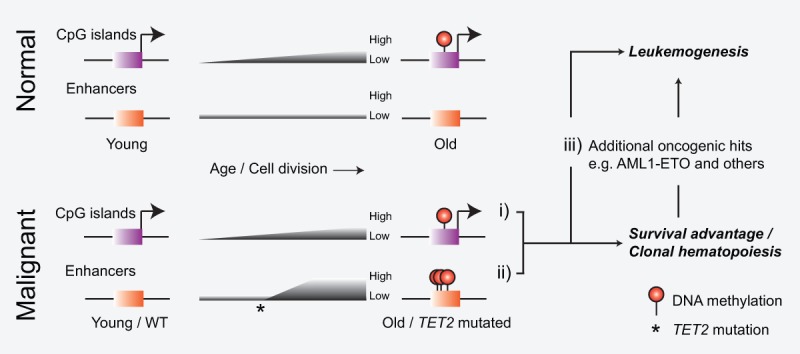
Model of DNA methylation changes associated with aging and leukemogenesis. Normal hematopoietic cells experience age-dependent accumulation of DNA methylation on CpG islands (i). Mutation of TET2 leads to rapid hypermethylation of enhancers (ii) that, together with additional oncogenic aberrations (iii [as well as i]), can collaborate to induce malignant transformation.
We and others have shown that Tet1 is preferentially localized to CpG islands and CpG-rich promoters through binding of its CXXC domain to unmethylated CpGs (Williams et al. 2011; Wu and Zhang 2011). In contrast, the lack of reliable antibodies for reproducible ChIP of Tet2 has made it difficult to establish a similar direct chromatin interaction map. However, several studies examining the genome-wide localization of 5hmC in ES cells have suggested that Tet proteins can exert their activity on regions outside CpG islands. In fact, using a range of different techniques for 5hmC enrichment and sequencing, it was shown that 5hmC is specifically enriched at lowly methylated regions associated with enhancer elements and CTCF insulators (Pastor et al. 2011; Stadler et al. 2011; Stroud et al. 2011; Williams et al. 2011; Yu et al. 2012; Hon et al. 2014). Here, using primary hematopoietic cells, we report widespread 5hmC depletion on enhancers upon Tet2 disruption, thus supporting a direct functional role of Tet2 on these elements. This was further confirmed by eRRBS analysis that found 467 out of 2107 covered regions in enhancers to be hypermethylated, suggesting that ∼25% of all enhancers in hematopoietic cells become hypermethylated upon Tet2 disruption. Interestingly, during the performance of this study, two studies investigating the effect of knocking out Tet proteins in mouse ES cells were published that found a similar proportion of enhancers to be hypermethylated (Hon et al. 2014; Lu et al. 2014). Thus, considering that increased methylation of enhancers has now been shown in both malignant (AE and MLL-AF9 transformed) and nonmalignant cell types (in vivo GMP and ES cells), it is likely that TET2 has a general role in regulating DNA methylation of enhancers in all cell types.
Along with promoters, associated enhancer elements are important determinants of context-dependent transcriptional output of genes (Plank and Dean 2014). Interestingly, several recent studies suggest that DNA methylation correlates negatively with enhancer activity and gene transcription, possibly by interfering with transcription factor binding (Stadler et al. 2011; Wiench et al. 2011; Thurman et al. 2012). In addition, Aran et al. (2013) examined both promoter and enhancer methylation with respect to gene expression and found that methylation of enhancers was a significantly better predictor of gene expression. Using our data sets of enhancer DNA methylation, 5hmC enrichment, and H3K27ac abundance (surrogate marker of enhancer activity) as well as expression of adjacent genes, we could show that enhancer hypermethylation correlates negatively with levels of the H3K27ac histone mark (Fig. 5C; Supplemental Fig. S5C). Thus, our data suggest that increased DNA methylation levels on enhancers decrease their activity by changing the chromatin environment and cause the down-regulation of gene expression of nearby genes. It should be noted that it was not possible to find a statistically significant correlation between expression of the nearest gene and changes in either enhancer DNA methylation or 5hmC abundance (an effect likely masked due to combinatorial and redundant enhancer usage of individual genes as well as subtle gene expression changes) (Supplemental Fig. S5C; Kasowski et al. 2013). However, when considering all genes within 100 kb of the enhancer, increased enhancer DNA methylation correlated negatively with gene expression, albeit only for enhancers with the strongest gain of methylation (Fig. 5D).
Given these findings, it is tempting to speculate that increase of DNA methylation at one or a few enhancers can be directly linked to deregulation of specific genes that in turn results in oncogenic transformation. In our study, we identified numerous such loci, including down-regulated putative tumor suppressor genes (e.g., Mtss1, Las2, Lxn, Ctdspl, Grap2, etc.) and up-regulated putative oncogenes (e.g., Aff3, Pim2, Nepn, Notch3, Igf1r, etc.) that constitute interesting candidates for functional testing in future studies. However, as previously mentioned, our results also suggest that mutations of TET2 lead to genome-wide enhancer hypermethylation affecting thousands (up to 25%) of active enhancers. Hence, it is likely that leukemogenesis in TET2 mutated cells is not due to deregulation of a single gene (or a few) but rather proceeds through a complex interplay between DNA methylation of multiple enhancers and a subtle destabilization of genetic networks. In addition, it should also be noted that only limited evidence suggests a direct instructive role of DNA methylation on CpG-poor regulatory regions such as enhancers (for review, see Schübeler 2015). Accordingly, it is rather the global and simultaneous perturbation of DNA methylation on numerous loci, although modest when observing only a single locus, that might account for the strong phenotype observed upon disruption of TET2 function. Finally, we believe that such a global mechanism could explain why TET2 mutations are not limited to a specific subtype of hematopoietic disease but co-occur frequently with a broad range of genetic aberrations in various hematopoietic disorders (Solary et al. 2014).
For decades, analysis of DNA methylation in cancer patients has been focused on promoter-proximal CpG islands and gene bodies. Instead, our results now suggest that analysis of TET2 mutated patients should be aimed at deciphering the effect on DNA methylation levels at enhancer elements. It is interesting to note that the frequency of somatic TET2 mutations is strongly age-related and that these have been described in otherwise healthy elderly individuals with clonal hematopoiesis (Busque et al. 2012). In addition, several studies have pointed to TET2 mutations as an early, if not the first, event in the clonal evolution of hematological disorders (Jan et al. 2012; Itzykson et al. 2013). Therefore, it is tempting to speculate that inactivating somatic TET2 mutations can exist latently in hematopoietic cells for years if not decades. During this period, enhancer methylation patterns may progressively deteriorate and result in aberrant gene expression and clonal dominance. Moreover, in some individuals, these premalignant cells can acquire a cooperating mutation whose characteristics drive the development of one of the wide range of hematological malignancies associated with TET2 mutations. Thus, careful examination of the epigenetic state of enhancer elements in these patients can potentially unravel the first molecular events that lead to initiation of the disease state and potentially lead to the identification of druggable candidates for targeting the full clonal hierarchy of malignant hematopoietic cells.
Materials and methods
Mice and in vitro cell culture systems
The conditional Tet2fl/fl mouse line described previously (Quivoron et al. 2011) and derived Cre lines were housed according to institutional guidelines. All animal work was performed under the approval of the Danish Animal Ethical Committee. Primary hematopoietic cell lines expressing MLL-AF9 in the Tet2fl/fl;CreER background were generated and maintained in suspension medium as previously described (Somervaille and Cleary 2006). Primary Tet2fl/fl;CreER HSPCs freshly transduced with MigR1-AE-IRES-GFP or MigR1-AE9a-IRES-GFP (Yan et al. 2006) were sorted for GFP and plated in methylcellulose medium (M3534, StemCell Technologies). After three rounds of replating (each lasting 5 d), the cells were shifted to grow in suspension in nontissue culture-treated plasticware in medium containing 1× StemPro-34 SFM (Invitrogen), 1× L-glutamine, with SCF (CHO producer cell line), 10 ng/mL IL-3 (Peprotech), 10 ng/mL IL-6 (Peprotech), and 0.1 mM 2-mercapto-ethanol. To induce the CreER-mediated excision of Tet2, the cells were treated with 200 nM 4-OHT (experimental) or EtOH (control) for two passages (4 d). The cells were subsequently washed to remove residual 4-OHT and passaged every 2 d for a total of 10–20 passages.
Gene expression analysis
Total RNA from FACS-sorted in vivo L-GMP cells as well as wild-type GMP cells (Lin−cKit+Sca1−CD16/32+CD150−) were labeled and hybridized to Agilent SurePrint G3 Mouse GE 8x60K arrays. Total RNA from FACS-sorted iGMP cells (GFP+, Gr1−, Mac1−, CD16/32+) from Tet2fl/fl;AE and Tet2−/−;AE cultures at passage 2 and passage 10 were hybridized to GeneChip Mouse Gene ST 2.0 arrays (Affymetrix).
ChIP-seq and 5hmC-DIP-seq
ChIP experiments (α-H3K4me1, α-H3K4me3, and α-H3K27ac) were performed essentially as described previously (Williams et al. 2011) on duplicate samples of Tet2fl/fl;AE and Tet2−/−;AE cells at passage 10 after deletion. 5hmC-DIP-seq was performed as described previously (Williams et al. 2011) on duplicate samples of Tet2fl/fl;AE and Tet2−/−;AE cells at passage 5 after Tet2 deletion (passage in which maximum global 5hmC depletion was achieved).
eRRBS
eRRBS libraries were generated as described (Akalin et al. 2012). In brief, ∼1 μg of genomic DNA was digested with MspI enzyme overnight followed by phenol/chloroform extraction. After library generation using preannealed methylcytosine-containing Illumina adaptors (TruSeq DNA sample preparation kit, Illumina) and gel-based size selection, the ligated fragments were treated with bisulfite (EZ DNA methylation kit, Zymo) to convert unmethylated cytosines. Finally, the converted fragments were PCR-amplified (15 PCR cycles) and sequenced on an Illumina HiSeq2000 using 100-bp single-end sequencing.
Enhancers
Active distal enhancers were defined as promoter-distal (nonoverlapping with a region extending ±2.5 kb from all transcription start sites), overlapping ChIP-seq-enriched regions of H3K27ac and H3K4me1 histone modifications with low mean H3K4me3 enrichment in duplicate samples. The resulting regions separated by <1 kb were merged to avoid redundant detection. This yielded a final high-confidence map of enhancers comprising a total of 8739 in Tet2fl/fl;AE cells and 9595 in Tet2−/−;AE cells.
Analysis of methylation state in TET2-deficient AML patients
TCGA Illumina Infinium 450k methylation array data and patient mutational analysis data were downloaded from the TCGA homepage (The Cancer Genome Atlas Research Network 2013). Each probe on the array was assigned to a category (gene body, promoter, CpG islands, and enhancers). Human predicted enhancers were defined as in Andersson et al. (2014). Probes that did not fall into any of the above categories were defined as intergenic.
Accession numbers
Raw and processed data sets are available for download at the Gene Expression Omnibus (GEO) database under the accession number GSE59591.
Supplementary Material
Acknowledgments
We thank Kirsten Grønbæk and the Helin laboratory for discussions. We thank Lotte Frederiksen and Anna Fossum for assistance with histology and flow cytometry, respectively. K.D.R. is supported by a post-doctoral fellowship from the Danish Medical Research Council (1333-00120B). This work was supported by the European Research Council (294666_DNAMET), the Danish Cancer Society, Danish National Research Foundation (DNRF 82), the Danish Council for Strategic Research, and the Novo Nordisk Foundation and through a center grant from the Novo Nordisk Foundation (the Novo Nordisk Foundation Section for Stem Cell Biology in Human Disease).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.260174.115.
References
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. 2012. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet 8: e1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T, et al. 2014. An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Sabato S, Hellman A. 2013. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol 14: R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P, Ralfkiaer E, et al. 2013. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 98: 1912–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. 2011. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer 11: 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, et al. 2012. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 44: 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Valle Della V, James C, Trannoy S, Massé A, Kosmider O, Le Couédic J-P, Robert F, Alberdi A, et al. 2009. Mutation in TET2 in myeloid cancers. N Engl J Med 360: 2289–2301. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné M-C, De Vos J, Hernández JM, Hofmann W-K, Mills KI, et al. 2010. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 28: 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Song C-X, Du T, Jin F, Selvaraj S, Lee AY, Yen C-A, Ye Z, Mao S-Q, Wang B-A, et al. 2014. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell 56: 286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa J-P. 2014. Aging and epigenetic drift: a vicious cycle. J Clin Invest 124: 24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C, Adès L, Fenaux P, Platzbecker U, Gagey O, et al. 2013. Clonal architecture of chronic myelomonocytic leukemias. Blood 121: 2186–2198. [DOI] [PubMed] [Google Scholar]
- Jan M, Snyder TM, Corces-Zimmerman MR, Vyas P, Weissman IL, Quake SR, Majeti R. 2012. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med 4: 149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, Boyle AP, Zhang QC, Zakharia F, Spacek DV, et al. 2013. Extensive variation in chromatin states across humans. Science 342: 750–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. 2010. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468: 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie J-N, Beyne-Rauzy O, et al. 2009. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 94: 1676–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein DA, Goldfinger N, Zundelevich A, et al. 2012. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet 44: 1207–1214. [DOI] [PubMed] [Google Scholar]
- Langemeijer SMC, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RAP, et al. 2009. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet 41: 838–842. [DOI] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, Xu M. 2011. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118: 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M-C, Peterson LF, Yan M, Cong X, Jin F, Shia W-J, Matsuura S, Ahn E-Y, Komeno Y, Ly M, et al. 2012. Combined gene expression and DNA occupancy profiling identifies potential therapeutic targets of t(8;21) AML. Blood 120: 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry C, Ntziachristos P, Ndiaye-Lobry D, Oh P, Cimmino L, Zhu N, Araldi E, Hu W, Freund J, Abdel-Wahab O, et al. 2013. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J Exp Med 210: 301–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liu Y, Jiang L, Yamaguchi S, Zhang Y. 2014. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev 28: 2103–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Gough SM, Watanabe-Okochi N, Lu Y, Zhang N, Castoro RJ, Estecio MRH, Jelinek J, Liang S, Kitamura T, et al. 2014. Age-related epigenetic drift in the pathogenesis of MDS and AML. Genome Res 24: 580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JHA, Mandoli A, Simmer F, Wierenga B-J, Saeed S, Singh AA, Altucci L, Vellenga E, Stunnenberg HG. 2012. ERG and FLI1 binding sites demarcate targets for aberrant epigenetic regulation by AML1-ETO in acute myeloid leukemia. Blood 120: 4038–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. 2008. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messerschmidt DM, Knowles BB, Solter D. 2014. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev 28: 812–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. 2011. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20: 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, et al. 2011. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473: 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. 2013. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14: 341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez C, Martínez-Calle N, Martín-Subero JI, Segura V, Delabesse E, Fernandez-Mercado M, Garate L, Alvarez S, Rifon J, Varea S, et al. 2012. TET2 mutations are associated with specific 5-methylcytosine and 5-hydroxymethylcytosine profiles in patients with chronic myelomonocytic leukemia. PLoS One 7: e31605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plank JL, Dean A. 2014. Enhancer function: mechanistic and genome-wide insights come together. Mol Cell 55: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptasinska A, Assi SA, Mannari D, James SR, Williamson D, Dunne J, Hoogenkamp M, Wu M, Care M, McNeill H, et al. 2012. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia 26: 1829–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronné L, Valle Della V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern M-H, et al. 2011. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20: 25–38. [DOI] [PubMed] [Google Scholar]
- Schoofs T, Berdel WE, Müller-Tidow C. 2014. Origins of aberrant DNA methylation in acute myeloid leukemia. Leukemia 28: 1–14. [DOI] [PubMed] [Google Scholar]
- Schübeler D. 2015. Function and information content of DNA methylation. Nature 517: 321–326. [DOI] [PubMed] [Google Scholar]
- Shide K, Kameda T, Shimoda H, Yamaji T, Abe H, Kamiunten A, Sekine M, Hidaka T, Katayose K, Kubuki Y, et al. 2012. TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia 26: 2216–2223. [DOI] [PubMed] [Google Scholar]
- Shih AH, Abdel-Wahab O, Patel JP, Levine RL. 2012. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 12: 599–612. [DOI] [PubMed] [Google Scholar]
- Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. 2014. The ten-eleven translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 28: 485–496. [DOI] [PubMed] [Google Scholar]
- Somervaille TCP, Cleary ML. 2006. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 10: 257–268. [DOI] [PubMed] [Google Scholar]
- Soucie E, Hanssens K, Mercher T, Georgin-Lavialle S, Damaj G, Livideanu C, Chandesris MO, Acin Y, Létard S, de Sepulveda P, et al. 2012. In aggressive forms of mastocytosis, TET2 loss cooperates with c-KITD816V to transform mast cells. Blood 120: 4846–4849. [DOI] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. 2011. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495. [DOI] [PubMed] [Google Scholar]
- Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 2011. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol 12: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, West J, Beck S. 2013. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet 22: R7–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S-I, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, et al. 2006. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11: 805–814. [DOI] [PubMed] [Google Scholar]
- Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, Dicker F, Fasan A, Haferlach C, Haferlach T, et al. 2012. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 26: 934–942. [DOI] [PubMed] [Google Scholar]
- Wiench M, John S, Baek S, Johnson TA, Sung M-H, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ, et al. 2011. DNA methylation status predicts cell type-specific enhancer activity. EMBO J 30: 3028–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, Helin K. 2011. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473: 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y. 2011. Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells. Cell Cycle 10: 2428–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L, Kantarjian HM, Estecio MR, Jelinek J, Godley LA, et al. 2012. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 7: 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M, Kanbe E, Peterson LF, Boyapati A, Miao Y, Wang Y, Chen I-M, Chen Z, Rowley JD, Willman CL, et al. 2006. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med 12: 945–949. [DOI] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song C-X, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149: 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.