

Abstract
Failure of accurate DNA damage sensing and repair mechanisms manifests as a variety of human diseases, including neurodegenerative disorders, immunodeficiency, infertility and cancer. The accuracy and efficiency of DNA damage detection and repair, collectively termed the DNA damage response (DDR), requires the recruitment and subsequent post-translational modification (PTM) of a complex network of proteins. Ubiquitin and the ubiquitin-like protein (UBL) SUMO have established roles in regulating the cellular response to DNA double-strand breaks (DSBs). A role for other UBLs, such as NEDD8, is also now emerging. This article provides an overview of the DDR, discusses our current understanding of the process and function of PTM by ubiquitin and NEDD8, and reviews the literature surrounding the role of ubiquitylation and neddylation in DNA repair processes, focusing particularly on DNA DSB repair.
Keywords: DNA damage response, double-strand break repair, ubiquitin, NEDD8, MLN4924
2. Introduction
Organisms have developed elaborate cellular pathways that encompass the sensing, signalling and repair of damaged DNA, collectively termed the DNA damage response (DDR), in order to protect themselves from the long-term adverse effects of DNA damage [1,2]. The cascade of events that takes place following an insult to DNA involves the recruitment and localization of DNA damage sensor and mediator proteins into visible sub-nuclear foci [3]. Signalling pathways link these, often chromatin-associated complexes, with transducer/effector proteins that move throughout the nucleus, serving to amplify the DNA damage signal and to coordinate global as well as more localized cellular changes. The cellular response to DNA damage largely depends on the nature of the insult and subsequent type of damage, with DNA double-strand breaks (DSBs) being the most cytotoxic. The whole DDR process is tightly controlled by reversible protein post-translational modifications (PTMs), including phosphorylation, poly(ADP-ribosyl)ation, ubiquitylation, sumoylation, methylation, acetylation and others, which regulate protein stability, localization and activity without the need for changes in de novo protein synthesis.
DNA damage comes in many different forms, which may arise in isolation, or occur as a complex mixture depending on the nature of the insult. In addition, spontaneously arising DNA lesions contribute to mutagenesis and ageing [4]. DNA damage can result from endogenous sources, such as reactive oxygen species or other by-products of cellular metabolism, DNA mismatches during replication or as a result of abortive topoisomerase activity. DNA DSBs can also arise through programmed cellular events, such as during chromosomal crossover and recombination in meiosis or through V(D)J and class-switch recombination in developing lymphocytes to generate immune receptor and antibody diversity [5–7]. Alternatively, exogenous sources of DNA damage include ionizing radiation (IR), ultraviolet light (UV) and environmental carcinogens, including those derived from tobacco smoke.
Clinical syndromes arising due to hereditary defects in DDR proteins are typified by immunodeficiency, infertility, neurodegeneration, cancer predisposition and, in some cases, accelerated ageing, highlighting some of the physiological processes that rely on functional DNA repair pathways [1,2]. Genomic instability in particular is a hallmark of cancer, and many tumours are deficient in one or more DNA repair pathways. This, along with the inherent replication stress in many tumours, provides a therapeutic window for cytotoxic chemotherapeutics that act through the generation of DNA damage and has also led to the clinical development of small molecule inhibitors of key DDR enzymes [8–10].
2.1. Sensing a DNA double-strand break
A DSB is detected very quickly by various DSB ‘sensor’ proteins that subsequently direct signalling and repair via one of two predominant DSB repair pathways in human cells: homologous recombination (HR) or non-homologous end-joining (NHEJ). One of these DSB sensors is the Ku protein, a heterodimer formed by two structurally related polypeptides of 70 and 83 kDa (Ku70 and Ku80, respectively) [11,12]. Ku is a highly abundant DNA-binding protein, capable of binding free DNA ends, and is essential for repair by NHEJ [13,14]. DNA binding of Ku occurs rapidly following a DSB and is independent of DNA sequence [15–17]. Ku is able to self-associate and the binding of two Ku molecules to either side of the DSB enables bridging of Ku and stabilization of the DNA ends, while maintaining access to the DNA ends by ligation enzymes [18–20]. In addition, Ku serves to recruit all other core components of the NHEJ complex, including DNA-PKcs [17,21,22], XRCC4/LIG4 [23,24], XLF [25] and the recently identified PAXX protein [26,27], to enable DNA end-ligation/repair.
Another DSB sensor is the (MRN) protein complex comprising MRE11 (meiotic recombination 11), RAD50 and NBS1 (Nijmegen breakage syndrome 1) [28–31]. MRE11 has intrinsic DNA-binding activity [32], as well as endo- and exonuclease activity [33,34]. It is important for the short-range stabilization of DNA ends and, together with its binding partner CtIP (also known as RBBP8; retinoblastoma binding protein 8), promotes initiation of DNA end resection to promote HR [35,36]. The MRE11–RAD50 components of MRN also partially unwind DNA ends and are believed to play a role in the long-range tethering of DNA molecules, whereas NBS1 contributes to recruitment and activation of ATM (ataxia-telangiectasia mutated) kinase, which mediates downstream signalling events [37–40].
The poly(ADP-ribose) polymerase proteins PARP1 and PARP2 also recognize both single- and double-stranded DNA lesions, with such binding triggering their enzymatic activities to synthesize poly (ADP)-ribose (PAR) chains attached to PARP1/2 themselves as well as other proteins in the vicinity of DNA breaks [41–43]. The best-described DDR function for PARP is in single-strand break (SSB) repair, where PAR chains promote recruitment of DNA repair factors such as XRCC1 (X-ray repair cross-complementing protein 1) and LIG3 (DNA ligase 3) [44,45]. PARP1 also promotes DSB repair by alternative NHEJ [46]. It is not yet clear how a particular DSB might promote the recruitment of one over another DNA damage sensing molecule and, indeed, studies have shown that both MRN and Ku co-localize at some DSBs, at least initially [16]. DSB repair pathway choice at various levels of the signalling cascade is an area of intense current research, with recent work highlighting how it is at least in part controlled by cell cycle status [47] and chromatin structure [48].
2.2. Signalling events following a double-strand break
Even a single DSB can evoke a complex cellular response that occurs not only in the vicinity of the break but also globally throughout the cell to coordinate the most appropriate outcomes. DNA DSB signalling events are largely coordinated by the apical phosphatidylinositol 3-kinase-related kinases (PIKKs): ATM, ATR (ataxia-telangiectasia and Rad3-related protein) and DNA-PKcs. These kinases preferentially phosphorylate serine or threonine residues followed by a glutamine residue (S/TQ) [49]. Hundreds of potential substrates have been identified for ATM and ATR [50], although the physiological relevance of many of these is still not known. Interestingly, DNA-PKcs itself is the only physiologically relevant DNA-PK substrate identified to date [51,52]. As described above, ATM is predominantly activated following DSB formation by its interaction with NBS1 [37–39], although ATM activation can be potentiated by other factors, particularly in the context of a damaged or disrupted chromatin state [53]. ATR, however, is activated by RPA (replication protein A)-bound single-stranded DNA (ssDNA), which can arise either as a result of replication stress (following uncoupling of the replication helicase and polymerase) or following DNA end resection, as associated with HR-mediated DSB repair [54]. ATR is recruited to RPA-ssDNA by its obligate partner ATRIP (ATR-interacting protein), where it is activated by TOPBP1 (topoisomerase binding partner 1) [55–57]. Germline mutations in ATM or ATR result in ataxia-telangiectasia [58,59] and Seckel syndrome, respectively [60,61]. The catalytic activity of DNA-PK is brought about via Ku-mediated DNA binding [21] and promotes NHEJ [62–64]. Germline mutations in DNA-PKcs result in severe combined immunodeficiency syndromes [65].
A critical early step in the cellular response to DNA DSBs is phosphorylation of histone H2AX on serine 139 (known as γH2AX) [66], largely by ATM in response to IR although functional redundancy exists with ATR and DNA-PK [67]. MDC1 (mediator of DNA damage checkpoint protein 1) directly binds γH2AX through its carboxyl-terminal BRCT repeats and potentiates the γH2AX signal, by both promoting its phosphorylation and curtailing its dephosphorylation [68]. Spreading of γH2AX to over a megabase from the site of the initial lesion [66] is required to effectively sustain the DNA damage signal sufficiently to recruit and retain mediator proteins such as 53BP1 at IR-induced foci. MDC1 also serves as a docking site for recruitment of other DDR proteins on the damaged chromatin and is the cornerstone molecule for crosstalk between phosphorylation and ubiquitylation signalling cascades in the DDR (figure 1).
Figure 1.

Simplified illustration of the major protein players involved in ubiquitin signalling following DSB induction. See text for details. Horizontal lines represent DNA. P, phosphorylation; Ub, ubiquitylation; Me, methylation. Protein X denotes unknown protein.
As well as coordinating the local recruitment of mediator proteins to DNA DSBs, the PIKKs also phosphorylate effector molecules that regulate more global cellular responses, including transcription, apoptosis, senescence and delayed cell cycle progression. CHK1 (checkpoint kinase 1) and CHK2 (checkpoint kinase 2) are two well-characterized substrates of ATR and ATM, respectively, that function throughout the nucleus [69]. CHK1 is important for activation and maintenance of the G2/M checkpoint, whereas CHK2 is considered to work mainly, but not exclusively, in the G1/S checkpoint [70]. CDC25A is a principal substrate of CHK1, phosphorylation of which leads to SCFβTRCP-mediated degradation and activation of the intra-S and G2/M checkpoints [71]. A critical substrate of CHK2 is p53, phosphorylation of which promotes stabilization and subsequent apoptosis when the number of DNA lesions exceeds the repair capacity of the cell [72,73]. A third checkpoint kinase, MAPKAP-K2 (MK2), has also been characterized, which is the effector kinase of the p38 SAPK pathway [74]. In response to DSBs, the p38MAPK/MK2 pathway is activated downstream of ATM/ATR and functions independently of CHK1 activation to maintain G2/M and intra-S phase arrest [75]. Major substrates of MK2 following DNA damage include CDC25 family members as well as protein complexes important for RNA biology [74–76]. Together, the signalling pathways described above are critical for cell survival following DNA damage as they regulate the kinetics of cell cycle progression, allowing sufficient time for DNA repair.
2.3. DNA repair pathways
As mentioned previously, in human cells, the predominant DSB repair pathways are classical NHEJ and HR. NHEJ is initiated by the binding of Ku to DNA ends, which subsequently recruits DNA-PKcs, end-processing and ligation factors to the break. It is a relatively rapid repair pathway that occurs in all cell cycle stages. In the absence of classical NHEJ proteins, DNA DSBs can still be ligated, albeit more slowly, by an alternative NHEJ pathway (alt-NHEJ; also known as MMEJ—microhomology mediated end-joining) [77]. While it is generally assumed that alt-NHEJ only plays a detectable role in DSB repair when classical NHEJ is compromised, it appears to function more predominantly in the repair of certain types of breaks, such as those generated during immunoglobulin gene class-switch recombination [78]. Alt-NHEJ is thought to contribute to the excessive genomic deletions and chromosomal translocations seen in various tumours. PARP1, XRCC1, LIG3, LIG1 and CtIP have all been shown to play a role in alt-NHEJ; however, the exact mechanism of repair has not been fully defined [79].
In contrast to NHEJ, DSB repair by HR requires significant DNA end-processing and is initiated following 3′–5′ DNA end resection coordinated by CtIP and the MRN complex [2]. The resulting ssDNA is stabilized through RPA (replication protein A) coating, leading to ATR activation. BRCA2 (breast cancer type 2 susceptibility protein), probably with the help of BRCA1 (breast cancer type 1 susceptibility protein) and PALB2 (partner and localizer of BRCA2), promotes the loading of RAD51 onto RPA-coated ssDNA, which then enables strand invasion of a homologous DNA sequence in a sister chromatid and formation of a complex DNA arrangement that is resolved by mechanisms that may or may not result in crossover of the two DNA molecules [80]. HR is restricted to S/G2 phases of the cell cycle and, compared with NHEJ, is a relatively slow process, sometimes taking hours to complete. Hereditary defects in genes for HR factors such as BRCA1 and BRCA2 lead to cancer predisposition syndromes and tumours that are reliant on alternative repair pathways for survival. This observation led to the idea of using the synthetic lethality concept to treat such cancers and the clinical development of PARP inhibitors for the treatment of BRCA1- and BRCA2-deficient tumours [81–83]. Inhibitors of PARP activity with molecules such as olaparib/LynparzaTM (AstraZeneca) inhibit SSB repair and also cause PARP to become trapped on DNA repair intermediates, resulting in DNA replication associated DSBs [84]. PARP inhibitors therefore generate one-ended DNA DSBs in cells that are normally repaired by HR processes; consequently, PARP inhibitors selectively kill HR deficient tumours [81,82].
Besides DNA DSBs, DNA damage can also consist of DNA SSBs, damaged DNA bases, DNA mismatches, as well as inter- and intrastrand DNA cross-links, which engage with specialized cellular pathways for their repair (table 1).
Table 1.
Brief description of DNA repair pathways in human cells. See text for details on repair by NR and NHEJ.
| DNA repair pathways | |
|---|---|
| mismatch repair (MMR) | DNA mismatches can arise during normal DNA replication and are repaired through MMR pathways involving the collective actions of a nuclease, polymerase and ligase [85]. Hereditary defects in MMR genes, such as occur in Lynch syndrome (also known as HNPCC, hereditary non-polyposis colorectal cancer) result in tumours with high levels of microsatellite instability |
| SSB repair | SSBs are recognized by PARP, which synthesizes PAR chains in the vicinity of the DNA break and promotes recruitment of DNA repair factors such as XRCC1 and LIG3 [86]. SSBs can occur as a result of IR or treatment with various chemical agents, and also arise as intermediates during BER and NER (see below) |
| base excision repair (BER) | involves the recognition, excision and replacement of damaged bases in cells, using enzymes that overlap with those required for SSB repair [87] |
| nucleotide excision repair (NER) | NER removes helix-distorting lesions from DNA, in particular the UV-induced photo lesions CPD (cyclobutane pyrimidine dimers) and 6-4PP (pyrimidine 6-4 pyrimidone photoproducts). Xeroderma pigmentosum (XP) is the archetypal human NER-deficiency syndrome, causing extreme sensitivity to UV light and very high incidences of skin malignancies. NER involves removal of a short oligonucleotide that includes the damaged lesion and subsequent restoration of the DNA sequence using the undamaged DNA as a template. Two sub-pathways of NER, global genome NER (GG-NER) and transcription coupled NER (TC-NER) use different mechanisms to recognize DNA lesions and promote either repair of DNA lesions throughout the genome or lesions encountered during active transcription, respectively [88] |
| trans-lesion synthesis (TLS) | TLS is a DNA damage bypass mechanism that protects against DSB break generation following replication fork stalling. It employs specialized DNA polymerases, principally from the Y-family, to replicate past the damaged DNA template and is inherently error-prone [89] |
| DNA interstrand cross-link (ICL) repair | ICLs can arise following exposure to a range of environmental mutagens, but are particularly abundant in cells following exposure to alkylating or platinum-based chemotherapeutics [90]. Fanconi anaemia is a rare genetic disorder causing aplastic anaemia, developmental defects and cancer predisposition, which is characterized by hypersensitivity to DNA interstrand cross-linking agents. It is caused by autosomal recessive mutations in one of 15 known genes that are required for ICL repair. The core Fanconi anaemia complex is made up of eight proteins (FANCA, B, C, E, F, G, L and M) required for the detection and repair of ICLs. ICLs can stall progression of the replication fork, causing replication fork collapse and the generation of a DNA DSB requiring coordination between translesion synthesis and homologous recombination mechanisms for repair [89] |
3. Post-translational modification with ubiquitin
3.1. Ubiquitin and ubiquitin conjugation to substrates
Ubiquitin is a highly evolutionarily conserved, small (76 amino acid residue) protein, originally identified through its ability to mediate ATP-dependent protein degradation in reticulocyte extracts [91–93]. Four genes encode ubiquitin in the human genome (UBC, UBB, UBA52 and UBA80), which are first transcribed either fused to ribosomal proteins (UBA52, UBA80), or as linear poly-ubiquitin chains that require processing to ubiquitin monomers (UBC, UBB) [94–96]. Full-length ubiquitin is a precursor peptide, requiring cleavage to expose a carboxyl-terminal di-glycine motif. Ubiquitin is then covalently conjugated via its carboxyl-terminus to target proteins, generally to the ε-amino group on a substrate lysine. This conjugation involves a three-step enzymatic process, first described in the 1980s [97,98], using an E1- (activating), E2- (conjugating) and E3- (ligase) enzyme (subsequently referred to as E1, E2 and E3, respectively; see figure 2 for further description of the enzymatic cascade).
Figure 2.

Illustration of ubiquitylation cascade. Ubiquitin is produced as a precursor polypeptide and cleaved to reveal a carboxyl-terminal GG- motif. In an ATP-dependent reaction, an E1 enzyme transforms this motif into a ubiquitin-adenylate intermediate, which reacts with a Cys in the catalytic domain of the E1 to form an E1∼Ub, thioester linkage. At least for UBA1 (the best-characterized ubiquitin E1), a second ubiquitin molecule is adenylated and remains non-covalently linked to the E1 adenylation active site. Double loading of the E1 with ubiquitin is believed to potentiate transfer of ubiquitin from the E1 to the E2 [99]. The ubiquitin-charged E1 is recognized by an E2 conjugating enzyme and ubiquitin is transferred to the catalytic cysteine of the E2 via a thioester linkage. Ubiquitin is subsequently conjugated to a substrate lysine, through E2 recognition of a substrate/E3 ligase complex. E1 and E3 binding sites to the E2 overlap, ensuring progression of the ubiquitylation cascade. RING E3s facilitate transfer of ubiquitin from the E2 to substrate without binding ubiquitin directly. Alternatively, ubiquitin is transferred to an active site cysteine in HECT/RBR E3s before forming an isopeptide linkage with the substrate lysine. Multiple cycles of substrate binding to ubiquitin-charged E2s lead to ubiquitin chain formation. Ubiquitylation can be reversed by de-ubiquitylating enzymes (DUBs).
A pyramid of enzymatic complexity exists to enable conjugation of ubiquitin to a plethora of substrates and the subsequent regulation of a wide range of biological processes. In humans, there are eight known E1s, two of which are specific for ubiquitin (UBA1 and UBA6) [100], 35 active E2s [101] and there are predicted to be more than 1000 E3s [102]. E3s can be divided into three major families: RING (really interesting new gene), HECT (homology to E6AP carboxyl-terminus) and RBR (ring between ring) [102,103]. The RING E3s bind simultaneously to the ubiquitin-charged E2 and substrate (either directly, or through E3-binding partners), facilitating the transfer of ubiquitin to the substrate, without the E3 binding ubiquitin directly. The RING domain of such E3s contains seven highly conserved cysteine residues and a highly conserved histidine residue that coordinate binding to two central Zn2+ ions, the structure of which is essential for E2 binding [104]. The term RING E3 ligase is rather a misnomer therefore, as RING E3s contain no catalytic activity per se, although they do catalyse the transfer of ubiquitin from the E2 to substrate by positioning the ubiquitin moiety into a favourable position for conjugation [105]. In the case of HECT and RBR E3s, however, ubiquitin is transferred from the E2 to an active site cysteine in the E3 and then to the substrate. U-box E3s are a smaller family of E3 enzymes, originally called E4s, as they were shown to fine-tune pre-formed ubiquitin chains [106]. They contain a U-box motif that has a similar three-dimensional structure to the RING domain but lacks the conserved Zn2+ binding residues; U-box containing proteins have subsequently been shown to have independent E3 ligase activity [107].
Unlike sumoylation and perhaps also neddylation, there is no target consensus motif for ubiquitin conjugation of substrates. In the majority of cases (especially for polyubiquitylation with K48 and K11 chains), ubiquitylation is not restricted to a particular substrate lysine, although examples of lysine-specific ubiquitylation do exist, for example K164 mono-ubiquitylation on PCNA (proliferating cell nuclear antigen). Depending on the E3 and substrate involved, lysine specificity can be determined by the E3, interacting partners of the E3 and/or the substrate itself [103].
Like all PTMs, ubiquitylation is dynamic and can be reversed by deubiquitylating enzymes (DUBs). There are five families of DUBs, comprising a total of 79 genes in the human genome (UCHs, ubiquitin carboxyl-terminal hydrolases; USPs, ubiquitin-specific proteases; OTUs, ovarian tumour proteases; MJDs, Machado-Josephin domain proteases; and JAMMs, JAB1/MPN/MOV34 metalloenzymes, also known as MPN+). Broadly speaking, the function of DUBs can be divided into three groups: processing of linear ubiquitin chains to generate free ubiquitin, editing of ubiquitin chains and reversal of ubiquitylation on substrates [108,109]. DUBs are generally highly specific for ubiquitin versus other UBLs, peptide versus isopeptide linkages, linkage chain type and different substrates. In addition, some DUBs have a preference for distal rather than proximal ubiquitin molecules, some preferentially cleave mono-ubiquitin and others have specially evolved for the recycling of ubiquitin following proteasomal degradation [108,109]. The balance between ubiquitylating and deubiquitylating activity is essential for regulating a wide range of cellular processes, encompassing all those described for ubiquitylation below.
3.2. Function of ubiquitylation and the ‘ubiquitin code’
Some substrates are modified by a single ubiquitin moiety on a single residue, termed mono-ubiquitylation, while others are multi-mono-ubiquitylated. Mono-ubiquitylation has been shown to regulate lysosomal degradation of proteins [110] and also mediates protein–protein interactions, such as regulating the recruitment of translesion synthesis polymerases following mono-ubiquitylation of PCNA after DNA damage [111]. Ubiquitin, however, contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63), enabling the formation of ubiquitin chains and thus the poly-ubiquitylation of substrates. In addition, linear ubiquitin chains (also known as M1 linkage) can be formed using the amino-terminal methionine of ubiquitin [112]. The linkage specificity is largely determined by the pairing of specific E2s and E3s [113]. K48 poly-ubiquitylation was the first described, and labels proteins for proteasome-mediated degradation [114]. With the exception of K63-linked chains, all other chains accumulate following proteasomal inhibition in yeast [115], suggesting at least some role in directing proteasomal degradation for all non-K63-linked chains. Like mono-ubiquitylation, mediating non-proteasomal protein–protein interactions is an important additional function for poly-ubiquitylation, particularly K63 chains, and a number of different ubiquitin-binding domains (UBDs) have been characterized on a wide range of proteins [116,117]. The non-degradative K63 chains are particularly important in regulating the DDR and are discussed in more detail below. Other described functions for poly-ubiquitylation include the regulation of protein activity and localization [118,119]. Although mixed and branched ubiquitin chains are readily formed in vitro, the prevalence and physiological relevance of such linkages in vivo is still not fully clear [118]. Given the heterogeneity of the ubiquitin code, it is perhaps not surprising that ubiquitylation regulates a wide range of biological processes, including cell cycle progression, transcription, apoptosis and inflammation, as well as DNA damage signalling and repair. As such, its function or dysfunction is associated with various diseases, especially neurodegenerative disorders, viral diseases and cancer, and targeting ubiquitin-system components is currently an active area for drug development.
3.3. Ubiquitin-like proteins
Since the discovery of ubiquitin, a number of UBLs have been described, based on their structural similarity and sequence homology to ubiquitin. As discussed in more detail elsewhere, excluding ubiquitin itself, nine UBL families have been defined: NEDD8 (neural precursor cell expressed developmentally downregulated protein 8), SUMO1–3 (small ubiquitin-related modifier-1, -2 or -3), ISG15 (interferon-stimulated gene 15), URM1 (ubiquitin-related modifier 1), UFM1 (ubiquitin-fold modifier 1), FAT1 (HLA-F adjacent transcript 10), FAU (Finkel-Biskis-Reilly murine sarcoma virus ubiquitously expressed, also known as FUB1 or MNSF-β), ATG12 (autophagy-related protein 12), ATG8 (autophagy-related protein 8) and paralogues MAP1LC3 (microtubule-associated protein 1 light chain 3)-A, -B and -C and GABARAPL (gamma-aminobutyric acid receptor-associated protein-like)-1, -2 and -3 [100,120,121]. Like ubiquitin, the UBLs rely on an E1–E2–E3 enzymatic cascade to mediate their conjugation to substrates, although the enzymes used and downstream effects of conjugation are specific to each UBL. Along with SUMO, NEDD8 is one of the best-characterized UBLs to date, and the specifics of the NEDD8 conjugation pathway along with published functions of neddylation in the DDR are discussed further in following sections.
4. Role of ubiquitylation in DNA double-strand break repair
A role for ubiquitylation in the DDR first came through the discovery that the post-replication repair protein yeast Rad6 is actually a ubiquitin E2 [122] which, in association with the E3 Rad18, mono-ubiquitylates the replication factor PCNA on lysine 164 in response to DNA damage [123]. Subsequently, mono-ubiquitylation of PCNA by human RAD18 was shown to regulate the switch from the use of replicative polymerases to damage-tolerant polymerases at the replication fork [124,125]. Notably, in vertebrate cells RAD18-independent ubiquitylation of PCNA K164 also occurs, suggesting some differences between the regulation of post-replication repair in yeast and higher eukaryotes [126]. Mono-, as well as poly-ubiquitylation of PCNA is therefore crucial for coordinating DNA damage tolerance events in both yeast and human cells [89]. Ubiquitylation has since been shown to regulate almost all DNA repair pathways and, in particular, is integral to the early signalling events following DNA DSBs (figure 1).
4.1. Double-strand break signalling by RNF8 and RNF168
DNA damage sites are enriched with K63 ubiquitin chains [127,128], and RNF8 provides a critical link between phosphorylation and ubiquitylation events in the DDR (figure 1). Three independent studies in 2007 identified the RING ubiquitin E3 ligase RNF8 as a predominant E3 regulating ubiquitylation at DSB sites [129–131]. These studies demonstrated that RNF8 binds ATM-phosphorylated MDC1 via its FHA (forkhead-associated) domain, mediates K63-linked ubiquitylation at DNA damage sites with the E2 UBE2N (also known as UBC13) and is required for the recruitment of both 53BP1 and the RAP80/BRCA1 complex to DSB sites. In addition to binding phosphorylated MDC1, the FHA domain of mammalian RNF8 was also shown to interact with ATM-phosphorylated HERC2 (also a RING E3 ligase) following DNA damage to form an MDC1–RNF8–HERC2 complex [132]. Although it is not yet known whether the E3 ligase activity of HERC2 is required for its function in the DDR, in human cells, HERC2 has been reported to stabilize the interaction between RNF8 and UBE2N and also maintains the level of RNF8, to promote RNF8-mediated ubiquitylation and subsequent downstream events at DNA damage sites [132]. Notably, however, knockout of the gene for HERC2 in the chicken DT40 cell line does not cause DNA damage hypersensitivity or defects in ubiquitin accumulation at DNA damage sites [133], suggesting that at least some functions for this very large protein are not conserved throughout vertebrates.
The ubiquitylation signal required for DDR signalling is not sufficiently maintained by RNF8 alone, but is highly dependent on the activity of a second RING E3 ubiquitin ligase, RNF168. RNF168 MIUs (motif interacting with ubiquitin domains) and UMI (UIM and MIU-related ubiquitin-binding domain) bind as-yet undefined, RNF8-ubiquitylated substrates flanking DSB sites [127,128,134]. RNF168 then promotes ubiquitylation of histone H2A K13/15, thereby promoting assembly of 53BP1 and BRCA1/RAP80 complexes at sites of DNA damage [135]. Recent work has shed light on how a hierarchy of ubiquitin E3 recruitment is achieved in the DDR and has demonstrated that regions outside ubiquitin-binding domains, named LR motifs, as well as properties of the substrates themselves determine recruitment and substrate specificity of the E3 [136,137]. Current models propose that RNF8 ubiquitylates an as-yet unidentified non-nucleosomal substrate that serves as a docking site for RNF168 recruitment and subsequent ubiquitylation of lysines 13/15 on histone H2A(X), enabling recruitment of 53BP1 and BRCA1 complexes to DNA damage sites [135].
Functionally, bi-allelic heterozygous nonsense mutations in the gene encoding RNF168 cause RIDDLE syndrome (radiosensitivity, immunodeficiency, dysmorphic features and learning difficulties) [127,138] and depletion of either RNF8 or RNF168 in cells causes hypersensitivity to DSB inducing agents. Other studies have also demonstrated a role for RNF8/168 in immunoglobulin class-switch recombination [139], telomere end-protection [140,141] and transcriptional repression at DNA damage sites [142]. Interestingly, the DDR is suppressed during mitosis at the level of RNF8 function [143,144], highlighting the importance of RNF8-mediated ubiquitylation in driving DSB repair and associated signalling events.
4.2. Ubiquitylation impacts on 53BP1 and BRCA1 functions
The recruitment of 53BP1 and BRCA1 to DSB sites is essentially triggered by the same signal: RNF8/168-mediated ubiquitylation (figure 1). However, 53BP1 and BRCA1 have seemingly opposing functions, with 53BP1 promoting NHEJ [145] and BRCA1 promoting HR [146]. Until recently, despite being dependent on histone H2A K13/15 ubiquitylation, the recruitment of 53BP1 to DNA damage sites appeared to be through its TUDOR domain binding of histone H4K20me2 [147]. The demonstration that 53BP1 also contains a ubiquitylation-dependent recruitment (UDR) motif, required for its interaction with histone H2A ubiquitylated on K15 (H2A K15ub), has now explained the RNF168 dependency of 53BP1 recruitment to sites flanking DNA DSBs [148], with the current model proposing that 53BP1 binds nucleosomes bivalently, through interacting with both H4K20me2 and H2A K15ub at DNA damage sites [148].
The seminal studies on RNF8 demonstrated that the maintenance of BRCA1 at DSB sites depends on RAP80 binding of the BRCA1-A complex, via its ubiquitin-binding modules (UIMs), to K63-linked ubiquitin chains on chromatin [129–131]. Early recruitment of BRCA1 to damaged sites occurs independently of RAP80 however [149], and may be explained by the direct binding of BRCA1 to DNA [150]. In addition to its UIMs, RAP80 binding to SUMO at DNA damage sites via a SUMO-interacting motif (SIM) is also important for its recruitment to DSB sites [151,152]. Notably, BRCA1 contains a RING domain and forms a stable heterodimer with the RING domain of BARD1 to function as a ubiquitin E3 ligase. While BRCA1/BARD1 has been implicated as the E3 for a range of substrates at DNA damage sites, including histone H2A and CtIP [153], several studies suggest that the ligase activity of BRCA1 is not critical for its tumour suppressive functions and role in promoting HR, although its ability to interact with BARD1 through the RING domain and other proteins in the BRCA1-A complex is important [154–156]. The mechanisms regulating the competition between 53BP1 and BRCA1 are still not well defined, although clearly an antagonistic relationship exists [157–159]. Further studies will help determine the complex relationship between these two proteins as well as the determinants for DSB pathway choice.
4.3. Other ubiquitin E3 ligases linked to the DNA damage response
A number of other ubiquitin E3 ligases have been implicated in the DDR. While negative regulation of ubiquitylation signalling events is a well-documented function of DUBs [160], RNF169 has been found to add an additional layer of regulation to the early ubiquitylation signalling cascade initiated by RNF8/168, by also acting as a negative regulator of the pathway. RNF169 is closely related to RNF168, with a very similar overall domain architecture [137]. Recent studies have shown that RNF169 competes with 53BP1 and BRCA1/RAP80 binding to RNF168-ubiquitylated histone H2A and consequently affects the balance of repair between HR and NHEJ [137,161,162]. In addition, the two HECT-E3 ligases TRIP12 (thyroid hormone receptor interactor 12) and UBR5 (ubiquitin protein ligase E3 component amino-recognin 5) function to limit RNF168 and ubiquitin accumulation at DSB sites by regulating steady-state levels of RNF168 [163]. Another ubiquitin E3, the RNF20/RNF40 heterodimer, is responsible for histone H2B mono-ubiquitylation on transcriptionally active chromatin and has also been shown to cause histone H2B K120 mono-ubiquitylation following DNA damage, which appears important for timely repair by both HR and NHEJ [164,165]. FANCL is the RING E3 subunit of the Fanconi anaemia complex [166], which functions with the E2 UBE2T [167] to mono-ubiquitylate FANCD2 and FANCI following interstrand cross-link (ICL) detection, and is therefore crucial for ICL repair. FANCD2/FANCI mono-ubiquitylation promotes retention of the core Fanconi anaemia complex on damaged chromatin and has been shown to be an essential step in coordinating recruitment of a number of key proteins to ICL repair sites, including the nuclease FAN1 [168–170] and CtIP [171]. Both PRP19 (pre-mRNA processing factor 19) and RFWD3 (RING finger and WD repeat domain 3) have been implicated in ATR activation following the generation of RPA-coated ssDNA [172–176]. RFWD3 is a RING ubiquitin E3 that has been shown to promote p53 stability following DNA damage [177]. RFWD3 also physically interacts with RPA and appears important for DNA DSB repair [175,176]. PRP19 is a U-box ubiquitin E3, which in a complex with CDC5L, PRL1 and SPF27 has an established role in pre-mRNA splicing [178]. Recent studies have shown that PRP19 also interacts with RPA and suggest that PRP19 is important for the ubiquitylation of RPA following DNA damage, which promotes ATRIP accumulation and subsequent ATR activation at damaged sites [173,174]. How the functions of PRP19 and RFWD3 are coordinated in respect to DNA damage is not currently known.
STUBLs (SUMO-targeted ubiquitin ligases) are SIM (SUMO-interacting motif)-containing ubiquitin E3 ligases that bind and ubiquitylate sumoylated proteins. They play important roles in the dissociation of highly sumoylated complexes at DSB sites and link sumoylation to proteasomal degradation. The first described STUBL was Slx8-Rfp in Schizosaccharomyces pombe (Slx5–Slx8 in Saccharomyces cerevisiae), the lack of which was shown to cause an accumulation of sumoylated proteins and yeast DNA damage hypersensitivity [179]. A similar level of DNA damage sensitivity is seen in human cells deficient in the Slx5–Slx8 homologue RNF4, which is important for DSB repair by both HR and NHEJ as well as for the release/turnover of several key DSB repair proteins from DNA damage sites, including MDC1, 53BP1, RPA, RAD51, FANCI and FANCD [180–182]. RNF4 accumulation at DNA DSB sites is dependent on its tandem SIM domains, and its recruitment most probably occurs in response to a collection of sumoylated proteins at DNA damage sites, where it functions to ubiquitylate and target proteins for removal via the proteasome and/or the p97/VCP segregase [181,182]. In fact, it has recently been demonstrated that the ubiquitin E3 ligase activity of RNF4 depends on RNF4 binding to SUMO2/3 polymeric chains and subsequent RNF4 dimerization [183]. In addition to its role in promoting the turnover of proteins, RNF4 might also be important for the formation of hybrid SUMO/ubiquitin chains at DNA damage sites, which mediate the recruitment of proteins containing both SIMs and UBDs, such as RAP80 [151]. A second human STUBL with a role in the DDR has now also been described in the literature—RNF111 (also known as Arkadia)—although in this case, the function of ubiquitylation is not to target the substrate for proteasomal degradation; rather, RNF111 preferentially ubiquitylates sumoylated XPC following UV damage to promote its recruitment to damaged DNA [184].
4.4. Links between the ubiquitin–proteasome system and VCP/p97 in the DNA damage response
The proteasome is a more than 2.5 megadalton protease that functions to degrade ubiquitylated proteins [185], and consists of two major components: the 28-subunit core particle (also known as the 20S subunit) and the regulatory particle (also known as the 19S subunit). Substrate entry into the central chamber of the barrel-shaped 20S subunit, where the proteolytic active sites reside, is regulated by the 19S subunit, which recognizes ubiquitin tagged proteins. Proteasomal subunits can be detected at DNA DSB sites [181,186] and a functional proteasome is important for DNA DSB repair, being required for the turnover of DNA damage checkpoint proteins such as CDC25A [187] as well as the turnover of DNA repair proteins such as MDC1, BRCA1 and RPA [181,188,189].
VCP (valosin-containing protein; also known as p97 or Cdc48 in budding yeast) is a hexameric AAA (ATPases-associated with various cellular activities)-ATPase, that uses ATP hydrolysis to structurally unfold or remodel proteins [190]. VCP associates with a number of different ubiquitin-binding domain-containing cofactors that regulate the activity and functions of VCP and of which UFD1 and NPL4 are the most studied. The segregase/chaperone functions of VCP are the best described, where VCP-UFD1-NPL4 binds misfolded and/or polyubiquitylated proteins, extracts them from protein complexes, cellular surfaces or chromatin and delivers them to the proteasome for degradation. By its association with ubiquitin chain-editing proteins and DUBs, VCP can also modify ubiquitin chains to facilitate protein turnover. A growing amount of evidence demonstrates that chromatin extraction of DNA repair proteins by VCP is a general mechanism for removing ubiquitylated proteins from DNA damage sites [191]. In global genome nucleotide excision repair (GG-NER), there is strong evidence to suggest that VCP is important for the removal of CRL4DDB1-ubiquitylated DDB2 and XPC from DNA damage sites [192], as well as for the extraction of chromatin/PCNA-associated CDT1 following DNA damage [193]. VCP is also important for DSB repair and is recruited to DNA DSB sites, where it most probably functions to facilitate the clearance of K48-linked ubiquitylated proteins [191,194]. DVC1 (also known as SPRTN and C1orf124) has recently been shown to be a VCP adaptor protein, required for the recruitment of VCP to stalled replication forks [195,196]. DVC1 depletion in cells enhances UV-induced mutagenesis, and it has been proposed that VCP might facilitate extraction of translesion synthesis polymerases during post-replication repair [195,196]. In vivo, DVC1 deficiency causes genomic instability and progerioid phenotypes [197,198]. In addition, patients with biallelic germline mutations in DVC1 develop early onset hepatocellular carcinoma [198].
Interestingly, UFD1 and NPL4 also bind SUMO through their SIMs [199], and in budding yeast have been shown to extract sumoylated Rad52 from chromatin, thereby restricting Rad51-dependent HR [200]. The function of VCP may therefore extend beyond the ubiquitylation system and most probably plays critical but as-yet relatively undefined roles in DSB repair.
4.5. Deubiquitylating enzymes and double-strand break repair
Various DUBs have now been directly implicated in DNA DSB repair, and the role of DUBs in the DDR has been more extensively reviewed elsewhere [160]. OTUB1 promotes p53 stabilization and also functions as a negative regulator of RNF168-dependent ubiquitylation at sites of DNA damage [201,202]. OTUB1 inhibits the RNF168 E2 enzyme UBE2N (and probably also other E2s) independently of its de-ubiquitylating activity [203,204]. USP44 is recruited to sites of DNA damage and has been shown to antagonize RNF168 recruitment to such sites [205]. It is also important for the regulation of the mitotic spindle checkpoint [206,207]. USP1 functions in DNA ICL repair by regulating FANCD2 mono-ubiquitylation [208], and in its absence cells are hypersensitive to DNA cross-linking agents because of constitutive chromatin association of FANCD2 [209]. USP28 is recruited to DNA DSBs in a 53BP1-dependent manner and was reported to be important for the stability of NBS1, CHK2, 53BP1, CLSPN and TOPBP1 after DNA damage [210]. It has been subsequently demonstrated, however, that USP28 depletion has little effect on DNA repair, and therefore USP28 is unlikely to play a major role in regulating DNA DSB responses [211]. POH1 (also known as PSMD14) is a proteasome associated DUB which has been shown to negatively regulate K63 ubiquitin and 53BP1 accumulation at DNA DSBs as well as to facilitate HR through promoting RAD51 loading [186]. BAP1 (BRCA1-associated protein 1; also known as UCHL2) is encoded by a tumour suppressor gene, which is frequently mutated in a range of cancers [212]. Depletion of BAP1 results in hypersensitivity to DNA damaging agents, and it has been proposed that BAP1 is important for the recruitment of HR effector proteins and the polycomb deubiquitylase complex to sites of DNA damage [213,214]. In addition to the above, a recently published siRNA screen has systematically characterized all known human DUBs against a panel of DDR assays, thereby highlighting likely DSB repair functions for various DUBs [215]. One of the top hits from this screen, UCHL5, was subsequently shown to promote DNA resection and HR through stabilization of NFRKB (nuclear factor related to kappa-B-binding protein), a component of the INO80 chromatin-remodelling complex [215].
5. Post-translational modification with NEDD8
In 1992, 10 genes were identified that showed developmental downregulation of their expression in the mouse brain and were named NEDD1–10 (neural precursor cell expressed, developmentally downregulated 1–10) [216]. NEDD8 was later shown to be a UBL, and of the UBLs that is the most highly related to ubiquitin at the sequence and secondary structure levels (figure 3a) [217,218]. NEDD8 is conjugated to target proteins by an enzymatic neddylation cascade that is extremely well conserved from yeast to human, and is an essential pathway in all organisms studied, with the curious and currently unexplained exception of Saccharomyces cerevisiae [219,220].
Figure 3.

NEDD8 sequence homology and the neddylation cascade. (a) Sequence alignment of human NEDD8, ubiquitin and the NEDD8 homologues in Saccharomyces pombe (ubl1) and Saccharomyces cerevisiae (rub1) using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Lysines critical for ubiquitin chain formation are outlined in blue. Residue 72 (Arg in ubiquitin and Ala in NEDD8) critical for E1 specificity is outlined in red. Colours of amino acids relate to their physiochemical properties: red, small (small + hydrophobic, including aromatic Y); blue, acidic; magenta, basic; green, hydroxyl + sulfhydryl + amine + G. Asterisk (*) denotes fully conserved residue, the symbols :/. denote conservation between groups of strongly/weakly similar properties. (b) Representation of the major neddylation pathway components. NEDD8 (N8) is conjugated in an ATP-dependent cascade involving an E1 (NAE1-UBA3), E2 (UBE2M or F) and E3 (RBX1 or -2) to cullin substrates (sub). The enzymatic pathway is analogous to ubiquitylation (see figure 2 for details). DCUN1D1–5 are cofactors for the NEDD8 E3s. Neddylation is reversed by the CSN complex. MLN4924 inhibits UBA3. See text for details.
Like ubiquitin, NEDD8 is first synthesized as a precursor (from a single NEDD8 gene however) that is processed at the conserved carboxyl-terminal Gly76 residue by the hydrolase activity of deneddylating enzymes, exposing a di-glycine motif that is required for NEDD8 covalent attachment to target substrates. Both NEDP1 (also known as DEN1 or SENP8) and UCHL3 have been described as NEDD8 processing enzymes [221,222]. NEDD8 is conjugated to a substrate lysine (predominantly on cullin proteins) in a three-step enzymatic process analogous to ubiquitylation, using E1, E2 and E3 enzymes specific for neddylation (figure 3b). The NEDD8 E1, NAE (NEDD8 activating enzyme), is composed of the NAE1-UBA3 heterodimer, with NAE1 and UBA3 being homologues of the amino- and carboxyl-terminal regions, respectively, of the ubiquitin E1 UBE1. Given the extent of homology between both NEDD8 and ubiquitin, as well as between NAE and UBE1, an interesting question is how specificity between the UBL and E1 is achieved. Site-directed mutagenesis studies identified Arg72 in ubiquitin (Ala72 in NEDD8) as being critical for E1 specificity [223], as Arg190 on UBA3 repels Arg72 on ubiquitin [224]. A NEDD8 Ala72Arg mutant and ubiquitin Arg72Ala mutant is capable of binding the ubiquitin and NEDD8 E1, respectively [218,225,226], highlighting the importance of this residue. Specificity between the respective ubiquitin and NEDD8 pathway enzymes is maintained through a unique interaction between an amino-terminal sequence on UBE2M and a binding groove of UBA3 [227]. Despite these features, however, NEDD8 activation by the ubiquitin E1 UBE1 is possible and has been demonstrated in circumstances when the NEDD8:ubiquitin balance in cells is perturbed—either through overexpression of NEDD8 or following chemical or physiological depletion of ubiquitin [228,229].
There are two known NEDD8 E2s: UBE2M and UBE2F [219,230–232], which preferentially partner with the E3 RBX1 (also known as ROC1) or RBX2 (also known as ROC2 or RNF7), respectively [232]. RBX1 and -2 are RING E3s which simultaneously bind their substrate and NEDD8∼UBE2M/F to catalyse NEDD8 transfer [233]. Interestingly, RBX1 and RBX2 also function as the ubiquitin E3s for cullin-RING complexes [234,235]. An additional group of proteins—Dcn1 (defective in cullin neddylation 1) in budding yeast and DCUN1D1–5 (defective in cullin neddylation 1, domain-containing 1–5, also known as DCNL1–5) in humans—have been described that act as cofactors for RBX1 and RBX2, potentiating their neddylation activity [233,236–240]. The DCUN1D proteins contain a UBD and a conserved, unique PONY (potentiating neddylation) domain that binds amino-terminally acetylated UBE2M/F and the cullin substrate [238,240]. DCUN1D1–5 do not appear to affect E2/E3 substrate specificity and are each able to bind all of the cullins via their PONY domains. However, they potentiate the neddylation activity of different E3-cullin complexes to varying degrees [240]. Currently, substrate specificity of NEDD8 is only loosely defined at the level of the E2, with UBE2M/RBX1 and UBE2F/RBX2 preferentially neddylating CUL1–4 or CUL5, respectively [232]. Why five human orthologues of DCN1 exist remains unknown, and subcellular localization or varying expression levels of the DCUN1D proteins may prove to be important for spatio-temporal regulation of cullin-RING ligase (CRL) activity [239–241].
Like ubiquitin, NEDD8 also contains internal lysine residues suitable for chain formation, although, at least in yeast, these conserved residues do not appear to be functionally essential [242]. Although NEDD8 chain formation has been reported in the literature, in vivo it has only been described on non-cullin substrates when NEDD8 is overexpressed, so the physiological relevance of NEDD8 chains remains unknown [228,243,244].
As for other protein PTMs, neddylation is a reversible process, with the CSN (COP9 signalosome) being the predominant de-neddylase. The CSN is a conserved complex of eight proteins (CSN1–8), named after a group of COP (constitutive photomorphogenesis) mutants that showed constitutive photomorphogenesis, pigmented seed coats and premature death in plants [245]. The JAMM motif (Jab1/MPN domain) of CSN5 was later shown to be critical for the de-neddylating activity of the CSN and linked the CSN to the regulation of the CRLs [246]. A complete CSN complex is required for full enzymatic activity in vitro [247], with CSN5 being constitutively inactive until binding of the CSN to the cullin substrate triggers conformational changes that result in activation of isopeptidase activity [248]. The CSN is the only isopeptidase that efficiently cleaves NEDD8 from cullin substrates in vivo, although a number of other proteins have been shown to possess NEDD8 isopeptidase activity in vitro [249].
5.1. Neddylation substrates: the cullins
5.1.1. Cullin-RING ligases
The cullins are the best-defined NEDD8 substrates, of which there are eight encoded by the human genome (CUL1, 2, 3, 4A, 4B, 5, 7 and PARC), and they all share an evolutionarily conserved cullin homology domain and carboxyl-terminal neddylation site. The APC2 subunit of the anaphase promoting complex/cyclosome (APC/C) also contains cullin homology domains, but lacks the carboxyl-terminal NEDD8 consensus sequence (IVRIMKMR) and is therefore functionally distinct from the other cullins. Cullins are molecular scaffolds for the CRLs, which are E3 ubiquitin ligases responsible for up to 20% of all ubiquitin-mediated proteasomal degradation [250–252]. CUL1-containing CRL1 is the archetypal CRL and is also commonly known as SCF (SKP, cullin, F-box containing complex). The CRLs contain three major components: a cullin scaffold, a RING finger protein (RBX1 or RBX2; that recruits a ubiquitin-charged E2 enzyme, as well as the NEDD8 loaded E2) and a substrate recognition protein, that may or may not be associated with a substrate adaptor protein, which places substrates in proximity to the ubiquitin E2 enzyme to facilitate ubiquitin transfer (figure 4). Cdc34 is the only known E2 to associate with CRLs in yeast, although both CDC34a/b (also known as UBE2R1 and UBE2R2) and UBCH5 (also known as UBE2D1) have been described as ubiquitin E2s for the CRLs in humans, and others may also exist [250,253].
Figure 4.
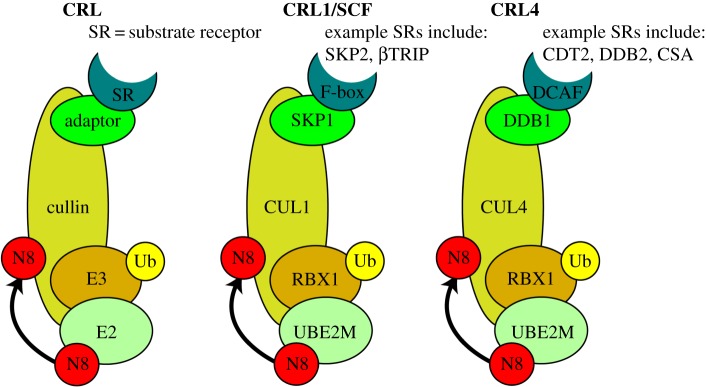
Cullin-RING ligases (CRLs). Simplified diagram of CRL structure. The cullin forms the backbone of the CRL complex. Cullin homology domains at the carboxyl-terminus are required for binding to the E3 and the amino-terminus interacts with the substrate adaptor proteins where required (there is no known substrate adaptor for CUL3) [242]. Neddylation of the cullins on a conserved carboxyl-terminal lysine induces conformational changes to promote ubiquitylation of CRL substrates. SKP1 and DDB1 are the substrate adaptor proteins for CRL1 and CRL4 complexes, respectively. F-box proteins and DCAFs are the substrate receptors for CRL1 and CRL4 complexes, respectively. See text for more details.
SKP1 (S-phase kinase-associated protein 1) and DDB1 (damage-specific DNA-binding protein 1) are the best-described substrate adaptor proteins and link CUL1 and CUL4A/B, respectively, to their substrate receptors. The substrate receptors for CUL1, 2, 3, 5 and 7 are structurally related FBX (F-box-like) proteins, although CUL4A and CUL4B use distinct substrate receptors known as DCAFs (DDB1- and CUL4-associated factors). Many of the DCAFs contain WD40-like repeats [254] of which CDT2 (chromatin licensing and DNA replication factor 2) and DDB2 (damage-specific DNA-binding protein 2) are examples. The WD40 domain is one of the most abundant domains encoded by the eukaryotic genome. They commonly mediate protein–protein interactions, and many WD40 domain-containing proteins have been found as interactors of CUL4A and CUL4B in proteomic studies [255,256]. A single substrate receptor can bind multiple substrates however. For example, CRL4CDT2 binds CDT1, p21 and SETD8, among others. Therefore, the substrate specificity of the CRLs depends on both the combination of its components and also the cellular context of the substrate itself; for example, CRL4CDT2 only binds CDT1 when it is associated with chromatin-bound PCNA (see later sections for more details). Proteomics studies have revealed the complexity of the CRL network [255] and diversity of CRL substrates [257,258], culminating in more than 200 unique CRL complexes that play key roles in cell cycle regulation, embryogenesis, DNA replication and repair [251].
5.1.2. Regulation of cullin-RING ligase activity
CRL activity towards its substrate is tightly controlled, and regulation is multi-factorial. Neddylation of the cullins, on a conserved carboxyl-terminal lysine, serves to increase the ubiquitylating activity of the CRLs by triggering conformational changes within the cullin and by preventing association with the CRL inhibitor CAND1 (cullin-associated NEDD8-dissociated 1) [259]. CAND1 binds to de-neddylated cullins, promotes dissociation of the cullin from the substrate receptor and inhibits CRL activity in vitro [260,261]. As well as showing inhibitory CRL effects, however, genetic studies demonstrated that CAND1 also promotes CRL activity, with this functional paradox being explained by the importance of CAND1 for maintaining an available pool of substrate receptors and therefore promoting the dynamics of CRL formation and dissociation [262–264].
CSN regulation of CRL activity is also more complex than initially thought. CSN association with the CRL maintains the cullin in a de-neddylated state. This, along with its association with the de-ubiquitylating enzyme USP15 [265], inhibits the ubiquitylation of CRL substrates and protects substrate receptor proteins from auto-degradation by the CRL. In some cases, the CSN may also physically block substrate access. Taken together, CSN dissociation from the CRL has therefore become a recognized mechanism for promoting substrate ubiquitylation [266–269].
Independently of cullin association with the negative regulators CSN and CAND1, cullin neddylation and thus activation also appears to be dependent on the formation of a complete CRL unit, including cullin binding to adaptor proteins and substrate receptor subunits, possibly through inducing a conformational change or dimerization of the CRL, which promotes neddylation [270]. PTMs of the substrate binding motif or ‘degron’ also regulate CRL interaction with its target and subsequent activation in some cases [271]. Further structural and biochemical studies will better determine the mechanisms of CRL activation. Clearly, however, spatio-temporal regulation of CRL activity is critical and is achieved through a fine balance of promoting and inhibitory mechanisms.
5.2. The neddylation inhibitor MLN4924
Many CRL components are either overexpressed or mutated in human cancers [272,273], making CRL inhibition an attractive anti-cancer strategy. In addition, success of the proteasome inhibitor bortezomib (Velcade®; Takeda Oncology) for the treatment of multiple myeloma and mantle cell lymphoma has paved the way for novel therapeutics that inhibit ubiquitin-mediated protein degradation pathways.
MLN4924 (Takeda Oncology) is an irreversible inhibitor of UBA3 that potently and specifically inhibits UBA3 activation of NEDD8 in cells, and subsequently inhibits global NEDD8 conjugation and CRL activity [252]. MLN4924 is an AMP analogue which is catalysed by UBA3 to form a covalent UBA3-MLN4924 adduct that can no longer be used by the NEDD8 machinery [274]. Therefore, treatment of cells with MLN4924 rapidly leads to the accumulation of CRL substrates, including CDT1, p27 and NRF2, among others [252]. The predominant cellular phenotype, in a range of cell lines tested, following prolonged (8 h) MLN4924 treatment is S-phase arrest and DNA re-replication, thought to be largely due to CRL4CDT2 inhibition [275], although CRL1 is also likely to contribute. CRL4CDT2 inhibition causes stabilization of CDT1 and SETD8 (also known as SET8 or PR-SET7), both of which contribute to the re-replication observed. CDT1 is required for loading of the DNA replication helicase complex (MCM2–7) onto chromatin in the G1 phase of the cell cycle, and CRL4CDT2/SCFSKP2-mediated degradation of CDT1 during S-phase prevents DNA re-replication [276]. SETD8 catalyses histone monomethylation on Lys 20 of histone H4 (H4K20me1), and PCNA-dependent degradation of SETD8 by CRL4CDT2 during S-phase inhibits DNA re-replication (although the mechanism by which SETD8 promotes loading of the replication complex onto chromatin remains unknown) [277,278]. DNA re-replication is a recognized cause of DNA damage [279] and prolonged MLN4924 treatment causes replication-dependent activation of ATM- and ATR-mediated DDR signalling, DSB formation as measured by γH2AX formation and eventual apoptosis or cellular senescence [279–281]. In addition to re-replication itself being a cause of DNA damage, recently it has been shown that CRL-dependent ubiquitylation is required for dissociation of the DNA replisome in Saccharomyces cerevisiae and Xenopus [282,283]. It will be interesting to determine how failure to remove the MCM complex on completion of DNA replication might contribute to DSB generation following treatment with MLN4924. Regardless of mechanism, cell survival in yeast mutants that undergo DNA re-replication or in human cells following MLN4924 treatment is reduced in DSB repair deficient backgrounds [284,285], clearly suggesting that cell death following MLN4924 treatment is at least in part due to the generation of DNA DSBs.
Pre-clinical data suggest that inhibiting neddylation might be an effective anti-cancer strategy for solid tumours and haematological malignancies [286]. MLN4924 is well tolerated and has shown anti-tumour activity in phase I oncology trials involving haematological and solid tumours [287] (see also http://meetinglibrary.asco.org). Phase I combination trials are on-going (http://www.cancer.gov/clinicaltrials).
5.3. Non-cullin neddylation substrates
A number of non-cullin substrates for NEDD8 have been described in the literature, including p53 [288], the ribosomal protein L11 [289] and histone H4 [290]. In addition, proteomics studies have identified neddylation targets both within and outside the known CRL network [243,244]. However, these studies have largely relied on the overexpression of tagged-NEDD8 constructs as their bait, and as conjugation of NEDD8 to target proteins using the ubiquitin machinery can occur in these circumstances [229], it is difficult to be convinced that proteins other than cullins are true NEDD8 substrates in vivo. It is also worth noting that, while MLN4924 specifically inhibits neddylation, it also leads to potent inhibition of CRL ubiquitylating activity. Consequently, demonstrating that neddylation of a substrate is MLN4924 dependent does not discriminate between conjugation being mediated by the NEDD8 or ubiquitin machineries. Furthermore, histones are heavily ubiquitylated by CRLs and are often identified as neddylation substrates when NEDD8 is overexpressed. A recent excellent review by leading laboratories in the neddylation field has suggested criteria for identifying physiological neddylation substrates and, to date, only the cullins fulfil all these criteria [291].
6. Role of neddylation in the DNA damage response
6.1. CUL4A and CUL4B
Of all the cullins, CUL4A and CUL4B are the most strongly linked to the DDR. Higher eukaryotes such as Homo sapiens, Mus musculus and Xenopus tropicalis have both types of CUL4, which share approximately 85% sequence identity and are derived from a common CUL4 ancestor found in Caenorhabditis elegans, Drosophila melanogaster and S. pombe. At a cellular level, a large amount of functional redundancy exists between CUL4A and CUL4B. Some non-overlapping functions of the two proteins have been revealed by mouse genetic studies however, because although Cul4A−/− mice are viable with no overt developmental defects [292], presumably due to functional compensation by Cul4B, Cul4A−/− male mice are sterile, which is thought to be due to a failure to resolve recombination crossover intermediates during meiosis [293]. In addition, mutations in CUL4B are linked to an X-linked mental retardation syndrome in man [294] and Cul4B−/− mice display embryonic lethality due to developmental failure of extra-embryonic tissues [295]. Beyond these limited examples, CUL4A and CUL4B appear to be functionally redundant with one another, and the roles described below for CRL4 in the DDR cannot be linked to either one of the proteins in isolation.
6.2. CRL4 and nucleotide excision repair
Both GG-NER and TC-NER (table 1) use mechanisms involving CRL4 to recognize DNA lesions and promote repair [88]. DDB2 is the DNA damage sensor protein for GG-NER and is the substrate receptor of the CUL4–DDB1 CRL complex (CRL4DDB2), which in the nuclear fraction of unperturbed cells is closely associated with the CSN [296]. In the current model, upon binding to UV-damaged sites, CRL4DDB2 dissociates from the CSN, leading to activation of the CRL and ubiquitylation of XPC and DDB2 [266,296]. Interestingly, the inhibitory effects of the CSN on CRL4DDB2 appear to be independent of CSN5 de-neddylase activity [266]. Dimerization of CRL4DDB2 upon binding to DNA may also be important for facilitating ubiquitylation of its substrates [297]. Ubiquitylation of DDB2 reduces its affinity for DNA and causes its proteasome-mediated degradation, whereas ubiquitylation of XPC potentiates its association with the DNA, enabling repair [298]. In addition, CRL4DDB2 ubiquitylates histone H3 and H4 in the vicinity of the DNA lesions, facilitating access of the NER repair machinery [299]. Mutations in the DNA-binding region of DDB2 give rise to the archetypal NER syndrome xeroderma pigmentosum [300,301].
CSA (Cockayne syndrome type A protein) is another substrate receptor for CUL4–DDB1, and CRL4CSA is recruited to TC-NER sites where it is believed to ubiquitylate the SWI/SNF ATPase CSB (Cockayne syndrome type B protein) and is important for repair and ensuing transcriptional re-start [266,302]. Although CSA and DDB2 are quite distinct at the sequence level, they are structurally very similar in their complex formation with DDB1, and like CRL4DDB2, CRL4CSA activity appears to be regulated by its association/dissociation with the CSN [266].
6.3. Cullin-RING ligase regulation of DNA damage cell cycle checkpoint responses
CRLs play prominent roles in cell cycle regulation [276] and are also important for cell cycle checkpoint responses following DNA damage. As described above, CDT1 degradation by CRL4CDT2/SCFSKP2 during normal S-phase is essential in preventing DNA re-replication. In addition, following DNA damage, CRL4CDT2 promotes CDT1 degradation in a chromatin-bound PCNA-dependent manner. CDT1 contains a ‘PIP degron’, which is made up of a canonical PCNA binding motif (the PIP box; through which CDT1 binds PCNA) and a basic patch of four amino acids downstream (B+4), which mediates the interaction between CDT1 and CRL4CDT2. CRL4CDT2 interacting with both PCNA and CDT1 via the PIP degron is a common mechanism by which CRL4CDT2 interacts with its substrates [303], although how this interaction is regulated to occur only in the context of chromatin-bound PCNA is not known [304].
CDC25A is a phosphatase required for activation of cyclin-dependent kinases, enabling cell cycle progression. Following DNA damage, CDC25A is phosphorylated in a predominantly CHEK1/CHEK2-dependent manner, leading to its ubiquitylation by SCFβTRCP, degradation and subsequent cell cycle arrest [305]. Similarly, a phospho-degron signal on CLASPIN (a protein required for ATR-mediated CHEK1 activation following stalled replication forks and DNA end resection during HR) triggers its ubiquitylation by SCFβTRCP and subsequent degradation, enabling checkpoint recovery [306,307].
6.4. Neddylation and double-strand break repair
In vitro studies using Xenopus laevis egg extracts have shown that, upon binding DNA, Ku80 is polyubiquitylated with K48 chains by the cullin 1 containing complex SCFFBXL12 and removed from the DNA [308,309]. Truncation mutants of Ku80 that are proficient in DNA binding, but not DNA repair, are efficiently ubiquitylated, suggesting that in this system DNA binding rather than the completion of repair is the signal for Ku ubiquitylation and release [309]. The mechanism by which Ku is extracted from the DNA remains unclear, however. Postow et al. [309] showed that Ku removal is independent of the proteasome and hypothesized that VCP unfolds Ku, thereby promoting its removal from DNA [310]. Ku forms a highly stable ring-like structure that threads onto DNA ends [311], and failure to remove Ku from repaired DNA will likely interfere with cellular processes such as DNA replication and transcription [312,313], highlighting the need for an active mechanism to release DNA bound Ku [310]. It has not yet been determined whether a CRL-dependent mechanism of Ku release is conserved in human cells.
In human cells, inhibition of NEDD8 conjugation has been shown to hypersensitize cells to DNA damaging agents such as mitomycin C, cisplatin and IR [314–321]. Synergy between MLN4924 and DNA cross-linking agents also clearly exists across a range of cell lines [314,316–319]. The mechanisms behind this synergy are not fully elucidated and have not been consistent between studies [316] but may include inhibitory effects on assembly of the Fanconi complex [317], promotion of reactive oxygen species (ROS) generation [318] and increased replication fork collapse due to BRCA2/RAD51 stabilization [314]. Given the wide-ranging cellular effects of neddylation inhibition, it is likely that the synergy observed is multi-factorial and potentially cell-type specific. Further studies will refine our understanding of the mechanisms involved.
Recent studies have linked neddylation with DSB repair more directly and NEDD8 has been shown to localize to DNA damage sites [290]. In this study by Ma et al., it was concluded that RNF111 functions as the NEDD8 E3 ligase responsible for NEDD8 accumulation at sites of DNA damage. More specifically, the authors concluded that RNF111-mediated neddylation of histone H4 is essential for RNF168 recruitment to DSB sites [290]. RNF111, also known as Arkadia, is a ubiquitin E3 ligase with an established role in TGF-β signalling [322], and more recently has been shown to function as a STUBL in PML degradation and in NER [184,323]. Whether the effects of RNF111 depletion on DSB repair reported by Ma et al. are due to the ubiquitylation rather than the suggested neddylation activity of RNF111 has not been established. In addition, as recently published, like most ubiquitin E3s, the RING structure of RNF111 does not favour interaction with the NEDD8 E2 UBE2M [291,324], and as a consequence, it would be interesting to determine the NEDD8 E2 responsible for the proposed neddylation function of RNF111.
Other studies linking neddylation and DSB repair include a published report by Wu et al. [325] that the CUL1 substrate receptor SKP2 is important for NBS1 K63 ubiquitylation and subsequent ATM activation. Interestingly, it has also been demonstrated that NBS1 ubiquitylation by RNF8 is important for efficient NBS1 recruitment to DNA damage sites [326], and both pathways appear to be important for repair by HR. More recently, it has been proposed that RNF111-mediated neddylation regulates the extent of DNA end resection and neddylation inhibition increases rates of HR [327]. RNF168 has also been reported to function as a NEDD8 E3 ligase in response to DNA damage, where it has been reported to neddylate histone H2A and subsequently negatively regulate the ubiquitylation of H2A and the downstream recruitment of BRCA1 [328]. As discussed above, the existence of non-cullin neddylation substrates is a contentious issue in the field [291], and further studies are required to substantiate H2A as a neddylation substrate and RNF168 as a NEDD8 E3 ligase.
7. Summary
The importance of PTMs in the DDR has been appreciated for many years, and the role of ubiquitin and the UBLs in DSB repair has been an area of active research over the last decade. A major unanswered question in the field is the identity of the RNF8 substrate responsible for RNF168 recruitment. In addition, it is likely that the full spectrum of ubiquitin-system proteins important for the DDR has not yet been fully established. Indeed, it seems likely that further work in this area will uncover new mechanistic insights into DDR processes as well as provide promising new drug targets.
Given the complexity of the crosstalk between SUMO and ubiquitin systems during cellular responses to DNA DSBs [329], it would probably not be surprising if similar levels of crosstalk exist between the neddylation and ubiquitylation systems. As the predominant function of neddylation in the literature to date is to regulate the ubiquitylating activity of the CRLs, we feel that it will be particularly interesting to determine how CRL activity is regulated in the context of responses to DNA damage. While MLN4924 is an extremely useful tool for investigating the cellular consequences of neddylation inhibition, prolonged treatment with MLN4924 has wide-ranging cellular effects and generates high levels of DNA DSBs [252]. Potent inhibition of neddylation is achieved within minutes in cells treated with MLN4924, so short treatment times should be used wherever possible and appropriate experimentally. Likewise, given the potential utilization of NEDD8 by the ubiquitin machinery, caution should be exercised when interpreting results using overexpressed NEDD8 systems, and reagents should be validated as being specific for NEDD8. Inhibitors specific for individual CRL complexes are not available, but would be extremely helpful for determining the fine-tuning of the complex CRL network.
Studying the mechanisms by which ubiquitin and the UBLs regulate DSB repair and associated processes is a technical challenge, not least because of the functional redundancy in these systems. A better understanding of how these PTMs interconnect, as well as the development of more small molecule inhibitors such as MLN4924 will enable the cellular functions of these and connected enzymatic pathways to be determined through approaches that until now have not been feasible. In particular, further advances in proteomics techniques will better define the substrates of various PTMs, and evolving gene-targeting and cell engineering approaches [330,331] will complement the use of inhibitors in determining the functions of PTM pathway members. Being frequently mutated or overexpressed in human cancers, the ubiquitin and UBL pathways offer a spectrum of attractive drug targets, and with the recent approval of PARP inhibitor olaparib/LynparzaTM (AstraZeneca) by the European Medicines Agency and the US Food and Drug Administration for the treatment of BRCA-mutated, high-grade serous epithelial ovarian cancer, the precedent is now set for DDR inhibitors in the clinic.
Acknowledgements
We thank Natasha Lukashchuk and Yaron Galanty for useful discussions and also Kate Dry for critical reading of the manuscript.
Funding statement
J.S.B. is funded by the Wellcome Trust Clinical Fellowship (grant no. 094794/Z/10/Z). Research in the Jackson laboratory is funded by Cancer Research UK programme grant C6/A11224, the European Research Council and the European Community Seventh Framework Programme grant agreement no. HEALTH-F2-2010-259893 (DDResponse). Core funding is provided by CRUK (C6946/A14492) and the Wellcome Trust (WT092096). S.P.J. receives his salary from the University of Cambridge UK, supplemented by CRUK.
Author contributions
Both J.S.B. and S.P.J. contributed to drafting and revising the manuscript. Both authors gave final approval before submission.
Conflict of interests
J.S.B. has no competing interests. S.P.J. has no conflicts of interest but discloses that he is a founder and shareholder of MISSION Therapeutics Ltd.
References
- 1.Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (doi:10.1038/nature08467) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 (doi:10.1016/j.molcel.2010.09.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433 (doi:10.1101/gad.2021311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T. 1993. Instability and decay of the primary structure of DNA. Nature 22, 709–715 (doi:10.1038/362709a0) [DOI] [PubMed] [Google Scholar]
- 5.Bassing CH, Alt FW. 2004. The cellular response to general and programmed DNA double strand breaks. DNA Repair 3, 781–796 (doi:10.1016/j.dnarep.2004.06.001) [DOI] [PubMed] [Google Scholar]
- 6.Schlissel MS, Kaffer CR, Curry JD. 2006. Leukemia and lymphoma: a cost of doing business for adaptive immunity. Genes Dev. 20, 1539–1544 (doi:10.1101/gad.1446506) [DOI] [PubMed] [Google Scholar]
- 7.Richardson C, Horikoshi N, Pandita TK. 2004. The role of the DNA double-strand break response network in meiosis. DNA Repair 3, 1149–1164 (doi:10.1016/j.dnarep.2004.05.007) [DOI] [PubMed] [Google Scholar]
- 8.Basu B, Yap TA, Molife LR, de Bono JS. 2012. Targeting the DNA damage response in oncology: past, present and future perspectives. Curr. Opin. Oncol. 24, 316–324 (doi:10.1097/CCO.0b013e32835280c6) [DOI] [PubMed] [Google Scholar]
- 9.Curtin NJ. 2012. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 12, 801–817 (doi:10.1038/nrc3399) [DOI] [PubMed] [Google Scholar]
- 10.Furgason JM, Bahassi EM. 2013. Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 137, 298–308 (doi:10.1016/j.pharmthera.2012.10.009) [DOI] [PubMed] [Google Scholar]
- 11.Griffith AJ, Blier PR, Mimori T, Hardin JA. 1992. Ku polypeptides synthesized in vitro assemble into complexes which recognize ends of double-stranded DNA. J. Biol. Chem. 267, 331–338. [PubMed] [Google Scholar]
- 12.Mimori T, Hardin JA, Steitz JA. 1986. Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J. Biol. Chem. 261, 2274–2278. [PubMed] [Google Scholar]
- 13.Smider V, Rathmell WK, Lieber MR, Chu G. 1994. Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science 266, 288–291 (doi:10.1126/science.7939667) [DOI] [PubMed] [Google Scholar]
- 14.Taccioli GE, et al. 1994. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science 265, 1442–1445 (doi:10.1126/science.8073286) [DOI] [PubMed] [Google Scholar]
- 15.Mari P-O, et al. 2006. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl Acad. Sci. USA 103, 18 597–18 602 (doi:10.1073/pnas.0609061103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Britton S, Coates J, Jackson SP. 2013. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 202, 579–595 (doi:10.1083/jcb.201303073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uematsu N, et al. 2007. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J. Cell Biol. 177, 219–229 (doi:10.1083/jcb.200608077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramsden DA, Gellert M. 1998. Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J. 17, 609–614 (doi:10.1093/emboj/17.2.609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cary RB, Peterson SR, Wang J, Bear DG, Bradbury EM, Chen DJ. 1997. DNA looping by Ku and the DNA-dependent protein kinase. Proc. Natl Acad. Sci. USA 94, 4267–4272 (doi:10.1073/pnas.94.9.4267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Vries E, van Driel W, Bergsma WG, Arnberg AC, van der Vliet PC. 1989. HeLa nuclear protein recognizing DNA termini and translocating on DNA forming a regular DNA-multimeric protein complex. J. Mol. Biol. 208, 65–78 (doi:10.1016/0022-2836(89)90088-0) [DOI] [PubMed] [Google Scholar]
- 21.Gottlieb T, Jackson S. 1993. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72, 131–142 (doi:10.1016/0092-8674(93)90057-W) [DOI] [PubMed] [Google Scholar]
- 22.Smith GC, Jackson SP. 1999. The DNA-dependent protein kinase. Genes Dev. 13, 916–934 (doi:10.1101/gad.13.8.916) [DOI] [PubMed] [Google Scholar]
- 23.Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. 2007. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair 6, 712–722 (doi:10.1016/j.dnarep.2006.12.007) [DOI] [PubMed] [Google Scholar]
- 24.Hsu HL, Yannone SM, Chen DJ. 2002. Defining interactions between DNA-PK and ligase IV/XRCC4. DNA Repair 1, 225–235 (doi:10.1016/S1568-7864(01)00018-0) [DOI] [PubMed] [Google Scholar]
- 25.Yano K, Morotomi-Yano K, Wang S-Y, Uematsu N, Lee K-J, Asaithamby A, Weterings E, Chen DJ. 2008. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 9, 91–96 (doi:10.1038/sj.embor.7401137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochi T, et al. 2015. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 347, 185–188 (doi:10.1126/science.1261971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing M, et al. 2015. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat. Commun. 6, 6233 (doi:10.1038/ncomms7233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolganov GM, Maser RS, Novikov A, Tosto L, Chong S, Bressan DA, Petrini JH. 1996. Human Rad50 is physically associated with human Mre11: identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol. Cell. Biol. 16, 4832–4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogawa H, Johzuka K, Nakagawa T, Leem SH, Hagihara AH. 1995. Functions of the yeast meiotic recombination genes, MRE11 and MRE2. Adv. Biophys. 31, 67–76 (doi:10.1016/0065-227X(95)99383-Z) [DOI] [PubMed] [Google Scholar]
- 30.Trujillo KM, Yuan SS, Lee EY, Sung P. 1998. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 273, 21 447–21 450 (doi:10.1074/jbc.273.34.21447) [DOI] [PubMed] [Google Scholar]
- 31.Usui T, Ohta T, Oshiumi H, Tomizawa J, Ogawa H, Ogawa T. 1998. Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell 95, 705–716 (doi:10.1016/S0092-8674(00)81640-2) [DOI] [PubMed] [Google Scholar]
- 32.De Jager M, Dronkert ML, Modesti M, Beerens CE, Kanaar R, van Gent DC. 2001. DNA-binding and strand-annealing activities of human Mre11: implications for its roles in DNA double-strand break repair pathways. Nucleic Acids Res. 29, 1317–1325 (doi:10.1093/nar/29.6.1317) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paull TT, Gellert M. 1998. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1, 969–979 (doi:10.1016/S1097-2765(00)80097-0) [DOI] [PubMed] [Google Scholar]
- 34.Paull TT, Gellert M. 1999. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 13, 1276–1288 (doi:10.1101/gad.13.10.1276) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GCM, Lukas J, Jackson SP. 2006. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 8, 37–45 (doi:10.1038/ncb1337) [DOI] [PubMed] [Google Scholar]
- 36.Sartori A, Lukas C, Coates J, Mistrik M, Fu S. 2007. Human CtIP promotes DNA end resection. Nature 450, 509–514 (doi:10.1038/nature06337) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611 (doi:10.1038/nature03442) [DOI] [PubMed] [Google Scholar]
- 38.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. 2003. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22, 5612–5621 (doi:10.1093/emboj/cdg541) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J-H, Paull TT. 2005. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308, 551–554 (doi:10.1126/science.1108297) [DOI] [PubMed] [Google Scholar]
- 40.Lamarche BJ, Orazio NI, Weitzman MD. 2010. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 584, 3682–3695 (doi:10.1016/j.febslet.2010.07.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinfeld M, Chaudhry MA, D'Amours D, Pelletier JD, Poirier GG, Povirk LF, Lees-Miller SP. 1997. Interaction of DNA-dependent protein kinase and poly(ADP-ribose) polymerase with radiation-induced DNA strand breaks. Radiat. Res. 148, 22–28 (doi:10.2307/3579534) [PubMed] [Google Scholar]
- 42.Bramson J, Prévost J, Malapetsa A, Noë AJ, Poirier GG, DesNoyers S, Alaoui-Jamali M, Panasci L. 1993. Poly(ADP-ribose) polymerase can bind melphalan damaged DNA. Cancer Res. 53, 5370–5373. [PubMed] [Google Scholar]
- 43.D'Silva I, Pelletier JD, Lagueux J, D'Amours D, Chaudhry MA, Weinfeld M, Lees-Miller SP, Poirier GG. 1999. Relative affinities of poly(ADP-ribose) polymerase and DNA-dependent protein kinase for DNA strand interruptions. Biochim. Biophys. Acta 1430, 119–126 (doi:10.1016/S0167-4838(98)00278-7) [DOI] [PubMed] [Google Scholar]
- 44.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. 2003. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533 (doi:10.1093/nar/gkg761) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mortusewicz O, Rothbauer U, Cardoso MC, Leonhardt H. 2006. Differential recruitment of DNA ligase I and III to DNA repair sites. Nucleic Acids Res. 34, 3523–3532 (doi:10.1093/nar/gkl492) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. 2006. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 34, 6170–6182 (doi:10.1093/nar/gkl840) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chapman JR, Taylor MRG, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 47, 497–510 (doi:10.1016/j.molcel.2012.07.029) [DOI] [PubMed] [Google Scholar]
- 48.Jha DK, Pfister SX, Humphrey TC, Strahl BD. 2014. SET-ting the stage for DNA repair. Nat. Struct. Mol. Biol. 21, 655–657 (doi:10.1038/nsmb.2866) [DOI] [PubMed] [Google Scholar]
- 49.Kim S-T, Lim D-S, Canman CE, Kastan MB. 1999. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 274, 37 538–37 543 (doi:10.1074/jbc.274.53.37538) [DOI] [PubMed] [Google Scholar]
- 50.Matsuoka S, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 (doi:10.1126/science.1140321) [DOI] [PubMed] [Google Scholar]
- 51.Mahaney BL, Meek K, Lees-Miller SP. 2009. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 417, 639–650 (doi:10.1042/BJ20080413) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang C, Lees-Miller SP. 2013. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int. J. Radiat. Oncol. Biol. Phys. 86, 440–449 (doi:10.1016/j.ijrobp.2013.01.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaidi A, Jackson SP. 2013. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 498, 70–74 (doi:10.1038/nature12201) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. 2003. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell 11, 203–213 (doi:10.1016/S1097-2765(02)00799-2) [DOI] [PubMed] [Google Scholar]
- 55.Zou L, Elledge SJ. 2003. Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300, 1542–1548 (doi:10.1126/science.1083430) [DOI] [PubMed] [Google Scholar]
- 56.Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 (doi:10.1038/nrm2450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nam EA, Cortez D. 2011. ATR signalling: more than meeting at the fork. Biochem. J. 436, 527–536 (doi:10.1042/BJ20102162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. 1997. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene 15, 159–167 (doi:10.1038/sj.onc.1201319) [DOI] [PubMed] [Google Scholar]
- 59.Shiloh Y. 2014. ATM: expanding roles as a chief guardian of genome stability. Exp. Cell Res. 329, 154–161 (doi:10.1016/j.yexcr.2014.09.002) [DOI] [PubMed] [Google Scholar]
- 60.O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501 (doi:10.1038/ng1129) [DOI] [PubMed] [Google Scholar]
- 61.O'Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA. 2004. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair 3, 1227–1235 (doi:10.1016/j.dnarep.2004.03.025) [DOI] [PubMed] [Google Scholar]
- 62.Finnie N, Gottlieb T. 1995. DNA-dependent protein kinase activity is absent in xrs-6 cells: Implications for site-specific recombination and DNA double-strand break repair. Proc. Natl Acad. Sci. USA 92, 320–324 (doi:10.1073/pnas.92.1.320) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blunt T, et al. 1995. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80, 813–823 (doi:10.1016/0092-8674(95)90360-7) [DOI] [PubMed] [Google Scholar]
- 64.Kurimasa A, Kumano S. 1999. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol. 19, 3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van der Burg M, et al. 2009. A DNA-PKcs mutation in a radiosensitive T–B–SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Invest. 119, 91–98 (doi:10.1172/JCI37141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rogakou E, Pilch D, Orr A, Ivanova V, Bonner W. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (doi:10.1074/jbc.273.10.5858) [DOI] [PubMed] [Google Scholar]
- 67.Stiff T, Driscoll MO, Rief N, Iwabuchi K, Lo M, Jeggo PA. 2004. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 64, 2390–2396 (doi:10.1158/0008-5472.CAN-03-3207) [DOI] [PubMed] [Google Scholar]
- 68.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. 2005. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226 (doi:10.1016/j.cell.2005.09.038) [DOI] [PubMed] [Google Scholar]
- 69.Stracker TH, Usui T, Petrini JHJ. 2009. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 8, 1047–1054 (doi:10.1016/j.dnarep.2009.04.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bartek J, Lukas J. 2003. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429 (doi:10.1016/S1535-6108(03)00110-7) [DOI] [PubMed] [Google Scholar]
- 71.Reinhardt H, Yaffe M. 2009. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255 (doi:10.1016/j.ceb.2009.01.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirao A, et al. 2002. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol. Cell. Biol. 22, 6521–6532 (doi:10.1128/MCB.22.18.6521-6532.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takai H, et al. 2002. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 21, 5195–5205 (doi:10.1093/emboj/cdf506) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AEH, Yaffe MB. 2005. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17, 37–48 (doi:10.1016/j.molcel.2004.11.021) [DOI] [PubMed] [Google Scholar]
- 75.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. 2007. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11, 175–189 (doi:10.1016/j.ccr.2006.11.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blasius M, Wagner S. 2014. A quantitative 14-3-3 interaction screen connects the nuclear exosome targeting complex to the DNA damage response. Genes Dev. 28, 1977–1982 (doi:10.1101/gad.246272.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deriano L, Roth DB. 2013. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 47, 433–455 (doi:10.1146/annurev-genet-110711-155540) [DOI] [PubMed] [Google Scholar]
- 78.Yan CT, et al. 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449, 478–482 (doi:10.1038/nature06020) [DOI] [PubMed] [Google Scholar]
- 79.Frit P, Barboule N, Yuan Y, Gomez D, Calsou P. 2014. Alternative end-joining pathway(s): bricolage at DNA breaks. DNA Repair 17, 81–97 (doi:10.1016/j.dnarep.2014.02.007) [DOI] [PubMed] [Google Scholar]
- 80.Park J-Y, Zhang F, Andreassen PR. 2014. PALB2: the hub of a network of tumor suppressors involved in DNA damage responses. Biochim. Biophys. Acta 1846, 263–275 (doi:10.1016/j.bbcan.2014.06.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Farmer H, et al. 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (doi:10.1038/nature03445) [DOI] [PubMed] [Google Scholar]
- 82.Bryant HE, et al. 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (doi:10.1038/nature03443) [DOI] [PubMed] [Google Scholar]
- 83.Fong P, et al. 2009. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 (doi:10.1056/NEJMoa0900212) [DOI] [PubMed] [Google Scholar]
- 84.Helleday T. 2011. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393 (doi:10.1016/j.molonc.2011.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiricny J. 2006. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 7, 335–346 (doi:10.1038/nrm1907) [DOI] [PubMed] [Google Scholar]
- 86.Caldecott KW. 2008. Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (doi:10.1038/nrg2380) [DOI] [PubMed] [Google Scholar]
- 87.Dianov GL, Hübscher U. 2013. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 41, 3483–3490 (doi:10.1093/nar/gkt076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fousteri M, Mullenders LHF. 2008. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 18, 73–84 (doi:10.1038/cr.2008.6) [DOI] [PubMed] [Google Scholar]
- 89.Sale JE. 2012. Competition, collaboration and coordination—determining how cells bypass DNA damage. J. Cell Sci. 125, 1633–1643 (doi:10.1242/jcs.094748) [DOI] [PubMed] [Google Scholar]
- 90.Deans AJ, West SC. 2011. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480 (doi:10.1038/nrc3088) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ciehanover A, Hod Y, Hershko A. 1978. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. 81, 1100–1105 (doi:10.1016/0006-291X(78)91249-4) [DOI] [PubMed] [Google Scholar]
- 92.Hershko A, Ciechanover A. 1980. Proposed role of ATP in protein breakdown: conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl Acad. Sci. USA 77, 1783–1786 (doi:10.1073/pnas.77.4.1783) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wilkinson K, Urban M, Haas A. 1980. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes. J. Biol. Chem. 1, 7529–7532. [PubMed] [Google Scholar]
- 94.Ozkaynak E, Finley D. 1987. The yeast ubiquitin genes: a family of natural gene fusions. EMBO J. 6, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baker RT, Board PG. 1987. The human ubiquitin gene family: structure of a gene and pseudogenes from the Ub B subfamily. Nucleic Acids Res. 15, 443–463 (doi:10.1093/nar/15.2.443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wiborg O, Pedersen M, Wind A. 1985. The human ubiquitin multigene family: some genes contain multiple directly repeated ubiquitin coding sequences. EMBO J. 4, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hershkos A, Heller H, Elias S, Ciechanover A. 1983. Components of ubiquitin-protein ligase system resolution, affinity purification. J. Biol. Chem. 258, 8206–8214. [PubMed] [Google Scholar]
- 98.Ciechanover A, Elias S, Heller H, Hershko A. 1982. ‘Covalent affinity’ purification of ubiquitin-activating enzyme. J. Biol. Chem. 257, 2537–2542. [PubMed] [Google Scholar]
- 99.Pickart CM, Kasperek EM, Beal R, Kim A. 1994. Substrate properties of site-specific mutant ubiquitin protein (G76A) reveal unexpected mechanistic features of ubiquitin-activating enzyme (E1). J. Biol. Chem. 269, 7115–7123. [PubMed] [Google Scholar]
- 100.Schulman BA, Harper JW. 2009. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 (doi:10.1038/nrm2673) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van Wijk SJL, Timmers HTM. 2010. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J. 24, 981–993 (doi:10.1096/fj.09-136259) [DOI] [PubMed] [Google Scholar]
- 102.Berndsen CE, Wolberger C. 2014. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307 (doi:10.1038/nsmb.2780) [DOI] [PubMed] [Google Scholar]
- 103.Mattiroli F, Sixma TK. 2014. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 21, 308–316 (doi:10.1038/nsmb.2792) [DOI] [PubMed] [Google Scholar]
- 104.Deshaies RJ, Joazeiro CAP. 2009. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 (doi:10.1146/annurev.biochem.78.101807.093809) [DOI] [PubMed] [Google Scholar]
- 105.Plechanovová A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. 2012. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 (doi:10.1038/nature11376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. 1999. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644 (doi:10.1016/S0092-8674(00)80574-7) [DOI] [PubMed] [Google Scholar]
- 107.Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI. 2001. U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276, 33 111–33 120 (doi:10.1074/jbc.M102755200) [DOI] [PubMed] [Google Scholar]
- 108.Komander D, Clague MJ, Urbé S. 2009. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 (doi:10.1038/nrm2731) [DOI] [PubMed] [Google Scholar]
- 109.Turcu F, Ventii K, Wilkinson K. 2009. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 78, 363–397 (doi:10.1146/annurev.biochem.78.082307.091526) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mukhopadhyay D, Riezman H. 2007. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205 (doi:10.1126/science.1127085) [DOI] [PubMed] [Google Scholar]
- 111.Bienko M, et al. 2005. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310, 1821–1824 (doi:10.1126/science.1120615) [DOI] [PubMed] [Google Scholar]
- 112.Kirisako T, et al. 2006. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 25, 4877–4887 (doi:10.1038/sj.emboj.7601360) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rieser E, Cordier SM, Walczak H. 2013. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem. Sci. 38, 94–102 (doi:10.1016/j.tibs.2012.11.007) [DOI] [PubMed] [Google Scholar]
- 114.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (doi:10.1146/annurev.biochem.67.1.425) [DOI] [PubMed] [Google Scholar]
- 115.Xu P, et al. 2009. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 (doi:10.1016/j.cell.2009.01.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hurley JH, Lee S, Prag G. 2006. Ubiquitin-binding domains. Biochem. J. 399, 361–372 (doi:10.1042/BJ20061138) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Husnjak K, Dikic I. 2012. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322 (doi:10.1146/annurev-biochem-051810-094654) [DOI] [PubMed] [Google Scholar]
- 118.Komander D. 2009. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953 (doi:10.1042/BST0370937) [DOI] [PubMed] [Google Scholar]
- 119.Komander D, Rape M. 2012. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 (doi:10.1146/annurev-biochem-060310-170328) [DOI] [PubMed] [Google Scholar]
- 120.Hochstrasser M. 2009. Origin and function of ubiquitin-like proteins. Nature 458, 422–429 (doi:10.1038/nature07958) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Van der Veen AG, Ploegh HL. 2012. Ubiquitin-like proteins. Annu. Rev. Biochem. 81, 323–357 (doi:10.1146/annurev-biochem-093010-153308) [DOI] [PubMed] [Google Scholar]
- 122.Jentsch S, McGrath J, Varshavsky A. 1986. The yeast DNA repair gene RAD6 encodes a ubiquitin-conjugating enzyme. Nature 329, 131–134 (doi:10.1038/329131a0) [DOI] [PubMed] [Google Scholar]
- 123.Hoege C, Pfander B, Moldovan G-L, Pyrowolakis G, Jentsch S. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 (doi:10.1038/nature00991) [DOI] [PubMed] [Google Scholar]
- 124.Kannouche PL, Wing J, Lehmann AR. 2004. Interaction of human DNA polymerase η with monoubiquitinated PCNA. Mol. Cell 14, 491–500 (doi:10.1016/S1097-2765(04)00259-X) [DOI] [PubMed] [Google Scholar]
- 125.Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. 2004. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 23, 3886–3896 (doi:10.1038/sj.emboj.7600383) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Simpson LJ, Ross A-L, Szüts D, Alviani CA, Oestergaard VH, Patel KJ, Sale JE. 2006. RAD18-independent ubiquitination of proliferating-cell nuclear antigen in the avian cell line DT40. EMBO Rep. 7, 927–932 (doi:10.1038/sj.embor.7400777) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stewart GS, et al. 2009. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434 (doi:10.1016/j.cell.2008.12.042) [DOI] [PubMed] [Google Scholar]
- 128.Doil C, et al. 2009. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446 (doi:10.1016/j.cell.2008.12.041) [DOI] [PubMed] [Google Scholar]
- 129.Kolas NK, et al. 2007. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640 (doi:10.1126/science.1150034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huen MSY, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. 2007. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914 (doi:10.1016/j.cell.2007.09.041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. 2007. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900 (doi:10.1016/j.cell.2007.09.040) [DOI] [PubMed] [Google Scholar]
- 132.Bekker-Jensen S, Danielsen JR, Fugger K, Gromova I, Nerstedt A, Lukas C, Bartek J, Lukas J, Mailand N. 2010. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 12, 80–86 (doi:10.1038/ncb2008) [DOI] [PubMed] [Google Scholar]
- 133.Oestergaard VH, Pentzold C, Pedersen RT, Iosif S, Alpi A, Bekker-Jensen S, Mailand N, Lisby M. 2012. RNF8 and RNF168 but not HERC2 are required for DNA damage-induced ubiquitylation in chicken DT40 cells. DNA Repair 11, 892–905 (doi:10.1016/j.dnarep.2012.08.005) [DOI] [PubMed] [Google Scholar]
- 134.Pinato S, Gatti M, Scandiuzzi C, Confalonieri S, Penengo L. 2011. UMI, a novel RNF168 ubiquitin binding domain involved in the DNA damage signaling pathway. Mol. Cell. Biol. 31, 118–126 (doi:10.1128/MCB.00818-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mattiroli F, Vissers JHA, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. 2012. RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195 (doi:10.1016/j.cell.2012.08.005) [DOI] [PubMed] [Google Scholar]
- 136.Mattiroli F, Uckelmann M, Sahtoe DD, van Dijk WJ, Sixma TK. 2014. The nucleosome acidic patch plays a critical role in RNF168-dependent ubiquitination of histone H2A. Nat. Commun. 5, 3291 (doi:10.1038/ncomms4291) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Panier S, Ichijima Y, Fradet-Turcotte A, Leung CCY, Kaustov L, Arrowsmith CH, Durocher D. 2012. Tandem protein interaction modules organize the ubiquitin-dependent response to DNA double-strand breaks. Mol. Cell 47, 383–395 (doi:10.1016/j.molcel.2012.05.045) [DOI] [PubMed] [Google Scholar]
- 138.Stewart GS, et al. 2007. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc. Natl Acad. Sci. USA 104, 16 910–16 915 (doi:10.1073/pnas.0708408104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kracker S, Durandy A. 2011. Insights into the B cell specific process of immunoglobulin class switch recombination. Immunol. Lett. 138, 97–103 (doi:10.1016/j.imlet.2011.02.004) [DOI] [PubMed] [Google Scholar]
- 140.Rai R, Li J, Zheng H, Lok G. 2011. The E3 ubiquitin ligase RNF8 stabilizes TPP1 to promote telomere end protection. Nat. Struct. 18, 1400–1407 (doi:10.1038/nsmb.2172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Peuscher MH, Jacobs JJL. 2011. DNA-damage response and repair activities at uncapped telomeres depend on RNF8. Nat. Cell Biol. 13, 1139–1145 (doi:10.1038/ncb2326) [DOI] [PubMed] [Google Scholar]
- 142.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. 2010. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981 (doi:10.1016/j.cell.2010.04.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Giunta S, Belotserkovskaya R, Jackson SP. 2010. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 190, 197–207 (doi:10.1083/jcb.200911156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. 2014. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193 (doi:10.1126/science.1248024) [DOI] [PubMed] [Google Scholar]
- 145.Noon AT, Goodarzi AA. 2011. 53BP1-mediated DNA double strand break repair: insert bad pun here. DNA Repair 10, 1071–1076 (doi:10.1016/j.dnarep.2011.07.012) [DOI] [PubMed] [Google Scholar]
- 146.Li M, Greenberg R. 2012. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem. Sci. 37, 418–424 (doi:10.1016/j.tibs.2012.06.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huyen Y, Zgheib O. 2004. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411 (doi:10.1038/nature03114) [DOI] [PubMed] [Google Scholar]
- 148.Fradet-Turcotte A, et al. 2013. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (doi:10.1038/nature12318) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. 2011. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 25, 685–700 (doi:10.1101/gad.2011011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. 2006. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 173, 195–206 (doi:10.1083/jcb.200510130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. 2012. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci. Signal. 5, ra88 (doi:10.1126/scisignal.2003485) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hu X, Paul A, Wang B. 2012. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J. Biol. Chem. 287, 25 510–25 519 (doi:10.1074/jbc.M112.374116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Huen MSY, Sy SMH, Chen J. 2010. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 11, 138–148 (doi:10.1038/nrm2831) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng J-T, Jasin M, Baer R, Ludwig T. 2008. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl Acad. Sci. USA 105, 20 876–20 881 (doi:10.1073/pnas.0811203106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shakya R, et al. 2011. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 334, 525–528 (doi:10.1126/science.1209909) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Drost R, et al. 2011. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 20, 797–809 (doi:10.1016/j.ccr.2011.11.014) [DOI] [PubMed] [Google Scholar]
- 157.Bunting SF, et al. 2010. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (doi:10.1016/j.cell.2010.03.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chapman JR, et al. 2013. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell 49, 858–871 (doi:10.1016/j.molcel.2013.01.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Escribano-Díaz C, et al. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (doi:10.1016/j.molcel.2013.01.001) [DOI] [PubMed] [Google Scholar]
- 160.Jacq X, Kemp M, Martin NMB, Jackson SP. 2013. Deubiquitylating enzymes and DNA damage response pathways. Cell Biochem. Biophys. 67, 25–43 (doi:10.1007/s12013-013-9635-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Poulsen M, Lukas C, Lukas J, Bekker-Jensen S, Mailand N. 2012. Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J. Cell Biol. 197, 189–199 (doi:10.1083/jcb.201109100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen J, Feng W, Jiang J, Deng Y, Huen MSY. 2012. Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J. Biol. Chem. 287, 27 715–27 722 (doi:10.1074/jbc.M112.373530) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gudjonsson T, et al. 2012. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709 (doi:10.1016/j.cell.2012.06.039) [DOI] [PubMed] [Google Scholar]
- 164.Nakamura K, et al. 2011. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 41, 515–528 (doi:10.1016/j.molcel.2011.02.002) [DOI] [PubMed] [Google Scholar]
- 165.Moyal L, et al. 2011. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 41, 529–542 (doi:10.1016/j.molcel.2011.02.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Meetei AR, et al. 2003. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat. Genet. 35, 165–170 (doi:10.1038/ng1241) [DOI] [PubMed] [Google Scholar]
- 167.Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D'Andrea AD, Dutta A. 2006. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol. Cell 23, 589–596 (doi:10.1016/j.molcel.2006.06.024) [DOI] [PubMed] [Google Scholar]
- 168.MacKay C, et al. 2010. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76 (doi:10.1016/j.cell.2010.06.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kratz K, et al. 2010. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142, 77–88 (doi:10.1016/j.cell.2010.06.022) [DOI] [PubMed] [Google Scholar]
- 170.Smogorzewska A, et al. 2010. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell 39, 36–47 (doi:10.1016/j.molcel.2010.06.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Murina O, von Aesch C, Karakus U, Ferretti LP, Bolck HA, Hänggi K, Sartori AA. 2014. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. 7, 1030–1038 (doi:10.1016/j.celrep.2014.03.069) [DOI] [PubMed] [Google Scholar]
- 172.Zhang N, Kaur R, Akhter S, Legerski RJ. 2009. Cdc5L interacts with ATR and is required for the S-phase cell-cycle checkpoint. EMBO Rep. 10, 1029–1035 (doi:10.1038/embor.2009.122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Marechal A, et al. 2014. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 53, 235–246 (doi:10.1016/j.molcel.2013.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wan L, Huang J. 2014. The PSO4 protein complex associates with replication protein A (RPA) and modulates the activation of ataxia telangiectasia-mutated and RAD3-related (ATR). J. Biol. Chem. 289, 6619–6626 (doi:10.1074/jbc.M113.543439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gong Z, Chen J. 2011. E3 ligase RFWD3 participates in replication checkpoint control. J. Biol. Chem. 286, 22 308–22 313 (doi:10.1074/jbc.M111.222869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu S, Chu J, Yucer N, Leng M, Wang SY, Chen BPC, Hittelman WN, Wang Y. 2011. RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J. Biol. Chem. 286, 22 314–22 322 (doi:10.1074/jbc.M111.222802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fu X, et al. 2010. RFWD3-Mdm2 ubiquitin ligase complex positively regulates p53 stability in response to DNA damage. Proc. Natl Acad. Sci. USA 107, 4579–4584 (doi:10.1073/pnas.0912094107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chan S-P, Kao D-I, Tsai W-Y, Cheng S-C. 2003. The Prp19p-associated complex in spliceosome activation. Science 302, 279–282 (doi:10.1126/science.1086602) [DOI] [PubMed] [Google Scholar]
- 179.Prudden J, Pebernard S, Raffa G, Slavin DA, Perry JJP, Tainer JA, McGowan CH, Boddy MN. 2007. SUMO-targeted ubiquitin ligases in genome stability. EMBO J. 26, 4089–4101 (doi:10.1038/sj.emboj.7601838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yin Y, Seifert A, Chua JS, Maure J-F, Golebiowski F, Hay RT. 2012. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 26, 1196–1208 (doi:10.1101/gad.189274.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. 2012. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 26, 1179–1195 (doi:10.1101/gad.188284.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gibbs-Seymour I, et al. 2015. Ubiquitin-SUMO circuitry controls activated Fanconi anemia ID complex dosage in response to DNA damage. Mol. Cell 57, 150–164 (doi:10.1016/j.molcel.2014.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rojas-Fernandez A, Plechanovová A, Hattersley N, Jaffray E, Tatham MH, Hay RT. 2014. SUMO chain-induced dimerization activates RNF4. Mol. Cell 53, 880–892 (doi:10.1016/j.molcel.2014.02.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Poulsen SL, et al. 2013. RNF111/Arkadia is a SUMO-targeted ubiquitin ligase that facilitates the DNA damage response. J. Cell Biol. 201, 797–807 (doi:10.1083/jcb.201212075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 (doi:10.1146/annurev.biochem.78.081507.101607) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Butler LR, et al. 2012. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 31, 3918–3934 (doi:10.1038/emboj.2012.232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF. 2000. Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep. 1, 71–79 (doi:10.1093/embo-reports/kvd018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wu W, Sato K, Koike A, Nishikawa H, Koizumi H, Venkitaraman AR, Ohta T. 2010. HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 70, 6384–6392 (doi:10.1158/0008-5472.CAN-10-1304) [DOI] [PubMed] [Google Scholar]
- 189.Shi W, Ma Z, Willers H, Akhtar K, Scott SP, Zhang J, Powell S, Zhang J. 2008. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J. Biol. Chem. 283, 31 608–31 616 (doi:10.1074/jbc.M801082200) [DOI] [PubMed] [Google Scholar]
- 190.Meyer H, Bug M, Bremer S. 2012. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 (doi:10.1038/ncb2407) [DOI] [PubMed] [Google Scholar]
- 191.Dantuma NP, Acs K, Luijsterburg MS. 2014. Should I stay or should I go: VCP/p97-mediated chromatin extraction in the DNA damage response. Exp. Cell Res 329, 9–17 (doi:10.1016/j.yexcr.2014.08.025) [DOI] [PubMed] [Google Scholar]
- 192.Puumalainen M-R, Lessel D, Rüthemann P, Kaczmarek N, Bachmann K, Ramadan K, Naegeli H. 2014. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat. Commun. 5, 3695 (doi:10.1038/ncomms4695) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Raman M, Havens CG, Walter JC, Harper JW. 2011. A genome-wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1 destruction. Mol. Cell 44, 72–84 (doi:10.1016/j.molcel.2011.06.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Meerang M, et al. 2011. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 13, 1376–1382 (doi:10.1038/ncb2367) [DOI] [PubMed] [Google Scholar]
- 195.Davis EJ, Lachaud C, Appleton P, Macartney TJ, Näthke I, Rouse J. 2012. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat. Struct. Mol. Biol. 19, 1093–1100 (doi:10.1038/nsmb.2394) [DOI] [PubMed] [Google Scholar]
- 196.Mosbech A, et al. 2012. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 19, 1084–1092 (doi:10.1038/nsmb.2395) [DOI] [PubMed] [Google Scholar]
- 197.Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, Machida YJ. 2014. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 5, 5744 (doi:10.1038/ncomms6744) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lessel D, et al. 2014. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 46, 1239–1244 (doi:10.1038/ng.3103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nie M, Aslanian A, Prudden J, Heideker J, Vashisht AA, Wohlschlegel JA, Yates JR, Boddy MN. 2012. Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem. 287, 29 610–29 619 (doi:10.1074/jbc.M112.379768) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bergink S, Ammon T, Kern M, Schermelleh L, Leonhardt H, Jentsch S. 2013. Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51-Rad52 interaction. Nat. Cell Biol. 15, 526–532 (doi:10.1038/ncb2729) [DOI] [PubMed] [Google Scholar]
- 201.Sun X-X, Challagundla KB, Dai M-S. 2011. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J. 31, 576–592 (doi:10.1038/emboj.2011.434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Nakada S, et al. 2010. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 466, 941–946 (doi:10.1038/nature09297) [DOI] [PubMed] [Google Scholar]
- 203.Juang Y-C, et al. 2012. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol. Cell 45, 384–397 (doi:10.1016/j.molcel.2012.01.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wiener R, Zhang X, Wang T, Wolberger C. 2012. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483, 618–622 (doi:10.1038/nature10911) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. 2013. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J. Biol. Chem. 288, 16 579–16 587 (doi:10.1074/jbc.M113.459917) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, van Deursen J, Galardy PJ. 2012. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J. Clin. Invest. 122, 4362–4374 (doi:10.1172/JCI63084) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Stegmeier F, et al. 2007. Anaphase initiation is regulated by antagonistic ubiquitination and deubiquitination activities. Nature 446, 876–881 (doi:10.1038/nature05694) [DOI] [PubMed] [Google Scholar]
- 208.Nijman SMB, Huang TT, Dirac AMG, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R. 2005. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 17, 331–339 (doi:10.1016/j.molcel.2005.01.008) [DOI] [PubMed] [Google Scholar]
- 209.Oestergaard VH, et al. 2007. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol. Cell 28, 798–809 (doi:10.1016/j.molcel.2007.09.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang D, Zaugg K, Mak TW, Elledge SJ. 2006. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 126, 529–542 (doi:10.1016/j.cell.2006.06.039) [DOI] [PubMed] [Google Scholar]
- 211.Knobel PA, Belotserkovskaya R, Galanty Y, Schmidt CK, Jackson SP, Stracker TH. 2014. USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double-strand break metabolism. Mol. Cell. Biol. 34, 2062–2074 (doi:10.1128/MCB.00197-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. 2013. BAP1 and cancer. Nat. Rev. Cancer 13, 153–159 (doi:10.1038/nrc3459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ismail IH, Davidson R, Gagné JP, Xu ZZ, Poirier GG, Hendzel MJ. 2014. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res. 74, 4282–4294 (doi:10.1158/0008-5472.CAN-13-3109) [DOI] [PubMed] [Google Scholar]
- 214.Yu H, et al. 2014. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl Acad. Sci. USA 111, 285–290 (doi:10.1073/pnas.1309085110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nishi R, Wijnhoven P, le Sage C, Tjeertes J, Galanty Y, Forment JV, Clague MJ, Urbé S, Jackson SP. 2014. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 16, 1016–1026 (doi:10.1038/ncb3028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kumar S, Tomooka Y, Noda M. 1992. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem. Biophys. Res. 185, 1155–1161 (doi:10.1016/0006-291X(92)91747-E) [DOI] [PubMed] [Google Scholar]
- 217.Kamitani T, Kito K, Nguyen HP, Yeh ETH. 1997. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J. Biol. Chem. 272, 28 557–28 562 (doi:10.1074/jbc.272.45.28557) [DOI] [PubMed] [Google Scholar]
- 218.Whitby FG, Xia G, Pickart CM, Hill CP. 1998. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J. Biol. Chem. 273, 34 983–34 991 (doi:10.1074/jbc.273.52.34983) [DOI] [PubMed] [Google Scholar]
- 219.Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. 1998. A novel protein modification pathway related to the ubiquitin system. EMBO J. 17, 2208–2214 (doi:10.1093/emboj/17.8.2208) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lammer D, Mathias N, Laplaza JM, Jiang W, Liu Y, Callis J, Goebl M, Estelle M. 1998. Modification of yeast Cdc53p by the ubiquitin-related protein Rub1p affects function of the SCFCdc4 complex. Genes Dev. 12, 914–926 (doi:10.1101/gad.12.7.914) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mendoza HM, Shen L-N, Botting C, Lewis A, Chen J, Ink B, Hay RT. 2003. NEDP1, a highly conserved cysteine protease that deNEDDylates cullins. J. Biol. Chem. 278, 25 637–25 643 (doi:10.1074/jbc.M212948200) [DOI] [PubMed] [Google Scholar]
- 222.Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. 1998. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem. Biophys. Res. Commun. 251, 688–692 (doi:10.1006/bbrc.1998.9532) [DOI] [PubMed] [Google Scholar]
- 223.Burch TJ, Haas AL. 1994. Site-directed mutagenesis of ubiquitin. Differential roles for arginine in the interaction with ubiquitin-activating enzyme. Biochemistry 33, 7300–7308 (doi:10.1021/bi00189a035) [DOI] [PubMed] [Google Scholar]
- 224.Souphron J, Waddell MB, Paydar A, Tokgöz-Gromley Z, Roussel MF, Schulman BA. 2008. Structural dissection of a gating mechanism preventing misactivation of ubiquitin by NEDD8′s E1. Biochemistry 47, 8961–8969 (doi:10.1021/bi800604c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL, Holton JM, Schulman BA. 2003. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 12, 1427–1437 (doi:10.1016/S1097-2765(03)00452-0) [DOI] [PubMed] [Google Scholar]
- 226.Bohnsack RN, Haas AL. 2003. Conservation in the mechanism of Nedd8 activation by the human AppBp1-Uba3 heterodimer. J. Biol. Chem. 278, 26 823–26 830 (doi:10.1074/jbc.M303177200) [DOI] [PubMed] [Google Scholar]
- 227.Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. 2005. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8′s E1. Mol. Cell 17, 341–350 (doi:10.1016/j.molcel.2004.12.020) [DOI] [PubMed] [Google Scholar]
- 228.Leidecker O, Matic I, Mahata B, Pion E, Xirodimas DP. 2012. The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle 11, 1142–1150 (doi:10.4161/cc.11.6.19559) [DOI] [PubMed] [Google Scholar]
- 229.Hjerpe R, Thomas Y, Chen J, Zemla A, Curran S, Shpiro N, Dick LR, Kurz T. 2012. Changes in the ratio of free NEDD8 to ubiquitin triggers NEDDylation by ubiquitin enzymes. Biochem. J. 441, 927–936 (doi:10.1042/BJ20111671) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Gong L, Yeh ETH. 1999. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J. Biol. Chem. 274, 12 036–12 042 (doi:10.1074/jbc.274.17.12036) [DOI] [PubMed] [Google Scholar]
- 231.Osaka F, Kawasaki H, Aida N, Saeki M, Chiba T, Kawashima S, Tanaka K, Kato S. 1998. A new NEDD8-ligating system for cullin-4A. Genes Dev. 12, 2263–2268 (doi:10.1101/gad.12.15.2263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Huang DT, et al. 2009. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol. Cell 33, 483–495 (doi:10.1016/j.molcel.2009.01.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Scott DC, Monda JK, Grace CRR, Duda DM, Kriwacki RW, Kurz T, Schulman BA. 2010. A dual E3 mechanism for Rub1 ligation to Cdc53. Mol. Cell 39, 784–796 (doi:10.1016/j.molcel.2010.08.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kamura T, Conrad MN, Yan Q, Conaway RC, Conaway JW. 1999. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. 13, 2928–2933 (doi:10.1101/gad.13.22.2928) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Petroski MD, Deshaies RJ. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 (doi:10.1038/nrm1547) [DOI] [PubMed] [Google Scholar]
- 236.Kurz T, Ozlü N, Rudolf F, O'Rourke SM, Luke B, Hofmann K, Hyman AA, Bowerman B, Peter M. 2005. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature 435, 1257–1261 (doi:10.1038/nature03662) [DOI] [PubMed] [Google Scholar]
- 237.Kim AY, et al. 2008. SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. J. Biol. Chem. 283, 33 211–33 220 (doi:10.1074/jbc.M804440200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kurz T, Chou Y-C, Willems AR, Meyer-Schaller N, Hecht M-L, Tyers M, Peter M, Sicheri F. 2008. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol. Cell 29, 23–35 (doi:10.1016/j.molcel.2007.12.012) [DOI] [PubMed] [Google Scholar]
- 239.Meyer-Schaller N, et al. 2009. The human Dcn1-like protein DCNL3 promotes Cul3 neddylation at membranes. Proc. Natl Acad. Sci. USA 106, 12 365–12 370 (doi:10.1073/pnas.0812528106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Monda JK, Scott DC, Miller DJ, Lydeard J, King D, Harper JW, Bennett EJ, Schulman BA. 2013. Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure 21, 42–53 (doi:10.1016/j.str.2012.10.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wu K, Yan H, Fang L, Wang X, Pfleger C, Jiang X, Huang L, Pan Z-Q. 2011. Mono-ubiquitination drives nuclear export of the human DCN1-like protein hDCNL1. J. Biol. Chem. 286, 34 060–34 070 (doi:10.1074/jbc.M111.273045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Girdwood D, Xirodimas DP, Gordon C. 2011. The essential functions of NEDD8 are mediated via distinct surface regions, and not by polyneddylation in Schizosaccharomyces pombe. PLoS ONE 6, e20089 (doi:10.1371/journal.pone.0020089) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Xirodimas DP, Sundqvist A, Nakamura A, Shen L, Botting C, Hay RT. 2008. Ribosomal proteins are targets for the NEDD8 pathway. EMBO Rep. 9, 280–286 (doi:10.1038/embor.2008.10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jones J, Wu K, Yang Y, Guerrero C. 2008. A targeted proteomic analysis of the ubiquitin-like modifier Nedd8 and associated proteins. J. Proteome 7, 1274–1287 (doi:10.1021/pr700749v) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wei N, Deng XW. 1992. COP9: a new genetic locus involved in light-regulated development and gene expression in arabidopsis. Plant Cell 4, 1507–1518 (doi:10.1105/tpc.4.12.1507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Cope GA, Suh GSB, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ. 2002. Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298, 608–611 (doi:10.1126/science.1075901) [DOI] [PubMed] [Google Scholar]
- 247.Sharon M, Mao H, Boeri Erba E, Stephens E, Zheng N, Robinson CV. 2009. Symmetrical modularity of the COP9 signalosome complex suggests its multifunctionality. Structure 17, 31–40 (doi:10.1016/j.str.2008.10.012) [DOI] [PubMed] [Google Scholar]
- 248.Lingaraju GM, Bunker RD, Cavadini S, Hess D, Hassiepen U, Renatus M, Fischer ES, Thomä NH. 2014. Crystal structure of the human COP9 signalosome. Nature 512, 161–165 (doi:10.1038/nature13566) [DOI] [PubMed] [Google Scholar]
- 249.Rabut G, Peter M. 2008. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 9, 969–976 (doi:10.1038/embor.2008.183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Lydeard JR, Schulman BA, Harper JW. 2013. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 14, 1050–1061 (doi:10.1038/embor.2013.173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sarikas A, Hartmann T, Pan Z-Q. 2011. The cullin protein family. Genome Biol. 12, 220 (doi:10.1186/gb-2011-12-4-220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Soucy TA, et al. 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (doi:10.1038/nature07884) [DOI] [PubMed] [Google Scholar]
- 253.Shibata E, Abbas T, Huang X, Wohlschlegel JA, Dutta A. 2011. Selective ubiquitylation of p21 and Cdt1 by UBCH8 and UBE2G ubiquitin-conjugating enzymes via the CRL4Cdt2 ubiquitin ligase complex. Mol. Cell. Biol. 31, 3136–3145 (doi:10.1128/MCB.05496-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee J, Zhou P. 2007. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 26, 775–780 (doi:10.1016/j.molcel.2007.06.001) [DOI] [PubMed] [Google Scholar]
- 255.Bennett EJ, Rush J, Gygi SP, Harper JW. 2010. Dynamics of cullin-RING ubiquitin ligase network revealed by systematic quantitative proteomics. Cell 143, 951–965 (doi:10.1016/j.cell.2010.11.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Xu C, Min J. 2011. Structure and function of WD40 domain proteins. Protein Cell 2, 202–214 (doi:10.1007/s13238-011-1018-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Emanuele MJ, et al. 2011. Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 (doi:10.1016/j.cell.2011.09.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Larance M, Kirkwood KJ, Xirodimas DP, Lundberg E, Uhlen M, Lamond AI. 2011. Characterization of MRFAP1 turnover and interactions downstream of the NEDD8 pathway. Mol. Cell. Proteomics 11, M111.014407 (doi:10.1074/mcp.M111.014407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. 2008. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006 (doi:10.1016/j.cell.2008.07.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zheng J, et al. 2002. CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol. Cell 10, 1519–1526 (doi:10.1016/S1097-2765(02)00784-0) [DOI] [PubMed] [Google Scholar]
- 261.Liu J, Furukawa M, Matsumoto T, Xiong Y, Hill C, Carolina N. 2002. NEDD8 modification of CUL1 dissociates p120, an inhibitor of CUL1-SKP1 binding and SCF ligases. Mol. Cell 10, 1511–1518 (doi:10.1016/S1097-2765(02)00783-9) [DOI] [PubMed] [Google Scholar]
- 262.Zemla A, Thomas Y, Kedziora S, Knebel A, Wood NT, Rabut G, Kurz T. 2013. CSN- and CAND1-dependent remodelling of the budding yeast SCF complex. Nat. Commun. 4, 1641 (doi:10.1038/ncomms2628) [DOI] [PubMed] [Google Scholar]
- 263.Pierce NW, et al. 2013. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 153, 206–215 (doi:10.1016/j.cell.2013.02.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Wu S, Zhu W, Nhan T, Toth JI, Petroski MD, Wolf DA. 2013. CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nat. Commun. 4, 1642 (doi:10.1038/ncomms2636) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Zhou C, Wee S, Rhee E, Naumann M, Dubiel W, Wolf DA. 2003. Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol. Cell 11, 927–938 (doi:10.1016/S1097-2765(03)00136-9) [DOI] [PubMed] [Google Scholar]
- 266.Fischer ES, et al. 2011. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039 (doi:10.1016/j.cell.2011.10.035) [DOI] [PubMed] [Google Scholar]
- 267.Emberley ED, Mosadeghi R, Deshaies RJ. 2012. Deconjugation of Nedd8 from Cul1 is directly regulated by Skp1-F-box and substrate, and the COP9 signalosome inhibits deneddylated SCF by a noncatalytic mechanism. J. Biol. Chem. 287, 29 679–29 689 (doi:10.1074/jbc.M112.352484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Enchev RI, Scott DC, da Fonseca PCA, Schreiber A, Monda JK, Schulman BA, Peter M, Morris EP. 2012. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2, 616–627 (doi:10.1016/j.celrep.2012.08.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Schmidt MW, McQuary PR, Wee S, Hofmann K, Wolf DA. 2009. F-box-directed CRL complex assembly and regulation by the CSN and CAND1. Mol. Cell 35, 586–597 (doi:10.1016/j.molcel.2009.07.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chew E-H, Hagen T. 2007. Substrate-mediated regulation of cullin neddylation. J. Biol. Chem. 282, 17 032–17 040 (doi:10.1074/jbc.M701153200) [DOI] [PubMed] [Google Scholar]
- 271.Duda DM, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, Schulman BA. 2011. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 21, 257–264 (doi:10.1016/j.sbi.2011.01.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Watson IR, Irwin MS, Ohh M. 2011. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell 19, 168–176 (doi:10.1016/j.ccr.2011.01.002) [DOI] [PubMed] [Google Scholar]
- 273.Melchor L, et al. 2009. Comprehensive characterization of the DNA amplification at 13q34 in human breast cancer reveals TFDP1 and CUL4A as likely candidate target genes. Breast Cancer Res. 11, R86 (doi:10.1186/bcr2456) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Brownell JE, et al. 2010. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell 37, 102–111 (doi:10.1016/j.molcel.2009.12.024) [DOI] [PubMed] [Google Scholar]
- 275.Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. 2010. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 70, 10 310–10 320 (doi:10.1158/0008-5472.CAN-10-2062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Rizzardi LF, Cook JG. 2012. Flipping the switch from G1 to S phase with E3 ubiquitin ligases. Genes Cancer 3, 634–648 (doi:10.1177/1947601912473307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. 2010. CRL4Cdt2 regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21 (doi:10.1016/j.molcel.2010.09.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 12, 1086–1093 (doi:10.1038/ncb2113) [DOI] [PubMed] [Google Scholar]
- 279.Zhu W, Dutta A. 2006. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol. Cell. Biol. 26, 4601–4611 (doi:10.1128/MCB.02141-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. 2011. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 71, 3042–3051 (doi:10.1158/0008-5472.CAN-10-2122) [DOI] [PubMed] [Google Scholar]
- 281.Jia L, Li H. 2011. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia (New York, NY) 13, 561–569 (doi:10.1593/neo.11420) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Priego Moreno S, Bailey R, Campion N, Herron S, Gambus A. 2014. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 346, 477–481 (doi:10.1126/science.1253585) [DOI] [PubMed] [Google Scholar]
- 283.Maric M, Maculins T, De Piccoli G, Labib K. 2014. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346, 1253596 (doi:10.1126/science.1253596) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Archambault V, Ikui A, Drapkin B, Cross F. 2005. Disruption of mechanisms that prevent rereplication triggers a DNA damage response. Mol. Cell 25, 6707–6721 (doi:10.1128/MCB.25.15.6707-6721.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Blank JL, et al. 2013. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 73, 225–234 (doi:10.1158/0008-5472.CAN-12-1729) [DOI] [PubMed] [Google Scholar]
- 286.Zhao Y, Morgan MA, Sun Y. 2014. Targeting neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid. Redox Signal. 21, 2383–2400 (doi:10.1089/ars.2013.5795) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Swords RT, et al. In press. Pevonedistat (MLN4924), a First-in-Class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br. J. Haematol. (doi:10.1111/bjh.13323) [DOI] [PubMed] [Google Scholar]
- 288.Xirodimas DP, Saville MK, Bourdon J-C, Hay RT, Lane DP. 2004. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 118, 83–97 (doi:10.1016/j.cell.2004.06.016) [DOI] [PubMed] [Google Scholar]
- 289.Sundqvist A, Liu G, Mirsaliotis A, Xirodimas DP. 2009. Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO Rep. 10, 1132–1139 (doi:10.1038/embor.2009.178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Ma T, Chen Y, Zhang F, Yang C, Wang S, Yu X. 2013. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol. Cell 49, 897–907 (doi:10.1016/j.molcel.2013.01.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Enchev RI, Schulman BA, Peter M. 2014. Protein neddylation: beyond cullin–RING ligases. Nat. Rev. Mol. Cell Biol. 16, 30–44 (doi:10.1038/nrm3919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Liu L, et al. 2009. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell 34, 451–460 (doi:10.1016/j.molcel.2009.04.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yin Y, et al. 2011. The E3 ubiquitin ligase cullin 4A regulates meiotic progression in mouse spermatogenesis. Dev. Biol. 356, 51–62 (doi:10.1016/j.ydbio.2011.05.661) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Tarpey PS, et al. 2007. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 80, 345–352 (doi:10.1086/511134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Liu L, Yin Y, Li Y, Prevedel L, Lacy EH, Ma L, Zhou P. 2012. Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Res. 22, 1258–1269 (doi:10.1038/cr.2012.48) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. 2003. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357–367 (doi:10.1016/S0092-8674(03)00316-7) [DOI] [PubMed] [Google Scholar]
- 297.Yeh JI, et al. 2012. Damaged DNA induced UV-damaged DNA-binding protein (UV-DDB) dimerization and its roles in chromatinized DNA repair. Proc. Natl Acad. Sci. USA 109, E2737–E2746 (doi:10.1073/pnas.1110067109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Sugasawa K, et al. 2005. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387–400 (doi:10.1016/j.cell.2005.02.035) [DOI] [PubMed] [Google Scholar]
- 299.Wang H, Zhai L, Xu J, Joo H-Y, Jackson S, Erdjument-Bromage H, Tempst P, Xiong Y, Zhang Y. 2006. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 22, 383–394 (doi:10.1016/j.molcel.2006.03.035) [DOI] [PubMed] [Google Scholar]
- 300.Tang J, Chu G. 2002. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair 1, 601–616 (doi:10.1016/S1568-7864(02)00052-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Scrima A, et al. 2008. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 135, 1213–1223 (doi:10.1016/j.cell.2008.10.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Tornaletti S. 2009. DNA repair in mammalian cells: transcription-coupled DNA repair: directing your effort where it's most needed. Cell. Mol. Life Sci. 66, 1010–1020 (doi:10.1007/s00018-009-8738-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Havens CG, Walter JC. 2011. Mechanism of CRL4(Cdt2), a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 25, 1568–1582 (doi:10.1101/gad.2068611) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Ulrich HD. 2014. Two-way communications between ubiquitin-like modifiers and DNA. Nat. Struct. Mol. Biol. 21, 317–324 (doi:10.1038/nsmb.2805) [DOI] [PubMed] [Google Scholar]
- 305.Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. 2004. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 (doi:10.1146/annurev.biochem.73.011303.073723) [DOI] [PubMed] [Google Scholar]
- 306.Mailand N, Bekker-Jensen S, Bartek J, Lukas J. 2006. Destruction of Claspin by SCFβTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol. Cell 23, 307–318 (doi:10.1016/j.molcel.2006.06.016) [DOI] [PubMed] [Google Scholar]
- 307.Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. 2006. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol. Cell 23, 319–329 (doi:10.1016/j.molcel.2006.06.013) [DOI] [PubMed] [Google Scholar]
- 308.Postow L, Funabiki H. 2013. An SCF complex containing Fbxl12 mediates DNA damage-induced Ku80 ubiquitylation. Cell Cycle 12, 587–595 (doi:10.4161/cc.23408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Postow L, Ghenoiu C, Woo EM, Krutchinsky AN, Chait BT, Funabiki H. 2008. Ku80 removal from DNA through double strand break-induced ubiquitylation. J. Cell Biol. 182, 467–479 (doi:10.1083/jcb.200802146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Postow L. 2012. Destroying the ring: freeing DNA from Ku with ubiquitin. FEBS Lett. 585, 2876–2882 (doi:10.1016/j.febslet.2011.05.046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Walker JR, Corpina RA, Goldberg J. 2001. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412, 607–614 (doi:10.1038/35088000) [DOI] [PubMed] [Google Scholar]
- 312.Frit P, Li RY, Arzel D, Salles B, Calsou P. 2000. Ku entry into DNA inhibits inward DNA transactions in vitro. J. Biol. Chem. 275, 35 684–35 691 (doi:10.1074/jbc.M004315200) [DOI] [PubMed] [Google Scholar]
- 313.Ono M, Tucker PW, Capra JD. 1996. Ku is a general inhibitor of DNA-protein complex formation and transcription. Mol. Immunol. 33, 787–796 (doi:10.1016/0161-5890(96)00030-2) [DOI] [PubMed] [Google Scholar]
- 314.Garcia K, et al. 2014. Nedd8-activating enzyme inhibitor MLN4924 provides synergy with mitomycin C through interactions with ATR, BRCA1/BRCA2 and chromatin dynamics pathways. Mol. Cancer Ther. 13, 1625–1635 (doi:10.1158/1535-7163.MCT-13-0634) [DOI] [PubMed] [Google Scholar]
- 315.Guihard S, Ramolu L, Macabre C, Wasylyk B, Noël G, Abecassis J, Jung AC. 2012. The NEDD8 conjugation pathway regulates p53 transcriptional activity and head and neck cancer cell sensitivity to ionizing radiation. Int. J. Oncol. 41, 1531–1540 (doi:10.3892/ijo.2012.1584) [DOI] [PubMed] [Google Scholar]
- 316.Jazaeri A, et al. 2013. Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor MLN4924. Mol. Cancer Ther 12, 1958–1967 (doi:10.1158/1535-7163.MCT-12-1028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kee Y, Huang M, Chang S, Moreau LA, Park E, Smith PG, D'Andrea AD. 2012. Inhibition of the Nedd8 system sensitizes cells to DNA interstrand cross-linking agents. Mol. Cancer Res. 10, 369–377 (doi:10.1158/1541-7786.MCR-11-0497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Nawrocki ST, et al. 2013. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin. Cancer Res. 19, 3577 (doi:10.1158/1078-0432.CCR-12-3212) [DOI] [PubMed] [Google Scholar]
- 319.Pan W-W, Zhou J-J, Yu C, Xu Y, Guo L-J, Zhang H-Y, Zhou D, Song F-Z, Fan H-Y. 2013. Ubiquitin E3 ligase CRL4CDT2/DCAF2 as a potential chemotherapeutic target for ovarian surface epithelial cancer. J. Biol. Chem. 288, 29 680–29 691 (doi:10.1074/jbc.M113.495069) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, Smith PG, Morgan MA, Sun Y. 2011. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 72, 282–293 (doi:10.1158/0008-5472.CAN-11-2866) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Yang D, Tan M, Wang G, Sun Y. 2012. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS ONE 7, e34079 (doi:10.1371/journal.pone.0034079) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Koinuma D, Shinozaki M, Komuro A. 2003. Arkadia amplifies TGF-β superfamily signalling through degradation of Smad7. EMBO J. 22, 6458–6470 (doi:10.1093/emboj/cdg632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Erker Y, Neyret-Kahn H, Seeler JS, Dejean A, Atfi A, Levy L. 2013. Arkadia, a novel SUMO-targeted ubiquitin ligase involved in PML degradation. Mol. Cell. Biol. 33, 2163–2177 (doi:10.1128/MCB.01019-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Scott DC, Sviderskiy VO, Monda JK, Lydeard JR, Cho SE, Harper JW, Schulman BA. 2014. Structure of a RING E3 trapped in action reveals ligation mechanism for the ubiquitin-like protein NEDD8. Cell 157, 1671–1684 (doi:10.1016/j.cell.2014.04.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Wu J, et al. 2012. Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol. Cell 46, 1–11 (doi:10.1016/j.molcel.2012.03.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Lu C-S, et al. 2012. The RING finger protein RNF8 ubiquitinates Nbs1 to promote DNA double-strand break repair by homologous recombination. J. Biol. Chem. 287, 43 984–43 994 (doi:10.1074/jbc.M112.421545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Jimeno S, Fernandez-Avila MJ, Cruz-Garcia A, Cepeda-Garcia C, Gomez-Cabello D, Huertas P. 2015. Neddylation inhibits CtIP-mediated resection and regulates DNA double strand break repair pathway choice. Nucleic Acids Res 43, 987–999 (doi:10.10.1093/nar/gku1384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Li T, Guan J, Huang Z, Hu X, Zheng X. 2014. RNF168-mediated H2A neddylation antagonizes its ubiquitination and regulates DNA damage repair. J. Cell Sci. 127, 2238–2248 (doi:10.1242/jcs.138891) [DOI] [PubMed] [Google Scholar]
- 329.Jackson SP, Durocher D. 2013. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 (doi:10.1016/j.molcel.2013.01.017) [DOI] [PubMed] [Google Scholar]
- 330.Doudna JA, Charpentier E. 2014. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (doi:10.1126/science.1258096) [DOI] [PubMed] [Google Scholar]
- 331.Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (doi:10.1016/j.cell.2014.05.010) [DOI] [PMC free article] [PubMed] [Google Scholar]