

Abstract
Cancer is a disease arising from both genetic and epigenetic modifications of DNA that contribute to changes in gene expression in the cell. Genetic modifications include loss or amplification of DNA, loss of heterozygosity (LOH) as well as gene mutations. Epigenetic changes in cancer are generally thought to be brought about by alterations in DNA and histone modifications that lead to the silencing of tumour suppressor genes and the activation of oncogenic genes. Other consequences that result from epigenetic changes, such as inappropriate expression or repression of some genes in the wrong cellular context, can also result in the alteration of control and physiological systems such that a normal cell becomes tumorigenic. Excessive levels of the enzymes that act as epigenetic modifiers have been reported as markers of aggressive breast cancer and are associated with metastatic progression. It is likely that this is a common contributor to the recurrence and spread of the disease. The emphasis on genetic changes, for example in genome-wide association studies and increasingly in whole genome sequencing analyses of tumours, has resulted in the importance of epigenetic changes having less attention until recently. Epigenetic alterations at both the DNA and histone level are increasingly being recognised as playing a role in tumourigenesis. Recent studies have found that distinct subgroups of poor-prognosis tumours lack genetic alterations but are epigenetically deregulated, pointing to the important role that epigenetic modifications and/or their modifiers may play in cancer. In this review, we highlight the multitude of epigenetic changes that can occur and will discuss how deregulation of epigenetic modifiers contributes to cancer progression. We also discuss the off-target effects that epigenetic modifiers may have, notably the effects that histone modifiers have on non-histone proteins that can modulate protein expression and activity, as well as the role of hypoxia in epigenetic regulation.
Keywords: Epigenetics, Hypoxia, Cancer, DNA methylation, Histone modifications, Acetylation, Demethylation, Transcription
Introduction
Cancer initiation and progression have been recognised for many years to be secondary to the accumulation of genetic mutations which lead to changes in cellular function. While inherited or sporadic mutations may result in the activation of oncogenes or the inactivation of tumour suppressor genes, changes in modification of both DNA and histones (collectively the epigenome) can also contribute to the initiation and the progression of cancer. Although epigenetics is formally defined as a heritable change in gene expression or chromosomal stability by utilising DNA methylation, covalent modification of histones or non-coding RNAs without a change in DNA sequence, it is increasingly used to define long term changes that alter the physiology of a subset of cells in a tissue independent of a change in the DNA sequence. It should be noted that epigenetic marks are dynamic and can respond to changes in physiological conditions and hence, in addition to gene mutations, can be drivers of the development of the cancer. Global reprogramming of epigenetic marks, including alterations in DNA methylation and histone modifications, is known to occur in malignancy [1].
Epigenetic regulation
Epigenetic modification of chromatin plays an important role in the regulation of gene expression. DNA is methylated post-synthetically on cytosine residues predominantly in the sequence CpG and in vitro methylated promoters are known to be generally inactive when transfected into eukaryotic cells [2]. DNA methylation is catalysed by a family of DNA methyltransferases (DNMTs): DNMT1 is the methyltransferase that maintains reciprocal methylation of the new DNA strand complementary to hemi-methylated DNA that is produced as a result of semi-conservative DNA replication. DNMT3a and DNMT3b are known as de novo methyltransferases, being able to methylate the completely unmethylated DNA duplex in vivo[3,4]. More recently it has been shown that 5-methylcytosine can be oxidised to 5-hydroxymethylcytosine by a family of Fe2+, 2-oxoglutarate dependent methylcytosine dioxygenases known as TET proteins [5], effectively resulting in the subsequent removal of the repressive methyl group by a mechanism that appears to include base excision repair processes. Other DNA modifications are also described such as methylation at sites other than CpG [6,7] and the generation of formyl and carboxyl derivatives of DNA [8].
Earlier discussions that derived from those that studied transgenerational phenomena focused on the classical set of DNMTs. However, epigenetic modifications go beyond DNA methylation. The histone proteins in chromatin are also modified on their N-terminal residues and transcriptional states are frequently associated with particular histone modifications [9]. The number and complexity of the potential combinations of these has grown very rapidly in recent years [10] but a simplified generalisation could be that acetylation of histones H3 and H4 and methylation of the lysine-4 residue of histone H3 (H3K4) are associated with active genes. Inactive genes are frequently hypoacetylated and may also be methylated on the lysine-9 (H3K9) or lysine-27 (H3K27) residues of histone H3 (reviewed in [11]). Clearly there are possibilities for more complex situations when, for example, both H3K4 and H3K27 are methylated as occurs at bivalent domains in embryonic stem cells [12]. Although most studies tend to focus attention on either the DNA or histone modifications, it is clear that in order for a gene to be transcribed there is interplay between the methylated DNA and the modified histones. Both the DNA and the histones should be in an open or “unlocked” configuration, as shown in Figure 1, to be in a permissible state for transcription. If the epigenetic marks on the DNA or histones are in a closed or “locked” state, the gene of interest will not be transcribed. This is a concept that we term the “Double Lock Principle” as both the DNA methylation status and histone modifications are critical to the expression of a gene. In addition, the required transcriptional activator must be present and the necessity to have it and the “double lock” correctly aligned explains a lot of data where genes are not expressed despite what could be considered to be tolerant conditions.
Figure 1.
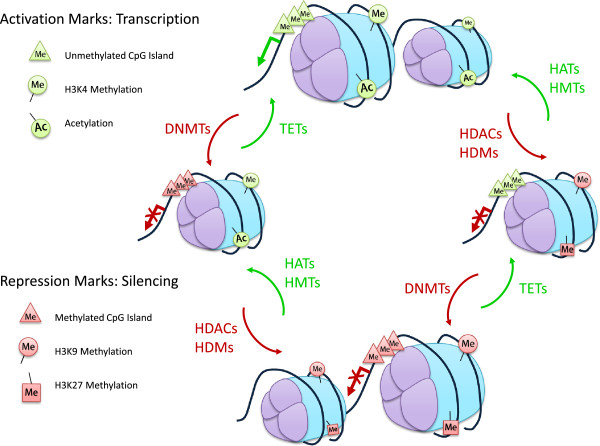
Double Lock Principle. A gene will be transcribed when it is in the open or “unlocked” state. The promoter region is demethylated, histones acetylated and H3K4me marked. If the gene is silenced, in a closed or “locked” state, DNA methyltransferases (DNMTs), histone deacetylases (HDACs), histone methyltransferases (HMTs) and histone demethyltransferases (HDMs) have modified the promoter region, removing the histone acetylation and modifying methylation accordingly. For the gene to be transcribed, the repression marks will need to be lifted to confer the open, “unlocked” state, by the TETs (removal of methylation on the promoter), histone acetyltransferases (HATs) and the HMTs/HDMs. If the DNA exists in any in-between state, with only partial silencing or activation marks, the gene remains repressed, hence the term “Double Lock”.
Many enzymes have been identified that methylate, demethylate, acetylate, deacetylate, phosphorylate, ubiquitinate or sumoylate histones. There is redundancy and specificity in these enzymes that is required to deliver the full range of potential histone post-translational modifications. Additionally these enzymes may modify non-histone proteins such as Reptin and p53, contributing to their post-translational regulation (Table 1).
Table 1.
Classification of epigenetic modifiers
| Class | Enzymes | ||
|---|---|---|---|
| Histone Acetyltransferases (HATs) | ELP3/KAT9 | PCAF/KAT2B | MORF/MYST4/KAT6B |
| GTF3C4 | CBP/KAT3A | HBO1/MYST2/KAT7 | |
| HAT3 | p300/KAT3B | MOF/MYST1/KAT8 | |
| HAT1/KAT1 | Tip60/KAT5 | KAT10 | |
| GCN5/KAT2A | MOZ/MYST3/KAT6A | TFIIIC90/KAT12 | |
| Histone Deacetylases (HDACs) | HDAC1 | HDAC7 | SIRT2 |
| HDAC2 | HDAC8 | SIRT3 | |
| HDAC3 | HDAC9 | SIRT4 | |
| HDAC4 | HDAC10 | SIRT5 | |
| HDAC5 | HDAC11 | SIRT6 | |
| HDAC6 | SIRT1 | SIRT7 | |
| Histone Methyltransferases (HMTs) | ASH1 | NSD1/KMT3B | SETD1A |
| Clr4/KMT1 | PRMT1 | SETD8/Pr-SET7/KMT5A | |
| Dot1L/KMT4 | PRMT3 | SETDB1 | |
| EZH2/KMT6 | PRMT4/CARM1 | SETDB2/KMT1F/CLL8 | |
| G9a/EHMT2 | PRMT5/JBP1 | SMYD2/KMT3C | |
| GLP/EHMT1 | PRMT6 | SUV39H1 | |
| KMT5B/KMT5C | Riz1/Riz2/KMT8 | SUV39H2 | |
| MLL1 | NF20 | SUV4-20H2/KMT5C | |
| MLL2 | RNF40 | TRX/ KMT2a | |
| MLL3 | SET1A | HIF-1/ SET2/HYPB/KMT3A | |
| MLL4 | SET1B | ||
| MLL5 | SET7/9 | ||
| Histone Demethylases (HDMs) | ARID1A | JHDM1b/FBXL10/KDM2B | JMJD2D/KDM4D |
| ARID5B | JHDM2A/KDM3A | JMJD3/KDM6B | |
| JARID1A/RBBP2/KDM5A | JHDM3A/JMJD2A/KDM4A | LSD1/KDM1 | |
| JARID1B/PLU1/KDM5B | JMJD1A | LSD2 | |
| JARID1C/SMCX/KDM5C | JMJD1B/KDM3B | PHF2 | |
| JARID1D/SMCY/KDM5D | JMJD2A | PLU1 | |
| JHD1/KDM2 | JMJD2B/KDM4B | UTX/KDM6A | |
| JHDM1a/FBXL11/KDM2A | JMJD2C/GASC1/KDM4C | ||
| DNA Methyltransferases (DNMTs) | DNMT1 | DNMT3b | DNMT1o |
| DNMT3a | DNMT3L | ||
| DNA Demethylases | TET1 | TET2 | TET3 |
DNA methylation patterns and histone modifications have been found to be different when normal tissues and tumours derived from them are compared. All gene expression is ultimately controlled by their epigenetic status and it is not surprising therefore that epigenetic changes may play an important role in tumorigenesis. However, why these changes occur is unknown nor is it always clear if these changes are the causes of the tumour growth or if they are responding to altered environments (e.g. hypoxia). It is most likely that both sequences of events occur and irrespective of whether these are causes or consequences the epigenetic status is crucial to the cellular outcomes.
The enzymes mediating epigenetic modifications have been found to be mutated in cancers, which adds to an indirect manner in which tumours develop as the change in the modifier can affect the gene expression patterns. This suggests also that epigenetic modifiers may act as novel targets for therapy. Mutations of DNMT3a have been observed in 22% of cases of acute myeloid leukaemia (AML) where they are associated with a poor outcome [13]. Similarly, the methylcytosine dioxygenase TET2 is mutated in ~15% of myeloid cancers [14]. Tet2-deficiency in mutant mice causes myeloproliferation, suggesting a role in stem cell function [15]. The H3K27 demethylase UTX is mutated in multiple human cancers, the highest frequency (~10%) being in multiple myeloma [16]. The discovery of mutations in genes that modify chromatin suggests that the disruption of epigenetic control has a very significant role in the promotion of cancers. There are also secondary roles where specific proteins bind to correctly modified histones. Alteration in their structure can also drive the development of tumours. For example ASXL1 (additional sex comb-like 1) is a member of the Polycomb group of proteins that bind modified histones and is mutated in 11% of myelodysplastic syndromes and 43% of chronic myelomonocytic leukaemias [16,17].
DNA methylation
The status of DNA methylation is crucial as one part of the “double lock” of gene expression. As a generalisation, promoters with methylated DNA tend not to be expressed. Clusters of CpGs (the predominant target for DNA methylation) are known as CpG islands and are located at the 5′ ends of many human genes. In tissues, most CpG islands are unmethylated, even when the associated genes are not expressed [18]. However in cancer, DNA hypermethylation occurs at many CpG islands, as well as global DNA hypomethylation (discussed in DNA demethylation section). Promoter methylation is almost always associated with gene-silencing, raising the possibility that aberrant methylation might cause silencing and be part of the transforming process. A potential role in tumorigenesis with a strong mechanistic pathway is suggested when methylation is shown to occur at known tumour suppressor genes. DNA hypermethylation of the cell cycle control gene RB (retinoblastoma) was one of the first epigenetic lesions to be implicated in carcinogenesis. Aberrant methylation occurs in approximately 10% of cases of sporadic unilateral retinoblastoma [19] and is associated with the loss of RB expression [20]. The case of DNA methylation in RB remains one of the strongest arguments in favour of a causal role for aberrant methylation in carcinogenesis as the RB gene is usually active in the precursor cells of tumours and promoter methylation appears to have the same effect as genetic mutation of the gene [21]. Another tumour type in which this occurs is microsatellite unstable colon cancer. Inherited forms of the disease are frequently caused by germline mutation of the DNA mismatch repair (MMR) protein MLH1 [22]. However, approximately 15% of cases of sporadic colon cancer lack MMR gene mutations yet still exhibit microsatellite instability [23]. These cases have methylated MLH1 promoters and lack expression of the gene [24]. In cell lines showing this abnormality, the MLH1 repression is reported to be reversed by treatment with the demethylating agent 5-aza-2′-deoxycytidine [25]. Another in a growing number of examples is the aberrant methylation of the p16INK4a/CDKN2A promoter which has been shown to be present in both human squamous cell carcinomas and their precursor lesions [26], indicating that it occurs in the early stages of neoplastic transformation. Similarly, methylation of GSTP1 (π-class glutathione S-transferase) is an early event in prostate carcinogenesis as it is also found in premalignant lesions [27]. In colorectal carcinogenesis, hypermethylation of a region of chromosome 17p corresponding to the location of the tumour suppressor p53 has been demonstrated to precede its allelic loss, suggesting that methylation may non-randomly mark chromosome regions that are altered during the development of specific tumours [28]. Because of these examples, it has been assumed that aberrant methylation plays a role in malignant transformation [1], particularly when methylation has been demonstrated to occur early in the tumorigenic process. The methylation-induced silencing of tumour suppressor genes may provide cells with a selective advantage over others, either by causing their increased proliferation or resistance to apoptosis. The clonal expansion of these premalignant cells could result in the hyperproliferative phenotype that is characteristic of the early stages of tumorigenesis [29]. Genes such as RB, MLH1 and VHL are methylated in the tumour types in which they are also commonly mutated, suggesting that CpG island hypermethylation may be selected for during tumorigenesis [30].
DNA hypermethylation has been used to subdivide tumour types and distinguish them from non-malignant tissue [31]. Tumour subgroups with high levels of DNA methylation have been designated as having a CpG island methylator phenotype (CIMP) and are predominantly associated with worse prognosis. CIMP was first identified in colorectal tumours where they encompass the majority of sporadic colorectal cancers with MMR-deficiency and MLH1 hypermethylation [32] and are specifically associated with the BRAFV600E mutation [33]. CIMP has subsequently been found to define a subset of glioblastomas [34], acute myeloid leukaemias [35], gastric cancers [36] and ependymomas [37]. CIMP tumours may thus represent distinct subgroups of tumours which otherwise have few genetic alterations, suggesting that drugs targeting the epigenetic machinery may offer novel approaches for therapy.
DNA demethylation
DNA demethylation has also been postulated to contribute to cancer development as despite evidence for regional hypermethylation, global levels of 5-methylcytosine have actually been found to be 5-10% less in tumours compared to normal cells [38,39]. The methylation changes have been suggested to occur specifically between the stages of hyperplasia and benign neoplasia as DNA was found to be significantly hypomethylated in both benign polyps and malignant tissues when compared to normal tissue [40]. Methylation patterns were therefore altered before the lesions became malignant, suggesting that they could be a key event in tumour evolution. The cause of global hypomethylation in cancer is unknown but the outcome, in due course, may be that oncogene expression is increased or other genes important for growth control are deregulated.
Several mechanisms have been proposed for the demethylation of DNA; passive demethylation may occur due to the inability of the maintenance methyltransferase to complete the methylation step that would normally be guided by hemi-methylated DNA post-replication. This is thought to be the case for the maternal pronucleus which undergoes passive demethylation during pre-implantation development, most likely due to sequestration of the oocyte-specific form of DNMT1 (DNMT1o) in the cytoplasm throughout most of cleavage [41]. Conversely, rapid demethylation of the paternal pronucleus appears to be due to the oxidation of 5-methylcytosine to 5-hydroxymethylcytosine by TET3 [42]. There is evidence that the maintenance methyltransferase DNMT1 does not restore methylation to cytosines in the newly synthesised daughter strand if the diagonally opposite cytosine on the parent strand is hydroxymethylated [43], resulting in replication-dependent passive dilution of 5-methylcytosine. Active DNA demethylation in cultured human cells and the adult mouse brain has been demonstrated to involve TET1-catalysed hydroxymethylation, followed by AID/APOBEC-mediated deamination of 5-hydroxymethylcytosine, with the resulting base mismatch being removed by the base excision repair pathway [44]. TET proteins are also able to further oxidise 5-hydroxymethylcytosine to 5-formylcytosine and 5-carboxylcytosine which can be excised by TDG (thymine DNA glycosylase) and repaired by the base excision repair pathway [45,46]. In a study that examined the methylation status of a number of genes when the cells were released from a synchronising block, DNA methylation and demethylation have been shown to cycle approximately every hour ([47,48]. This was a surprise discovery that permits different possibilities including a dynamic, replication-independent response to changes in physiological conditions such as hypoxia. One mechanism that has been proposed is that TDG and components of the base excision repair pathway were recruited to the promoter at the beginning of each transcriptionally productive cycle and a reduction in TDG expression impaired demethylation and reduced transcriptional activity [48].
Contrary to expectations, loss-of-function of the methylcytosine dioxygenase TET2 is predominantly associated with loss of DNA methylation [49]. TET2 is mutated in ~15% of myeloid cancers [14], resulting in impaired hydroxylation [49]. TET2 function is also inhibited by the oncometabolite 2-hydroxyglutarate generated by mutant IDH1 in acute myeloid leukaemias [35]. Downregulation of TET expression has been reported in breast and liver cancers, with reduced levels of 5-hydroxymethylcytosine [50]. DNA methylation patterns may thus be modified by altered expression or activity of epigenetic regulators such as TET.
Histone modifications
Chromatin remodelling is by the so called “histone-code” involving various covalent modifications of the histones such as acetylation, phosphorylation and methylation which have been subject to many studies and their importance is now well accepted [51]. However, the transcriptional state can also be regulated by many chromatin-associated protein complexes that are either involved in enhancing or fine-tuning of the promoter activity and some of these respond to the altered contexts that arise from the histone and DNA modifications. The histone methylation balance on specific residues in particular is crucial for maintaining genome integrity, gene expression and evasion of cancer [10,52,53].
Misregulation of the histone methyltransferases (HMTs) and the histone demethylases (HDMs) has been associated with a variety of cancer types including breast, prostate, lung and brain [54-58]. Specifically, the HMTs and the HDMs play important roles in multiple tissues regulating the methylation status of four lysine residues K4, K9, K27 and K36 on histone H3. Similar to DNA methylation patterns, histone modification patterns have also been used to predict prognosis in multiple cancers. Reduced levels of H3K9ac, H3K9me3 and H4K16ac correlated with recurrence of non-small cell lung cancer [59]. In prostate cancer, lower levels of H3K4me2 and H3K18ac were associated with poor prognosis [60]. Loss of H3K9me3 has been found in core promoter regions of genes in patients with acute myeloid leukaemia. Global H3K9me3 patterns were additionally able to independently predict patient prognosis in acute myeloid leukaemia [61]. These cancers have amplifications, deletions and somatic mutations which all lead to changes in the enzymatic activities of the HMTs and the HDMs. For example, the repressive histone mark trimethylated H3K27 (H3K27me3) is mediated by the catalytic SET domain of EZH2 (enhancer of zeste homologue 2), a protein that forms part of PRC2 (Polycomb repressive complex 2). EZH2 has been reported to be up-regulated in metastatic prostate cancer relative to localised disease or benign prostatic hypertrophy, suggesting a potential involvement in prostate cancer progression [57], and its over-expression also correlates with breast cancer aggressiveness and poor prognosis [56]. The H3K9 methyltransferase G9a reportedly promotes lung cancer invasion and metastasis by silencing Ep-CAM [55]. It is also known that hypoxia in tumours can influence methylation of the histone H3K9 as well as the chromatin remodelling factors by increasing G9a protein stability [62-64]. It should be noted that here, as was the case in consideration of the role of DNA methylation, it is the switching off of gene expression that drives tumour progression. Even though there is an equal possibility for genes that are deleterious to be switched on through changes in the enzymes that alter the epigenome, it would seem that the switching off of genes is the crucial trigger for the progression of tumours through altering the inherent stable balance in cells.
In order to maintain methylation balance, several “histone” demethylases exist which demethylate specific residues, i.e. the reverse of the action of the methyltransferases on different histone residues. There are two classes of HDM families identified which use distinct biochemical reactions to achieve demethylation. Lysine-specific demethylase 1 (LSD1) was the first enzyme identified to demethylate H3K4me1 and H3K4me2, and later found to also demethylate H3K9me1 and H3K9me2 [65,66]. LSD1 is known to utilise flavin adenine dinucleotide (FAD)-dependent amine oxidation reaction for demethylating its substrates and appears to be a very promiscuous protein, having the ability to interact with many proteins and to be involved in multiple biological functions. It should be noted that a potential linkage between metabolic state and gene expression arises from the use of this co-factor and this may be crucial to ensure that it does not destabilise the epigenome. The second class of demethylases includes several proteins that possess a catalytic JMJC domain. These enzymes demethylate histone residues through a dioxygenase reaction which depend on Fe (II) and α-ketoglutarate as cofactors. Again it is interesting to note the crucial role of a metabolite which suggests that the integration of diverse cellular processes and the environment in which the cell resides is decisive on defining the pattern of genes that will be expressed or repressed. It is self evident that some such process is a necessary integrator of cell physiology. Unlike LSD1, JMJC domain-containing demethylases such as JHDM3A have the ability to demethylate trimethylated histone H3K9 and H3K27 residues [67,68]. More recently, deregulation and mutations that affect the enzymatic activity have been found for the HDMs. The H3K27 demethylase JMJD3 is found to be down-regulated in liver and lung cancers [58] while inactivating somatic mutations in the UTX gene are frequently found in multiple tumour types [16]. Knock-out mouse models of some of these HDMs have been generated and result in distinct phenotypes [68,69] including many that are lethal, indicating that proper expression of HDMs is crucial for development [69,70].
Non-histone methylation
Although their name arises from the first substrate that was associated with them, several proteins other than histones have been identified to be methylated by the HMTs and also demethylated by the HDMs [71-73]. The tumour suppressor protein p53 was one of the first non-histone substrates identified to be methylated by several HMTs including Set9, smyd2 and G9a [71,72,74] and also demethylated by LSD1 [66,73]. Depending on which lysine residue is methylated, the transcriptional activity of p53 is specifically regulated. Methylation of non-histone proteins by HMTs has been shown to result in a range of outcomes ranging from functional activation [64,75] to repression [76] or degradation [77]. Hypoxia induces methylation of the chromatin remodelling protein Pontin by stabilising G9a. Methylated Pontin interacts with p300 histone acetyltransferase and HIF-α to hyperactivate a subset of HIF-α target genes [64] (Figure 2). G9a also increases methylation of another chromatin remodelling protein Reptin in a hypoxia-dependent manner. Unlike Pontin methylation, Reptin methylation results in negative regulation of a distinct subset of HIF-α target genes [63]. Two non-histone substrates of EZH2 have been reported recently both of which represses its transcriptional activity. GATA4 is methylated by EZH2 which reduces its interaction with its coactivator p300 [76]. Our group has shown that methylation of the nuclear receptor RORα by EZH2 results in increased polyubiquitination and proteasomal degradation leading to decreased transcriptional activity [77]. In turn this causes the loss of tumour suppressor activity of RORα, which ultimately leads to the development of more aggressive tumours.
Figure 2.
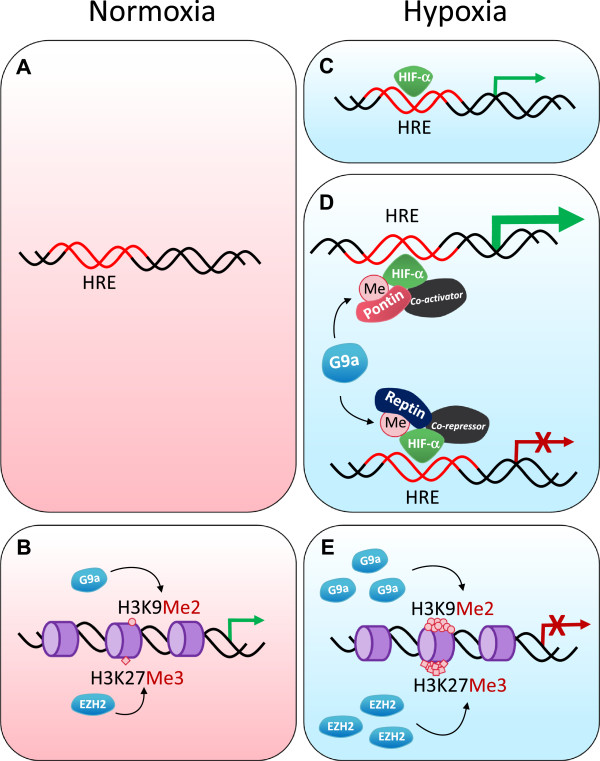
Transcriptional control in normoxia and hypoxia. (A) In normoxia, proteasomal degradation of HIFs prevents HIF-α binding to a hypoxia response element (HRE) and transcriptional activation does not occur. (B) The expression of other genes can be regulated by methylation at histones H3K9 and H3K27 by G9a and EZH2 respectively to maintain homeostasis. (C-E) In hypoxia, gene expression is regulated at multiple layers; (C) HIF-α is stabilised in hypoxia and is able to bind to HREs and activate transcription. (D) The transcriptional activity of HIF-α can be modulated by co-regulators; G9a methylates chromatin remodelling complex proteins such as Reptin and Pontin in hypoxia. Methylated Reptin negatively regulates transcriptional activation by HIF-α at a subset of HIF-α target genes by recruiting a transcriptional co-repressor. Conversely, Pontin methylation potentiates HIF-α-mediated transcription at another distinct subset of HIF-α target promoters by enhancing the recruitment of a transcriptional co-activator. (E) The expression of histone methyltransferases such as G9a and EZH2 is elevated in hypoxia which leads to silencing of tumour suppressors through the hypermethylation of histones H3K9 and H3K27.
It is not only the histone methyltransferases that interact with various non-histone proteins, we have also found that one of the HDMs (JMJD1A) interacts with several proteins, possibly targeting them for demethylation. Therefore, the net status of protein methylation appears to have a broad range of biological functions. Although the dynamic nature of this non-histone methylation appears to be important just as it is the case for histones, demethylation of these proteins has not been studied extensively.
Tumour hypoxia and regulation of gene expression
Tumour hypoxia is an example of how epigenetic reprogramming occurs in cancer progression. In solid tumours, hypoxia occurs as a result of the limitation of oxygen diffusion in avascular primary tumours or their metastases. Persistent hypoxia significantly reduces the efficacy of radiation and chemotherapy and leads to poor outcomes. This is mainly due to increases in pro-survival genes that suppress apoptosis such as c-myc, AMPK, GLUT1 and BNIP3 [78-81] and enhance tumour angiogenesis, EMT (epithelial-to-mesenchymal transition), invasiveness and metastasis [82,83].
Much of tumour hypoxia research has been centred on examining the transcriptional targets of HIFs (hypoxia-inducible factors). HIF-α is a heterodimeric transcription factor that is comprised of an oxygen-regulated α subunit (HIF-1α or HIF-2α) and a constitutively expressed β subunit (HIF-1β) [84,85]. HIF-α is an oxygen-responsive transcription factor that mediates adaptation to hypoxia. Under low oxygen concentrations, HIF-α is stabilised and translocates to the nucleus, leading to specific target gene expression through binding of HIF-1β to a hypoxia response element (HRE). HIF-α regulates hundreds of genes involved in many biological processes including tumour angiogenesis, glycolysis, invasion, metabolism and survival and hence dramatically changes the functioning of cells that reside in these conditions.
Hypoxia not only activates gene expression, but is also involved in gene repression. While some of these genes are known to be transcriptionally downregulated by the recruitment of specific repressors such as DEC1 and Snail [54,86], the contribution of hypoxia-driven epigenetic regulation to gene silencing remains unclear. It has been shown that the expression of G9a and EZH2 are elevated in hypoxic conditions, leading to global hypermethylation of H3K9 and H3K27 respectively. These repressive modifications were increased by hypoxia in the promoter regions of tumour suppressor genes such as RUNX3 and MLH1 which correlated with their silencing, potentially promoting tumour progression [62,87]. We have found that the activity of G9a is deregulated in a tumour setting; methylation of the non-histone proteins Reptin or Pontin in hypoxic conditions negatively or positively regulates the transcription of a particular set of genes involved in tumour metastasis [63,64] (Figure 2).
Conclusions
There has been significant attention in the literature to the accumulated changes in DNA sequences that ultimately give rise to tumours being formed. This has resulted in a rather simple model of tumourgenesis based on accumulated random mutations. In this article we focus on the role of the epigenome as an alternative mode of acquiring dysfunctional cells that result in cancers. Having indicated the necessity to have both the DNA and histone modifications correctly aligned such that the expression of a gene occurs, we point to the plethora of modifying enzymes that can have roles to play. These enzymes with their ability to switch on or off genes have every possibility to change a benign cell into one that is cancerous. Indeed their normal function is to ensure that the correct genes are expressed and that the level of this expression and its timing are all coordinated such that a physiologically normal cell exists. It is clear that any perturbation from this state can have the effect of either making the cell non-viable or to grow to an excessive level and hence become a tumour. A systems-based approach is hence needed to fully integrate all of the available information. What is clear is that both DNA and histone hypermethylation and hypomethylation (and in the case of histones the acetylation state) are associated with malignancy, indicating that balanced epigenetic control is required. Targeting epigenetic modifiers presents novel strategies for cancer therapy in both treating disease and delaying or even preventing resistance to other therapies such as aromatase inhibitors. A recent report found that extended use of aromatase inhibitors resulted in the recruitment of EZH2 and hence increased H3K27me3 of the homeobox gene HOXC10 in breast cancer cells, ultimately leading to HOXC10 methylation and silencing and resistance to aromatase inhibitors [88]. The DNA demethylating agents 5-azacytidine and 5-aza-2′-deoxycytidine (decitabine) and HDAC inhibitors SAHA (vorinostat) and romidepsin have been approved for clinical use with the aim of reversing gene silencing mediated by the DNA methyltransferases or histone deacetylases. These growing numbers of examples point to great complexity and crossover mediated by epigenetic changes between the different inhibitors in clinical use. Given the close interplay between DNA methylation and histone modifications, dual therapy targeting both types of epigenetic modifications may be required. Selected novel drugs targeting components of the epigenetic machinery are currently in pre-clinical or clinical development. Care should, however, be taken in inhibiting epigenetic modifiers due to their off-target effects as illustrated by the non-histone targets for histone modifying enzymes.
Competing interests
The authors declare no financial or non-financial competing interests.
Authors’ contributions
JSL, EB and FG wrote the manuscript. KW generated the figures. All authors read and approve the final manuscript.
Contributor Information
Eva Baxter, Email: Eva.Baxter@qimrberghofer.edu.au.
Karolina Windloch, Email: Karolina.Windloch@qimrberghofer.edu.au.
Frank Gannon, Email: Frank.Gannon@qimrberghofer.edu.au.
Jason S Lee, Email: Jason.Lee@qimrberghofer.edu.au.
References
- Jones PA, Laird PW. Cancer-epigenetics comes of age. Nat Genet. 1999;21(2):163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- Stein R, Razin A, Cedar H. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc Natl Acad Sci. 1982;79(11):3418–3422. doi: 10.1073/pnas.79.11.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JE, Ramsahoye BH, Wo ZG, Okano M, Li E. De novo methylation of MMLV provirus in embryonic stem cells: CpG versus non-CpG methylation. Gene. 2002;289(1–2):41–48. doi: 10.1016/s0378-1119(02)00469-9. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell W, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci. 2000;97(10):5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333(6047):1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications — miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10(7):457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Trends Biochem Sci. 2010;35(11):618–626. doi: 10.1016/j.tibs.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ. et al. DNMT3A mutations in acute myeloid leukemia. New Engl J Med. 2010;363(25):2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Valle VD, James C, Trannoy S, Massé A, Kosmider O, Le Couedic J-P, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, Romana SP, Dessen P, Soulier J, Viguié F, Fontenay M, Vainchenker W, Ber OA. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, Perna F, Pandey S, Madzo J, Song C, Dai Q, He C, Ibrahim S, Beran M, Zavadil J, Nimer SD, Melnick A, Godley LA, Aifantis I, Levine RL. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O’Meara S, Teague J, Butler A, Hinton J, Latimer C, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Cole J, Forbes S, Jia M, Jones D, Kok CY, Leroy C, Lin M-L, McBride DJ, Maddison M, Maquire S, McLay K, Menz A. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41(5):521–523. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsi-Boyer V, Trouplin V, Adélaïde J, Bonansea J, Cervera N, Carbuccia N, Lagarde A, Prebet T, Nezri M, Sainty D, Olschwang S, Xerri L, Chaffanet M, Mozziconacci M-J, Vey N, Birnbaum D. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol. 2009;145(6):788–800. doi: 10.1111/j.1365-2141.2009.07697.x. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Greger V, Debus N, Lohmann D, Hopping W, Passarge E, Horsthemke B, Debus N, Lohmann D, Hopping W, Passarge E, Horsthemke B. Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum Genet. 1994;94(5):491–496. doi: 10.1007/BF00211013. [DOI] [PubMed] [Google Scholar]
- Ohtani-Fujita N, Fujita T, Aoike A, Osifchin N, Robbins P, Sakai T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene. 1993;8(4):1063–1067. [PubMed] [Google Scholar]
- Stirzaker C, Millar DS, Paul CL, Warnecke PM, Harrison J, Vincent PC, Frommer M, Clark SJ, Millar DS, Paul CL, Warnecke PM, Harrison J, Vincent PC, Frommer M, Clark SJ. Extensive DNA methylation spanning the Rb promoter in retinoblastoma tumors. Cancer Res. 1997;57(11):2229–2237. [PubMed] [Google Scholar]
- Papadopoulos N, Nicolaides N, Wei Y, Ruben S, Carter K, Rosen C, Haseltine W, Fleischmann R, Fraser C, Adams M, Venter C, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomäki P, Jukka-Pekka Mecklin J, de la Chapell A. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263(5153):1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Konishi F, Masubuchi S, Shitoh K, Nagai H, Tsukamoto T. Densely methylated MLH1 promoter correlates with decreased mRNA expression in sporadic colorectal cancers. Genes Chr Cancer. 2002;35(1):1–10. doi: 10.1002/gcc.10100. [DOI] [PubMed] [Google Scholar]
- Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–811. [PubMed] [Google Scholar]
- Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa J-PJ, Markowitz S, Willson JKV, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci. 1998;95(12):6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, Baylin SB, Herman JG. Aberrant methylation of p16INK4a is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci. 1998;95(20):11891–11896. doi: 10.1073/pnas.95.20.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci. 1994;91(24):11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makos M, Nelkin BD, Lerman MI, Latif F, Zbar B, Baylin SB. Distinct hypermethylation patterns occur at altered chromosome loci in human lung and colon cancer. Proc Natl Acad Sci. 1992;89(5):1929–1933. doi: 10.1073/pnas.89.5.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Nelkin BD, Lerman MI, Latif F, Zbar B, Baylin SB. Genetic alterations during colorectal-tumor development. New Engl J Med. 1988;319(9):525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Melki J. DNA methylation and gene silencing in cancer: which is the guilty party? Oncogene. 2002;21:5380–5387. doi: 10.1038/sj.onc.1205598. [DOI] [PubMed] [Google Scholar]
- Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S, Yeh R-F, Wrensch MR, Wiemels JL, Karagas MR, Bueno R, Sugarbaker DJ, Nelson HH, Wiencke JK, Kelsey KT. Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res. 2009;69(15):6315–6321. doi: 10.1158/0008-5472.CAN-09-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa J-PJ. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci. 1999;96(15):8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RGW, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJM, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouridis H, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Löwenberg B, Licht JD, Godley LA, Delwel R, Valk PJM, Thompson CB, Levine RL, Melnick A. Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci Transl Med. 2012;4(156):156ra140. doi: 10.1126/scitranslmed.3004504. [DOI] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jager N, Jones DTW, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJH, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–450. doi: 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988;48(5):1159–1161. [PubMed] [Google Scholar]
- Gama-Sosa M, Slagel V, Trewyn R, Oxenhandler R, Kuo K, Gehrke C, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucl Acids Res. 1983;11(19):6883–6894. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goelz S, Vogelstein B, Hamilton S, Feinberg A. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228(4696):187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- Mertineit C, Yoder JA, Taketo T, Laird DW, Trasler JM, Bestor TH. Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development. 1998;125(5):889–897. doi: 10.1242/dev.125.5.889. [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67(3):946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming G-l, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song C-X, Zhang K, He C, Xu G-L. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286(41):35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature. 2008;452(7183):112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452(7183):45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu J, Xu ZD, Zhu HG, Ling ZQ, Ye D, Guan KL, Xiong Y. Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene. 2013;32(5):663–669. doi: 10.1038/onc.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12(6):1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP. G9a interacts with snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Invest. 2012;122(4):1469–1486. doi: 10.1172/JCI57349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M-W, Hua K-T, Kao H-J, Chi C-C, Wei L-H, Johansson G, Shiah S-G, Chen PS, Jeng Y-M, Cheng T-Y, Lai T-C, Chang J-S, Jan Y-H, Chien M-H, Yang C-J, Huang M-S, Hsiao M, Kuo M-L. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010;70(20):7830–7840. doi: 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RGAB, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci. 2003;100(20):11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Agger K, Cloos PAC, Rudkjær L, Williams K, Andersen G, Christensen J, Helin K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23(10):1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JS, Kim YS, Kim DK, Park SI, Jang SJ. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathol Intl. 2012;62(3):182–190. doi: 10.1111/j.1440-1827.2011.02776.x. [DOI] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435(7046):1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- Müller-Tidow C, Klein H-U, Hascher A, Isken F, Tickenbrock L, Thoennissen N, Agrawal-Singh S, Tschanter P, Disselhoff C, Wang Y, Becker A, Thiede C, Ehninger G, Zur Stadt U, Koschmieder S, Seidl M, Müller FU, Schmitz W, Schlenke P, McClelland M, Berdel WE, Dugas M, Serve H. Study A L. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood. 2010;116(18):3564–3571. doi: 10.1182/blood-2009-09-240978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M. Stress induces dimethylated histone cells. Cancer Res. 2006;66(18):9009–9016. doi: 10.1158/0008-5472.CAN-06-0101. [DOI] [PubMed] [Google Scholar]
- Lee JS, Kim Y, Kim IS, Kim B, Choi HJ, Lee JM, Shin H-JR, Kim JH, Kim J-Y, Seo S-B, Lee H, Binda O, Gozani O, Semenza GL, Kim M, Kim KI, Hwang D, Baek SH. Negative regulation of hypoxic responses via induced reptin methylation. Mol Cell. 2010;39(1):71–85. doi: 10.1016/j.molcel.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Kim Y, Bhin J, Shin H-JR, Nam HJ, Lee SH, Yoon J-B, Binda O, Gozani O, Hwang D, Baek SH, Kim Y, Bhin J, Shin H-JR, Nam HJ, Lee SH, Yoon J-B, Binda O, Gozani O, Hwang D, Baek SH. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci. 2011;108(33):13510–13515. doi: 10.1073/pnas.1106106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Huang Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci. 2007;104(19):8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BMR, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448(7149):87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature. 2007;450(7166):119–123. doi: 10.1038/nature06236. [DOI] [PubMed] [Google Scholar]
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, Chen T. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41(1):125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Yamazaki Y, Katoh-Fukui Y, Tsuchiya R, Kondo S, Motoyama J, Higashinakagawa T. Gene trap capture of a novel mouse gene, jumonji, required for neural tube formation. Genes Dev. 1995;9(10):1211–1222. doi: 10.1101/gad.9.10.1211. [DOI] [PubMed] [Google Scholar]
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D. Regulation of p53 activity through lysine methylation. Nature. 2004;432(7015):353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444(7119):629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449(7158):105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Huang J, Dorsey J, Chuikov S, Zhang X, Jenuwein T, Reinberg D, Berger SL. G9a and Glp methylate Lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285(13):9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, Lee C, Joo KM, Rich JN, Nam D-H, Lee J. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, Pu WT. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26(1):37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Lee JS, Kim H, Kim K, Park H, Kim J-Y, Seung H, Lee SH, Kim LKS, Kim J, Lee M, Chung CH, Seo S-B, Yoon J-B, Ko E, Noh D-Y, Keun I, Kim KI, Kim KK, Baek SH. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell. 2012;48(4):572–586. doi: 10.1016/j.molcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borger DR, Gavrilescu LC, Bucur MC, Ivan M, Decaprio JA. AMP-activated protein kinase is essential for survival in chronic hypoxia. Biochem Biophys Res Commun. 2008;370(2):230–234. doi: 10.1016/j.bbrc.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci. 1997;94(9):4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov SV, Salnikow K, Ivanova AV, Bai L, Lerman MI. Hypoxic repression of STAT1 and its downstream genes by a pVHL/HIF-1 target DEC1/STRA13. Oncogene. 2007;26(6):802–812. doi: 10.1038/sj.onc.1209842. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim J, Kim WH, Lee YM. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene. 2008;28(2):184–194. doi: 10.1038/onc.2008.377. [DOI] [PubMed] [Google Scholar]
- Pathiraja TN, Nayak SR, Xi Y, Jiang S, Garee JP, Edwards DP, Lee AV, Chen J, Shea MJ, Santen RJ, Gannon F, Kangaspeska S, Jelinek J, Issa J-PJ, Richer JK, Elias A, McIlroy M, Young LS, Davidson NE, Schiff R, Li W, Oesterreich S. Epigenetic reprogramming of HOXC10 in endocrine-resistant breast cancer. Sci Transl Med. 2014;6(229):229ra41. doi: 10.1126/scitranslmed.3008326. [DOI] [PMC free article] [PubMed] [Google Scholar]