

Abstract
Purpose of review
Nephronophthisis (NPHP) represents an autosomal recessive cystic kidney disease and is one of the most common genetic disorders causing end-stage renal disease (ESRD) in children and adolescents. NPHP is a genetically heterogenous disorder with twenty identified genes. NPHP occurs as an isolated kidney disease but approxmiately 15% of NPHP patients have additional extrarenal symptoms affecting other organs (e.g. eyes, liver, bones, and CNS). The pleiotropy in NPHP is explained by the finding that almost all NPHP gene products share expression in primary cilia, a sensory organelle present in most mammalian cells. If extrarenal symptoms are present in addition to NPHP, these disorders are classified as NPHP-related ciliopathies (NPHP-RC). This review provides an update about the recent advances in the field of NPHP-RC.
Recent findings
The identification of novel disease-causing genes has improved our understanding of pathomechanisms contributing to NPHP-RC. Multiple interactions between different NPHP-RC gene products have been published and outline the interconnectivity of the affected proteins and shared pathways.
Summary
The significance of recently identified genes for NPHP-RC is discussed and the complex role and interaction of NPHP proteins in ciliary function and cellular signaling pathways is highlighted.
Keywords: Nephronophthisis, ciliopathy, Jeune syndrome, Joubert syndrome
INTRODUCTION
Nephronophthisis (NPHP) is a tubulo-interstitial, autosomal recessive cystic kidney disease (1–3). The name derives from the Greek and means “vanishing of the kidney” which relates to the progressively smaller kidney size with advancing kidney disease (4, 5). NPHP is characterized by polyuria, polydipsia, secondary enuresis, and anemia. Clinically, we distinguish three different forms of NPHP. Juvenile NPHP or NPHP type 1 is the most common form of NPHP, and is characterized by end-stage renal disease (ESRD) at a mean age of 13 years (6). Symptoms in juvenile NPHP may occur as early as 6 years of age. Infantile NPHP or NPHP type 2 is very rare with onset of ESRD prior to 4 years of age (7). Adolescent NPHP or NPHP type 3 has a mean age of onset of ESRD of 19 years (8). In early stages of chronic kidney disease (CKD), symptoms with NPHP are subtle. Later, severe anemia, growth retardation and hypertension are seen as part of end-stage renal disease (ESRD) (9). In the early stages of NPHP, renal ultrasound is normal or shows unspecific changes with increased renal echogenicity (10, 11). With advancing kidney disease smaller, hyperechogenic kidneys with cortico-medullary cysts and poor cortico-medullary differentiation are described (Fig. 1). The histopathology in NPHP is not disease-specific and is characterized by tubulo-interstitial alterations such as tubular atrophy, thickening or thinning of the tubular membrane, interstitial fibrosis and inflammation (12) (Fig. 2). In contrast to other forms of NPHP, infantile or type 2 NPHP has enlarged kidneys in renal ultrasound and histopathology shows an overlap of features of NPHP (interstitial fibrosis, tubular atrophy) and polycystic kidney disease (widespread cyst development and enlarged kidneys) (7, 13). Clinical presentation of NPHP is frequently unspecific and other kidney diseases (e.g. primary hyperoxaluria) were shown to result in similar phenotypes as NPHP (14).
Fig. 1. Imaging findings by renal ultrasound in NPHP.
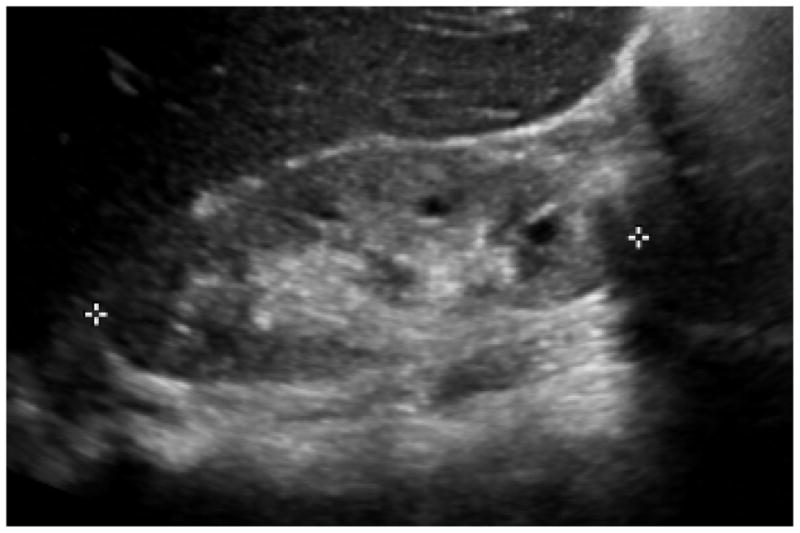
With advancing kidney disease small kidneys are detected bilaterally. There is development of hyperechogenic kidneys (compare to lower echogenicity of the liver) with cysts along the cortico-medullary border.
Fig. 2. Renal histopathology in NPHP.

Characteristically, histopathology in NPHP is characterized by tubular cysts, tubular atrophy, interstitial and periglomerular fibrosis, tubulo-interstitial infiltration, and tubular basement membrane disruption. Periodic acid-Schiff (PAS) staining, magnification 20x.
The diagnosis of NPHP is made either by renal biopsy or genetic testing (4). NPHP is genetically heterogenous. Since the discovery of the first NPHP1 gene in 1997, twenty different NPHP genes, which encode Nephrocystins, have been identified (Table 1) (15–38).
Table 1.
Summary of NPHP1-NPHP18 and NHPH1L and NPHP2L genes, gene products, chromosomal localization, phenotypes, extrarenal symptoms, and interaction partners.
| Gene (protein) | Chromosome | Phenotype (median age at ESRD) | Extrarenal symptoms | Interaction partners |
|---|---|---|---|---|
| NPHP1 (Nephrocystin-1) | 2q13 | NPHP (13yrs) | RP (10%), OMA (2%), JBTS (rarely) | Inversin, nephrocystin-3, nephrocystin-4, filamin A and B, tensin, β-tubulin, PTK2B |
| NPHP2/INVS (Inversin) | 9q31 | Infantile NPHP (<4yrs) | RP (10%), LF, situs inversus, CHD | Nephrocystin-1, calmodulin, catenins, β-tubulin, APC2 |
| NPHP3 (Nephrocystin-3) | 3q22 | Infantile and adolescent NPHP | LF, RP (10%), situs inversus, MKS, CHD | Nephrocystin-1 |
| NPHP4 (Nephrocystin-4) | 1p36 | NPHP (21 yrs) | RP (10%), OMA, LF | Nephrocystin-1, BCAR1, PTK2B |
| NPHP5/IQCB1 (Nephrocystin-5) | 3q21 | NPHP (13 years) | Early-onset RP | Calmodulin, RPGR, nephrocystin-6 |
| NPHP6/CEP290 (Nephrocystin-6/CEP290) | 12q21 | NPHP | JBTS, MKS | ATF4, nephrocystin-5, CC2D2A |
| NPHP7/GLIS2 (Nephrocystin-7/GLIS2) | 16p | NPHP | – | – |
| NPHP8/RPGRIP1L (Nephrocystin-8/RPGRIP1L) | 16q | NPHP | JBTS, MKS | Nephrocystin-1 |
| NPHP9/NEK8 (Nephrocystin-9/NEK8) | 17q11 | Infantile NPHP | – | – |
| NPHP10/SDCCAG8 (Nephrocystin-10/SDCCAG8) | 1q43 | Juvenile NPHP | RP (SLS), BBS-like | OFD1 |
| TMEM67/MKS3/NPHP11 (Nephrocystin-11/Meckelin) | 8q22.1 | NPHP | JBTS, MKS, LF | MKS1, nephrocystin-1, nephrocystin-4, nephrocystin-6, nesprin-2, TMEM216 |
| TTC21B//JBTS11/NPHP12 (Nephrocystin-12/IFT139) | 2q24.3 | Early onset NPHP, juvenile NPHP | JATD, MKS, JBTS, BBS- like | ciliopathy modifier |
| WDR19/NPHP13 (Nephrocystin-13/IFT144) | 4p14 | NPHP | JATD, SBS, CED, RP, Caroli, BBS-like | |
| ZNF423/NPHP14 (Nephrocystin-14/ZNF423) | 16q12.1 | Infantile NPHP, PKD | JBTS, situs inversus | PARP1, nephrocystin-6, |
| CEP164/NPHP15 (Nephrocystin-15 centrosomal protein 164 kDa) | 11q23.3 | NPHP (8 years) | RP, JBTS, LF, obesity | ATRIP, CCDC92, TTBK2, nephrocystin-3, nephrocystin-4, Dvl3 |
| ANKS6/NPHP16 (Nephrocystin-16/ANKS6) | 9q22.33 | NPHP, PKD | LF, situs inversus, cardiovascular abnorm. | INVS, nephrocystin-3, NEK8, HIF1AN, NEK7, BICC1 |
| IFT172/NPHP17 (Nephrocystin-17/IFT172) | 2p23.3 | NPHP | JATD, MZSDS, JBTS | IFT140, IFT80 |
| CEP83/NPHP18 (Nephrocystin-18/centrosomal protein 83 kDa) | 12q22 | Early-onset NPHP (3 years) | learning disability, hydrocephalus, LF | CEP164, IFT20 |
| NPHP1L/XPNPEP3 (nephrocystin-1L/XPNPEP3) | 22q13 | NPHP | Cardiomyopathy, seizures | cleaves LRRC50, ALMS1, nephrocystin-6 |
| NPHP2L/SLC41A1 (nephrocystin-2L/SLC41A1) | 1q32.1 | NPHP | bronchiectasis |
ATF4, activating transcription factor 4; APC2, anaphase-promoting complex 2; BCAR1, breast cancer anti-estrogen resistance 1; CAD, Cranioectodermal dysplasia; CC2D2A, coiled-coil and C2 domain containing 2A; CHD, congenital heart disease; JATD, Jeune asphyxiating thoracic dysplasia; JBTS, Joubert syndrome; LF, liver fibrosis; MKS, Meckel-Gruber syndrome; OMA, oculomotor apraxia; PTK2B, protein tyrosine kinase 2B; RP, retinitis pigmentosa; RPGR, retinitis pigmentosa GTPase regulator; SBS, Sensenbrenner syndrome
The most definitive diagnosis is made with the detection of compound heterozygous or homozygous mutations in a gene contributing to NPHP. Unfortunately, NPHP gene mutations are only detected in about one third of affected patients, while the responsible genes for the remaining two thirds are still unknown (39). The most common NPHP mutation is a homozygous deletion of NPHP1, which is identified in approximately 20% of NPHP patients with a NPHP gene mutation (39). About 10–15% of NPHP patients have additional extrarenal symptoms (40). Mutations in specific NPHP genes predispose to a higher risk of extrarenal NPHP manifestations (Table 1) (3, 40).
EXTRARENAL PHENOTYPES WITH NEPHRONOPHTHISIS
A summary of extrarenal phenotypes associated with NPHP is listed in Table 2. Ophthalmological phenotypes
Table 2.
Extrarenal manifestations associated with NPHP and resulting syndromes associated with NPHP mutations. (Reprinted with permission from 3).
| Ophthalmologic disorder | Syndrome |
| Retinitis pigmentosa | Senior-Løken syndrome (SLSN) |
| Arima syndrome (cerebro-oculo-hepato-renal syndrome) | |
| Alstrom (RP, obesity, DM type 2, hearing impairment) | |
| RHYNS (RP, hypopituitarism, skeletal dysplasia) | |
| Oculomotor apraxia | Cogan syndrome |
| Nystagmus | Joubert syndrome/Joubert syndrome related disorders |
| Coloboma | Joubert syndrome/Joubert syndrome related disorders |
| Neurological disorder | |
| Encephalocele | Meckel-Gruber syndrome (occipital encephalocele, NPHP) |
| Vermis aplasia | Joubert syndrome/Joubert syndrome related disorders |
| Hypopituitarism | RHYNS (RP, hypopituitarism, skeletal dysplasia) |
| Hepatic disorder | |
| Liver fibrosis | Boichis syndrome |
| Meckel-Gruber syndrome (occipital encephaolocele, NPHP) | |
| Arima syndrome (cerebro-oculo-hepato-renal syndrome) | |
| Joubert syndrome/Joubert syndrome related disorders | |
| Skeletal disorder | |
| Short ribs | Jeune syndrome/asphyxiating thoracic dystrophy |
| Cone-shaped epiphysis | Mainzer-Saldino syndrome |
| Postaxial polydactyly | Joubert syndrome/Joubert syndrome related disorders |
| Bardet-Biedl syndrome (NPHP, RP, obesity, deafness) | |
| Ellis van Creveld | |
| Skeletal abnormalities | Sensenbrenner syndrome/cranioectodermal dysplasia |
| Ellis van Creveld | |
| Others | |
| Situs inversus | |
| Cardiac malformation | |
| Bronchiectasis | |
| Ulcerative colitis | |
RP, retinitis pigmentosa/retinal degeneration; DM, diabetes mellitus; NPHP, nephronophthisis
The eyes are the most frequently affected extrarenal organ with NPHP with two different ophthalmological phenotypes (40). The less common form of eye involvement is called oculomotor apraxia type Cogan [OMIM%257550], which is characterized by impaired horizontal gaze and nystagmus (41). The more frequent form of eye involvement is named retinitis pigmentosa (40). Early-onset retinitis pigmentosa resembles Leber’s congenital amaurosis, whereas late-onset retinitis pigmentosa is characterized by progressive vision loss and night blindness (42, 43). About 10–15% of NPHP patients have retinal degeneration. NPHP associated with retinitis pigmentosa is called Senior-Løken syndrome [OMIM#2669000].
Joubert syndrome
Features of Joubert syndrome [OMIM%213300] are developmental delay, neonatal breathing abnormalities, muscular hypotonia, and ataxia (44). MRI imaging reveals the characteristic molar tooth sign which is due to cerebellar vermis aplasia/hypoplasia (Fig. 3). Prevalence of Joubert syndrome is approximately 1 in 100,000 livebirths and about 20–30% of these patients develop NPHP. In addition to pure Joubert syndrome other clinical subgroups are described with involvement of other organs as liver (congenital liver fibrosis), eye (coloboma, retinal dystrophy), and bones (polydactyly, bone shortening). A combination of multiple organ involvement is possible as for example with cerebello-ocolo-renal (COR) presentation. More than 20 genes have been identified which contribute to Joubert syndrome and about a third of them also cause NPHP (44).
Fig. 3. Magnetic resonance imaging (MRI) of the brain in Joubert syndrome.
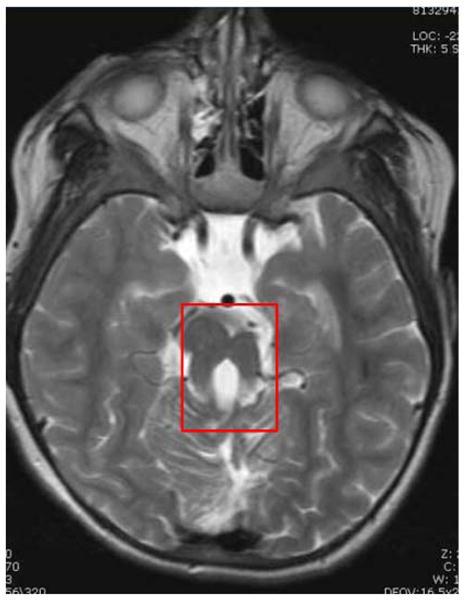
The “molar tooth sign” develops due to cerebellar vermis hypoplasia and can easily be detected by MRI (boxed). The “molar tooth sign” has become the classical neuroradiological indicator for Joubert syndrome.
Meckel-Gruber syndrome
Meckel-Gruber syndrome [OMIM#249000] is an extreme form of multiorgan involvement with NPHP. The affected patients frequently die in the perinatal period and are characterized by an occipital encephalocele, polydactyly, liver fibrosis and cystic kidney disease (45).
Liver fibrosis
Liver fibrosis occurs together with different syndromes (e.g. Arima, Meckel and Boichis syndrome) which may also have cystic kidney disease or isolated with NPHP (46).
Skeletal abnormalities
The most common forms of bone involvement with NPHP are Jeune syndrome [OMIM%208500], cranio-ectodermal dysplasia (aka Sennsenbrenner syndrome) [OMIM#218330] and Mainzer-Saldino syndrome [OMIM#266920] (47, 48). Jeune syndrome is characterized by rib cage narrowing, polydactyly, and brachydactyly; and can include cystic kidney disease, liver disease and retinal degeneration. The rib cage narrowing in these patients can be quite severe thus causing respiratory failure. Jeune syndrome is also known as asphyxiating thoracic dystrophy (49). Jeune syndrome and cranio-ectodermal dysplasia have overlapping phenotypes with rib cage narrowing (usually milder than in Jeune syndrome), polydactyly, and brachydactyly (48). More prominent in patients with cranio-ectodermal dysplasia are dolichocephaly, pectus excavatum, and in particular ectodermal involvement with delayed tooth eruption, skin laxity, sparse, fine hair and slow growing nails. Features of Mainzer-Saldino syndrome are phalangeal cone-shaped epiphyses, retinal dystrophy and NPHP.
Situs inverus and congential heart defects
Situs invertus and congential heart defects occur mostly in patients with infantile NPHP. The most common congenital heart defect in this setting is ventricular septal defect (17).
So how can all these different organs be associated with NPHP? Below, I will outline some of the different mechanisms which may explain the variety of associated symptoms with NPHP.
Primary cilia and ciliopathies
With the identification of Nephrocystin-2/Inversin and its colocalization with Nephrocystin-1 and β-tubulin in primary cilia, the pathogenesis of NPHP was linked for the first time to ciliary dysfunction. NPHP was included in a newly formed and growing class of diseases categorized as ciliopathies which also includes autosomal-dominant and autosomal-recessive polycystic kidney disease, Bardet-Biedl, Jeune, Joubert and Meckel-Gruber syndromes (50). They are caused by defects in ciliary function or structure. As these patients frequently present with NPHP, they are classified as NPHP-related ciliopathies (NPHP-RC).
Almost all Nephrocystins so far are expressed in primary cilia of renal epithelial cells and other cell types (40, 50). This led to the unifying theory of cystogenesis which states that most proteins altered in cystic kidney disease are expressed in primary cilia, basal body or centrosomes of renal epithelial cells (50). In contrast to motile cilia which contribute to locomotion and fluid movements, primary cilia are involved in sensation. The structure of the ciliary axoneme for primary cilia contains nine outer microtubule doublets (9+0) and motile cilia contain an additional inner microtubule doublet (9+2) (51). Primary cilia are present in almost every vertebrate cell. Primary cilia are highly evolutionary conserved sensory organelles which can detect flow, optic, osmotic, chemo, or olfactory stimuli among others (50, 52). Nephrocystin-1 and 4 homologues were described in ciliated neurons of C. elegans (53). In the mammalian kidney, primary cilia protrude antenna-like from the apical membrane of renal tubular cells into the lumen where they are thought to function as flow-, osmo- or chemosensors. Cilia are dependent on intra-ciliary protein transport as there is no protein synthesis inside cilia (Fig. 4) Required proteins are synthesized in the cytoplasm and pass the transition zone (TZ), which separates the cytoplasm from the ciliary axoneme. Inside the axoneme, they are transported along microtubules inside the cilium, a mechanism called intraflagellar transport (IFT) (48, 54). Cargo-rafts are moved either by kinesin motors (anterograde transport) or dynein motors (retrograde transport) (55). At the root of the cilium is the basal body from which the cilium is assembled (50). The basal body derives from the mother centriole.
Fig. 4. Model of ciliary transport and architecture.
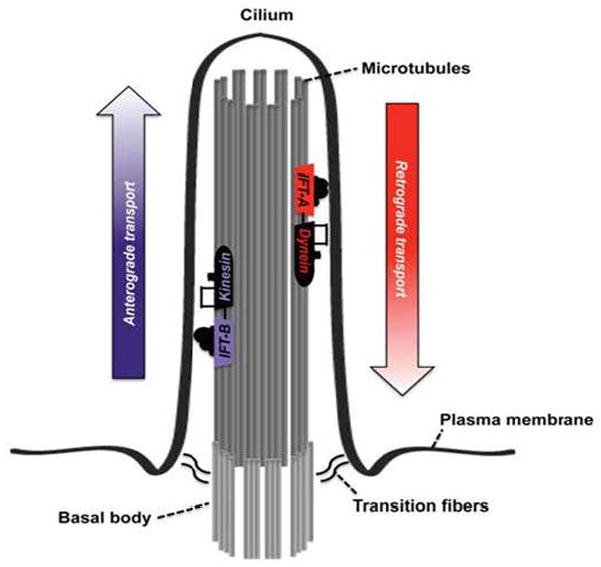
Primary cilia protrude from the apical plasma membrane of the cell into the tubular lumen. At the base of the primary cilium is the basal body located from which the cilium is initially assembled. The inside of the cilium contains the ciliary axoneme. Assembly and function of primary cilia depend on ciliary transport, also called intraflagellar transport (IFT). Transport occurs in both directions of the axoneme along microtubules. IFT from the ciliary base to the ciliary tip is called anterograde transport and is driven by the kinesin-2 motor and IFT-B complex. IFT from the ciliary tip back to the base is called retrograde IFT and is controlled by dynein-2 motor and IFT-A complex. The IFT-A complex consists of six different proteins whereas the IFT-B complex contains at least 14 different proteins. Mutations in all members of the IFT-A complex result in skeletal dysplasia with NPHP (reprinted with permission from 48).
Primary cilia are thought to link the extracellular stimuli via the primary cilium to a variety of different cellular mechanisms including epithelial cell polarity, planar cell polarity, cell-cycle control, and different signaling pathways (40, 50). Primary cilia are present in nearly all human cells, thus explaining the diverse organ involvement. For example, Inversin and Nephrocystin-3 are expressed during embryogenesis in the ventral node, which is critical for left-right axis determination, thus providing an association with situs inversus (17).
Another prominent organ with a cilia-related structure is the photoreceptor thus possibly explaining the association of retinitis pigmentosa with NPHP (42, 43). The photoreceptor contains the rod outer and rod inner segments which are connected by a structure called the connecting cilium (Fig. 5). The connecting cilium of the photoreceptor represents the structural counterpart of the primary cilium, and some authors call the rod outer segment the photosensitive cilium. The rod outer segment shares features with the primary cilium: both have sensory function and lack biosynthetic activity (43). Rhodopsin is synthesized in the rod inner segment and transported via the connecting cilium to the rod outer segment where it is sequestered in the light-sensing membranes. Accumulation of rhodopsin, transducin and other photo-transducing proteins in the rod inner segment is associated with apoptosis of the photoreceptor. Nephrocystin-5, nephrocystin-6 and nephrocystin-10 are the most prominent Nephrocystins being expressed in the connecting cilium (21, 27, 56). Dysfunctional Nephrocystin expression in the connecting cilium of these patients may contribute to the high frequency of retinitis pigmentosa in nephrocystin-5 and nephrocystin-10 patients with 100% and 80%, respectively. Nephrocystin-5 and nephrocystin-6 were also found to directly interact and both interact with another crucial protein named retinitis pigmentosa GTPase regulator (GPGR) (21, 56). Mutations in GPGR are responsible for X-linked retinitis pigmentosa. Hypomorphic NPHP6/CEP290 mutations were found in approximately 20% of humans with Leber’s congenital amaurosis (57). Other Nephrocystins detected in the photoreceptor include Nephrocystin-1, -8, -11 and -12 (29, 58–60). The sequence variant A229T in NPHP8/RPGRIP1L was also found to be associated with Leber’s congenital amaurosis (61).
Fig. 5. The photoreceptor contains the connecting cilium.
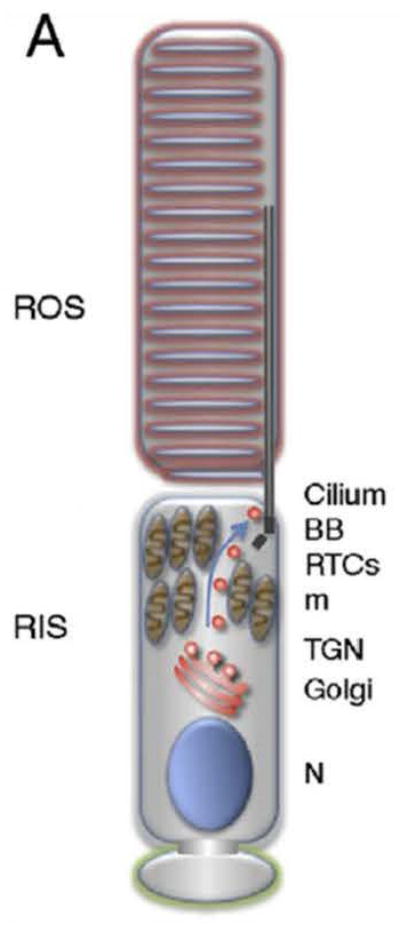
The photoreceptor consist of the rod outer (ROS) and rod inner segments (RIS), connected by the connecting cilium. Rhodopsin and other phototransducing substances are synthesized in the RIS as the ROS lacks any biosynthetic activity. Rhodopsin is sequestered in the light-sensing membranes of the ROS. Several Nephrocystins are localized in the ROS and the connecting cilium. BB, basal body; m, mitochrondria; N, nucleus; RTC, rhodopsin transport carrier; TGN, trans-golgi network (reprinted with permission from 43).
Other extrarenal phenotypes such as hepatic fibrosis, could also possibly be explained by expression of primary cilia in cholangiocytes (46). Nephrocystin-3 is expressed in the biliary tract and liver during development and cilia based signaling was found to be required for normal biliary and portal tract development (18). NPHP11/TMEM67 is the most commonly involved nephrocystin in patients with liver fibrosis (28). Liver fibrosis is very prevalent in patients carrying two missense mutations in NPHP11/TMEM67.
Over the last few years, exciting insights were gained regarding the pathogenesis of skeletal disorders associated with NPHP (47, 48). The identified genes in these multiorgan disorders involving bone and kidney were consistent with the ciliary hypothesis. Primary cilia were also found in chondrocytes (50, 62). Interestingly, all identified genes in patients with skeletal and renal involvement were members of the intraflagellar transport. Mutations in two components of the ciliary anterograde (Complex B: IFT80, Nephrocystin-17/IFT172) and all six components of the retrograde transport (Complex A: IFT43, IFT121, IFT122, IFT139, IFT140, IFT144) were published for Jeune, cranio-ectodermal and Mainzer-Saldino syndrome (Fig. 4, Table 3) (29, 31, 35, 63–69). It is widely understood that, NPHP-RC is mostly caused by Nephrocystins located in the TZ, whereas skeletal abnormalities with NPHP are caused by defective IFT.
Table 3.
Summary of affected members of the IFT in ciliary skeletopathies including anterograde transport (complex B) and retrograde transport (complex A). Affected protein is shown, followed by corresponding gene name in parenthesis. Mutated genes for JADT, CED and MZSDS are shown.
| Retrograde transport (IFT-A complex) | JADT | other SRP | CED | MZSDS | Renal involvement (NPHP) | References |
|---|---|---|---|---|---|---|
| IFT43 | + | + | 64 | |||
| IFT121 (WDR35) | + | + | 65 | |||
| IFT122 (WDR10) | + | + | 66 | |||
| IFT139 (TTC21B/NPHP12) | + | + | 29 | |||
| IFT140 | + | + | + | 67,68 | ||
| IFT144 (WDR19/NPHP13) | + | + | + | 30,31 | ||
| Anterograde transport (IFT-B complex) | ||||||
| IFT80 | + | − | 63 | |||
| IFT172 (NPHP17) | + | + | + | 35 |
CED, Cranioectodermal dysplasia; JATD, Jeune asphyxiating thoracic dysplasia; MZSDS, Mainzer-Saldino syndrome; other SRP, other short rib polydactyly syndromes except Jeune syndrome
Protein modules
Different nephrocystins are known to interact which each other and they are known to share colocalization in the primary cilium. In addition, there is awareness that other proteins involved with other ciliopathies are organized in large functional protein modules, as for example the BBSome (70). Recent data from an extensive proteomics approach applying copurification showed that at least three different Nephrocystin modules were identified with Nephrocystins-1-4-8, Nephrocystins-5-6 and MKS-1-6 modules (71) (Fig. 6). The Nephrocystin-1-4-8 module is mostly expressed in the ciliary TZ and accumulates at cell-cell contacts. This module is thought to organize specialized structures at the apical surface and may participate in epithelial morphogenesis. The Nephrocystin-5-6 module also binds to the gene product of ATXN10, which was found to be mutated in JBTS with NPHP (71). This module is localized in the basal body and is crucial for ciliogenesis. The MKS-1-6 module binds to Tectonic 2 (TCTN2) (71). This module contains gene products that are involved in MKS or JBTS by causing impaired ciliogenesis and neural tube defects. Dysfunctional ciliogenesis disturbs Hh signaling which results in embryonic neural tube dorsalization and polydactyly. The last module consists of Nephrocystins-2, -3 and -9, is expressed along the entire ciliary axoneme and bridges the three previous modules.
Fig. 6. Nephrocystins form different protein modules are expressed at different sites along the ciliary axoneme.
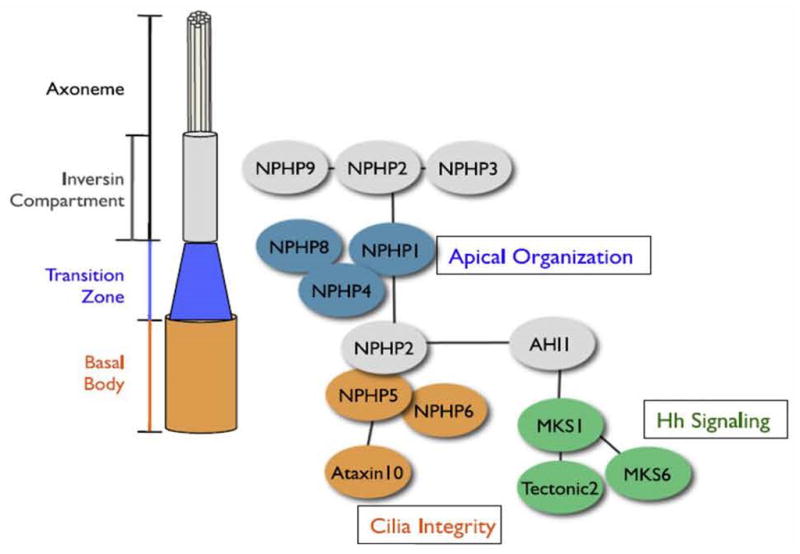
In contrast to other proteins involved in cystic kidney disease (e.g. the BBSome) Nephrocystins form multiple smaller protein modules. The module Nephrocystin-1-4-8 is mostly expressed in the transition zone and is involved in apical organization of epithelial cells. The Nephrocystin-5-6 complex is localized in the basal body and is crucial for proper ciliogenesis. The MKS1-6 module links ciliary function to Sonic Hedgehog (Hh) signaling. Dysfunctional ciliogenesis disturbs Hh signaling which results in embryonic neural tube dorsalization and polydactyly. The bridging component consists of Nephrocystin-2-3-9 and is expressed along the entire axoneme. One may speculate if some of these protein modules correlate with extrarenal phenotypes: Nephrocystins-5 and -6 cause retinitis pigmentosa, altered MKS1 and 6 result in Meckel-Gruber and Joubert syndromes, and the components of the bridging component result in infantile NPHP and congenital heart defects (reprinted with permission from 71).
It is tempting to think about these modules from the phenotype standpoint: NPHP5/IQCB1 and NPHP6/CEP290 mutations both cause in 100% of patients retinitis pigmentosa, the MKS1-6 module causes MKS and JBTS, and the bridging components Nephrocystins-2-3-9 can all result in infantile NPHP and congenital heart disease. Recently, a novel interaction partner of the bridging component was found with NPHP16/ANKS6. Nephrocystin-16 also results in infantile NPHP with congenital heart defects (33, 34).
Signaling pathways
At least four different signaling pathways are associated with the pathogenesis of NPHP (5). Inversin, Nephrocystin-3 and Nephrocystin-4 are thought to interfere with Wnt signaling (72–74).
Groundbreaking studies by Simons et al. in cell culture and zebrafish showed that Wnt signaling is modified by tubular flow (72). Tubular fluid flow increased Inversin levels, which reduced cytoplasmic Dishevelled levels due to degradation and resulted in a switch from canonical to non-canonical Wnt signaling. In case of Inversin mutations the balance of canonical to non-canonical Wnt signaling is disturbed towards a dominating canonical Wnt signaling pathway which disrupts apical-basolateral polarity of the renal tubular cells. Similar to Inversin, Nephrocystin-3 inhibits canoncical Wnt signaling, and Nephrocystin-4 also colocalizes with the final effector of the Wnt signaling pathway called β-catenin (73, 75). Nephrocystin-4 is required for proper pronephros formation in zebrafish by surpressing the Wnt/β-catenin pathway (74).
Another signaling pathway involved in the pathogenesis of NPHP is Hedgehog signaling (23). Nephrocystin-7/GLIS2 was found to alter Hedgehog signaling. Altered Hedgehog signaling was also detected in a mutant nephrocystin-6/cep290 mouse which was found to be a viable model for Joubert syndrome (76). The Hedgehog pathway seems to be of particular importance regarding skeletal disorders. While Indian hedgehog (Ihh) is crucial for endochondral ossification, Sonic Hedgehog (Shh) is required for patterning of the forming skeleton (77).
The Hippo pathway was found to be altered by Nephrocystin-4 and Nephrocystin-9/NEK8 (78–80). Finally, NPHP9/NEK8, NPHP14/ZNF423, and NPHP15/CEP164 were identified to alter a highly conserved signaling pathway called DNA damage response signaling (DDR) (32, 81).
Oligogenicity
Single locus allelism has remained insufficient to explain the variability in penetrance and expressivity in ciliopathies. Digenic and triallelic inheritance could possibly provide an explanation. Triallelic inheritance was first demonstrated for BBS (82). Here, a third mutated allele in a second NPHP gene may modify disease severity and extrarenal phenotypes. Similar to this scenario a broad range of other diseases were found with homozygous NPHP6/CEP290 mutations and an additional heterozygous NPHP mutation in other Nephrocystin genes: Isolated NPHP and SLSN with an additional heterozygous NPHP4 mutation, in BBS or MKS with a heterozygous TMEM67/NPHP11 mutation and in JBTS with a heterozgous AHI1 mutation, which is known to cause JBTS when both alleles are affected (22, 83–86).
Other Nephrocystins as NPHP1-4, NPHP8/RPGRIP1L, NPHP12/TTC21B, but also BBS4, and AHI1 were shown to function as genetic modifiers of disease severity (29, 61, 87–91). NPHP12/TTC21B may function as a genetic modifier of disease in approximately 5% of ciliopathy patients by increasing the mutational load.
CONCLUSION
The involvement of extrarenal organs with NPHP may be due to shared protein expression in primary cilia of multiple tissues and shared Nephrocystin modules. Multiple Nephrocystins may interfere with the same signaling pathway as the Wnt, Hedgehog, Hippo and DDR signaling. These pathways are important for the development and maintenance of different organ systems. Current NPHP mutations account for approximately 30–40% of all NPHP-RC cases. This means that many other genes will be identified which will provide us with novel insights in the future.
KEY BULLET POINTS.
In 15–20% of NPHP patients extrarenal symptoms are identified.
As NPHP is a ciliopathy, these extrarenal manifestations are called NPHP-related ciliopathies.
Many of the extrarenal symptoms may be due to shared expression of primary cilia in multiple tissues and different cell types (e.g. renal epithelial cells, ventral node, cholangiocytes and chondrocytes).
In contrast to motile cilia, primary cilia are thought to function as flow-, osmo- or chemosensors.
Several Nephrocystins work together in different protein modules and a variety of different signaling pathways which also may contribute to the variable extrarenal organ involvement.
Acknowledgments
I thank Dr. Sandy Cope-Yokoyama (Department of Pathology, Children’s Medical Center Dallas and UT Southwestern Medical Center, Dallas) for her contribution regarding the images of renal pathology in nephronophthisis and Dr. Michael Craig Morris (Department of Radiology, Children’s Medical Center Dallas and UT Southwestern Medical Center, Dallas) for his contribution of the Joubert syndrome image. I thank Helen Manning, Drs. Gattineni and Attanassio, and Dorothy Buckely for critical review of the manuscript.
FINANCIAL SUPPORT AND SPONSORSHIP
MT Wolf is supported by grants from National Institutes of Health (K08-DK095994-03), American Society of Nephrology and Children’s Clinical Research Advisory Committee (CCRAC), Children’s Medical Center, Dallas.
Footnotes
CONFLICT OF INTEREST
None
Disclosure: The author is supported by NIH funding (K08-DK095994-03), the American Society of Nephrology, and the Children’s Clinical Research Advisory Committee (CCRAC), Children’s Medical Center, Dallas.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the last 18 months, have been highlighted as:
▪ of special interst
▪▪ of outstanding interest
- 1.Fanconi G, Hanhart E, von AA, et al. Familial, juvenile nephronophthisis (idiopathic parenchymal contracted kidney) Helv Paediatr Acta. 1951;6:1–49. [PubMed] [Google Scholar]
- 2.Hurd TW, Hildebrandt F. Mechanisms of nephronophthisis and related ciliopathies. Nephron Exp Nephrol. 2011;118:e9–14. doi: 10.1159/000320888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolf MT, Hildebrandt F. Nephronophthisis. Pediatr Nephrol. 2011;26:181–194. doi: 10.1007/s00467-010-1585-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simms RJ, Hynes AM, Eley L, Sayer JA. Nephronophthisis: a genetically diverse ciliopathy. Int J Nephrol. 2011;2011:527137. doi: 10.4061/2011/527137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benzing T, Schermer B. Clinical spectrum and pathogenesis of nephronophthisis. Curr Opin Nephrol Hypertens. 2012;21:272–278. doi: 10.1097/MNH.0b013e3283520f17. [DOI] [PubMed] [Google Scholar]
- 6.Hildebrandt F, Strahm B, Nothwang HG, et al. Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. APN Study Group. Arbeitsgemeinschaft fur Padiatrische Nephrologie. Kidney Int. 1997;51:261–269. doi: 10.1038/ki.1997.31. [DOI] [PubMed] [Google Scholar]
- 7.Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol. 1989;3:50–55. doi: 10.1007/BF00859626. [DOI] [PubMed] [Google Scholar]
- 8.Omran H, Fernandez C, Jung M, et al. Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet. 2000;66:118–127. doi: 10.1086/302705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ala-Mello S, Kivivuori SM, Ronnholm KA, et al. Mechanism underlying early anaemia in children with familial juvenile nephronophthisis. Pediatr Nephrol. 1996;10:578–581. doi: 10.1007/s004670050164. [DOI] [PubMed] [Google Scholar]
- 10.Blowey DL, Querfeld U, Geary D, et al. Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol. 1996;10:22–24. doi: 10.1007/BF00863431. [DOI] [PubMed] [Google Scholar]
- 11.Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol. 2009;24:2333–2344. doi: 10.1007/s00467-008-0840-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waldherr R, Lennert T, Weber HP, et al. The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch A Pathol Anat Histol. 1982;394:235–254. doi: 10.1007/BF00430668. [DOI] [PubMed] [Google Scholar]
- 13.Haider NB, Carmi R, Shalev H, et al. A Bedouin kindred with infantile nephronophthisis demonstrates linkage to chromosome 9 by homozygosity mapping. Am J Hum Genet. 1998;63:1404–1410. doi: 10.1086/302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪14.Gee HY, Otto EA, Hurd TW, et al. Whole-exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014;85:880–887. doi: 10.1038/ki.2013.450. The authors outline other kidney diseases which can mimick the symptoms of NPHP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hildebrandt F, Otto E, Rensing C, et al. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet. 1997;17:149–153. doi: 10.1038/ng1097-149. [DOI] [PubMed] [Google Scholar]
- 16.Saunier S, Calado J, Heilig R, et al. A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet. 1997;6:2317–2323. doi: 10.1093/hmg/6.13.2317. [DOI] [PubMed] [Google Scholar]
- 17.Otto EA, Schermer B, Obara T, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olbrich H, Fliegauf M, Hoefele J, et al. Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet. 2003;34:455–459. doi: 10.1038/ng1216. [DOI] [PubMed] [Google Scholar]
- 19.Mollet G, Salomon R, Gribouval O, et al. The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet. 2002;32:300–305. doi: 10.1038/ng996. [DOI] [PubMed] [Google Scholar]
- 20.Otto E, Hoefele J, Ruf R, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet. 2002;71:1161–1167. doi: 10.1086/344395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otto EA, Loeys B, Khanna H, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 22.Sayer JA, Otto EA, O’Toole JF, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 23.Attanasio M, Uhlenhaut NH, Sousa VH, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–1024. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- 24.Delous M, Baala L, Salomon R, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 25.Wolf MT, Saunier S, O’Toole JF, et al. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int. 2007;72:1520–1526. doi: 10.1038/sj.ki.5002630. [DOI] [PubMed] [Google Scholar]
- 26.Otto EA, Trapp ML, Schultheiss UT, et al. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol. 2008;19:587–592. doi: 10.1681/ASN.2007040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto EA, Hurd TW, Airik R, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42:840–850. doi: 10.1038/ng.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otto EA, Tory K, Attanasio M, et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11) J Med Genet. 2009;46:663–670. doi: 10.1136/jmg.2009.066613. [DOI] [PubMed] [Google Scholar]
- 29.Davis EE, Zhang Q, Liu Q, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–196. doi: 10.1038/ng.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fehrenbach H, Decker C, Eisenberger T, et al. Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies. Pediatr Nephrol. 2014;29:1451–1456. doi: 10.1007/s00467-014-2762-2. [DOI] [PubMed] [Google Scholar]
- 31.Bredrup C, Saunier S, Oud MM, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;895:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaki M, Airik R, Ghosh AK, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪33.Taskiran EZ, Korkmaz E, Gucer S, et al. Mutations in ANKS6 cause a nephronophthisis-like phenotype with ESRD. J Am Soc Nephrol. 2014;25:1653–1661. doi: 10.1681/ASN.2013060646. The authors confirm ANKS6 as a gene contributing to NPHP and demonstrate upregaulted Wnt signaling with ANKS6 mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪34.Hoff S, Halbritter J, Epting D, et al. ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat Genet. 2013;45:951–956. doi: 10.1038/ng.2681. This is an outstanding study which identified ANKS6 as interaction partner of NEK8/NPHP9. In addition, patients with ANKS6/NPHP16 mutations have frequently congenital heart defects and ANKS6 has been linked to oxygen-sensing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪35.Halbritter J, Bizet AA, Schmidts M, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013;93:915–925. doi: 10.1016/j.ajhg.2013.09.012. This interesting study describes the first member of the anterograde IFT complex responsible for NPHP with skeletal disorders. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪36.Failler M, Gee HY, Krug P, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet. 2014;94:905–914. doi: 10.1016/j.ajhg.2014.05.002. This exciting publication describes mutations in a protein member of the distal appendages causing NPHP with intellectual disabilities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Toole JF, Liu Y, Davis EE, et al. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J Clin Invest. 2010;120:791–802. doi: 10.1172/JCI40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪38.Hurd TW, Otto EA, Mishima E, et al. Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis-like phenotype. J Am Soc Nephrol. 2013;24:967–977. doi: 10.1681/ASN.2012101034. The authors identify mutations in a magnesium transporter as a reason for a NPHP-like phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪39.Halbritter J, Porath JD, Diaz KA, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132:865–884. doi: 10.1007/s00439-013-1297-0. This study represents the most comprehensive mutation analysis in NPHP-RC and yielded insight in some unexpected phenotypes with specific gene mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20:23–35. doi: 10.1681/ASN.2008050456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Betz R, Rensing C, Otto E, et al. Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr. 2000;136:828–831. [PubMed] [Google Scholar]
- 42.Ronquillo CC, Bernstein PS, Baehr W. Senior-Loken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012;75:88–97. doi: 10.1016/j.visres.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪43.Wang J, Deretic D. Molecular complexes that direct rhodopsin transport to primary cilia. Prog Retin Eye Res. 2014;38:1–19. doi: 10.1016/j.preteyeres.2013.08.004. Excellent review how primary cilia may relate to photoreceptor degeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪44.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12:894–905. doi: 10.1016/S1474-4422(13)70136-4. Very good summary of the current known genes and concepts contributing to Joubert syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪▪45.Barker AR, Thomas R, Dawe HR. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis. 2014;10:96–107. doi: 10.4161/org.27375. Interesting, current review of pathogenesis of Meckel-Gruber syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:296–306. doi: 10.1002/ajmg.c.30225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C:165–74. doi: 10.1002/ajmg.c.31336. [DOI] [PubMed] [Google Scholar]
- 48.Arts H, Knoers N. NCBI Bookshelf. Seattle: 1993–2014. Cranioectodermal Dysplasia. GeneReviews. [Google Scholar]
- 49.Baujat G, Huber C, El Hokayem J, et al. Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J Med Genet. 2013;50:91–98. doi: 10.1136/jmedgenet-2012-101282. [DOI] [PubMed] [Google Scholar]
- 50.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 52.Omran H. NPHP proteins: gatekeepers of the ciliary compartment. J Cell Biol. 2010;190:715–717. doi: 10.1083/jcb.201008080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf MT, Lee J, Panther F, et al. Expression and phenotype analysis of the nephrocystin-1 and nephrocystin-4 homologs in Caenorhabditis elegans. J Am Soc Nephrol. 2005;16:676–687. doi: 10.1681/ASN.2003121025. [DOI] [PubMed] [Google Scholar]
- ▪54.Buisson J, Chenouard N, Lagache T, et al. Intraflagellar transport proteins cycle between the flagellum and its base. J Cell Sci. 2013;126:327–338. doi: 10.1242/jcs.117069. The authors studied the recycling of intraflagellar transport in Trypanosoma brucei. [DOI] [PubMed] [Google Scholar]
- 55.Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83:S12–22. doi: 10.1016/j.diff.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang B, Khanna H, Hawes N, et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum Mol Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fliegauf M, Horvath J, von Schnakenburg C, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol. 2006;17:2424–2433. doi: 10.1681/ASN.2005121351. [DOI] [PubMed] [Google Scholar]
- 59.Murga-Zamalloa CA, Desai NJ, Hildebrandt F, Khanna H. Interaction of ciliary disease protein retinitis pigmentosa GTPase regulator with nephronophthisis-associated proteins in mammalian retinas. Mol Vis. 2010;16:1373–1381. [PMC free article] [PubMed] [Google Scholar]
- 60.Collin GB, Won J, Hicks WL, et al. Meckelin is necessary for photoreceptor intraciliary transport and outer segment morphogenesis. Invest Ophthalmol Vis Sci. 2012;53:967–974. doi: 10.1167/iovs.11-8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khanna H, Davis EE, Murga-Zamalloa CA, et al. A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. Nat Genet. 2009;41:739–745. doi: 10.1038/ng.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scherft JP, Daems WT. Single cilia in chondrocytes. J Ultrastruct Res. 1967;19:546–555. doi: 10.1016/s0022-5320(67)80080-7. [DOI] [PubMed] [Google Scholar]
- 63.Beales PL, Bland E, Tobin JL, et al. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- 64.Arts HH, Bongers EM, Mans DA, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 65.Mill P, Lockhart PJ, Fitzpatrick E, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet. 2011;88:508–515. doi: 10.1016/j.ajhg.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walczak-Sztulpa J, Eggenschwiler J, Osborn D, et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪67.Schmidts M, Frank V, Eisenberger T, et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney Disease. Hum Mutat. 2013;34:714–724. doi: 10.1002/humu.22294. The authors confirm mutations in IFT140 in patients with NPHP and Mainzer-Saldino syndrome and expanded the phenotype to patients with NPHP and Jeune syndrome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perrault I, Saunier S, Hanein S, et al. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet. 2012;90:864–870. doi: 10.1016/j.ajhg.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilissen C, Arts HH, Hoischen A, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–423. doi: 10.1016/j.ajhg.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seo S, Baye LM, Schulz NP, et al. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci U S A. 2010;107:1488–1493. doi: 10.1073/pnas.0910268107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sang L, Miller JJ, Corbit KC, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simons M, Gloy J, Ganner A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bergmann C, Fliegauf M, Bruchle NO, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008;82:959–970. doi: 10.1016/j.ajhg.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burckle C, Gaude HM, Vesque C, et al. Control of the Wnt pathways by nephrocystin-4 is required for morphogenesis of the zebrafish pronephros. Hum Mol Genet. 2011;20:2611–2627. doi: 10.1093/hmg/ddr164. [DOI] [PubMed] [Google Scholar]
- 75.Mollet G, Silbermann F, Delous M, et al. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet. 2005;14:645–656. doi: 10.1093/hmg/ddi061. [DOI] [PubMed] [Google Scholar]
- ▪76.Hynes AM, Giles RH, Srivastava S, et al. Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis. Proc Natl Acad Sci U S A. 2014;111:9893–9898. doi: 10.1073/pnas.1322373111. The authors provide interesting data regarding the phenotype in a Cep290 gene trap mouse model as a model for Joubert syndrome. They demonstrate abnormal Hedgehog signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whitfield JF. The solitary (primary) cilium--a mechanosensory toggle switch in bone and cartilage cells. Cell Signal. 2008;20:1019–1024. doi: 10.1016/j.cellsig.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 78.Habbig S, Bartram MP, Muller RU, et al. NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J Cell Biol. 2011;193:633–642. doi: 10.1083/jcb.201009069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Habbig S, Bartram MP, Sagmuller JG, et al. The ciliopathy disease protein NPHP9 promotes nuclear delivery and activation of the oncogenic transcriptional regulator TAZ. Hum Mol Genet. 2012;21:5528–5538. doi: 10.1093/hmg/dds408. [DOI] [PubMed] [Google Scholar]
- ▪80.Frank V, Habbig S, Bartram MP, et al. Mutations in NEK8 link multiple organ dysplasia with altered Hippo signalling and increased c-MYC expression. Hum Mol Genet. 2013;22:2177–2185. doi: 10.1093/hmg/ddt070. This is an intriguing paper demonstrating increased c-myc and altered Hippo signaling in patients with NPHP9/NEK8 mutations. [DOI] [PubMed] [Google Scholar]
- ▪81.Choi HJ, Lin JR, Vannier JB, et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol Cell. 2013;51:423–439. doi: 10.1016/j.molcel.2013.08.006. This group provides a link between NPHP9/NEK8 and DNA damage response by demonstrating accumulation of DNA damage in Nek8 mutant mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259. doi: 10.1126/science.1063525. [DOI] [PubMed] [Google Scholar]
- 83.Baala L, Audollent S, Martinovic J, et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frank V, den Hollander AI, Bruchle NO, et al. Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel-Gruber syndrome. Hum Mutat. 2008;29:45–52. doi: 10.1002/humu.20614. [DOI] [PubMed] [Google Scholar]
- 85.Helou J, Otto EA, Attanasio M, et al. Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Loken syndrome. J Med Genet. 2007;44:657–663. doi: 10.1136/jmg.2007.052027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leitch CC, Zaghloul NA, Davis EE, et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 87.Louie CM, Caridi G, Lopes VS, et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat Genet. 2010;42:175–180. doi: 10.1038/ng.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ▪88.Zhang Y, Seo S, Bhattarai S, et al. BBS mutations modify phenotypic expression of CEP290-related ciliopathies. Hum Mol Genet. 2014;23:40–51. doi: 10.1093/hmg/ddt394. This is an interesting study indicating genetic interactions between BBSome components and CEP290/NPHP6, thus explaining overlapping phenotypes and variable expression of ciliopathies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoefele J, Wolf MT, O’Toole JF, et al. Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 90.Badano JL, Kim JC, Hoskins BE, et al. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12:1651–1659. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 91.Tory K, Lacoste T, Burglen L, et al. High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol. 2007;18:1566–1575. doi: 10.1681/ASN.2006101164. [DOI] [PubMed] [Google Scholar]