

Abstract
Genetically modified organisms (GMOs) are increasingly deployed at large scales and in open environments. Genetic biocontainment strategies are needed to prevent unintended proliferation of GMOs in natural ecosystems. Existing biocontainment methods are insufficient either because they impose evolutionary pressure on the organism to eject the safeguard, because they can be circumvented by environmentally available compounds, or because they can be overcome by horizontal gene transfer (HGT). Here we computationally redesign essential enzymes in the first organism possessing an altered genetic code to confer metabolic dependence on nonstandard amino acids for survival. The resulting GMOs cannot metabolically circumvent their biocontainment mechanisms using environmentally available compounds, and they exhibit unprecedented resistance to evolutionary escape via mutagenesis and HGT. This work provides a foundation for safer GMOs that are isolated from natural ecosystems by reliance on synthetic metabolites.
Genetically modified organisms (GMOs) are rapidly being deployed for large-scale use in bioremediation, agriculture, bioenergy, and therapeutics1. In order to protect natural ecosystems and address public concern it is critical that the scientific community implements robust biocontainment mechanisms to prevent unintended proliferation of GMOs. Current strategies rely on integrating toxin/antitoxin “kill switches”2,3, establishing auxotrophies for essential compounds4, or both5,6. Toxin/antitoxin systems suffer from selective pressure to improve fitness through deactivation of the toxic product7,8, while metabolic auxotrophies can be circumvented by scavenging essential metabolites from nearby decayed cells or cross-feeding from established ecological niches. Effective biocontainment strategies must protect against three possible escape mechanisms: mutagenic drift, environmental supplementation and horizontal gene transfer (HGT). Here we introduce “synthetic auxotrophy” for non-natural compounds as a means to biological containment that is robust against all three mechanisms. Using the first genomically recoded organism (GRO)9 we assigned the UAG stop codon to incorporate a nonstandard amino acid (NSAA) and computationally redesigned the cores of essential enzymes to require the NSAA for proper translation, folding and function. X-ray crystallography of a redesigned enzyme shows atomic-level agreement with the predicted structure. Combining multiple redesigned enzymes resulted in GROs that exhibit dramatically reduced escape frequencies and readily succumb to competition by unmodified organisms in nonpermissive conditions. Whole-genome sequencing of viable escapees revealed escape mutations in a redesigned enzyme and also disruption of cellular protein degradation machinery. Accordingly, reducing the activity of the NSAA aminoacyl-tRNA synthetase (aaRS) in nonpermissive conditions produced double- and triple-enzyme synthetic auxotrophs with undetectable escape when monitored for 14 days (detection limit: 2.2 × 10−12 escapees/c.f.u.). We additionally show that while bacterial lysate supports growth of common metabolic auxotrophs, the environmental absence of NSAAs prevents such natural products from sustaining synthetic auxotrophs. Further, distributing redesigned enzymes throughout the genome reduces susceptibility to horizontal gene transfer. When our GROs incorporate sufficient foreign DNA to overwrite the NSAA-dependent enzymes, they also revert UAG function, thereby preserving biocontainment by deactivating recoded genes. The general strategy developed here provides a critical advance in biocontainment as GMOs are considered for broader deployment in open environments.
Computational design of synthetic auxotrophs
We focused on the NSAA L-4,4′-biphenylalanine (bipA), which has a size and geometry unlike any standard amino acid, and a hydrophobic chemistry expected to be compatible with protein cores. We introduced a plasmid containing a codon-optimized version of the bipA aminoacyl-tRNA synthetase (bipARS)/tRNAbipA system10 into a GRO (genomically recoded E. coli strain C321.ΔA9), thereby assigning UAG as a dedicated codon for bipA incorporation. Using a model of bipA in the Rosetta software for macromolecular modeling11 we applied our computational design protocol to 13,564 core positions in 112 essential proteins12 with X-ray structures, refining designs for cores that tightly pack bipA while maximizing neighboring compensatory mutations (Methods) predicted to destabilize the proteins in the presence of standard amino acid suppressors at UAG positions (Fig. 1a). We further required that candidate enzymes produce products that cannot be supplemented by environmentally available compounds. For example, we rejected glmS designs because glucosamine supplementation rescues growth of glmS mutants13. We selected designs of six essential genes for experimental characterization: adenylate kinase (adk), alanyl-tRNA synthetase (alaS), DNA polymerase III subunit delta (holB), methionyl-tRNA synthetase (metG), phosphoglycerate kinase (pgk), and tyrosyl-tRNA synthetase (tyrS). For all cases we designed oligonucleotides (Supplementary Table 1) encoding small libraries suggested by the computational models (Supplementary Table 2) and used them to directly edit the target essential gene in C321.ΔA using co-selection multiplex automatable genome engineering (CoS-MAGE)14. Since tyrS featured the greatest number of compensatory mutations, we additionally synthesized eight computational tyrS designs and used them to replace the endogenous tyrS gene (Supplementary Table 3). We screened our CoS-MAGE populations for bipA-dependent clones by replica plating from permissive media (containing bipA and arabinose for bipARS induction) to nonpermissive media (lacking bipA and arabinose) and validated candidates by monitoring kinetic growth in the presence and absence of bipA (Methods, Extended Data Fig. 1). Mass spectrometry confirmed the specific incorporation of bipA in redesigned enzymes (Methods, Extended Data Fig. 2). X-ray crystallography of a redesigned enzyme to 2.65 Å resolution (PDB code 4OUD, Extended Data Table 1) shows atomic level agreement with computational predictions (Fig. 1b–d, Extended Data Fig. 3, Supplementary Discussion). Selectivity for bipA in a redesigned core was further confirmed by measuring soluble protein content when bipA is mutated to leucine (wild-type residue) or tryptophan (most similar natural residue to bipA by mass) (Methods, Extended Data Fig. 4).
Figure 1. Computational design of NSAA-dependent essential proteins.
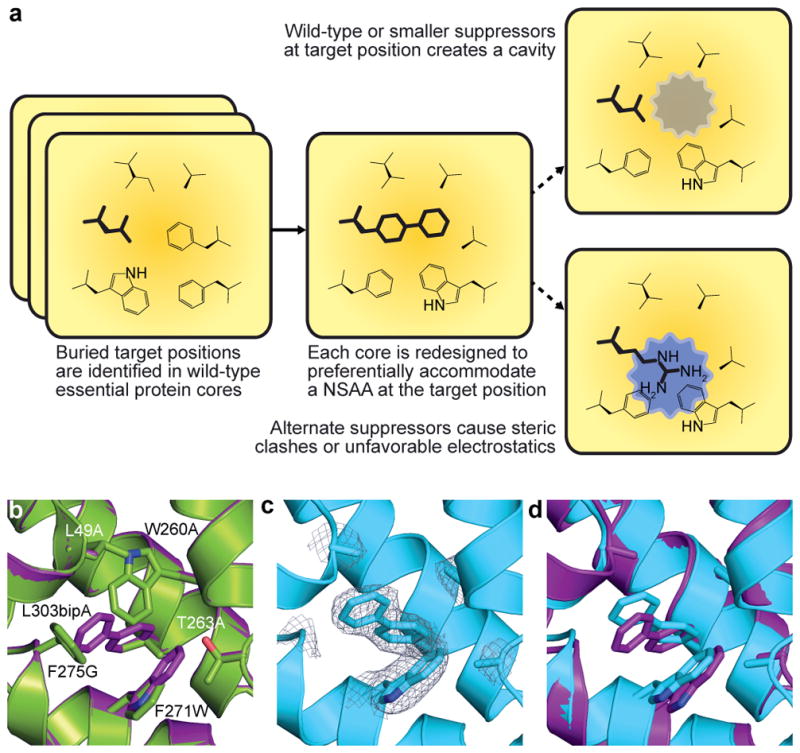
a, Overview of the computational second-site suppressor strategy. b, Computational design of a NSAA-dependent tyrosyl-tRNA synthetase (purple) overlaid on the wild-type structure (green; PDB code 2YXN). Six substituted residues are shown as sticks. c, X-ray crystallography of the redesigned synthetase with an electron density map (2Fo-Fc contoured at 1.0 σ) for substituted residues; substitution F236A is on a disordered loop and is not observed. d, The crystal structure of the redesigned enzyme (cyan) superimposed onto the computationally predicted model (purple).
Characterization of synthetic auxotrophs
We characterized the escape frequencies of eight strains by plating on nonpermissive media with and without bipARS inducer arabinose (Fig. 2, Supplementary Tables 4, 5, Methods). Escapees exhibiting varying fitness were detected by the emergence of colonies in the absence of bipA. Two tyrS variants (tyrS.d6 and tyrS.d7) and two adk variants (adk.d4 and adk.d6) show robust growth in permissive conditions and low escape frequencies in the absence of bipA. Strain alaS.d5 shows only minor impairment in the absence of bipA, suggesting that near-cognate suppression of the UAG codon by endogenous tRNA or mischarging of natural amino acids by BipARS is adequate to support growth. Consistent with this hypothesis, inserting a UAG immediately after the start codon (strain alaS.d5.startUAG) further impairs growth in the absence of bipA, although bipA dependence is readily overcome by mutational escape. HolB recombinants presented only the designed bipA mutation (holB.d1) and none of the compensatory mutations, suggesting that the intended compensation may be too destabilizing, or that the native amino acids at those positions may be critical for function. The lack of compensation for bipA results in a strong and continuous selective pressure to incorporate standard amino acids at the bipA position, so holB.d1 was not carried forward.
Figure 2. Escape frequencies and doubling times of auxotrophic strains.

Escape frequencies for engineered auxotrophic strains calculated as colonies observed per colony forming unit (c.f.u.) plated over 3 technical replicates on solid media lacking arabinose and bipA. Assay limit is calculated as 1/(total c.f.u. plated) for the most conservative detection limit of a cohort, with a single-enzyme auxotroph limit of 3.5 × 10−9 escapees/c.f.u., a double-enzyme auxotroph limit of 8.3 × 10−11 escapees/c.f.u. and a triple-enzyme auxotroph limit of 6.41 × 10−11 escapees/c.f.u. Positive error bars are standard error of the mean (s.e.m.) of the escape frequency over three technical replicates (Methods). The top panel presents the doubling times for each strain in the presence of 10 μM or 100 μM bipA, with the parental strain doubling times represented by the dashed horizontal lines. MetG.d3 growth was undetectable in 100 μM bipA. Positive and negative error bars are s.e.m.
We hypothesized that since the designed proteins have structurally distinct cores, each variant may favor different standard amino acids at the bipA position. Therefore, viable UAG suppressors for one enzyme may be deleterious for another. We sought to determine the distribution of standard amino acids accommodated at the UAG position in each variant in order to identify combinations of redesigned enzymes that could drive escape frequencies even lower. We grew up the top seven dependent strains in permissive media and used multiplex automatable genome engineering (MAGE)15 to introduce all 64 codons at the UAG positions (Methods). For tyrS.d7 we collaterally introduced the V307A mutation observed in tyrS.d6, since the same oligo containing V307A was used to encode degeneracy for both strains at the UAG position, producing the eighth strain tyrS.d8. Immediately following electroporation, cells were shifted to nonpermissive media so that recombinants with canonical amino acids replacing bipA would overtake the population according to their relative fitness.
We sampled the eight populations at 1 hour and 4 hour time points, at confluence, and at 2 subsequent passages to confluence (100-fold dilution in each passage), by which point the preferred genotypes emerged. Using next-generation sequencing we determined the relative abundance of all standard amino acid codons at the UAG positions for each time point (Extended Data Fig. 5, Supplementary Table 6) and computed the “flatness” (Shannon entropy16) of each amino acid frequency distribution (Fig. 3). The two strains showing the greatest escape frequencies, alaS.d5 and metG.d3, also have the flattest amino acid frequency distributions. Correspondingly, the strains with the lowest escape frequencies exhibit peaked amino acid frequency distributions. These amino acid preference profiles show a strong relationship between structural selectivity for bipA and escape frequency, supporting the rationale underlying our computational strategy. Furthermore, they confirm our hypothesis that different redesigned protein cores will favor different standard amino acids. In particular, phenylalanine and tryptophan (aromatics) dominate tyrS.d7 and tyrS.d8 populations, while the other recombinants tend towards valine, leucine, isoleucine and methionine (aliphatics) (Fig. 3a). In agreement with these observations, we were able to isolate viable recombinants of adk.d6 containing leucine but not tryptophan at the bipA position, while also isolating viable recombinants of tyrS.d8 containing tryptophan but not leucine at the bipA position (Supplementary Table 7). In considering candidates for combination, we omitted alaS and metG due to their susceptibility for near-cognate suppression. We also determined that pgk mutants can grow robustly in the presence of pyruvate and/or succinate (Extended Data Fig. 6) even though they do not grow in LB-Lennox (LBL)12. Since these carbon sources are environmentally available, pgk violates our definition of essentiality and we removed pgk.d4 from consideration. Finally, we excluded adk.d4 due to its poor survival at stationary phase (Supplementary Discussion). We therefore focused on combinations of tyrS.d6, tyrS.d7 and tyrS.d8 with adk.d6, all of which maintain robust growth in permissive media, show strong dependence for bipA, and are metabolically isolated from environmental compounds.
Figure 3. Structural specificity at designed UAG positions in eight NSAA-dependent enzymes correlates with escape frequencies.
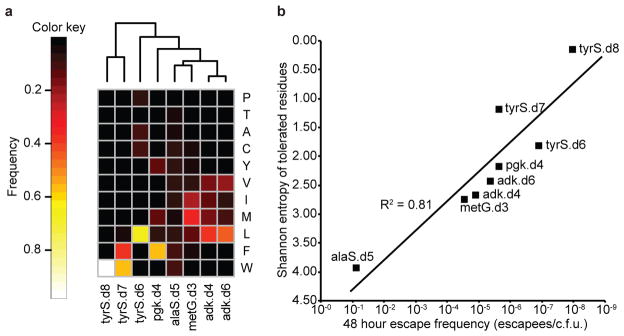
a, Amino acid preferences at UAG positions in eight synthetic auxotrophs were determined by replacing the UAG codon with full NNN degeneracy and then sequencing the resulting populations with an Illumina MiSeq. Frequencies of each amino acid as a fraction of total sequences observed after three 1:100 passages to confluence are shown (top 11 most frequent amino acids only). Samples are clustered by Euclidean distance between amino acid frequencies. The frequency of an amino acid reports on the fitness conferred by the corresponding natural amino acid suppressor at the UAG position relative to all other amino acids. b, Shannon entropy was computed over the distributions of amino acids preferred at the UAG positions of the eight single-enzyme auxotrophs and plotted against the 48 hour escape frequency for each strain. Entropy correlates log-linearly with escape frequency, suggesting that enzyme cores with high structural specificity for bipA at the UAG position have less fit evolutionary routes to escape. Strains alaS.d5 and metG.d3 have a deactivated mutS gene.
Combining top tyrS designs with adk.d6 yielded three strains with no detectable escapees after 24 hours, including adk.d6_tyrS.d8, which has undetectable growth after > 72 hours (detection limit 7.44 × 10−11 escapees/c.f.u., Fig. 2, Supplementary Table 4). Colonies bearing the adk.d6_tyrS.d8 genotype were observed between four and seven days of incubation, but show severely impaired fitness when grown in nonpermissive liquid culture and are readily outcompeted by prototrophic E. coli (Fig. 4, Supplementary Discussion). The relative reductions in escape frequencies support the hypothesis that combining variants with distinct amino acid preferences at the UAG position decreases fitness of escapees. Even though tyrS.d6 and tyrS.d8 exhibit similar escape frequencies as single-enzyme auxotrophs, strain adk.d6_tyrS.d6 produces faster growing escapees (adk.d6 and tyrS.d6 share a preference for leucine) than strain adk.d6_tyrS.d8 (tyrS.d8 prefers tryptophan). Although tyrS.d7 and tyrS.d8 both prefer aromatic residues, tyrS.d7 exhibits a broader amino acid preference profile (Fig. 3a) and produces faster-growing escapees than tyrS.d8 (Fig. 2). Accordingly, adk.d6_tyrS.d8 yields the lowest escape frequency of the combined tyrS and adk variants.
Figure 4. Competition between synthetic auxotroph escapees and prototrophic E. coli.
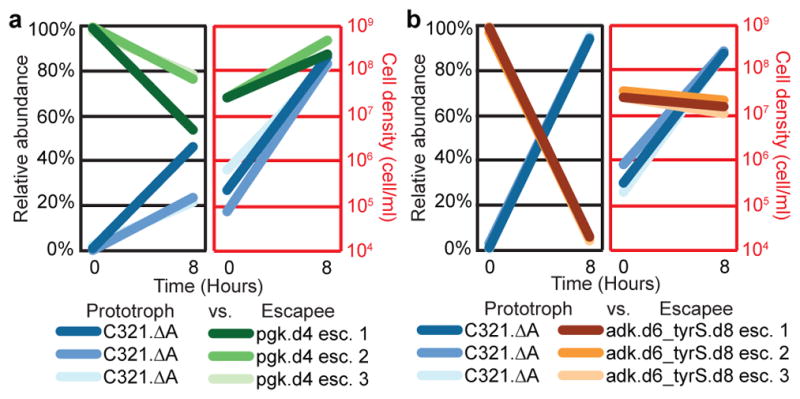
C321.ΔA was competed in the absence of bipA against escapees from a single-enzyme bipA auxotroph (pgk.d4, moderate NSAA dependence), or from a double-enzyme bipA auxotroph (adk.d6_tyrS.d8, strong bipA-dependence). Populations were seeded with 100-fold excess escapees and grown for 8 hours in nonpermissive conditions. The populations were evaluated using flow cytometry for episomally expressed fluorescent proteins at t = 0 and t = 8 hours. Results from separate competition experiments against 3 different escapees are shown for each synthetic auxotroph. a, Pgk.d4 escapees continue to expand in a mixed population with C321.ΔA after 8 hours. b, Adk.d6_tyrS.d8 escapees are rapidly outcompeted by C321.ΔA, which overtakes the population after 8 hours.
Prevention of mutagenic escape
The appearance of escapee colonies from adk.d6_tyrS.d8 after > 72 hours suggests the emergence of rare genotypes conferring weak viability (doubling time ≥ 348 min) in the absence of bipA. To uncover mutagenic routes to escape we performed whole-genome sequencing on escapees of adk.d6, tyrS.d8 and adk.d6_tyrS.d8 (Methods, four escapees for each single-enzyme auxotroph and three escapees for the double-enzyme auxotroph were sequenced). We observed no mutations in the ribosome or tRNAs that could account for UAG translation in the absence of bipA, nor did we observe mutations to any designed amino acid positions. However, we identified a point mutation (A70V) to tyrS in all four tyrS.d8 escapees sequenced (Supplementary Table 9). The A70V mutation may improve packing of the tyrS.d8 catalytic domain in the context of a destabilized neighboring helical bundle lacking bipA (Extended Data Fig. 7a). To validate this escape mechanism we produced strain tyrS.d8.A70V and performed an escape assay on nonpermissive media. Within 5 days of plating, we observed colony formation from all plated cells (Extended Data Fig. 7b), confirming that A70V is an escape mechanism for tyrS.d8. The A70V mutant of tyrS.d8 does not impair fitness in permissive conditions (Supplementary Table 10), so the genotype is easily visited as a neutral mutation within the fitness landscape by replication errors. Still, targeted sequencing of the tyrS gene in eight additional tyrS.d8 escapees did not reveal the A70V mutation, suggesting that A70V is not the only escape mechanism for tyrS.d8.
Whole-genome sequencing of adk.d6 and adk.d6_tyrS.d8 escapees revealed disruptive mutations to Lon protease in all seven cases. One clone contained a frame shift and another contained a non-synonymous substitution (L611P) within the lon gene. The remaining five cases had a transposable element inserted within the promoter of lon. Targeted sequencing characterized the insertion sequence in at least one clone as IS186, exactly recapitulating the Lon protease deficiency of E. coli BL2117. We validated Lon disruption as an escape mechanism using λ Red-mediated recombination to replace lon with a kanamycin resistance gene (kanR) in adk.d6, tyrS.d8 and adk.d6_tyrS.d8. Recombinants were replica plated from permissive to nonpermissive media containing kanamycin. Colony PCR confirmed that 27 of 27 non-bipA-dependent colonies screened (9 escapees per dependent strain) had Lon deleted by kanR.
Since the Lon protease is the primary apparatus for bulk degradation of misfolded proteins in the E. coli cytoplasm18, we hypothesized that its disruption would allow persistence of poorly folded adk.d6 and tyrS.d8 proteins when standard amino acids are incorporated in place of bipA. We further hypothesized that basal UAG suppression from promiscuous activity of pEVOL-BipARS produced sufficient full-length protein to support viability in the absence of Lon-mediated degradation. To test this hypothesis and safeguard against Lon-mediated escape we pursued two independent strategies to reduce activity of BipARS in nonpermissive conditions. First, we reduced the gene copy number approximately 10-fold by moving bipARS and tRNAbipA from the p15A pEVOL plasmid to the genome of adk.d6, producing the strain adk.d6_int (Methods). Second, we applied our computational second-site suppressor strategy to residue V291 in bipARS (homologous to L303bipA in our tyrS designs) and reintroduced it into adk.d6 on the pEVOL vector, producing strain adk.d6_bipARS.d7 (Methods). This latter strategy produced a BipARS variant that requires bipA for folding and function, thereby abrogating residual activity towards standard amino acids in the absence of bipA. Both strategies resulted in a > 200-fold reduction in 7 day escape frequency (Supplementary Table 4). Introducing tyrS.d8 to these strains produced double- and triple-enzyme synthetic auxotrophs adk.d6_tyrS.d8_int and adk.d6_tyrS.d8_bipARS.d7 that exhibit undetectable escape when monitored for 14 days (Fig. 2 and Supplementary Table 4, detection limit 2.2 × 10−12 escapees/c.f.u.). Both strains also show undetectable escape in the presence of arabinose, and present no fitness impairment relative to the parental adk.d6_tyrS.d8 synthetic auxotroph (Supplementary Table 4, doubling times of 57 and 55 min).
Protection from natural supplementation
To compare our synthetic auxotrophs to current biocontainment practices we generated natural metaboblic auxotrophs by knocking out asd and thyA genes from an MG1655-derived E. coli strain (EcNR1). The asd knockout renders the strain dependent on diaminopimelic acid (DAP) for cell wall biosynthesis4, while the thyA knockout deprives the cell of thymine, an essential nucleobase19. These well-studied auxotrophies are commonly incorporated into biocontainment strategies4,6. In agreement with previous studies, the asd knockout shows strong dependence on its requisite metabolite, with a 7 day escape frequency of 8.97 × 10−9 escapees/c.f.u. (Supplementary Table 11). Knocking out thyA from this strain to produce a double-enzyme auxotroph did not reduce the 7 day escape frequency (8.79 × 10−9 escapees/c.f.u.). Nevertheless, metabolic strategies could complement synthetic auxotrophies to improve escape frequencies in defined ecological niches. To test this principle we knocked out asd from the double-enzyme synthetic auxotrophs of adk and tyrS resulting in three triple-enzyme auxotrophs (adk.d6_tyrS.d6_asd, adk.d6_tyrS.d7_asd and adk.d6_tyrS.d8_asd) that grow robustly in permissive conditions but show undetectable escape after 7 days on media lacking bipA and DAP (Fig. 2, detection limit 6.4 × 10−11 escapees/c.f.u., Supplementary Tables 4, 5).
While bacterial growth assays are often carried out in variations of media enriched with yeast extract, GMOs are increasingly deployed among a diversity of ecosystems that may provide opportunities for scavenging or cross-feeding essential metabolites. To compare metabolic and synthetic auxotroph strategies in an environment mimicking endogenous bacterial communities we grew engineered variants of both natural and synthetic auxotrophs in LBL containing E. coli lysate (Methods). We hypothesized that since DAP is an essential component of the bacterial cell wall, the Δasd strains may scavenge sufficient DAP from E. coli lysate to complement the auxotrophy. As anticipated, metabolic auxotrophs obtained sufficient nutrients from the yeast/tryptone (LBL) and the bacterial remnants (lysate) to support exponential growth (Extended Data Fig. 6e–h), while the synthetic auxotrophs failed to circumvent their dependencies. These results highlight the importance of establishing auxotrophies for compounds that are not environmentally available, and of ensuring the metabolic essentiality of enzymes intended to confer dependence.
Resistance to horizontal gene transfer
Horizontal gene transfer is an important mechanism of evolution in any genetically rich environment20. We developed a conjugation escape assay to assess how DNA transfer within an ecosystem enables a GMO to escape biocontainment. Whereas any recombination event that replaces an inactivated gene could overcome metabolic auxotrophies21, we hypothesized that conjugal escape would be disfavored in GROs because donor DNA replacing bipA-dependent genes would also overwrite crucial genetic elements involved in genetic code reassignment (Fig. 5a). For example, reintroducing UAG stop codons into essential genes without restoring RF1-mediated translational termination could be deleterious9 or lethal22. Furthermore, reintroducing RF1 would result in competition between bipA incorporation and translational termination, undermining the recoded functions of the GRO.
Figure 5. Synthetic auxotrophy and genomic recoding reduce HGT-mediated escape.
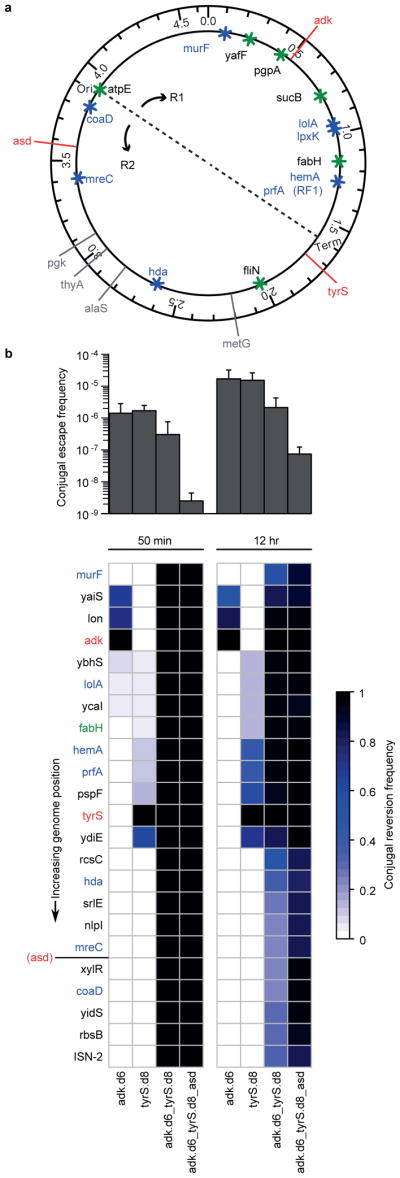
a, The positions of key alleles are plotted to scale on the genome schematic. Red lines indicate auxotrophies used in the multi-enzyme auxotrophs and gray lines indicate other auxotrophies that were not included in this assay. Asterisks indicate important alleles associated with the reassignment of UAG translation function (blue are essential genes and green are potentially important genes9). Conjugation-mediated reversion of the UAA codons back to the wild-type UAG is expected to be deleterious unless the natural UAG translational termination function is reverted. b, Combining multiple synthetic auxotrophies in a single genome requires a large portion of the genome to be overwritten by wild-type donor DNA, reducing the frequency of conjugal escape (top panel) and increasing the likelihood of overwriting the portions of the genome (bottom panel) that provide expanded biological function (e.g., prfA encodes RF1, which mediates translational termination at UAG codons). Positive error bars indicate standard deviation.
In order to simulate a worst-case scenario in ecosystems containing a rich source of conjugal donors, we used Tn5 transposition to integrate an origin of transfer (oriT) into a population of E. coli MG1655 conjugal donor strains. We isolated a population of ~450 independent clones (one oriT for every ~10kb portion of the 4.6 megabase pair genome) and sequenced the flanking genomic regions of 96 donor colonies to confirm that oriT integration was well-distributed throughout the population. We then conjugated this donor population into our auxotrophic strains at a ratio of 1 donor to 100 recipients to increase the probability that conjugal transfer will initiate from one oriT position per recipient. Conjugation was performed for durations of 50 minutes and 12 hours (average conjugation times predicted to transfer 0.5 and 7.2 genomes) to simulate a single conjugal interaction and an ecological worst-case scenario, respectively. Conjugal escapees were selected on nonpermissive media, and 23 alleles distributed throughout the genome (Fig. 5a) were screened using multiplex allele-specific colony PCR (mascPCR) to assess how much of the recoded genome is replaced by wild-type donor DNA.
Conjugal escape frequency decreases as the number of auxotrophic gene variants increases (Fig. 5b, top panel; Extended Data Fig. 8), consistent with larger portions of the genome that must be overwritten for conjugal escape of the multi-enzyme auxotrophs (Fig. 5b, bottom panel). The 12 hour conjugations effect higher escape frequencies than do the 50 minute conjugations, and the 12 hour conjugations produce a larger diversity of conjugal escape genotypes, consistent with an increased opportunity to initiate new conjugal transfers during the mating period. Encouragingly, all 50 minute conjugal escapees from multi-enzyme auxotrophs exhibit the wild-type donor sequence at all 23 assayed alleles (Fig. 5b, bottom panel; Supplementary Table 12), resulting in the reintroduction of release factor 1 and its UAG-mediated translational termination function. This collateral replacement of recombinant genomic DNA could be extended to other recombinant payloads such as antibiotic resistance genes, recombinases, catabolic enzymes, toxins, and orthogonal aaRS/tRNA pairs used for NSAA incorporation.
Discussion
Effective biocontainment mechanisms for GMOs should place high barriers between modified organisms and the natural environment. Our NSAA design strategy produces organisms with an altered chemical language that isolates them from natural ecosystems. By conferring dependence on synthetic metabolites at the level of protein translation, folding and function, synthetic auxotrophy addresses the need for GMOs that are refractory to mutational escape, metabolic supplementation and horizontal gene transfer. Because our NSAAs are incorporated into essential enzymes with second-site mutations, escapees are rare and too sick to outcompete prototrophic microbial communities. In part, robustness emerges from simplicity: our most escape-resistant synthetic auxotrophs contain only 32 (adk.d6_tyrS.d8_int) and 49 (adk.d6_tyrS.d8_bipARS.d7) base pair substitutions across the 4.6 Mbp parental genome and bipARS, with no essential genes deleted or toxic products added. Further, NSAA-based biocontainment with bipA only modestly increases the cost per liter of proliferating culture (Extended Data Table 2).
This work highlights the delicate balance required to engineer essential proteins that are conditionally stabilized by a single NSAA. The design must confer sufficient instability in nonpermissive conditions to deactivate the protein, while providing functional stability in the presence of the correct NSAA. Future design strategies could include polar or charged NSAAs to engineer hydrogen bonds requiring exquisite geometric specificity23 for folding, enzyme-substrate interactions, or macromolecular associations. This approach may reduce susceptibility to suppressors, although fewer protein microenvironments may accommodate burial of charged or polar residues. Reassigning additional codons would permit the incorporation of multiple NSAAs that confer dependence either in different structural motifs or in participation of a joint chemistry. Eventually, organisms with orthogonal genomic chemistries including expanded genetic alphabets24 and their associated replication machinery could provide additional layers of isolation25.
Our results demonstrate that mutational escape frequency under laboratory growth conditions is a necessary but insufficient metric to evaluate biocontainment strategies. Many genes considered to be essential have functions that can be complemented by environmental compounds, as demonstrated here for auxotrophies of natural (asd, thy) and designed (pgk.d4) enzymes. Further, localizing biocontainment mechanisms to a small portion of the genome increases susceptibility to escape by uptake of foreign DNA. Distributing multiple NSAA-dependent enzymes throughout a recoded genome acts as a genomic safeguard against escape by HGT, and demands that conjugal escape replaces large portions of the recipient genome. This collateral replacement of GMO genomic DNA could be exploited to delete recombinant payloads upon exposure to conjugal donors in the environment. Further, by recoding restricted payloads with essential UAG codons, they can be prevented from functioning in natural organisms. Therefore, the expanded genetic code of GROs can be exploited both to prevent their undesired survival in natural ecosystems and to block incoming and outgoing horizontal gene transfer with natural organisms.
METHODS
Essential protein selection and buried residue determination
Candidate genes were selected by searching the Keio collection12 of comprehensive single-gene E. coli K-12 knockouts for genes classified as essential. X-ray structures were identified by mapping essential gene GenBank26 PIDs to Protein Data Bank27 (PDB) entries through the UniProtKB28. In cases of multiple PDB entries the highest-resolution structure was selected. 112 high-resolution X-ray structures (resolution <= 2.8 Å) were analyzed. Structures were preprocessed to remove alternative side-chain conformers (the first listed conformer was kept), to remove atoms without occupancy, to remove heteroatoms, to convert selenomethionines to methionines, and to remove chains other than the first listed chain of the essential protein. The solvent accessible surface area (SASA) of each position in each candidate structure was calculated using the PyRosetta29 interface to the Rosetta SasaCalculator class with a 1.0 Å probe radius (a radius smaller than 1.4 Å allowed finer sampling of spaces around candidate positions). Positions were considered buried if their SASA was not more than 20% of the residue-specific average SASA value from a 30-member random ensemble of Gly-X-Gly peptides, where X is the residue type, as determined by the GETAREA method30,31. The average SASA values are Ala: 64.9, Arg: 195.5, Asn: 114.3, Asp: 113.0, Cys: 102.3, Gln: 143.7, Glu: 141.2, His: 154.6, Ile: 147.3, Gly: 87.2, Leu: 146.2, Lys: 164.5, Met: 158.3, Phe: 180.1, Pro: 105.2, Ser: 77.4, Thr: 106.2, Trp: 224.6, Tyr: 193.1, Val: 122.3. By these criteria 13,564 residues in the dataset were considered buried.
Design and refinement of NSAA-dependent proteins
The side chains of each structure were relaxed into local minima of the Rosetta forcefield by the Rosetta sidechain_min application (Rosetta command lines, below). Three separate design simulations were then carried out for each target buried position using Rosetta Design32. The first simulation sets the target position to L-4,4′-biphenylalanine (bipA), as implemented in Rosetta (residue type B30)11, and sets the surrounding residues to either redesign (varies both amino acid identity and side-chain conformation) or repack (varies only the conformation of the wild-type amino acid). Residues with Cα atoms within 6 Å of the target position, or with Cα atoms within 8 Å of the target position and Cβ atoms closer than the Cα atom to the target position were set to redesign. Residues with Cα atoms within 10 Å of the target position, or with Cα atoms within 12 Å of the target position and Cβ atoms closer than the Cα atom to the target postion were set to repack. All other side-chains were fixed at their minimized coordinates, together with all backbone atoms. The resulting energy terms were appended with the target position SASA as calculated by the PyRosetta SasaCalculator with a 1.0 Å probe radius. We term the Rosetta scores of these designs “compensated scores”. In the second simulation, the same calculation is performed, except all positions previously set to redesign are restricted only to repack. We term the resulting scores “uncompensated scores”. The difference between the “compensated score” and the “uncompensated score” reports on the extent to which the target site must change in order to accommodate bipA. In the third simulation, the target position maintains its wild-type identity, all coordinates are fixed at the positions output by the sidechain_min application, and the structure is rescored using the same scoring parameters as the other two simulations (Rosetta command lines, below). We term the resulting scores “wild-type scores”. The difference between the “compensated score” and the “wild-type score” reports on the predicted stability of the redesigned core relative to the wild-type structure.
The design goal was to obtain variants that were functionally stable with bipA at the designed position, while being maximally destabilized with a natural amino acid at the bipA position. Accordingly, designs were filtered for the following criteria:
The minimized “wild-type score” must be less than 10 Rosetta energy units (REU) to ensure that the starting structure can be reasonably modeled with the Rosetta forcefield
The “compensated score” must be less than or equal to the “wild-type score”, to select for redesigned cores that do not destabilize the protein relative to the wild-type sequence
The “uncompensated score” must be greater than the “wild-type score”, to ensure that compensatory mutations are necessary to accommodate bipA
The “compensated score” must be less than the “uncompensated score”, to select for compensatory mutations that improve the stability of the core in the presence of bipA relative to the uncompensated mutant. This requirement also selects for sequences that reduce the fitness of suppressors at the compensatory positions
The SASA score must be < 0.75, to select for cores that tightly pack around bipA, both to select for stability in the presence of bipA and to reduce the fitness of standard amino acids at the bipA or compensatory positions
The designs for 16 engineered UAG sites in 12 enzymes meeting these criteria were then ranked by the difference between “compensated score” and “uncompensated score”, as a key metric for bipA dependence, and were further filtered by the following criteria based on known structural and functional data from the literature:
The redesigned residues must not participate in ligand binding, catalysis or be required for allosteric signal transduction via conformational rearrangements
The product of the reaction must not be environmentally available
The product of the reaction must not be complementable by another environmentally available molecule
Using these criteria 10 designs were subject to refinement. Positions to design, repack, or revert to wild-type were selected by visual inspection. A second round of fixed backbone design was then applied to generate 100 designs from each unrefined structure (Rosetta command lines, below). Designs from six enzymes were carried forward for experimental characterization. Frequently occurring mutations in the refined designs assessed by visual inspection were included in MAGE oligos (Supplementary Table 1). For tyrS, eight additional all-atom designs were encoded by PCR primers for gene assembly (Supplementary Table 3).
Rosetta command lines. All Rosetta calculations were performed with Rosetta version 48561.
-
Example command line for preparative side-chain minimization of scaffold structures
sidechain_min.linuxgccrelease -database __ROSETTA_DATABASE__ -loops::input_pdb __PREFIX__.pdb -output_tag __PREFIX__ -ex1 -ex2 -overwrite
-
Example command line for “wild-type score”
score.linuxgccrelease -database __ROSETTA_DATABASE__ -l *.pdb -score:hbond_His_Phil_fix -in:file:fullatom -no_optH -no_his_his_pairE -score:weights mm_std
-
Example command line for initial design for “compensated score” and “uncompensated score”
fixbb.linuxgccrelease -database __ROSETTA_DATABASE__ -ex1 -ex2 -s __PREFIX__.pdb -resfile __PREFIX__.resfile -minimize_sidechains -score:weights mm_std -score::hbond_His_Phil_fix -no_his_his_pairE -nstruct 1 -out:pdb_gz -overwrite
-
Example command line for refinement of dependent designs
fixbb.linuxgccrelease -database __ROSETTA_DATABASE__ -s 2YXNA_min.pdb -minimize_sidechains -score::hbond_His_Phil_fix -no_his_his_pairE -ex1 -ex2 -nstruct 100 -resfile __PREFIX__.resfile -overwrite
Culture and selection conditions
Growth media consisted of LB-Lennox (LBL, 10 g/L bacto tryptone, 5 g/L sodium chloride, 5 g/L yeast extract). Permissive growth media for bipA-dependent auxotrophs was LBL supplemented with sodium dodecyl sulfate (SDS), chloramphenicol, bipA, and arabinose. Nonpermissive media lacked bipA and arabinose. The following selective agents, nutrients, and inducers were used when indicated: chloramphenicol (20 μg/ml), kanamycin (30 μg/ml), spectinomycin (95 μg/ml), tetracycline (12 μg/ml), zeocin (10 μg/ml), gentamycin (5 μg/ml), SDS (0.005% w/v), vancomycin (64 μg/ml), Colicin E1 (ColE1; ~10 μg/ml), DAP (75 μg/ml), thymidine (100 μg/ml), bipA (10 μM), glucose (0.2% w/v), pyruvate (0.2% w/v), succinate (0.2% w/v), arabinose (0.2% w/v), anhydrotetracycline (30 ng/μL). For strains adk.d6_tyrS.d8_bipARS.d7 and adk.d6_tyrS.d8_int permissive media contained 100 μM bipA. Permissive media for metabolic auxotrophs is LBL supplemented with 75 μg/ml DAP and 100 μg/ml thymidine. TolC selections (SDS) and counterselections (colicin E1) were performed as previously described33. Tdk selections used LBL supplemented with 20 μg/ml 2′-deoxy-5-fluorouridine and 100 μg/ml deoxythymidine, and counterselections used LBL supplemented with 5μM azidothymidine.
Strain engineering
Two strategies were undertaken to engineer redesigned essential proteins. Strains adk.d4, adk.d6, alaS.d5, holB.d1, metG.d3, and pgk.d4 were generated by performing CoS-MAGE14 with designed single stranded oligonucleotide pools (Supplementary Table 1) and tolC co-selection14. Recombined populations were plated on permissive media, and then replica plated on nonpermissive media to screen for bipA-dependent clones. Top candidates were identified by kinetic growth monitoring (Biotek H1 or H4 plate reader) of 10–40 bipA-dependent clones in permissive and nonpermissive liquid growth media. Strains showing robust growth in permissive media and little to no growth in nonpermissive media were carried forward. The tyrS.d6, tyrS.d7, and tyrS.d8 gene variants were constructed by PCR amplification of the E. coli MG1655 tyrS gene with mutagenic primers, followed by full-length Gibson assembly34 (Supplementary Tables 1 and 3) and recombination onto the genome using λ Red recombineering35,36. Strains tyrS.d6, tyrS.d7, tyrS.d8, adk.d6_tyrS.d6, adk.d6_tyrS.d7, and adk.d6_tyrS.d8 were produced by (1) deleting the endogenous tdk gene from C321.ΔA, (2) replacing the endogenous adk and tyrS genes with their codon-shuffled variants (adk(recode)-tdk and tyrS(recode)-tdk, Supplementary Table 3) transcriptionally fused to tdk, and (3) replacing the fusion cassettes with the adk.d6, tyrS.d6, tyrS.d7, or tyrS.d8 variants. Variants of adk.d6, tyrS.d7 and tyrS.d8 containing leucine and tryptophan at the bipA position were constructed by MAGE with oligos containing the appropriate mutations and clonal populations were isolated on LBL plates lacking bipA and arabinose. Triple-enzyme auxotrophs were created by replacing asd with a Δasd::specR cassette. We reactivated mismatch repair using mutS_null_revert-2* in the pgk, adk, and tyrS single-enzyme auxotrophs and all of the multi-enzyme auxotrophs. For construction of the quadruplet tRNAbipA (Supplementary Discussion) QuikChange was used to replace the CUA anticodon with UCUA. Quadruplet versions of adk.d6 and tyrS.d8 with UAGA at the bipA positions were constructed by PCR and Gibson assembly followed by λ Red-mediated recombination into the genome as described above. All genotypes (Supplementary Table 3) were confirmed using mascPCR37 and Sanger sequencing using primers from Supplementary Table 1.
Strain doubling time analysis
Strain doubling times were calculated as previously described9. Briefly, cultures were grown in flat-bottom 96-well plates (150 μL LBL, 34 °C, 300 rpm). Kinetic growth (OD600) was monitored on a Biotek H1 plate reader at 5 minute intervals. Doubling times were calculated by tdouble = Δt*ln(2)/m, where Δt = 5 minutes per time point and m is the maximum slope of ln(OD600) calculated from the linear regression through a sliding window of 5 contiguous time points (20 minutes). For escapee strains exhibiting growth rates that were too slow for this analysis doubling times were calculated by tdouble = Δt*ln(2)/ln(P2/P1), where Δt represents sliding windows of 15 minutes and P2/P1 represents initial/final OD600 values for the window. Strains that exhibited doubling times greater than 900 minutes and/or maximum OD600 values less than 0.2 after the specified culture duration were considered to exhibit no growth (“none observed”) for the given conditions. Improved aeration doubling times for strains adk.d6_tyrS.d8_int and adk.d6_tyrS.d8_bipARS.d7 were obtained by growing strains in 3 ml LBL in 28 ml culture tubes (3 tubes dedicated for each time point), measuring OD600 of three technical replicates in 1 cm cuvettes in a spectrophotometer (Beckman DU640) at 20 minute intervals for 3.66 hours, and determining the slope of the log transformed data over each 40 minute window. Least doubling time (20 min*ln(2)/slope) and corresponding R2 values are reported (Supplementary Table 4).
Expression and purification of tyrS.d7
C321.ΔA cells were grown to mid-log phase in LBL and co-electrotransformed with 5 ng each of plasmid pEVOL-bipA10 and an additional plasmid containing full-length tyrS.d7 as a N-terminal GST fusion under an anhydrotetracycline (aTc) inducible promoter. After 90 minutes of recovery cells were plated on LBL agar supplemented with Cm and Kan. Single colonies were used to inoculate 2 ml starter cultures of LBL supplemented with Cm and Kan that were grown overnight at 34 °C. Saturated overnight growths were diluted 1:100 into six 1 L cultures containing LBL supplemented with Cm and Kan, which were grown at 34 °C with shaking at 250 r.p.m. to an OD600 of 0.6. The temperature was then reduced to 18 °C, and bipA was added to a final concentration of 500 μM. After an additional 60–90 minutes aTc and arabinose were added to final concentrations of 30 ng/ml and 0.2%, respectively. After 24 hours of expression, cells were harvested by centrifugation at 10,000 × g and snap frozen in a dry ice and ethanol bath. Approximately 10 g thawed cell pellet was suspended in 100 ml of Buffer A (20 mM Tris-HCl (pH 7.2), 500 mM NaCl, and 5 % (v/v) glycerol) supplemented with 1 mg/ml lysozyme. After sonication (6 cycles of 30 seconds each), the cell lysate was centrifuged at 20,000 × g for 20 minutes at 4 °C, and the supernatant was mixed with 5 ml of polyethyleneimine (pH 7.9) on ice. After centrifugation again at 20,000 × g for 10 minutes at 4 °C, the supernatant was filtered through a 0.45 μm PVDF membrane, and suspended with 3 ml of glutathione sepharose 4B beads (GE Healthcare Life Sciences). The beads were extensively washed with Buffer A supplemented with 1 mM dithiothreitol (DTT) and incubated with 120 units of PreScission protease (GE Healthcare Life Sciences) at 5 °C for 16 hours. The untagged protein was eluted and dialyzed against Buffer B (20 mM Tris–HCl (pH 7.5), 50 mM NaCl, 10 mM 2-mercaptoethanol and 5 % (v/v) glycerol) at 4 °C. The redesigned enzyme was concentrated to approximately 5.5 mg/ml.
Determination of the tyrS.d7 crystallographic structure
One microliter drops of protein were mixed with an equal volume of a reservoir solution containing 0.1 M sodium malonate (pH 5.5) and 18 % (w/v) polyethylene glycol 3350. Crystals were grown at room temperature via hanging drop vapor phase diffusion, and then were transferred into 0.1 M sodium malonate (pH 5.5), 25 % (w/v) polyethylene glycol 3350 and 15 % ethylene glycol. Crystals were frozen in liquid nitrogen, and diffraction data were collected at the Advanced Light Source (ALS) beamline 5.0.1. The data were processed using the HKL2000 package38. The crystal structure of a truncated E. coli TyrS variant protein (PDB code, 2YXN) was used as a search model in molecular replacement. The crystallographic model was built using COOT39, refined using REFMAC5 and Crystallography and NMR system (CNS)40, and deposited in the RCSB Protein Data Bank (PDB ID code 4OUD). Statistics of the data collection and refinement are provided in Extended Data Table 1.
Mass spectrometry of NSAA-dependent enzymes
Strains adk.d6, tyrS.d7 and tyrS.d8 were grown to midlog phase in 10 ml of permissive media. Cell pellets were obtained and soluble lysate fractions were collected as above. Samples were normalized to 250 μg (adk.d6) or 50 μg (tyrS.d7 and tyrS.d8) total protein content and resolved by SDS-PAGE. Gel slices from each strain containing the enzyme (resolved by size comparison to a known standard) were digested with trypsin. Peptide sequence analysis of each digestion mixture was performed by microcapillary reversed-phase high-performance liquid chromatography coupled with nanoelectrospray tandem mass spectrometry (μLC-MS/MS) on an LTQ-Orbitrap Elite mass spectrometer (ThermoFisher Scientific, San Jose, CA). The Orbitrap repetitively surveyed an m/z range from 395 to 1600, while data-dependent MS/MS spectra on the twenty most abundant ions in each survey scan were acquired in the linear ion trap. MS/MS spectra were acquired with relative collision energy of 30%, 2.5-Da isolation width, and recurring ions dynamically excluded for 60 s. Preliminary sequencing of peptides was facilitated with the SEQUEST algorithm with a 30 ppm mass tolerance against the Uniprot Knowledgebase E. coli K12 reference proteome supplemented with a database of common laboratory contaminants, concatenated to a reverse decoy database. Using a custom version of Proteomics Browser Suite (PBS v.2.7, ThermoFisher Scientific), peptide-spectrum matches were accepted with mass error < 2.5 ppm and score thresholds to attain an estimated false discovery rate of ~1%.
Western blot analysis of tyrS.d7 variant GST fusions
Cell pellets for all variants were obtained as described above, except the expression culture volume was 10 ml. Cells were lysed using B-PER Bacterial Protein Extraction Reagent, lysozyme (100 mg/ml), DNAseI (5000 U/ml), and Halt Protease Inhibitor Cocktail (all Thermo Scientific) according to the manufacturer’s specifications. Lysates were centrifuged at 15,000 × g for 5 minutes and the soluble fractions were collected. Protein concentration was determined fluorometrically using the Qubit Protein Assay Kit (Life Technologies). Lysates were normalized to 5 ug samples, resolved by SDS-PAGE, and electro-blotted onto PVDF membranes (Life Technologies #IB24002). Western blotting was performed using an anti-GST mouse monoclonal primary antibody (Genscript #A00865-40) and anti-GAPDH mouse monoclonal loading control antibody (Thermo Scientific #MA5-15738) followed by secondary binding to a HRP-conjugated anti-mouse antibody (Thermo Scientific #35080). Samples were imaged by luminol chemiluminescence on a ChemiDoc system (BioRad) and protein content was quantified by densitometry and normalized to GAPDH.
Solid media escape assays for natural metabolic and synthetic auxotrophs
All strains were grown in permissive conditions and harvested in late exponential phase. Cells were washed twice in LBL and resuspended in LBL. Viable c.f.u. were calculated from the mean and standard error of the mean (s.e.m.) of 3 technical replicates of 10-fold serial dilutions on permissive media. Three technical replicates were plated on nonpermissive media and monitored for seven days. The order of magnitude of cells plated ranged from 102 to 109 depending on the escape frequency of the strain. Synthetic auxotrophs were plated on two different nonpermissive media conditions: “SC”, LBL with SDS and chloramphenicol (Supplementary Table 4), and “SCA”, LBL with SDS, chloramphenicol, and 0.2% arabinose (Supplementary Table 5). Metabolic auxotrophs were plated on LBL for nonpermissive conditions (Supplementary Table 11). If synthetic auxotrophs exhibited escape frequencies above detection limit (lawns) on SC at day 1, 2 or 7 (alaS.d5, metG.d3, tyrS.d7), escape frequencies for those days were calculated from additional platings at lower density. Additional platings at higher density were also used to obtain day 1 and day 2 escape frequencies for pgk.d4 on SC. The s.e.m. SX̄ across technical replicates of the cumulative escape frequency ν scored for a given day was calculated as: , where τ is the mean number of c.f.u. plated, SX̄τ is the s.e.m. of c.f.u. plated, n is the mean cumulative colony count up to the given day, and SX̄n is the s.e.m. of the cumulative colony count up to the given day. If synthetic auxotroph escapees emerged on SC, three clones were isolated, their growth rates were calculated as described above, and the doubling time of the fastest escapee was recorded (Supplementary Table 4).
Site saturation mutagenesis at designed UAG positions
To site-specifically replace UAG with all other codons, we used MAGE oligonucleotide pools that exactly matched the sequence of the bipA-dependent gene except that the UAG was replaced by all 64 “NNN” codons (Supplementary Table 1). This allowed us to assess which canonical amino acid substitutions resulted in the best survival of synthetic auxotroph escapees. Although some of these amino acid substitutions may be unlikely to be evolutionarily sampled (evolution will favor amino acids with many tRNA gene copies and whose cognate codons are a single mutation from UAG41), this unbiased strategy avoided missing mechanisms of tRNA suppression that are not yet characterized. Immediately after introducing NNN codon diversity via MAGE15, we recovered the cell populations in 1 ml of LBL without supplementing antibiotics, arabinose, or bipA. At this point, functional proteins utilizing bipA for proper expression, folding, and function are still present in the cell, but protein turnover eventually replaces the bipA-dependent proteins with bipA-independent variants in which the UAG codon is replaced by one of the 64 codons. This in turn provides a strong selection for canonical amino acids that can replace bipA and maintain protein function. Samples of the population were taken at five time points after electrotransformation to track the population dynamics—after 1 hour, 100 μL of culture was centrifuged at 16,000 × g, resuspended in 20 μL distilled water (dH2O), and frozen at −20 °C (time point 1); 2 ml of LBL was added to the culture and then growth was allowed to proceed for 3 more hours before 100 μL of culture was centrifuged at 16,000 × g, resuspended in 20 μL dH2O, and frozen at −20 °C (time point 2); the remaining culture was grown overnight to confluence after which 500 μL of culture was centrifuged at 16,000 × g, resuspended in 500 μL dH2O, and frozen at −20 °C (time point 3); 30 μL of confluent culture was diluted into 3 ml of fresh LBL and re-grown to confluence after which 500 μL of culture was centrifuged at 16,000 × g, resuspended in 500 μL dH2O, and frozen at −20 °C (time point 4); finally, 30 μL of confluent culture was diluted into 3 ml of fresh LBL and re-grown to confluence after which 500 μL of culture was centrifuged at 16,000 × g, resuspended in 500 μL dH2O, and frozen at −20 °C (time point 5). After sampling was complete, we had obtained five time points from eight strains amounting to 40 total samples. Population dynamics were analyzed by next-generation sequencing (Methods).
Next-generation sequencing of populations with degeneracy introduced at UAG-positions
We designed custom primers to amplify ~127–146 bp surrounding the UAG codon of each variant and to add Illumina adapters and barcodes for sequencing. In order to reduce primer dimers, we redesigned the P5 primer binding sequence (Sol-P5_alt-PCR, Supplementary Table 1). We used PCR to introduce Illumina sequencing primer binding sites separated from the target amplicon by a 4–6 bp “heterogeneity spacer” that allows low diversity Illumina libraries to be sequenced out of phase42 (Supplementary Table 1). We estimate that ~106 cells (1 μL of a confluent culture containing ~109 cells/ml) were assayed at each time point. This PCR was performed in 20 μL reactions containing 10 μL of KAPA HiFi HotStart ReadyMix, 9 μL of dH2O, 0.5 μL of each 20 μM primer, and 1 μL of template cells. Thermocycling (BioRad C1000 thermocycler) involved heat activation at 95 °C for 3 minutes, followed by 30 cycles of denaturation at 98 °C for 20 seconds, annealing at 62 °C for 15 seconds, and elongation at 72 °C for 30 seconds with a final elongation for 1 minute. PCR products (20 μL) were purified with magNA beads (40 μL)43 and eluted in 20 μL of dH2O. A second PCR amplification introduced Illumina adapters tagged with a unique 6 bp barcode (on the P7 adapter) for each sample and time point. The PCR and purification protocols were identical to those of PCR1 except that the products from PCR1 were used as template and different primers were used. The final DNA libraries were checked on a 1.5% w/v agarose gel and quantitated using a NanoDrop ND-1000 spectrophotometer. Equimolar samples of all 40 libraries were combined in a single tube and sequenced using a SE50 kit on an Illumina MiSeq (Dana Farber Cancer Institute Molecular Biology Core Facility). The P7 and Index1 reads were performed with standard sequencing primers, whereas the P5 read was sequenced with a custom primer (Sol-P5_alt-PCR, Supplementary Table 1).
Sequencing analysis of populations with NNN degeneracy at UAG positions
A simple python script was written to tally each of the 64 UAG→NNN codon mutations and 21 amino acid/translational stop substitutions. We discarded all reads that were too short to discern the NNN codon. For all other reads, a constant seed sequence was indexed within the read, and the NNN codon was located based on proximity to this known seed sequence. The NNN codon identity and translated amino acid identities were stored in dictionaries entitled “aas” and “codons”, respectively. The dictionaries and code are available together at GitHub (https://github.com/churchlab/NNN_sequencing_scripts).
Shannon entropy calculations
Shannon entropy was calculated using the standard relation H(X) = −Σi P(xi) log P(xi).
Whole genome sequencing analysis of mutagenic escapees
We performed whole genome sequencing on 20 escapees and their bipA-dependent parental strains. Sequencing libraries were prepared according to Rohland et al.43 and sequenced with 150 bp paired-end reads on an Illumina MiSeq. We used Millstone (http://churchlab.github.io/millstone/) to automatically call single nucleotide variants from raw fastq data with respect to our starting GRO C321.ΔA (NCBI Genbank Accession CP006698.1). Thus all variant positions are reported relative to the frame of this genome. All variant calls are available on Github (https://github.com/churchlab/dependence/tree/master/supplementary_materials). We then filtered these with custom scripts (https://github.com/churchlab/dependence) to identify alleles involved in hypothetical escape mechanisms: mutations in tRNAs that could lead to UAG suppression, mutations in translation machinery that could increase misincorporation of canonical amino acids, mutations in functionally related genes that could functionally complement the essential gene, and mutations in chaperones or proteases that could stabilize poorly folded Adk and TyrS proteins. Additionally, for strains with adequate coverage we performed de novo assembly of unmapped reads to uncover structural variants not reported by Millstone. We used Velvet44 with a hash length of 21 and the following parameters for the graphing step: -cov_cutoff 20 -ins_length 200 -ins_length_sd 90. We then systematically queried NCBI BLAST to identify each de novo sequence and biased the BLAST results to prefer hits against the canonical MG1655 genome so that we could later group contigs by position (https://github.com/churchlab/dependence/blob/master/supplementary_materials/velvet_contigs_and_BLAST_data_non_permissive_8_strains.csv). Putative Lon insertions were visually confirmed using Millstone’s JBrowse portal.
Integration of bipARS and tRNAbipA
Genomic integration of bipARS and tRNAbipA was achieved in two steps by first replacing the endogenous tdk gene with the ParaBAD-inducible bipARS gene from pEVOL. Subsequently, the pEVOL tRNA and chloramphenicol resistance gene were inserted immediately downstream of ParaBAD-bipARS. Kapa HiFi Ready Mix was used to amplify each PCR product (see Supplementary Table 1 for primer sequences), and λ Red recombination was used to introduce the PCR products into the genome. Proper insertion of the desired cassettes was confirmed by PCR using tdk.seq-f and tdk.seq-r. We observed that 10 μM bipA was not adequate to support growth of adk.d6 or tyrS.d8 when bipARS was integrated into the genome; however, 100 μM bipA accommodated robust growth of adk.d6_int, tyrS.d8_int, and adk.d6_tyrS.d8_int.
Design of the bipARS.d7 bipA-dependent synthetase
We applied our computational second-site suppressor strategy to position V290 of the bipyridylalanyl-tRNA synthetase X-ray structure (PDB code 2PXH, chain A). This position corresponds to bipA303 in our X-ray structure of tyrS.d7 when the structures are superimposed (alignable core backbone r.m.s.d. of 3.6 Å). We hypothesized that this position may be amenable to redesign in homologous structures. Six designs covering sequence variability observed in the computational models (Supplementary Table 2) were produced by PCR amplification of bipARS with mutagenic primers (Supplementary Table 1) and isothermal assembly into the pEVOL vector, maintaining only the arabinose-inducible copy of bipARS. We also included the D286R mutation previously shown to increase synthetase activity45 in all constructs. Since the bipARS designs should require bipA to translate, fold and function, all derived strains were initially co-transformed with a nonreplicating plasmid (R6γ origin of replication) containing a wild-type copy of bipARS to jumpstart production of tRNAbipA. Designs were co-transformed with the jumpstart plasmid into C321.ΔA, and transformants were then transformed with a previously described GFP reporter plasmid containing a single UAG codon9 to measure synthetase activity by GFP fluorescence. One design (bipARS.d7; T/A/G/G/A/bipA) produced > 5-fold bipA-dependent induction of fluorescence in permissive media but failed to induce any bipA-dependent fluorescence after passaging overnight 1:150 in nonpermissive media followed by an identical passage in permissive media. Since any functional synthetase remaining after nonpermissive passaging should facilitate exponential production of additional synthetase, this behavior suggests strong dependence of bipARS.d7 on bipA for translation and folding resulting in total clearance of bipARS.d7 and tRNAbipA after overnight growth in nonpermissive conditions. The bipARS.d7/tRNAbipA and jumpstart vectors were co-transformed into C321.ΔA and then adk.d6 and tyrS.d8 were introduced as described above.
Growth competition assays
The assayed single- and double-enzyme synthetic auxotroph escape strains (pgk.d4 esc. 1, 2, and 3; adk.d6_tyrS.d8 esc. 1, 2, and 3) were transformed with a pZE21 vector46 bearing mCFP under anhydrotetracycline (aTc) inducible control in the multiple cloning site. The parental prototrophic C321.ΔA strain was similarly transformed with an identical vector except that the fluorophore is YFP. Strains were grown up to late exponential phase in LBL supplemented with antibiotics (SDS, chloramphenicol, kanamycin), inducers (0.1% L-arabinose, 100 ng/ml aTc), and bipA. Cells were washed twice in M9 salts and adjusted to a cell concentration of roughly 1 × 109 cells/ml. Biological replicates of synthetic auxotroph escapees were mixed with the C321.ΔA strain at a ratio of 100:1 and diluted to a seeding concentration of roughly 2.5 × 107 cells/ml in nonpermissive media (LBL supplemented with SDS, chloramphenicol, kanamycin and aTc). Growth kinetics of the competition mixture were assayed in 200 μl sample volumes on microtiter plates incubated in a Biotek Synergy microplate reader at 34 °C. Cell mixtures were fixed in PBS with 1% paraformaldehyde at time 0 and at 8 hours. Fixed cells were run on a BD LSRFortessa and populations were binned based on YFP expression level. CFP was not used for species discrimination but rather to maintain consistent fitness costs associated with episomal DNA maintenance and fluorophore expression.
Bacterial lysate growth assays
All strains were grown up in permissive conditions and harvested in late exponential phase. Cells were washed twice in M9 salts (6 g/L Na2HPO4, 3 g/L KH2PO4, 1 g/L NH4Cl, 0.5 g/L NaCl) by centrifugation at 17,900 × g and then diluted 100-fold into LBL supplemented with 166.66 ml/L trypsin digested E. coli extract (Teknova Cat. No. 3T3900). Growth kinetics were assayed in 200 ul sample volumes on microtiter plates as described above. Three biological replicates were performed by splitting a single well-mixed initial seeding population.
Conjugal escape assays
The conjugal donor population was produced using the Epicentre EZ-Tn5 Custom Transposome kit to insert a mosaic-end-flanked kanR-oriT cassette into random positions of the E. coli MG1655 genome. The population of integrants was plated on LBL agar plates supplemented with kanamycin. Approximately 450 clones were lifted from the plate and pooled, which corresponds to one kanR-oriT per ~10 kb region of the genome, assuming an equal distribution of transposition across the 4.6 megabase E. coli MG1655 genome. The pRK24 conjugal plasmid was conjugated37 from E. coli strain 1100-247 into the kanR-oriT donor population. The kanR-oriT insertion sites were confirmed to be well-distributed. Briefly, the donor population was sheared on a Covaris E210, end repaired, and ligated to Illumina adapters as described by Rohland and Reich43. Genomic sequences flanking the insertion site were amplified using the Sol-P5-PCR primer and a series of nested primers (Supplementary Table 1) that hybridize within the kanR gene. PCR products corresponding to ~1 kb were gel purified from the smear and TOPO cloned (Invitrogen pCR™-Blunt II-TOPO®). Flanking genomic sequences were then identified by Sanger sequencing 96 TOPO clones. Conjugal escape assays were performed as described previously37 with 50 minute and 12 hour conjugal duration and a donor:auxotroph ratio of 1:100. Three technical replicates of two biological replicates were performed for all conjugation assays with the exception of the double-enzyme synthetic auxotroph experiments, which were performed with three biological replicates (3 technical replicates each) to produce enough escapees for mascPCR screening. To determine the proportion of the genome overwritten by donor DNA the following numbers of colonies were scored for the 50 minute/12 hour time points: adk.d6 n=51/6; tyrS.d8 n=44/7; adk.d6_tyrS.d8 n=8/59; adk.d6_tyrS.d8_asd:specR n=5/38. This set omits a small collection of clones that could not be scored due to polyclonality.
Extended Data
Extended Data Figure 1.

bipA dependence in synthetic auxotrophs.
Prototrophic and synthetic auxotrophic strains were grown in titrations of bipA and monitored in a microplate reader (Methods). Media for all bipA concentrations contained SDS, chloramphenicol, and arabinose. Doubling times for three technical replicates are shown. Positive and negative error bars are s.e.m. Growth was undetectable for synthetic auxotrophs at 0.00 μM, 0.01 μM and 0.10 μM bipA, as well as 0.50 μM bipA for adk.d6_tyrS.d8.
Extended Data Figure 2.

Mass spectrometry of NSAA-dependent enzymes.
Mass spectrometry was performed and peptide-spectrum matches (PSMs) were obtained as described in the Methods. Datasets were culled of minor contaminant PSMs and researched with SEQUEST against adk.d6 (X at position 178) or tyrS.d7 and tyrS.d8 (X at 303) sequences without taking into account enzyme specificity. To interrogate the sequences for bipA, tryptophan and leucine, the amino acid X was given the mass of leucine and searches were performed with differential modifications of +110.01565 and +72.99525 to account for the masses of bipA and tryptophan, respectively. In all samples, only bipA, and not leucine or tryptophan, was detected at these positions. The peptide spectrum match for adk.d6 is shown. Peptides observed to contain bipA are LVEYHQMTAP[bipA]IGYVSK (adk.d6), AQYV[bipA]AEQVTR (tyrS.d7) and AQYV[bipA]AEQATR (tyrS.d8).
Extended Data Figure 3.

Crystal structure of tyrS.d7.
a, Overall structure of the redesigned enzyme. The N-terminal domain (residues 4-330) that catalyzes tyrosine activation, the C-terminal tRNA binding domain (residues 350-424) and their connecting region are colored cyan, blue and yellow, respectively. The residues 232-241 are disordered (dash line). b, Comparison between the C-terminal tRNA recognition domains of tyrS.d7 (blue) and of T. thermophilus TyrS (orange; PDB code 1H3E). The residues 352-442 of the hyperthermophilic TyrS are shown. c, The N-terminal domain of the engineered protein is superposed on the crystal structure of its parental enzyme (green; PDB code 1X8X). The KMSKS loop of the parental enzyme is highlighted in magenta. d, Tyrosine molecule bound to tyrS.d7. An electron density map of L-tyrosine is shown as a gray mesh (2Fo-Fc contoured at 1.2 σ; upper panel). A tyrosine and the surrounding protein fold of tyrS.d7 (cyan) are very similar to those of the wild type TyrS structure (green; lower panel).
Extended Data Figure 4.

Western blot analysis of tyrS.d7 variants.
Variants of tyrS.d7 with leucine or tryptophan at the bipA position were expressed as GST fusions under identical conditions and analyzed by Western blot (Methods). Soluble protein content was quantified by densitometry and normalized to GAPDH. Mutating bipA to leucine or tryptophan reduced soluble TyrS levels by 2.5- or 2.1-fold, respectively (p < 0.05 by two-tailed unpaired student’s t-test with unequal variances). Three technical replicates were performed; a representative image is shown.
Extended Data Figure 5.
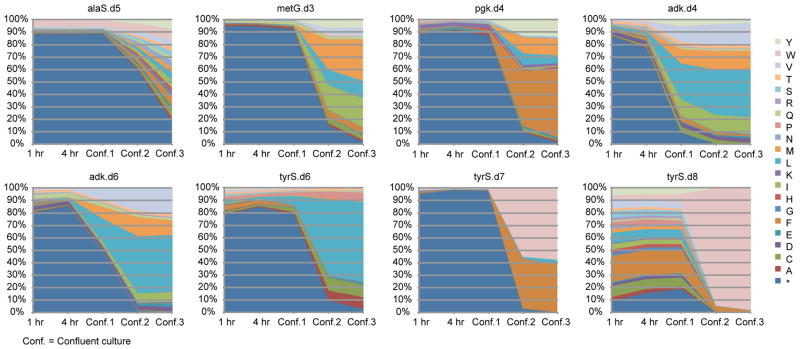
Population selection dynamics for canonical amino acid substitutions at designed UAG positions.
For each plot, degenerate MAGE oligos were used to create a population of cells in which the UAG codon was mutated to all 64 codons. Codon substitutions leading to survival in the absence of bipA were selected by growth in LBL media without bipA and arabinose supplementation. Aliquots of the culture population were taken at 1 hour, 4 hours, confluence 1 (once the culture reached confluence), confluence 2 (after regrowth of a 100-fold dilution of confluence 1), and confluence 3 (after regrowth of a 100-fold dilution of confluence 2). The amino acid identity at the bipA position was probed by targeted Illumina sequencing. Residual bipA-containing proteins were expected to remain active until intracellular protein turnover cleared them from the cell, making the 1 hour time point a reasonable representation of initial diversity present in the population. This data shows the relative fitness of amino acid substitutions in a given protein variant; relative fitness across multiple protein variants cannot be accurately assessed from this data.
Extended Data Figure 6.

Natural metabolites can circumvent auxotrophies.
(a–d) Synthetic auxotrophs of pgk can be complemented by pyruvate or succinate. Strains were cultured in LBL in the presence of pyruvate, succinate, glucose or bipA (10 μM) and monitored by kinetic growth. a, The single-enzyme synthetic auxotroph pgk.d4 grows similarly to prototrophic C321.ΔA (b) in the presence of pyruvate and succinate, but not glucose. Synthetic auxotrophs of adk (c) and tyrS (d) grow robustly in bipA but cannot be complemented by pyruvate or succinate. Growth of pgk.d4 and adk.d6 in glucose after 1000 minutes is due to mutational escape (loss of bipA dependence). e, The synthetic auxotroph parental strain (C321.ΔA), a second prototrophic MG1655-derived strain (EcNR1), and three natural auxotroph derivatives of EcNR1 were grown in LBL supplemented with 166.66 ml/L bacterial lysate (Teknova). Growth curves are shown with doubling times ± one standard deviation of three technical replicates next to the labels. The conditions fully complement the metabolic auxotrophy of EcNR1.ΔthyA, which doubles as robustly as prototrophic EcNR1. Strains lacking the asd gene (EcNR1.Δasd and the EcNR1.ΔasdΔthyA double knockout) show more impairment but enter exponential growth with doubling times of 91 to 137 minutes, respectively. f, (single-) and g, (double-)enzyme synthetic auxotrophies are not complemented by natural products in rich media or bacterial lysate. h, When the Δasd auxotrophy is combined with double-enzyme synthetic auxotrophies the natural products are no longer sufficient to support growth. No growth is indicated by * in f–h.
Extended Data Figure 7.
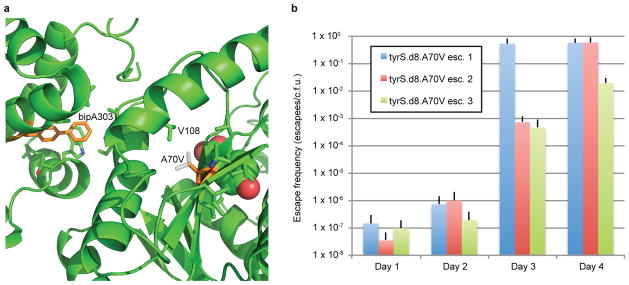
Analysis of the A70V mutation as an escape mechanism for tyrS.d8.
a, The X-ray structure of tyrS.d7 is shown; tyrS.d8 varies by the single mutation V307A. BipA303, A70 and their neighboring side chains are shown in sticks, with bipA303 and A70 colored orange. The bound tyrosine substrate is shown in space fill. The A70V mutation (white sticks) may stabilize the catalytic domain when bipA is replaced by natural amino acids by tightly packing with neighboring side chains including V108. b, Escape frequencies on nonpermissive media for three separately constructed tyrS.d8 A70V strains are shown for days 1 through 4. Although escapees are growth impaired in the absence of bipA (Supplementary Table 10), all cells form colonies after 5 days, suggesting that A70V confers 100% survival on nonpermissive media.
Extended Data Figure 8.
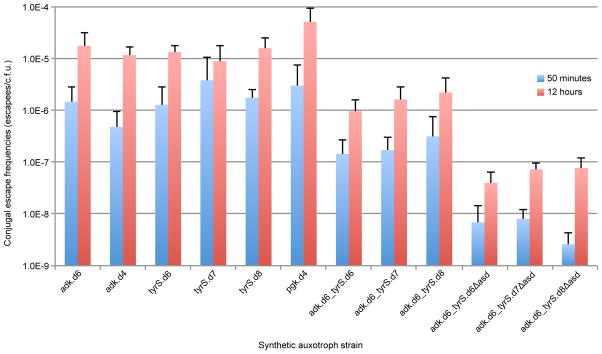
Conjugal escape frequencies of synthetic auxotrophs.
Single, double, and triple-enzyme auxotrophs were assayed to determine the frequency of escape by horizontal genetic transfer and recombination from a prototrophic donor as described in the methods. These results highlight the benefit of having multiple auxotrophies distributed throughout the genome. Notably, scaling from a single synthetic auxotroph to three distributed auxotrophies results in a reduction of conjugal escape by at least two orders of magnitude.
Extended Data Table 1.
Data collection and refinement statistics
| tyrS.d7 | |
|---|---|
| Data collection | |
| Space group | P 1211 |
| Cell dimensions | |
| a, b, c (Å) | 81.3, 67.2, 90.7 |
| a, b, g (°) | 90.0, 102.6, 90.0 |
| Resolution (Å) | 50.0 – 2.65 (2.74–2.65) * |
| Rsym or Rmerge | 0.074 (0.497) |
| I/sI | 29.2 (4.65) |
| Completeness (%) | 99.0 (98.4) |
| Redundancy | 7.6 (7.7) |
| Refinement | |
| Resolution (Å) | 45 – 2.65 |
| No. reflections | 26407 |
| Rwork/Rfree | 0.222/0.306 |
| No. atoms | |
| Protein | 6038 |
| Ligand/ion | 13 |
| Water | 57 |
| B-factors | |
| Protein | 58.66 |
| Ligand/ion | 52.10 |
| Water | 48.24 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.012 |
| Bond angles (°) | 1.530 |
The data was collected using a single crystal.
Highest resolution shell is shown in parenthesis.
Extended Data Table 2.
Cost per liter of culture for commonly used NSAAs
| NSAA | Vendor | Name at vendor | CAS# | MW | Cat# for 1g | Price of 1g | Optimal conc. (mM) | Cost per liter of culture |
|---|---|---|---|---|---|---|---|---|
| pAcF | peptech | L-4-Acetylphenylalanine | 122555-04-8 | 207.23 | AL624-1 | $500.00 | 1.0 | $103.62 |
| pAzF | Bachem | H-4-Azido-Phe-OH | 33173-53-4 | 206.2 | F-3075.0001 | $285.00 | 5.0 | $293.84 |
| pCNF | peptech | L-4-Cyanophenylalanine | 167479-78-9 | 190.2 | AL240-1 | $150.00 | 1.0 | $28.53 |
| bpa | peptech | L-4-Benzoylphenylalanine | 104504-45-2 | 269.3 | AL660-1 | $100.00 | 1.0 | $26.93 |
| napA | peptech | L-2-Naphthylalanine | 58438-03-2 | 215.25 | AL121-1 | $80.00 | 1.0 | $17.22 |
| bipA | peptech | L-4,4′-Biphenylalanine | 155760-02-4 | 241.29 | AL506-1 | $150.00 | 0.1 | $3.62 |
| pIF | peptech | L-4-Iodophenylalanine | 24250-85-9 | 291.09 | AL261-1 | $40.00 | 1.0 | $11.64 |
| bipyA | Asis Chem | Bipyridylalanine | custom synthesis | 245.282 | (25 g price) | $10,000/25g | 1.0 | $98.11 |
Supplementary Material
Acknowledgments
We thank Doug Renfrew for help with NSAA modeling in Rosetta, Dan Goodman and Raj Chari for sequence analysis assistance, Michael Napolitano for advice on Lon-mediated escape assays, Jun Teramoto and Barry Wanner for the pJTE2 jumpstart plasmid, and Farren Isaacs for manuscript comments. D.J.M. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation. M.J.L. was supported by a U.S. Department of Defense National Defense Science and Engineering Graduate Fellowship. M.T.M. was supported by a Doctoral Study Award from the Canadian Institutes of Health Research. The research was supported by Department of Energy Grant DE-FG02-02ER63445.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions D.J.M., M.J.L., M.T.M. and G.M.C. conceived the project and designed the study, with D.J.M. as computational lead and M.J.L. as experimental lead. D.J.M. computationally designed synthetic auxotrophs, performed strain engineering, characterized escape frequencies and fitness of synthetic auxotrophs, performed western blot analyses and prepared samples for mass spectrometry and X-ray crystallography. M.J.L. performed strain engineering, performed site-saturation mutagenesis at UAG positions, performed whole-genome sequencing of escapees, validated escape mechanisms and assessed HGT by conjugation. M.T.M. measured escape frequencies and fitness of natural metabolic auxotrophs, performed competition assays and assessed HGT by conjugation. R.T. and B.L.S. crystallized tyrS.d7 and determined the X-ray structure. G.K. analyzed whole-genome sequencing data of escapees. J.N. and C.J.G. developed the tdk selection protocol. D.J.M., M.J.L. and M.T.M. wrote the paper.
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data Bank under accession code 4OUD.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Moe-Behrens GH, Davis R, Haynes KA. Preparing synthetic biology for the world. Frontiers in microbiology. 2013;4:5. doi: 10.3389/fmicb.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molin S, et al. Conditional Suicide System for Containment of Bacteria and Plasmids. Nat Biotech. 1987;5:1315–1318. [Google Scholar]
- 3.Li Q, Wu Y-J. A fluorescent, genetically engineered microorganism that degrades organophosphates and commits suicide when required. Appl Microbiol Biotechnol. 2009;82:749–756. doi: 10.1007/s00253-009-1857-3. [DOI] [PubMed] [Google Scholar]
- 4.Curtiss R., 3rd Biological containment and cloning vector transmissibility. The Journal of infectious diseases. 1978;137:668–675. doi: 10.1093/infdis/137.5.668. [DOI] [PubMed] [Google Scholar]
- 5.Ronchel MC, Ramos JL. Dual system to reinforce biological containment of recombinant bacteria designed for rhizoremediation. Appl Environ Microbiol. 2001;67:2649–2656. doi: 10.1128/AEM.67.6.2649-2656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright O, Delmans M, Stan GB, Ellis T. GeneGuard: A Modular Plasmid System Designed for Biosafety. ACS synthetic biology. 2014 doi: 10.1021/sb500234s. [DOI] [PubMed] [Google Scholar]
- 7.Knudsen S, et al. Development and testing of improved suicide functions for biological containment of bacteria. Appl Environ Microbiol. 1995;61:985–991. doi: 10.1128/aem.61.3.985-991.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasotti L, Zucca S, Lupotto M, Cusella De Angelis MG, Magni P. Characterization of a synthetic bacterial self-destruction device for programmed cell death and for recombinant proteins release. Journal of biological engineering. 2011;5:8. doi: 10.1186/1754-1611-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lajoie MJ, et al. Genomically recoded organisms expand biological functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie J, Liu W, Schultz PG. A genetically encoded bidentate, metal-binding amino acid. Angewandte Chemie. 2007;46:9239–9242. doi: 10.1002/anie.200703397. [DOI] [PubMed] [Google Scholar]
- 11.Renfrew PD, Choi EJ, Bonneau R, Kuhlman B. Incorporation of noncanonical amino acids into Rosetta and use in computational protein-peptide interface design. PLoS One. 2012;7:e32637. doi: 10.1371/journal.pone.0032637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu HC, Wu TC. Isolation and characterization of a glucosamine-requiring mutant of Escherichia coli K-12 defective in glucosamine-6-phosphate synthetase. J Bacteriol. 1971;105:455–466. doi: 10.1128/jb.105.2.455-466.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carr PA, et al. Enhanced multiplex genome engineering through cooperative oligonucleotide co-selection. Nucleic Acids Research. 2012 doi: 10.1093/nar/gks455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang HH, et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon CE. A Mathematical Theory of Communication. Bell System Technical Journal. 1948;27:379–423. [Google Scholar]
- 17.saiSree L, Reddy M, Gowrishankar J. IS186 insertion at a hot spot in the lon promoter as a basis for lon protease deficiency of Escherichia coli B: identification of a consensus target sequence for IS186 transposition. J Bacteriol. 2001;183:6943–6946. doi: 10.1128/JB.183.23.6943-6946.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol. 2001;40:397–413. doi: 10.1046/j.1365-2958.2001.02383.x. [DOI] [PubMed] [Google Scholar]
- 19.Steidler L, et al. Biological containment of genetically modified Lactococcus lactis for intestinal delivery of human interleukin 10. Nat Biotechnol. 2003;21:785–789. doi: 10.1038/nbt840. [DOI] [PubMed] [Google Scholar]
- 20.Smillie CS, et al. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 21.Wollman EL, Jacob F, Hayes W. Conjugation and genetic recombination in Escherichia coli K-12. Cold Spring Harb Symp Quant Biol. 1956;21:141–162. doi: 10.1101/sqb.1956.021.01.012. [DOI] [PubMed] [Google Scholar]
- 22.Mukai T, et al. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010;38:8188–8195. doi: 10.1093/nar/gkq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kortemme T, Morozov AV, Baker D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J Mol Biol. 2003;326:1239–1259. doi: 10.1016/s0022-2836(03)00021-4. S0022283603000214 [pii] [DOI] [PubMed] [Google Scholar]
- 24.Malyshev DA, et al. A semi-synthetic organism with an expanded genetic alphabet. Nature. 2014;509:385–388. doi: 10.1038/nature13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt M, de Lorenzo V. Synthetic constructs in/for the environment: managing the interplay between natural and engineered Biology. FEBS Lett. 2012;586:2199–2206. doi: 10.1016/j.febslet.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2005;33:D34–38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhury S, Lyskov S, Gray JJ. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. Journal of Computational Chemistry. 1998;19:319–333. doi: 10.1002/(SICI)1096-987X(199802)19:3<319::AID-JCC6>3.0.CO;2-W. [DOI] [Google Scholar]
- 31.Zhu H, Fraczkiewicz R, Braun W. Solvent Accessible Surface Areas, Atomic Solvation Energies, and Their Gradients for Macromolecules. 2012 < http://curie.utmb.edu/area_man.html>.
- 32.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci U S A. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gregg CJ, et al. Rational optimization of tolC as a powerful dual selectable marker for genome engineering. Nucleic Acids Research. 2014 doi: 10.1093/nar/gkt1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 35.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu D, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Isaacs FJ, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. In: Methods in Enzymology. Carter Charles W., Jr, editor. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Eggertsson G, Soll D. Transfer ribonucleic acid-mediated suppression of termination codons in Escherichia coli. Microbiological reviews. 1988;52:354–374. doi: 10.1128/mr.52.3.354-374.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fadrosh DW, et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2:6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome research. 2012;22:939–946. doi: 10.1101/gr.128124.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 46.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1–I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tolonen AC, Chilaka AC, Church GM. Targeted gene inactivation in Clostridium phytofermentans shows that cellulose degradation requires the family 9 hydrolase Cphy3367. Mol Microbiol. 2009;74:1300–1313. doi: 10.1111/j.1365-2958.2009.06890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.