

Abstract
Objective
In rheumatoid arthritis (RA), fibroblast-like synoviocytes (FLS) that line joint synovial membranes aggressively invade the extracellular matrix, destroying cartilage and bone. As signal transduction in FLS is mediated through multiple pathways involving protein tyrosine phosphorylation, we sought to identify protein tyrosine phosphatases (PTPs) regulating the invasiveness of RA FLS. We describe that the transmembrane receptor PTPκ (RPTPκ), encoded by the transforming growth factor (TGF) β-target gene, PTPRK, promotes RA FLS invasiveness.
Methods
Gene expression was quantified by quantitative PCR. PTP knockdown was achieved using antisense oligonucleotides. FLS invasion and migration were assessed in transwell or spot assays. FLS spreading was assessed by immunofluorescence microscopy. Activation of signalling pathways was analysed by Western blotting of FLS lysates using phosphospecific antibodies. In vivo FLS invasiveness was assessed by intradermal implantation of FLS into nude mice. The RPTPκ substrate was identified by pull-down assays.
Results
PTPRK expression was higher in FLS from patients with RA versus patients with osteoarthritis, resulting from increased TGFB1 expression in RA FLS. RPTPκ knockdown impaired RA FLS spreading, migration, invasiveness and responsiveness to platelet-derived growth factor, tumour necrosis factor and interleukin 1 stimulation. Furthermore, RPTPκ deficiency impaired the in vivo invasiveness of RA FLS. Molecular analysis revealed that RPTPκ promoted RA FLS migration by dephosphorylation of the inhibitory residue Y527 of SRC.
Conclusions
By regulating phosphorylation of SRC, RPTPκ promotes the pathogenic action of RA FLS, mediating cross-activation of growth factor and inflammatory cytokine signalling by TGFβ in RA FLS.
INTRODUCTION
Invasiveness is a pathogenic phenotype of fibroblast-like synoviocytes (FLS) in rheumatoid arthritis (RA).1–4 FLS secrete components of synovial fluid and provide structural and dynamic support to the joint. In RA, however, FLS assume intrinsic invasive features (referred to as a tumour-like phenotype) and mediate destruction of cartilage and bone. FLS obtained from patients with RA and cultured ex vivo or implanted into immunodeficient mice display increased invasiveness compared with FLS from healthy subjects or patients with osteoarthritis (OA).4,5 Targeting of FLS is being considered an option for the development of new therapies for RA.1,2,6
FLS behaviour is controlled by a network of signalling pathways, many of which rely upon reversible phosphorylation of proteins on tyrosine residues.2 Tyrosine phosphorylation results from the balanced action of protein tyrosine kinases (PTKs) and phosphatases (PTPs). At least 50 PTPs are expressed in FLS;7 however, with the exception of only a few studies,7–9 the involvement of PTPs in FLS intracellular signalling remains unexplored. We recently profiled the expression of the PTPome in RA FLS and showed that PTPN11, encoding the SH2-domain-containing PTP 2 (SHP-2), is overexpressed in RA FLS compared with FLS from patients with OA.7 Functional studies revealed that SHP-2 mediates the aggressive phenotype of RA FLS by promoting both survival and invasiveness of these cells. With the objective of identifying other signalling mediators involved in promoting the unique aggressive phenotype of RA FLS, we explored the role of another PTP we found overexpressed in RA FLS compared with OA FLS, PTPRK.
METHODS
Further information is available in the online supplementary methods.
Preparation of FLS
FLS lines were obtained from the UCSD Clinical and Translational Research Institute Biorepository. Each line had been previously obtained from a different patient with OA or RA. Discarded synovial tissue from patients with OA and RA had been obtained at the time of total joint replacement or synovectomy, as previously described.10 The diagnosis of RA conformed to American College of Rheumatology 1987 revised criteria.11 FLS were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Manassas, Virginia, USA) with 10% fetal bovine serum (FBS, Omega Scientific, Tarzana, California, USA), 6 mM L-glutamine, 50 μg/mL gentamicin, 100 units/mL of penicillin and 100 μg/mL streptomycin (Life Technologies, Carlsbad, California, USA) at 37°C in a humidified 5% CO2 atmosphere. For all experiments in this study, FLS were used between passages 4 and 10, and cells were synchronised in 0.1% FBS (serum-starvation media) for 48 h prior to analysis or functional assays. Since it has been reported that human FLS maintain a very stable transcriptional phenotype between passages 4 and 7,12 for comparison of mRNA expression between OA and RA FLS, cells were used between passages 4 and 6.
Antibodies and other reagents
The rabbit antireceptor protein tyrosine phosphatase κ (RPTPκ) antibody was a kind gift from Axel Ullrich (Max Planck Institute of Biochemistry). The anti-cadherin-11 antibody was purchased from Life Technologies. All other primary antibodies were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA). Secondary antibodies were purchased from GE Healthcare Life Sciences (Pittsburgh, Pennsylvania, USA). Transforming growth factor (TGF)-β1, tumour necrosis factor (TNF)-α, interleukin-1β (IL-1β) and platelet-derived growth factor BB (PDGF-BB) were purchased from eBioscience (San Diego, California, USA). Chemical inhibitors PP2, U73122 and PF573228, and horseradish peroxidase-conjugated S-protein were purchased from EMD Millipore (Billerca, Massachusetts, USA). Unless otherwise specified, chemicals and all other reagents were purchased from Sigma-Aldrich.
FLS treatment with antisense oligonucleotides
Cells were treated with 2.5 μM antisense oligonucleotides (ASOs; Gene Tools, LLC, Philomath, Oregon, USA) for 7 days. ASO was replaced in fresh culture medium after 3 days and in cell synchronisation medium after 5 days.
Transwell invasion assays
In vitro invasion assays were performed in transwell systems as previously described.5,13 Following treatment with ASO, RA FLS (2.5–5×105) were resuspended in assay media (DMEM with 0.5% BSA) and allowed to invade through BD BioCoat GFR Matrigel chambers in response to 50 ng/mL PDGF-BB for 24 h. Cells were prestained with 2 μM CellTracker Green or stained post invasion with 2 μM Hoechst (Life Technologies) for 30 min at room temperature. Fluorescence of invading cells on each membrane was visualised using an Eclipse 80i microscope. Images were acquired from four non-overlapping fields per membrane, and invading cells in each field were counted using ImageJ software. Each experiment included three to four membranes per sample.
Transwell migration assays
The transwell migration assays were performed similar to the invasion assays. For experiments with chemical inhibitors, cells were pretreated with compound or dimethylsulfoxide for 30 min. FLS were allowed to migrate through uncoated trans-well chambers in response to 5% FBS for the times indicated in the figure legends. Fluorescence of migrating cells on each membrane was visualised as above. Each experiment included three to four membranes per sample.
Statistical analysis
Two-tailed statistical analyses were performed as indicated in the figure legends using GraphPad Prism software. A comparison was considered significant if p value was <0.05.
RESULTS
PTPRK expression is upregulated in RA FLS
Comparison of PTP expression in FLS from three patients with RA and three patients with OA revealed increased PTPRK mRNA in RA FLS (figure 1A). PTPRK encodes RPTPκ, which belongs to a transmembrane PTP subfamily, including RPTPμ (encoded by PTPRM), RPTPρ (encoded by PTPRT) and RPTPψ (encoded by PTPRU). This subfamily is characterised by an extracellular region of a Meprin-A5-protein PTPμ domain, an immunoglobulin-like domain and four fibronectin III-type repeats and an intracellular region containing a juxtamembrane region and two PTP domains, of which only the first has catalytic activity.14 PTPRK caught our attention because it is reported to regulate cell growth and migration through dephosphorylation of PTKs, cadherin proteins and β-catenin.15–17 We reasoned RPTPκ might play a role in migration and invasion of FLS. We thus retested the expression of PTPRK in a further set of FLS lines from 13 patients with RA and 12 patients with OA and confirmed significantly increased (1.86-fold; p<0.05) PTPRK expression in RA FLS (figure 1B). We also detected increased expression of RPTPκ protein in RA compared with OA FLS (figure 1C). To confirm that RPTPκ is expressed in the primary rheumatoid synovial lining, we performed immunohistochemistry on synovial sections obtained from biopsies from patients with RA and found prominent expression of RPTPκ in the synovial intimal lining (figure 1D and see online supplementary figure S1).
Figure 1.

Transforming growth factor (TGF)-β1-responsive PTPRK is overexpressed in rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS). (A and B) PTPRK mRNA expression in FLS was measured by quantitative PCR. (A) Median±range is shown. (B) Median±IQR is shown. *p<0.05, Mann–Whitney test. (C) Western blotting of lysates of RA and osteoarthritis (OA) FLS. (D) Immunohistochemical (IHC) staining of RA synovial section using antireceptor protein tyrosine phosphatase κ (RPTP κ) antibody. (E) PTPRK and PTPRM mRNA expression in RA FLS was measured following cell stimulation with 50 ng/mL TGFβ1 for 24 h. Median±IQR is shown. *p<0.05, Mann–Whitney test. (F) PTPRK and TGFB1 mRNA expression in RA and OA FLS was measured. Graph shows PTPRK vs TGFB1 expression for each line. (G) RA FLS were treated with 25 μM SB505124 or dimethylsulfoxide (DMSO) for 24 h. Median±IQR PTPRK expression is shown. *p<0.05, Mann–Whitney test.
PTPRK overexpression in RA FLS is TGFB1 dependent
RA FLS were reported to express higher levels of TGFB1 than OA FLS.18 As PTPRK is a reported TGFβ/SMAD (Sma and mothers against decapentaplegic protein homolog) target gene,15 we reasoned the increased PTPRK expression in RA FLS might be due to increased expression of TGFB1. As shown in figure 1E, the expression of PTPRK, but not PTPRM, was increased 1.90-fold (p<0.05) in response to treatment of cells with TGFβ1. PTPRK expression is OA FLS was similarly induced by TGFβ1 stimulation (1.70-fold, p<0.05; see online supplementary figure S2) Treatment of RA FLS with the inflammatory cytokines TNF or IL-1 did not affect PTPRK expression (see online supplementary figure S3). We next tested whether the expression of PTPRK correlated with the expression of TGFB1 in RA FLS. As shown in figure 1F, we confirmed the trend of increased expression of TGFB1 in RA FLS18 and found a significant positive correlation between the expression levels of PTPRK and TGFB1 in the RA (Spearman r=0.5824, p<0.05), but not in the OA FLS. We then tested whether this was due to TGFB1-mediated upregulation of PTPRK or due to PTPRK-mediated potentiation of TGFB1 expression. We subjected RA FLS to knockdown of PTPRK expression using a cell-permeable ASO (PTPRK ASO) and found that TGFB1 expression was unaffected by PTPRK deficiency (see online supplementary figure S4). In contrast, when TGFβ signalling was blocked by treatment of RA FLS with the TGFβ type 1 receptor chemical inhibitor SB505124, the basal levels of PTPRK were reduced (figure 1G). Taken together, these data suggest that PTPRK is a TGFβ1-target gene in RA FLS and that the increased expression of PTPRK in RA FLS is likely due to increased autocrine expression of TGFβ by these cells.
RPTPκ promotes invasiveness of RA FLS
We next tested whether the reduction in RPTPκ expression could inhibit the invasiveness of RA FLS. As ex vivo invasiveness of FLS has been shown to correlate with radiographic damage in RA,5 we subjected the ASO-treated RA FLS to transwell invasion assays through Matrigel in response to PDGF, a prominent growth factor in the RA synovium that promotes FLS invasiveness.2 As shown in figure 2A, RA FLS treated with PTPRK ASO, compared with control non-targeting (Ctl) ASO-treated cells, were significantly less invasive in response to PDGF (median % max cells per field, 27.91 and 15.79–46.35 for Ctl ASO; 12.57 and 6.424–27.04 for PTPRK ASO; p<0.05). The effect was replicated by treatment of RA FLS with a second PTPRK-targeted ASO (median % max cells per field and IQR, 36.59 and 19.63–50.00 for Ctl ASO; 8.211 and 4.878–14.31 for PTPRK_2 ASO; p<0.05), but not by treatment of cells with a PTPRM-targeted ASO (figure 2B and see online supplementary figure S5).
Figure 2.

Receptor protein tyrosine phosphatase κ (RPTPκ) is required for rheumatoid arthritis (RA) fibroblast-like synoviocyte (FLS) invasiveness. (A and B) Following treatment with control (Ctl) or PTPRK (A) or PTPRK_2 (B) antisense oligonucleotide (ASO) for 7 days, RA FLS invaded through Matrigel-coated transwell chambers in response to 50 ng/mL platelet-derived growth factor BB (PDGF-BB) for 24 h. Graphs show median±IQR % maximum number of cells per field. Data from four (A) or three (B) independent experiments in different FLS lines are shown. (C) ASO-treated RA FLS migrated through uncoated transwell chambers in response to 5% fetal bovine serum (FBS) for 24 h. Graph shows median±IQR % maximum number of cells per field. Data from five independent experiments in different FLS lines are shown. (A–C) *p<0.05, Mann–Whitney test. (D and E) ASO-treated RA FLS migrated out of a Matrigel spot for 2 days in response to 10 ng/mL PDGF or media alone. (D) Median±IQR cells per field. Data from three independent experiments in different FLS lines are shown. *p<0.05, Wilcoxon-matched pairs signed-rank test. (E) Representative image of cells from (D). (F) ASO-treated RA FLS were plated on fibronectin (FN)-coated coverslips in the presence of 5% FBS. Graph shows median±IQR cell area after 15, 30 and 60 min. Data from three independent experiments in different FLS lines are shown. *p<0.05, Wilcoxon-matched pairs signed-rank test.
We next assessed whether the effect of PTPRK ASO on RA FLS invasiveness was due to impaired cell motility. PTPRK ASO-treated cells showed significantly reduced migration in a transwell assay in response to 5% FBS (figure 2C; median % max cells per field and IQR, 57.07 and 44.57–78.87 for Ctl ASO; 39.10 and 23.19–51.79 for PTPRK ASO; p<0.05), and out of a spot of Matrigel in response to PDGF (figure 2D–E; median and IQR cells per field, 193 and 181–201 for Ctl ASO; 47 and 39–58 for PTPRK ASO). We hypothesised that this effect could be due to increased cell death or due to reduced cytoskeletal reorganisation following RPTPκ knockdown. We, therefore, assayed the effect of PTPRK ASO on cell apoptosis and necrosis, and on cell spreading. Treatment of RA FLS with PTPRK ASO did not increase cell apoptosis or necrosis (see online supplementary figure S6), but did significantly reduce cell spreading on an extracellular matrix in the presence of 5% FBS (figure 2F and see online supplementary figure S7; median cell area and IQR at 60 min, 582.7 μm2 and 350.0–657.0 for Ctl ASO; 263.9 μm2 and 195.2–399.6 for PTPRK ASO; p<0.05). Taken together, these data strongly support a role for RPTPκ in promoting RA FLS cytoskeletal reorganisation, migration and invasiveness in response to PDGF.
RPTPκ promotes RA FLS migration through dephosphorylation of SRC (tyrosine-protein kinase v-src avian sarcoma viral oncogene homolog)
We investigated the molecular mechanism by which RPTPκ knockdown impairs PDGF-induced invasion and migration of RA FLS. We found no reduction in the expression of the PDGF receptor (PDGFR) upon PTPRK ASO treatment (data not shown). We explored as potential substrates, cadherins and β-catenin, RPTPκ candidate substrates that have a known role in FLS migration.2,16,19,20 Cadherin-11 is highly expressed in FLS and is critical for FLS invasiveness and formation and maintenance of the synovial lining in vivo and in in vitro synovial organ cultures.1 We detected basal tyrosine phosphorylation of cadherin-11 in RA FLS, which was unaffected by RPTPκ knockdown (see online supplementary figure S8). Additionally, RPTPκ knockdown had no effect on RA FLS synovial lining layer formation in vitro (see online supplementary figure S8). β-catenin is an important mediator of cell migration,19 and β-catenin-dependent Wnt signalling is overactive in RA FLS.21 However, we found no increase in β-catenin tyrosine phosphorylation—which promotes β-catenin nuclear recruitment and transcriptional activity19—nor alterations in the ratio of β-catenin cytosolic/nuclear localisation, upon RPTPκ knockdown (see online supplementary figure S8 and data not shown).
In mammary cells, RPTPκ promotes receptor tyrosine kinase (RTK) signalling pathways through dephosphorylation of the C-terminal inhibitory tyrosine residue (Y527 of SRC) of SRC family kinases (SFKs).15 Dephosphorylation of this site promotes SRC activation and enhances signalling downstream RTKs, including growth factor-induced activation of mediators of cell motility and invasion, phospholipase C-γ 1 (PLCγ1) and focal adhesion kinase (FAK).22–24 RA FLS treated with PTPRK ASO showed increased basal phosphorylation of SRC Y527 (figure 3A) and reduced PDGF-induced phosphorylation of PLCγ1 (Y783) and FAK (Y925) (figure 3B). The essential roles of SFK and PLCγ1 activity in RA FLS migration were confirmed using pharmacological inhibitors of these enzymes. Treatment with the SFK inhibitor PP225 or the PLCγ1 inhibitor U7312226 abolished growth factor-induced migration of RA FLS (figure 3C, D) without affecting cell survival (see online supplementary figure S9).
Figure 3.

RPTPκ promotes RA FLS migration through dephosphorylation of SRC. (A) Western blotting of ASO-treated RA FLS lysates. Data are representative of three independent experiments in different FLS lines. (B) Western blotting of lysates of ASO-treated RA FLS stimulated with 50 ng/mL PDGF-BB for 30 min or left unstimulated. Data are representative of two independent experiments in different FLS lines. (C and D) RA FLS migrated through uncoated transwell chambers in response to 5% FBS in the presence of PP2 (C) or U73122 (D). Graphs show median±IQR % maximum number of cells per field. Data from two independent experiments in different FLS lines are shown. *p<0.05, Mann–Whitney test. (E) HA-tagged RPTPκ was immunoprecipitated from COS-1 cells and incubated in vitro with RA FLS lysates, and pull-down was subjected to Western blotting. Data are representative of two independent experiments. (F) Agarose-bound S-tagged-substrate trapping mutant iPTPκ-D1051A was incubated in vitro with RA FLS lysates, and pull-down was subjected to Western blotting and probed using HRP-conjugated S-protein. A similar result was obtained when RA FLS were stimulated with 100 μM pervanadate for 15 min immediately prior to lysis (data not shown). (G and H) Immunoprecipitated wild type (WT) or catalytically inactive C1100S (C/S) HA-tagged RPTPκ was incubated in vitro with SRC pY527 phosphopeptide for 30 min. The reaction was stopped by addition of Biomol Green. (G) Absorbance following subtraction of the blank reaction. (H) Western blotting of a fraction of the immunoprecipitation reaction. (G and H) Data are representative of two independent experiments.
ASO, antisense oligonucleotide; COS, cells Simian CV-1 in origin and carrying SV40 genetic material; DMSO, dimethylsulfoxide; FLS, fibroblast-like synoviocytes; FBS, fetal bovine serum; GADPH, glyceraldehyde 3-phosphate dehydrogenase; HA, haemagglutinin tag; HRP, horseradish peroxidase; IP, immunoprecipitation; PDGF-BB, platelet-derived growth factor BB; RA, rheumatoid arthritis; RPTPκ, receptor protein tyrosine phosphatase κ; WB, Western blotting.
We next investigated whether RPTPκ directly interacts with and/or dephosphorylates SFKs. Both full-length RPTPκ and a recombinant substrate-trapping mutant of the RPTPκ catalytic domain (iPTPk-D1051A) precipitated SRC from RA FLS lysates in pull-down assays (figure 3E, F), but not YES or FYN (SFKs also expressed in FLS) (data not shown). Additionally, full-length RPTPκ efficiently dephosphorylated a SRC Y527 phosphopeptide (figure 3G, H).
In line with previously reported data,15 we found no direct effect of RPTPκ on TGFβ-mediated signalling in RA FLS, as assessed by lack of effect of PTPRK ASO on TGFβ1-induced phosphorylation and nuclear recruitment of SMAD3 (see online supplementary figure S10), suggesting that the RA FLS phenotype induced by knockdown of RPTPκ is not due to direct inhibition of TGFβ signalling.
Taken together, these data indicate that TGFβ-responsive RPTPκ promotes PDGF-induced RA FLS migration by dephosphorylation of the inhibitory residue Y527 of SRC, allowing for enhanced activation of signalling downstream the PDGFR.
RPTPκ is required for production of pathogenic factors in RA FLS
As the rheumatoid synovium is characterised by pathogenic overexpression of TNF and IL-1,2 and SRC activation is known to promote signalling through inflammatory cytokine receptors,27 we assessed the effect of PTPRK ASO on TNF and IL-1-induced expression of genes encoding mediators of FLS invasiveness. RPTPκ deficiency significantly decreased TNF-induced and IL-1-induced expression of several genes critical for FLS invasion, CXCL10, VCAM1, MMP8 and MMP13 (figure 4A and see online supplementary figure S11).1,2,28,29 MMP2 expression was not induced by cytokine stimulation but was constitutively decreased following RPTPκ knockdown (figure 4A and see online supplementary figure S11). No effect was observed on the expression of IL6, IL8, MMP1 or MMP3 (data not shown).
Figure 4.

RPTPκ is required for the pathogenic action of RA FLS. (A) ASO-treated RA FLS were stimulated with 50 ng/mL TNFα for 24 h or left unstimulated. Graph shows median±IQR mRNA expression levels relative to the Ctl ASO-treated, TNFα-stimulated samples from the same FLS line. Data from four (MMP8 and MMP13) or five (CXCL10, VCAM1, MMP2) independent experiments in different FLS lines are shown. *p<0.05, Mann–Whitney test. (B) Western blotting of lysates from ASO-treated RA FLS stimulated with 50 ng/mL TNFα for 15 min or left unstimulated. Data are representative of four independent experiments in different FLS lines. (C and D) ASO-treated RA FLS were intradermally implanted into nude mice following subcutaneous injection of CFA. After 5 days, FLS invasion towards the inflammation site was measured by immunohistochemical staining of FLS in skin immediately adjacent the CFA injection site with an anti-human class I HLA antibody. (C) Graph shows median±IQR cells per field. Data from three independent experiments in different FLS lines are shown. *p<0.05, Wilcoxon-matched pairs signed-rank test. (D) Representative 40× images of mouse skin samples. Blue arrows indicate invading FLS, identified by anti-human class I HLA antibody positivity.
RPTPκ, receptor protein tyrosine phosphatase κ; RA, rheumatoid arthritis; FLS, fibroblast-like synoviocytes; ASO, antisense oligonucleotide; GADPH, glyceraldehyde 3-phosphate dehydrogenase; JNK, Jun N-terminal kinase; TNF, tumour necrosis factor; CFA, complete Freunds adjuvant; HLA, human leucocyte antigen; ERK, extracellular-signal-regulated kinases; WB, Western blotting.
As FAK has been reported to promote downstream activation of the mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase ( JNK) and production of matrix metalloproteinases,22,24 and since our findings suggest a role for RPTPκ upstream the activation of FAK, we examined whether RPTPκ promotes activation of JNK. As shown in figure 4B, PTPRK ASO reduced basal and TNF-induced phosphorylation of JNK, with only minimal effect on the phosphorylation of the MAPKs ERK and p38. Consistently, we found a similar effect when TNF-stimulated RA FLS were treated with the FAK chemical inhibitor PF573228 (see online supplementary figure S12).
We next examined whether PTPRK ASO inhibits the in vivo invasiveness of RA FLS in a recently reported invasion assay.30 We induced skin inflammation in athymic nude mice by subcutaneously injecting complete Freund’s adjuvant, and then intradermally implanted RA FLS pretreated with Ctl or PTPRK ASO. After 5 days, we monitored FLS invasion from the implantation site towards the inflammation site. As shown in figure 4C, D, PTPRK ASO treatment significantly reduced the in vivo invasiveness of RA FLS (median cells per field and IQR, 78 and 64–101 for Ctl ASO; 58 and 53–65 for PTPRK ASO; p<0.05).
Our data indicate a model (figure 5A) in which autocrine TGFβ upregulates RPTPκ expression in RA FLS, in turn leading to increased RA FLS invasiveness through activation of SRC and cross-activation of PDGF and TNF and IL-1 signalling pathways. Synergistic stimulation with TGFβ and PDGF strongly amplifies FLS responsiveness to TNF.31 We reasoned that if our model is correct, the contribution of TGFβ to TNF signalling in this system might be mediated by PTPRK. We stimulated ASO-treated RA FLS with TGFβ1 for 24 h to increase PTPRK expression (as in see online supplementary figure S4). We then costimulated cells with further TGFβ1, and TNF and PDGF for an additional 24 h and assessed the expression of two SMAD-dependent genes that are key mediators of RA FLS invasiveness and are known to be TGFβ dependent, MMP13 and MMP14.32–34 Costimulation with TGFβ1 dramatically induced expression of MMP13 and MMP14; however, this effect was completely abolished by treatment with PTPRK ASO (figure 5B; 97.2% reduction of MMP13 expression and 98.0% reduction of MMP14 expression with PTPRK ASO; p<0.05). These findings indicate that RPTPκ promotes the invasiveness of RA FLS and mediates the potentiation of growth factor and inflammatory cytokine signalling by TGFβ in RA FLS.
Figure 5.
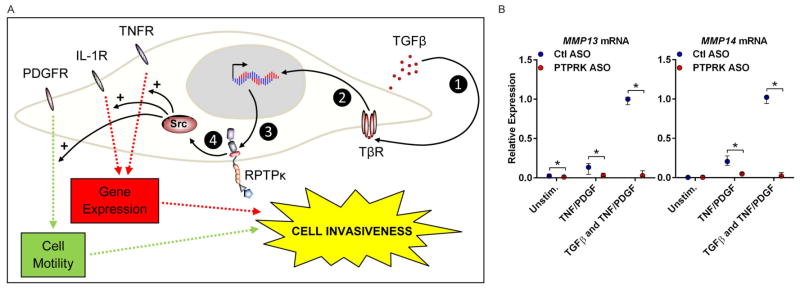
Receptor protein tyrosine phosphatase κ (RPTPκ) mediates cross-activation of platelet-derived growth factor (PDGF) and tumour necrosis factor (TNF) signalling by transforming growth factor (TGF)-β. (A) Model depicting the role of TGFβ-dependent RPTPκ in rheumatoid arthritis (RA) pathogenesis. Autocrine TGFβ binds to the TGFβ receptor complex (TβR) (1), inducing SMAD activation (2) and transcription of PTPRK (3). RPTPκ activates SRC (4), which promotes proinvasive signalling through the PDGF receptor (PDGFR), and the TNF and interleukin (IL)-1 receptors (TNFR and IL-1R). (B) Antisense oligonucleotide (ASO)-treated RA fibroblast-like synoviocytes (FLS) were prestimulated with 50 ng/mL TGFβ1 for 24 h (or left unstimulated) in the presence of ASO. Cells were then stimulated with 50 ng/mL PDGF/TNFα, or 50 ng/mL TGFβ1/PDGF/TNFα, or left unstimulated for 24 h. Graph shows median±IQR mRNA expression levels relative to the Ctl ASO-treated, TGFβ1/PDGF/TNFα-stimulated samples from the same FLS line. Data from three independent experiments in different FLS lines are shown. *p<0.05, Mann–Whitney test.
DISCUSSION
In this study, that is based entirely on human primary cells from patients with RA, we report the first characterisation of the role of a transmembrane PTP in RA FLS. We found that RPTPκ is overexpressed in FLS from patients with RA when compared with patients with OA, resulting from increased production of TGFβ by these cells. By regulating phosphorylation of the inhibitory Y527 of SRC, RPTPκ promotes RA FLS aggressiveness by enhancing responsiveness to PDGF, TNF and IL-1 stimulation. RPTPκ-deficient RA FLS display dramatically reduced spreading, migration, invasiveness and chemokine production. Furthermore, we found that RPTPκ is required for the cross-activation of growth factor and inflammatory cytokine signalling by TGFβ that has been reported to occur in RA FLS.31
Our observation that RPTPκ promotes RA FLS aggressiveness seems counter to previous reports of RPTPκ as a tumour suppressor, suggesting that RPTPκ controls signalling in RA FLS and cancer cells through different mechanisms. PTPRK has been reported to inhibit proliferation of several types of cancer cells, presumably through modulation of β-catenin function or epidermal growth factor receptor signalling.17,35 However, we found no evidence of a role for RPTPκ in regulation of β-catenin function nor RPTPκ inhibited migration or survival induced by growth factors in RA FLS.
We propose that inhibition of RPTPκ in patients with RA carrying high expression of PTPRK in FLS could mitigate disease severity. The transmembrane nature of RPTPκ suggests potential for modulation through its extracellular domains. Indeed inhibition by dimerisation has been suggested for other transmembrane PTPs36 and an anti-RPTPκ antibody targeted against the extracellular domain was reported to modulate RPTPκ activity.20 Although there is currently no mouse model available to examine the effect of Ptprk deficiency, deletion of Ptprk in the Long-Evans Cinnamon rat leads to immunodeficiency of T helper cells.37 This suggests that inhibition of RPTPκ could provide a dual means of affecting RA, targeting both T-cell-mediated and FLS-mediated pathogenesis. Functional evaluation of the role of RPTPκ in other RA-relevant cell types is certainly warranted and will elucidate if it holds value as a therapeutic target for RA.
Supplementary Material
Acknowledgments
The authors are grateful to the UCSD CTRI Biorepository for the FLS lines and to Dr Michela Locci for advice and assistance with reagents. This is manuscript #1717 from LJI.
Funding This work was supported by LJI Institutional funding (NB) and NIH (GSF; AR47825, AI070555 and Clinical and Translational Science Award UL1TR000100).
Footnotes
Additional material is published online only. To view please visit the journal online (http://dx.doi.org/10.1136/annrheumdis-2014-205790).
Contributors SMS, TM, DLB, GSF and NB contributed to study conception and design. SMS, GRAM, BB, CS, WBK, JS, MFM and MB contributed to acquisition of data. SMS, GRAM, BB, CS, WBK, JS, MFM, TM, DLB, GSF and NB contributed to analysis and interpretation of data.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–70. doi: 10.1111/j.1600-065X.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- 2.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ospelt C, Gay S. The role of resident synovial cells in destructive arthritis. Best Pract Res Clin Rheumatol. 2008;22:239–52. doi: 10.1016/j.berh.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Lefevre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–20. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tolboom TC, van der Helm-Van Mil AH, Nelissen RG, et al. Invasiveness of fibroblast-like synoviocytes is an individual patient characteristic associated with the rate of joint destruction in patients with rheumatoid arthritis. Arthritis Rheum. 2005;52:1999–2002. doi: 10.1002/art.21118. [DOI] [PubMed] [Google Scholar]
- 6.Niedermeier M, Pap T, Korb A. Therapeutic opportunities in fibroblasts in inflammatory arthritis. Best Pract Res Clin Rheumatol. 2010;24:527–40. doi: 10.1016/j.berh.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Stanford SM, Maestre MF, Campbell AM, et al. Protein tyrosine phosphatase expression profile of rheumatoid arthritis fibroblast-like synoviocytes: a novel role of SH2 domain-containing phosphatase 2 as a modulator of invasion and survival. Arthritis Rheum. 2013;65:1171–80. doi: 10.1002/art.37872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toh ML, Yang Y, Leech M, et al. Expression of mitogen-activated protein kinase phosphatase 1, a negative regulator of the mitogen-activated protein kinases, in rheumatoid arthritis: up-regulation by interleukin-1beta and glucocorticoids. Arthritis Rheum. 2004;50:3118–28. doi: 10.1002/art.20580. [DOI] [PubMed] [Google Scholar]
- 9.Pap T, Franz JK, Hummel KM, et al. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000;2:59–64. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvaro-Gracia JM, Zvaifler NJ, Brown CB, et al. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J Immunol. 1991;146:3365–71. [PubMed] [Google Scholar]
- 11.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 12.Nakano K, Boyle DL, Firestein GS. Regulation of DNA methylation in rheumatoid arthritis synoviocytes. J Immunol. 2013;190:1297–303. doi: 10.4049/jimmunol.1202572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laragione T, Brenner M, Mello A, et al. The arthritis severity locus Cia5d is a novel genetic regulator of the invasive properties of synovial fibroblasts. Arthritis Rheum. 2008;58:2296–306. doi: 10.1002/art.23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andersen JN, Mortensen OH, Peters GH, et al. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–36. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang SE, Wu FY, Shin I, et al. Transforming growth factor {beta} (TGF-{beta})-Smad target gene protein tyrosine phosphatase receptor type kappa is required for TGF-{beta} function. Mol Cell Biol. 2005;25:4703–15. doi: 10.1128/MCB.25.11.4703-4715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novellino L, De Filippo A, Deho P, et al. PTPRK negatively regulates transcriptional activity of wild type and mutated oncogenic beta-catenin and affects membrane distribution of beta-catenin/E-cadherin complexes in cancer cells. Cell Signal. 2008;20:872–83. doi: 10.1016/j.cellsig.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Shao Y, Zhou J, et al. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J Cell Biochem. 2009;107:873–80. doi: 10.1002/jcb.22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pohlers D, Beyer A, Koczan D, et al. Constitutive upregulation of the transforming growth factor-beta pathway in rheumatoid arthritis synovial fibroblasts. Arthritis ResTher. 2007;9:R59. doi: 10.1186/ar2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005;17:459–65. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Anders L, Mertins P, Lammich S, et al. Furin-, ADAM 10-, and gamma-secretase-mediated cleavage of a receptor tyrosine phosphatase and regulation of beta-catenin’s transcriptional activity. Mol Cell Biol. 2006;26:3917–34. doi: 10.1128/MCB.26.10.3917-3934.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen M. Wnt signalling in rheumatoid arthritis. Rheumatology. 2005;44:708–13. doi: 10.1093/rheumatology/keh553. [DOI] [PubMed] [Google Scholar]
- 22.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 23.Yu H, Fukami K, Itoh T, et al. Phosphorylation of phospholipase Cgamma1 on tyrosine residue 783 by platelet-derived growth factor regulates reorganization of the cytoskeleton. Exp Cell Res. 1998;243:113–22. doi: 10.1006/excr.1998.4132. [DOI] [PubMed] [Google Scholar]
- 24.Hanks SK, Ryzhova L, Shin NY, et al. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 2003;8:d982–96. doi: 10.2741/1114. [DOI] [PubMed] [Google Scholar]
- 25.Hanke JH, Gardner JP, Dow RL, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 26.Smith RJ, Sam LM, Justen JM, et al. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J Pharmacol Exp Ther. 1990;253:688–97. [PubMed] [Google Scholar]
- 27.Kant S, Swat W, Zhang S, et al. TNF-stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway. Genes Dev. 2011;25:2069–78. doi: 10.1101/gad.17224711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laragione T, Brenner M, Sherry B, et al. CXCL10 and its receptor CXCR3 regulate synovial fibroblast invasion in rheumatoid arthritis. Arthritis Rheum. 2011;63:3274–83. doi: 10.1002/art.30573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seemayer CA, Kuchen S, Kuenzler P, et al. Cartilage destruction mediated by synovial fibroblasts does not depend on proliferation in rheumatoid arthritis. Am J Pathol. 2003;162:1549–57. doi: 10.1016/S0002-9440(10)64289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You S, Yoo SA, Choi S, et al. Identification of key regulators for the migration and invasion of rheumatoid synoviocytes through a systems approach. Proc Natl Acad Sci USA. 2014;111:550–5. doi: 10.1073/pnas.1311239111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosengren S, Corr M, Boyle DL. Platelet-derived growth factor and transforming growth factor beta synergistically potentiate inflammatory mediator synthesis by fibroblast-like synoviocytes. Arthritis Res Ther. 2010;12:R65. doi: 10.1186/ar2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leivonen SK, Ala-Aho R, Koli K, et al. Activation of Smad signaling enhances collagenase-3 (MMP-13) expression and invasion of head and neck squamous carcinoma cells. Oncogene. 2006;25:2588–600. doi: 10.1038/sj.onc.1209291. [DOI] [PubMed] [Google Scholar]
- 33.Ottaviano AJ, Sun L, Ananthanarayanan V, et al. Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-beta1. Cancer Res. 2006;66:7032–40. doi: 10.1158/0008-5472.CAN-05-4421. [DOI] [PubMed] [Google Scholar]
- 34.Fu D, Yang Y, Xiao Y, et al. Role of p21-activated kinase 1 in regulating the migration and invasion of fibroblast-like synoviocytes from rheumatoid arthritis patients. Rheumatology. 2012;51:1170–80. doi: 10.1093/rheumatology/kes031. [DOI] [PubMed] [Google Scholar]
- 35.Julien SG, Dube N, Hardy S, et al. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 36.Majeti R, Weiss A. Regulatory mechanisms for receptor protein tyrosine phosphatases. Chem Rev. 2001;101:2441–8. doi: 10.1021/cr000085m. [DOI] [PubMed] [Google Scholar]
- 37.Asano A, Tsubomatsu K, Jung CG, et al. A deletion mutation of the protein tyrosine phosphatase kappa (Ptprk) gene is responsible for T-helper immunodeficiency (thid) in the LEC rat. Mamm Genome. 2007;18:779–86. doi: 10.1007/s00335-007-9062-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.