

Background: P1 is an adhesin on the surface of Streptococcus mutans.
Results: Adhesive P1 on the surface of S. mutans exhibits a macromolecular ultrastructure.
Conclusion:The architecture of P1 on the surface of S. mutans plays a critical role in adherence.
Significance: Recognizing the macromolecular assembly of P1 on the surface of S. mutans is critical to understanding the adhesive function of the molecule.
Keywords: Atomic Force Microscopy (AFM), Bacterial Adhesion, Electron Microscopy (EM), Streptococcus, Surface Plasmon Resonance (SPR), Bacterial Surface Architecture
Abstract
P1 (antigen I/II) is a sucrose-independent adhesin of Streptococcus mutans whose functional architecture on the cell surface is not fully understood. S. mutans cells subjected to mechanical extraction were significantly diminished in adherence to immobilized salivary agglutinin but remained immunoreactive and were readily aggregated by fluid-phase salivary agglutinin. Bacterial adherence was restored by incubation of postextracted cells with P1 fragments that contain each of the two known adhesive domains. In contrast to untreated cells, glutaraldehyde-treated bacteria gained reactivity with anti-C-terminal monoclonal antibodies (mAbs), whereas epitopes recognized by mAbs against other portions of the molecule were masked. Surface plasmon resonance experiments demonstrated the ability of apical and C-terminal fragments of P1 to interact. Binding of several different anti-P1 mAbs to unfixed cells triggered release of a C-terminal fragment from the bacterial surface, suggesting a novel mechanism of action of certain adherence-inhibiting antibodies. We also used atomic force microscopy-based single molecule force spectroscopy with tips bearing various mAbs to elucidate the spatial organization and orientation of P1 on living bacteria. The similar rupture lengths detected using mAbs against the head and C-terminal regions, which are widely separated in the tertiary structure, suggest a higher order architecture in which these domains are in close proximity on the cell surface. Taken together, our results suggest a supramolecular organization in which additional P1 polypeptides, including the C-terminal segment originally identified as antigen II, associate with covalently attached P1 to form the functional adhesive layer.
Introduction
Streptococcus mutans is an acidogenic Gram-positive oral bacterium that is a recognized etiological agent of human dental caries (cavities) (1). This ubiquitous infectious disease affects developed as well as non-developed countries with annual costs estimated by the American Dental Association to total over $40 billion annually in the United States alone. Additionally, S. mutans has been identified as a causative agent of infectious endocarditis (2–5). Identifying how S. mutans interacts with host components at the molecular level is essential for a comprehensive understanding of the virulence properties of the organism. The sucrose-independent adhesin P1 (also known as AgI/II,5 SpaP antigen B, and PAc) is localized on the surface of S. mutans as well as most other oral streptococci (6) and certain strains of Streptococcus pyogenes (7). The gene has also been detected in a subset of Streptococcus agalactiae (8). AgI/II family molecules are considered to mediate bacterial adhesion to mucosal glycoproteins (9–13) as well as to the extracellular matrix (14–17) and other bacteria (18–21). The contribution of P1 to bacterial adherence, colonization, and cariogenicity and its promise in clinical trials make it a therapeutic target and focus of immunization studies (22–26). In the oral environment within the salivary pellicle on tooth surfaces, S. mutans P1 interacts primarily with the glycoprotein salivary agglutinin complex (SAG) comprising predominantly the scavenger receptor gp340/DMBT1 (11–13, 22, 27–37). In contrast, the interaction of fluid-phase SAG with S. mutans P1 results in bacterial aggregation and represents an innate host defense clearance mechanism (38). The complete mechanisms by which P1 binds to host components, particularly how the architecture and assembly of this molecule on the bacterial surface facilitates S. mutans adherence to immobilized SAG, are not fully understood.
The primary sequence of the ∼185-kDa, 1561-amino acid P1 protein (see Fig. 1A) consists of several distinct regions: a 38-residue cleavable signal sequence that directs secretion followed by an N-terminal region, a series of three tandem alanine-rich repeats (A1–3), an intervening segment containing a variable region where strain differences are clustered (39), and a series of three tandem proline-rich repeats (P1–3) followed by three globular C-terminal domains (C1–3) that terminate in wall- and membrane-spanning sequences (6, 40). P1 is covalently attached to the bacterial cell wall by the transpeptidase sortase A (41), which recognizes an LPXTG consensus sequence at the C terminus of its substrate proteins (42, 43). X-ray crystallography studies have provided fundamental insights into the conformational properties of P1 (44–48). A current structural model of P1 based on recently elucidated crystal structures positions the globular head domain (i.e. apical head) intervening the A- and P-repeats away from the cell surface at the tip of a long (∼50 nm) and narrow extended stalk with the N-terminal region in close proximity to the C-terminal region (see Fig. 1B) (44, 45). The A-repeats form a long α-helix that intimately intertwines into a left-handed supercoiled structure with the helical polyproline P-repeats to form the stalk (45) The C-terminal region is also globular and comprises three structurally related β-sandwich domains stabilized by covalent isopeptide bonds (44, 47). The crystal structure of the N terminus of P1 in complex with its C-terminal intramolecular binding partner revealed that it forms a novel stabilizing scaffold at the base of the stalk that contributes to the stability and proper folding of the full-length molecule (48). Numerous monoclonal antibodies (mAbs) targeting different domains of P1 have been generated and characterized, providing powerful molecular probes of the highly complex protein structure (6, 49–51) (see Fig. 1C). However, despite the biological insight derived from recent crystal structures and the construction of an essentially complete tertiary model of the entire protein, a direct demonstration of the structural and biophysical properties of P1 on intact and live cells is still lacking.
FIGURE 1.
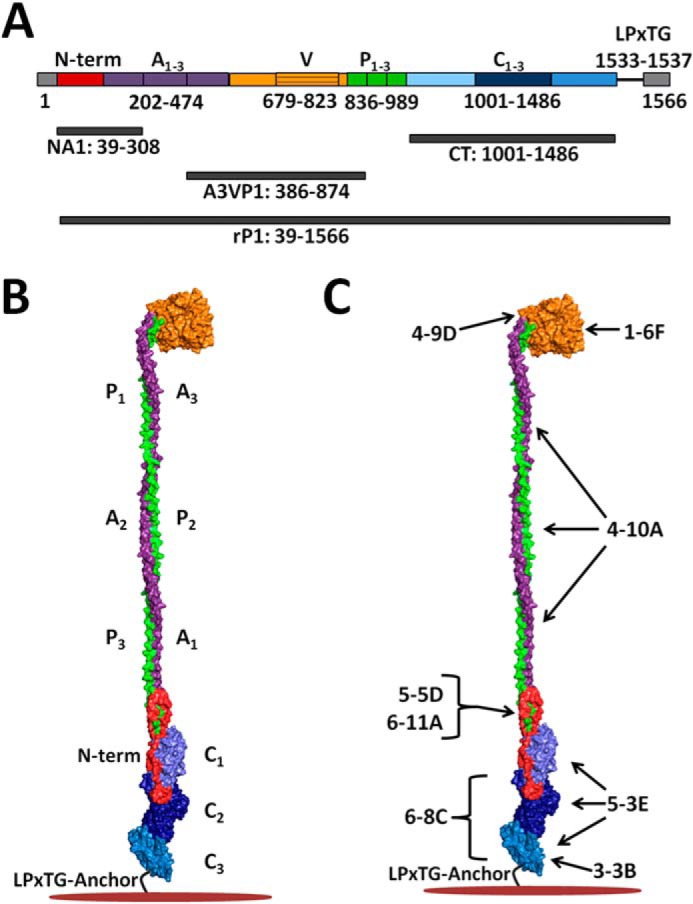
Schematic representations of S. mutans P1 primary and tertiary structures illustrating locations of polypeptides and approximate binding sites of anti-P1 monoclonal antibodies used in this study. A, primary structure of P1 and locations of the recombinant polypeptides used in this study. B, tertiary model of P1 based upon x-ray crystal structures of recombinant P1 polypeptides and velocity ultracentrifugation experiments (44–48). The adhesin is a highly elongated molecule ∼65 nm in length with globular adherent domains located on either side of a ∼50-nm hybrid helical stalk that is formed by the intertwined α- and polyproline type II helices of the A- and P-region tandem repeats. An intramolecular lock formed between the N terminus (red) and the C-terminal region facilitates folding and stabilizes the tertiary structure of the full-length molecule. C, approximate binding sites of the anti-P1 mAbs used in this study (49). A1–3, alanine-rich repeats; V, variable region; P1–3, proline-rich repeats; C1–3, C-terminal domains.
Upon discovery, P1 was thought to represent the product of two separate genes because of its initial identification as defined breakdown products (52). The adhesin was subsequently isolated as an ∼185-kDa dual antigen, dubbed AgI/II, with apparent molecular masses of ∼150 and ∼50 kDa for AgI and AgII, respectively (29, 53). The gene encoding the full-length P1 protein (spaP; pac) was later cloned and sequenced by two independent groups (54, 55). At that time, N-terminal sequencing identified AgI and AgII as corresponding to N- and C-terminal portions of the full-length protein (54). Early immunogold electron microscopy (EM) studies of S. mutans revealed P1 to be localized within a cell surface-associated “fuzzy coat” (50). Interestingly, anti-P1 mAbs 1-6F and 6-11A, which displayed similar distribution and reactivity patterns by immunogold EM (50), were mapped many years later to opposite ends of the folded molecule (49, 56) and found to have their cognate epitopes separated by ∼50 nm in the tertiary structure model of the full-length protein (see Fig. 1B). This perplexing finding was further confounded by the fact that a C terminus-specific mAb, 6-8C, which did not appear to bind to whole S. mutans cells by radioimmunoassay (57), was highly effective at inhibiting adherence of the organism to immobilized SAG (12). The C terminus of P1 has been demonstrated to be buried within the cell wall peptidoglycan (58); hence, it was not surprising that mAbs against this region would not be reactive with whole cells. However, it has also long been recognized that not all P1 is covalently linked to the cell wall because much of it, including the full-length ∼185-kDa protein and multiple breakdown products, can be removed by a variety of mechanisms, including boiling in SDS, mechanical agitation, and even incubating with anti-P1 antibodies (57, 59–63).
We used a combination of glutaraldehyde fixation, surface plasmon resonance, dot blot analysis, and immunogold electron microscopy as well as regeneration of adherence of postextracted cells with exogenously added P1 fragments to identify a critical functional role of non-covalently linked surface-associated P1 polypeptides in the adherence properties of the organism. Also, incubation of S. mutans with several different anti-P1 mAbs known to inhibit bacterial adherence to immobilized SAG caused the release of P1 fragments from the cell surface. These included a ∼50-kDa C-terminal fragment, likely corresponding to the previously identified AgII, suggesting an indirect mechanism for inhibition of P1-mediated adherence. In addition, we used atomic force microscopy (AFM)-based single molecule force spectroscopy (64–66) to characterize the supramolecular organization (cell surface density, distribution, conformation, orientation, and assembly) of P1 molecules on live S. mutans cells. Using AFM tips functionalized with specific mAbs (see Fig. 1C), we detected and stretched various domains (globular head, stalk, base of the stalk, and C-terminal region) of the protein, thereby providing direct information on the surface localization, extension, and orientation of the adhesin. Taken together, our findings suggest a previously unappreciated degree of quaternary architecture on the cell surface and indicate that a critical assembly of non-covalently linked P1 fragments in concert with the non-extractable covalently attached molecule is ultimately responsible for the adherence of S. mutans to immobilized SAG. This novel information increases our ability to interpret the distinct processes of adherence and aggregation in S. mutans and adds to our understanding of the role of macromolecular assemblies in streptococcal virulence.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Growth Conditions
S. mutans serotype c strain NG8 (67) was used in these studies. The isogenic spaP-negative mutant PC3370 (68) was used as the negative control. For the AFM experiments, S. mutans were grown by inoculating a single colony into 10 ml of tryptic soy casein broth (Bio-Rad) and incubated for 18–24 h at 37 °C. For all other experiments, S. mutans cultures were grown in Todd-Hewitt broth (BBL, Cockeysville, MD) supplemented with 0.3% yeast extract.
Anti-P1 Monoclonal Antibodies
Anti-P1 mAbs were obtained from previously established hybridomas made in our laboratory (50) and used as ascites fluid, or when indicated, the IgG fraction was purified from murine ascites fluid using Protein A affinity chromatography as described previously (69).
Construction of Subclones and Purification of P1 Polypeptides
DNA encoding the N terminus and first alanine-rich repeat of P1 (NA1, amino acids 39–308) (Fig. 1, A and B) was PCR-amplified from spaP of strain NG8 using chromosomal DNA as the template and CGCCATATGCACCATCATCATCATCATGATGAAACGACCACTACTAGT (NdeI restriction site in bold) and CGGAAGCTTTCAGTCAGTCATGCTTCGTTTGCGGCATTAGC (HindIII restriction site in bold and His6 tag underlined) as forward and reverse primers, respectively. Amplified DNA was cloned into pET-30(c) (EMD Millipore, Billerica, MA) and transformed into Escherichia coli BL21(DE3) cells (Life Technologies) as described previously (70). The NA1 polypeptide was purified by growing a 20-ml terrific broth culture overnight at 37 °C and the following day inoculating 1 liter of terrific broth with the entire culture. Bacteria were grown to an A600 of 0.7–0.8, induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, incubated overnight at 22 °C, harvested by centrifugation, and stored at −20 °C overnight. The cell pellet was suspended in 20 ml of TALON equilibration buffer (50 mm Tris-HCl, 300 mm NaCl, pH 7.4) supplemented with 20 μl of DNAse I (Thermo Scientific, Rockford, IL), 10 mg lysozyme, and 10 mm MgCl2 and passed through an Avestin EmulsiFlex-C5 high pressure homogenizer (Avestin Inc., Ottawa, Ontario, Canada) at a pressure of 20,000–25,000 p.s.i. three times. The cell lysate was centrifuged at 45,000 × g for 30 min, and the soluble supernatant fraction was filtered through a 0.22-μm syringe driven filter (Millipore). The filtered sample was applied to TALON metal affinity resin (Clontech), and bound protein was eluted with 150 mm imidazole and then polished over a HiLoad 16/600 Superdex 200 prep grade size exclusion column (GE Healthcare) using a Tris buffer (25 mm Tris, 150 mm NaCl, pH 7.4).
DNA encoding the third alanine-rich repeat, the variable region, and the first proline-rich repeat (A3VP1; amino acids 386–874) was subcloned into pET30(c) (EMD Millipore) and used to transform E. coli strain BL21 Star(DE3) (40). Following induction with isopropyl 1-thio-β-d-galactopyranoside, A3VP1 was purified as follows. Harvested cells were lysed in Bug Buster Master Mix (EMD Millipore), and insoluble debris was removed by centrifugation. Purification from the supernatant consisted first of immobilized metal affinity chromatography with a HisTrap HP column (GE Healthcare) using 30 mm Tris, 100 mm NaCl, 10 mm imidazole, pH 7.4 as binding buffer and 30 mm Tris, 100 mm NaCl, 300 mm imidazole, pH 7.4 as elution buffer followed by size exclusion chromatography with a HiLoad 16/600 Superdex 200 prep grade column and 30 mm Tris, 100 mm NaCl, pH 7.4 as the running buffer.
The C-terminal region of P1 (P1-CT) was generated by PCR amplification of base pairs 2998–4458 of spaP (amino acids 1000–1486) with primers CCAAATGCGGCCGCAGTTCAGCCGCAGGTTAACAAAG and GTACCGTCTCGAGTGAACTGTAAGTTACCCCATTG bearing NotI and XhoI restriction sites (bold), respectively. The pVS72 cloning vector (l-arabinose-inducible, ampicillin-resistant) (71) was modified to remove the XbaI site and to reintroduce an XhoI site. Ligated DNA (pVSP1-CT) was used to transform E. coli Top10 (Life Technologies) with selection on LB agar supplemented with 100 μg/ml ampicillin. To enable P1-CT purification from the cytoplasm, DNA encoding the CsgA signal sequence was deleted from a sequence-confirmed clone by circle PCR using the phosphorylated primers PO4GTTCAGCCGCAGGTTAACAAAG and PO4CATATGAATTCCTCCATCCAAAAAAACGG, and religated pVSP1-CTdss was used to transform E. coli VS39 (71). Expression was induced with 0.2% l-arabinose. Purification of P1-CT from the cell lysate consisted of three steps: immobilized metal affinity chromatography as described above followed by ion exchange chromatography using a HiTrap Q FF column with 15 mm Tris, 10 mm NaCl, pH 8.4 as the binding buffer and 15 mm Tris, 1 m NaCl, pH 8.4 as the elution buffer followed by size exclusion chromatography with a HiLoad 16/600 Superdex 200 prep grade column and 30 mm Tris, 100 mm NaCl, pH 7.0 as the running buffer.
DNA encoding full-length recombinant P1 (rP1) lacking the secretion signal sequence (amino acids 39–1561) was cloned into pQE-30 and used to transform E. coli M15-pREP4 as described previously (49, 72). Recombinant P1 was expressed and purified as described above for NA1.
Glutaraldehyde Fixation of S. mutans
S. mutans cells were harvested by centrifugation, washed two times in phosphate-buffered saline, pH 7.0 (PBS); resuspended in PBS containing 2.5% glutaraldehyde to the initial cell culture volume; and incubated for 1 h at room temperature. Following fixation, the cells were washed three times with PBS to remove residual glutaraldehyde and then used in further experiments.
Mechanical Extraction of P1 from S. mutans
S. mutans cells were harvested by centrifugation, washed once with PBS, and resuspended to the original culture volume in PBS. One milliliter of the cell suspension was added to a 1.5-ml microcentrifuge tube and vortexed at the maximum setting for 2 h at 4 °C. Following mechanical extraction, the supernatant was removed to analyze extracted proteins, and the cells were washed two times in PBS prior to use in further experiments.
Characterization of P1 Fragments Released by Mechanical Compared with SDS Extraction
Proteins released from S. mutans were analyzed by SDS-PAGE and Western blotting. Briefly, 20 μl of supernatant from the mechanically extracted cells was mixed with 5 μl of 5× SDS sample buffer and heated at 100 °C for ∼5 min prior to loading onto a 7.5% polyacrylamide gel. SDS extraction was performed by heating S. mutans whole cells harvested from 2.5 ml of an overnight culture in 500 μl of SDS sample buffer for ∼5 min at 100 °C. S. mutans cells are not lysed during this process, so only proteins extracted from the surface are released into the supernatant. Following heating, the cells were pelleted, and the upper 300 μl of SDS sample buffer was removed to avoid contamination with residual cells. Twenty microliters of the supernatant was loaded onto a 7.5% polyacrylamide gel. Following SDS-PAGE, the gel was electroblotted onto a Protran nitrocellulose membrane (Whatman), and membranes were stained with colloidal gold protein stain (Bio-Rad) or probed with mouse anti-P1 mAb 1-6F, 4-10A, or 6-8C ascites fluid (50) diluted 1:500 in PBS containing 0.03% Tween 20 (PBST) followed by HRP-labeled goat anti-mouse IgG (MP Biomedicals, LLC, Santa Ana, CA) diluted 1:1000 and developed as described previously (57).
Biacore Surface Plasmon Resonance
Adherence of S. mutans before and after Mechanical Extraction and Regeneration with P1 Polypeptides
SAG was prepared from pooled unstimulated saliva from healthy human volunteers as described previously (12). Adherence of S. mutans whole cells to SAG immobilized on a CM3 sensor chip was measured by Biacore surface plasmon resonance (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) using a Biacore 3000 instrument (GE Healthcare Bio-Sciences AB) as described previously (73). S. mutans strains NG8 and PC3370 functioned as positive and negative controls, respectively. Following mechanical extraction, cells were washed two times in adherence buffer (73) and then normalized to an A600 of 1.0 in adherence buffer prior to measuring adherence. Twenty microliters of each sample was injected over the chip surface at a flow rate of 10 μl/min. Ten microliters of regeneration buffer (73) was used to regenerate the chip surface between runs. Two uncoated surfaces with dextran only and no immobilized SAG were used as controls, and the background RU was subtracted from the observed signal for each injection. To determine whether exogenously added P1 could restore the loss of adherence capability of mechanically extracted cells, postextracted cells were washed two times with PBS, resuspended in PBS containing each purified recombinant P1 polypeptide (8 μm) under study or maltose-binding protein (MBP) as an irrelevant control, incubated for 1 h at room temperature on a rotary actuator, and washed again two times in PBS prior to use in further experiments. Strain PC3370 was used as a negative control to confirm that exogenous P1 polypeptides reacted with residual P1 present on the postextracted cells and not other S. mutans cell surface molecules.
Measurement of P1 Intermolecular Interactions
The interaction of immobilized purified A3VP1 with rP1 and P1 fragments was measured by surface plasmon resonance. A3VP1 (8 μm in PBS supplemented with 1 mm CaCl2) was immobilized on a CM3 sensor chip as described previously for other ligands (69, 73). Recombinant A3VP1, P1-CT, and full-length P1 were dialyzed overnight in PBS + CaCl2 and then diluted in the same buffer to 8, 4, and 2 μm concentrations. Eighty microliters of each sample was injected over the chip surface at a flow rate of 20 μl/min. Ten microliters of regeneration buffer (PBS containing 0.3% Tween 20, 10 mm EDTA, 100 mm NaCl, 100 mm NaOH) diluted 1:10 was used to regenerate the chip surface between injections. The uncoated surface with dextran only and no immobilized A3VP1 was used as a control surface, and the background RU was subtracted from the observed signal for each injection. Statistically significant differences were determined by one-way analysis of variance using GraphPad Prism 4.0. A p value of less than 0.05 was considered significant. Tukey's multiple comparison test determined differences among the groups. Experiments were repeated in triplicate.
SAG-mediated Aggregation of S. mutans
SAG-mediated aggregation of S. mutans was measured using a spectrophotometric assay as described previously (12). PBS without SAG served as a background control. NG8 and PC3370 were included as positive and negative controls, respectively.
Dot Blot Analysis
Dot blots were performed similarly as described previously (72, 74, 75) to characterize the antigenicity of cell surface P1 following various cell treatments (49). Briefly, S. mutans cells harvested from overnight cultures were washed two times in PBS and then resuspended in PBS to 8× the initial culture volume. 2-Fold serial dilutions of cell suspensions (50 μl) were spotted in duplicate onto a Protran nitrocellulose membrane using a 96-well manifold (MiniFold I, Whatman). The membrane was removed from the apparatus; washed and blocked with PBST; probed with mouse anti-P1 mAb 1-6F, 4-10A, or 6-8C ascites fluid (50) diluted 1:500 in PBST followed by HRP-labeled goat anti-mouse IgG diluted 1:1000 in PBST; and developed as described previously (57).
Antibody-mediated Release of P1 from the S. mutans Cell Surface
Untreated or glutaraldehyde-fixed S. mutans cells were incubated with anti-P1 murine mAbs, and the supernatant was subsequently analyzed by Western blotting using a rabbit polyclonal antiserum produced against the C-terminal 144 amino acids of P1 (kindly provided by Dr. Song Lee, Dalhousie University, Halifax, Nova Scotia) to detect release of fragments of P1 containing the C terminus. Briefly, cells were washed two times with PBS and resuspended in PBS to of the initial culture volume. The cell suspension was split into 500-μl aliquots, and 5 μl of murine ascites fluid containing each respective anti-P1 mAb, including irrelevant IgG and PBS as negative controls, was added to each sample. The cell-antibody mixtures were incubated for 1 h at room temperature on a rotary actuator, then cells were pelleted by centrifugation, and the upper 300 μl of supernatant fluid was removed. Twenty microliters of each cell-free supernatant fluid was mixed with 5 μl of 5× SDS sample buffer, heated at 100 °C for ∼5 min, and loaded onto a 7.5% polyacrylamide slab gel. Following electrophoresis, the gel was electroblotted onto a Protran nitrocellulose membrane, blocked with PBST, probed with the polyclonal rabbit anti-C terminus antiserum diluted 1:100 in PBST followed after washing with HRP-labeled goat anti-rabbit IgG diluted 1:1000 in PBST, and developed as described previously (57).
Trypsin Digestion of S. mutans Whole Cells and Detection of a C-terminal Fragment
S. mutans cell suspensions were prepared in PBS as described above and incubated with trypsin (Sigma-Aldrich) at a concentration of 250 μg/ml for 1 h at 37 °C. Cells were also incubated with buffer only as a negative control. Following digestion, soybean trypsin inhibitor was added to a final concentration of 500 μg/ml, cells were pelleted by centrifugation, and the upper 300 μl of the supernatant was removed. Western blot analysis of the cell-free supernatant was performed as described above using the polyclonal rabbit anti-C terminus antiserum.
Immunogold Electron Microscopy
Whole Cells
Immunogold transmission electron microscopy was used to visualize P1 on the surface of S. mutans whole cells. Cells were first washed two times with PBS and resuspended in ½ the initial cell culture volume. Ten microliters of the resuspended cells was spotted onto a 200 mesh carbon-Formvar copper grid (Ted Pella, Redding, CA) and incubated for 5 min. The cell droplet was then removed with Whatman filter paper, and 10 μl of anti-P1 mAb 1-6F or 6-8C IgG (50) diluted 1:50 in PBS containing 10% bovine serum albumin (PBS-BSA) was added to the grid and incubated at room temperature for 1 h. After incubation, the antibody solution was removed with filter paper, and the grids were washed three times with a 10-μl drop of PBS that was again removed with filter paper. Ten microliters of goat anti-mouse IgG conjugated to 15-nm gold particles (BBI Solutions, Cardiff, UK) diluted 1:50 in PBS-BSA was then spotted onto each grid and allowed to incubate at room temperature for 1 h. The grids were again washed three times with PBS. Finally, each grid was stained with 10 μl of 1% phosphotungstic acid, pH 7.4 for 1 min and washed three times with 10 μl of distilled water. Samples were examined with a Hitachi H-7000 TEM (Hitachi High Technologies America Inc., Schaumburg, IL), and digital images were acquired with a Veleta 2000 × 2000 megapixel camera and iTEM software (Olympus Soft Imaging Solutions Corp., Munster, Germany). All imaging was performed at the University of Florida Interdisciplinary Center for Biotechnology Research Electron Microscopy core facility.
Thin Sections
Cells were first washed two times and resuspended in PBS. They were then incubated with anti-P1 mAb 1-6F or 6-8C IgG (50) diluted 1:50 in PBS for 1 h at room temperature. After three washes in PBS, the cells were incubated with goat anti-mouse IgG conjugated to 15-nm gold particles diluted 1:20 in PBS, then spotted onto each grid, and allowed to incubate at room temperature for 1 h. The cells were again washed three times in PBS, then fixed in 2.5% glutaraldehyde for 1 h followed by 1% OsO4, dehydrated in ethanol, and embedded in LR White embedding medium (Electron Microscopy Sciences, Hatfield, PA). Once embedded into resin, each sample was sectioned into 80–100-nm-thick sections, mounted onto 200 mesh carbon-Formvar copper grid, and stained with 4% aqueous uranyl acetate followed by Reynolds' lead citrate (76). Samples were then examined with a Hitachi H-7000 TEM, and digital images were acquired with a Veleta 2000 × 2000 megapixel camera and iTEM software. All sample preparation and imaging were performed at the University of Florida Interdisciplinary Center for Biotechnology Research Electron Microscopy core facility.
Atomic Force Microscopy
Preparation of Bacterial Cells
S. mutans cells were harvested by centrifugation at 5000 × g for 10 min and resuspended in 10 ml of PBS. To obtain a working concentration of cells, the original suspension was further diluted in PBS to obtain an optical density of ∼0.01 at 540 nm. The cell suspension was gently vortexed to release cells from any aggregates that formed in the standing cultures and then filtered through a 0.6-μm Isopore polycarbonate membrane (DTTP 025 00, Millipore).
Preparation of Model P1 Surfaces
Purified histidine-tagged P1 (P1-His6) was immobilized onto gold-coated substrates via the specific binding between His tags and nitrilotriacetic acid (NTA)-terminated self-assembled monolayers. Briefly, glass coverslips were precoated with a ∼5-nm chromium layer followed by a ∼30-nm gold layer through thermal evaporation. Before use, the gold-coated surfaces were immersed overnight in a solution of 0.1 mm NTA-terminated (10%) and triethylene glycol-terminated (90%) alkane thiols in ethanol, then rinsed in ethanol, and incubated in 40 mm aqueous Ni2+ solution, pH 7.2 for 30 min. The surfaces were further incubated with 0.2 mg/ml purified P1 in PBS for 2 h at room temperature followed by rinsing with the same buffer. All surfaces were freshly prepared and used the same day.
Preparation of AFM Tips Bearing Anti-P1 mAbs
AFM tips were functionalized with monoclonal anti-P1 antibodies (mAbs 1-6F, 6-8C, 5-5D, and 4-10A; Fig. 1C) via a 6-nm-long polyethylene glycol (PEG) linker. Cantilevers were first washed with chloroform and ethanol, placed in a UV-ozone cleaner for 15 min, and immersed overnight in 5.6 m ethanolamine hydrochloride (in DMSO) to generate amino groups on the tip surface. The cantilevers were then incubated with PEG linkers carrying benzaldehyde groups on their free tangling end (77) and immersed further in 1% citric acid solution for 10 min. After rinsing with Milli-Q water (ELGA LabWater), the cantilevers were incubated in a 100–200-μl droplet of 0.2 mg/ml mAbs in PBS containing 10 mm sodium cyanoborohydride (NaCNBH3). After 50 min of incubation, 5 μl of a 1 m ethanolamine hydrochloride solution, pH 9.5 was added to block unreacted aldehyde groups for 10 min. Finally, the cantilevers were washed with PBS and stored in the same buffer with added 1% sodium azide (NaN3) until use (within 7 days).
Imaging and Force Spectroscopy
AFM images and force-distance curves were recorded at room temperature (25 °C) in PBS using a Nanoscope VIII Multimode AFM (Bruker Corp., Santa Barbara, CA) and oxide-sharpened Si3N4 cantilevers (MSCT, Bruker Corp.). Cells were immobilized onto 0.6-μm-diameter Isopore polycarbonate membranes, which are slightly smaller than the reported diameter of S. mutans (78, 79). The membrane filter was then gently rinsed with three baths of PBS, carefully cut into ∼1 × 1-cm2 square, attached to a steel sample puck (Bruker Corp.) using a double-sided adhesive (avoiding dewetting), and mounted onto the AFM liquid cell. The spring constants of the cantilevers were typically in the range of 0.02–0.04 newton/m as determined by a thermal noise method (80). Unmodified cantilevers were first used to image and localize individual cells. The unmodified cantilevers were then replaced with cantilevers bearing the antibodies for single molecule analysis of P1 on live S. mutans. Force mapping was performed by collecting a 32 × 32 array of force-distance curves on a 300–500-nm2 localized area of the cell, and the two-dimensional adhesion maps were reconstructed using the custom analysis described below. All force curves were recorded at ∼100-ms contact time with a maximum applied force of 250 pN and 1000 nm/s approach and retraction speeds unless specified otherwise.
Batch Analysis of the Force Curves
A custom batch analysis toolset written in IGOR Pro 6 (Wavemetrics, Portland, OR) was used to perform force data extraction, calculation, and reporting. For each force curve, the last peak adhesion force and the corresponding final rupture lengths were extracted or calculated using a rupture-finding algorithm that searches for the sections of each force curve with relatively abrupt drops in force that signify a rupture event. An additional distance threshold of 30 nm was implemented to eliminate <30-nm extensions, which could arise from nonspecific interactions. From this set of force ruptures, adhesion force maps, adhesion force histograms, and scatter plots were calculated and reported.
RESULTS
Adherence and Aggregation Properties of S. mutans following Mechanical Extraction of P1
Biacore surface plasmon resonance was used to evaluate adherence of S. mutans to immobilized SAG following mechanical extraction of surface-localized proteins, including P1, from the cell surface. Analysis of postextracted cells, compared with the non-extracted NG8 cells, demonstrated that removal of non-covalently linked P1 from the surface of the cell greatly diminished the ability of S. mutans to adhere to immobilized SAG (Fig. 2A). Mechanical extraction resulted in a ∼75% decrease in SAG binding compared with the untreated cells. Chemical fixation of non-covalently linked P1 to the cell surface with glutaraldehyde prior to the mechanical extraction process preserved the adherence capability of S. mutans. Although fixation modestly diminished the measured adherence of non-extracted cells by ∼20%, the fixed cells lost only ∼15% of their adherence capability compared with ∼75% for the unfixed cells following mechanical extraction. The ability of glutaraldehyde fixation to preserve adherence of cells subjected to the mechanical extraction process suggests that the fragments of P1 released during the extraction process mediate the interaction of S. mutans with immobilized SAG. A spectrophotometric assay that measures P1-mediated aggregation of S. mutans in the presence of fluid-phase SAG (12) showed that postextracted cells were aggregated to a similar degree as untreated cells (Fig. 2B). This demonstrates that residual cell surface-localized P1 remained following the extraction procedure and that it was fully capable of a functional interaction with fluid-phase SAG. Hence, extractable non-covalently linked P1 polypeptides appear to play a more prominent role in mediating S. mutans adherence compared with aggregation.
FIGURE 2.
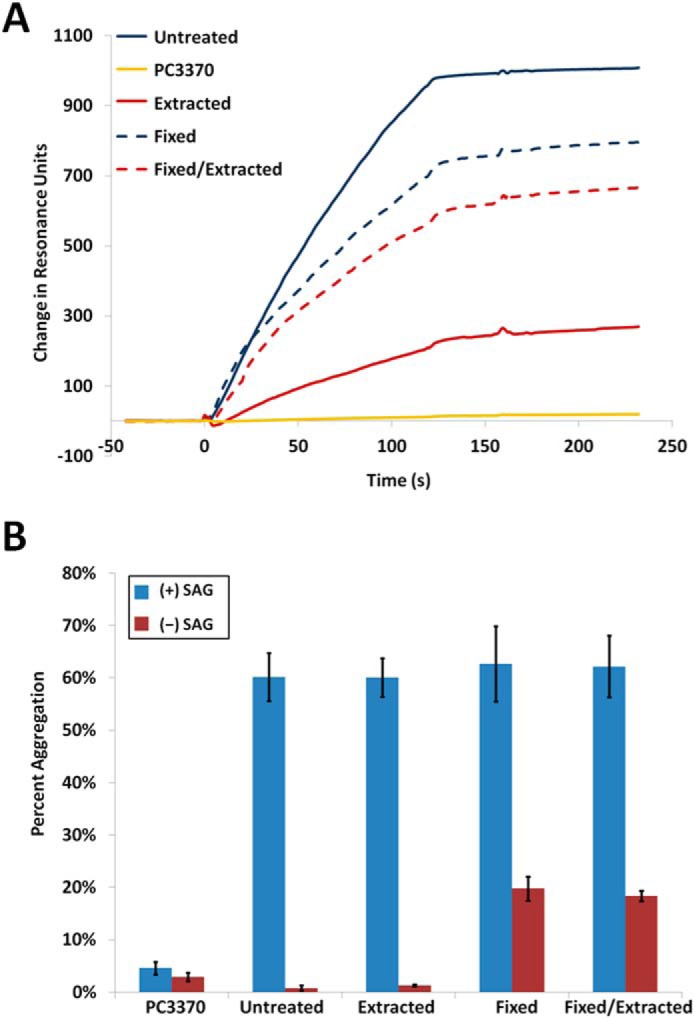
Adherence and aggregation of S. mutans to immobilized and fluid-phase SAG and antigenic properties following mechanical extraction and/or fixation. A, Biacore surface plasmon resonance sensorgram measuring the adherence of S. mutans strain NG8 to immobilized SAG. NG8 displays a substantial decrease in adherence to SAG following mechanical extraction of P1. Glutaraldehyde fixation of the bacterial cells prior to the mechanical extraction process preserved their ability to adhere to immobilized SAG. Both fixed and fixed/extracted cells displayed a modest decrease in measured adherence compared with untreated cells that may reflect either a diminution in adherence or a change in the refractive properties of the cells. PC3370 is an isogenic mutant of NG8 that lacks the spaP gene that encodes P1. B, S. mutans strain NG8 aggregation measured in the presence and absence of fluid-phase SAG. P1-mediated aggregation was unaffected by mechanical extraction of the protein from the surface of the cell. Chemical fixation of the cells resulted in a low level of self-aggregation in the absence of SAG that was unaffected by mechanical extraction. Bars represent the average S.E. (error bars) of three separate experiments.
P1 Antigenicity on the Surface of S. mutans before and after Mechanical Extraction
Despite the dramatic decrease observed in bacterial adherence to immobilized SAG following mechanical extraction, S. mutans cells displayed a large degree of residual reactivity with multiple different anti-P1 mAbs (49) (Fig. 3A). Postextracted cells displayed comparable reactivity as untreated cells with mAbs 1-6F and 4-9D. These antibodies recognize epitopes within the globular apical head of the molecule (49). Similar to what was observed in an earlier study (57), whole untreated cells did not appear to be reactive with mAbs 6-8C and 5-3E, which map to the globular C terminus of P1 (49). Previously, this result was attributed to the presumed inaccessibility of the C terminus to antibody because this portion of the adhesin was thought to lie exclusively at the base of the stalk immediately adjacent to the cell surface (44) or buried within the cell wall (58). However, following glutaraldehyde fixation, S. mutans displayed a notable change in the antigenicity of P1 on the cell surface (Fig. 3A). The reactivity of the C terminus-specific mAbs 6-8C and 5-3E was substantially enhanced. This suggests the additional presence of a C-terminal fragment on the surface of the fixed cells that is not retained on unfixed cells. In contrast, after glutaraldehyde treatment, the epitopes at the apex of the molecule recognized by mAbs 1-6F and 4-9D were not observed, and reactivity with mAb 4-10A, which maps to the helical stalk, was diminished. The discontinuous epitopes recognized by mAbs 6-11A and 5-5D, which depend on an A/P-region interaction at the base of the stalk, were also partially masked. The retention of a C-terminal fragment as a result of glutaraldehyde fixation appears to play a key role in preserving the adherence of S. mutans following this treatment. Loss of the non-covalently attached C-terminal polypeptide and potentially other adherent fragments as well therefore provides a likely explanation for the pronounced decrease in adherence observed following mechanical extraction.
FIGURE 3.

Dot blots of unfixed and glutaraldehyde-fixed S. mutans and comparison of P1 fragments released from S. mutans by mechanical and SDS extraction. A, the degree of reactivity of a panel of different anti-P1 mAbs, which map to distinct regions of the P1 tertiary structure (49) (Fig. 1C), was evaluated against fixed and unfixed cells both before and after mechanical extraction. The overall antibody reactivity profile was essentially unaffected by mechanical extraction. However, antigenicity following fixation was notably altered. The epitopes recognized by mAbs 1-6F, 4-9D, 4-10A, 6-11A, and 5-5D were masked following fixation, whereas the C-terminal epitopes recognized by mAbs 6-8C and 5-3E became apparent (49). B, SDS-PAGE and Western blots of proteins released from untreated or glutaraldehyde-fixed cells by mechanical extraction or boiling in SDS. The profiles of non-covalently linked P1 fragments released by both methods appear to be similar. P1 fragments recognized by mAbs 1-6F and 6-8C, which map to the globular head and C-terminal segments of the molecule, respectively (49) (Fig. 1C), are present in both samples. No detectable P1 fragments were released from the fixed cells by either method.
SDS-PAGE and Western Blot Analysis of P1 Released during Mechanical and SDS Extraction
Comparison of P1 fragments released from S. mutans during mechanical and SDS extraction demonstrated similar patterns of proteins released from the surface of the cell (Fig. 3B). Multiple C-terminal fragments were detected with the C terminus-specific antibody 6-8C in both samples. Additional and different polypeptides were recognized by mAb 1-6F, which maps to the apical head at the opposite end of the intact molecule, indicating that other P1 polypeptides are present and extractable from the cell surface as well. As expected, when the cells were fixed with glutaraldehyde prior to extraction, there was no detectable release of protein from the cell surface. The similarity in protein profiles following mechanical compared with SDS extraction demonstrates that the P1 fragments released during mechanical extraction were not the result of shear forces breaking molecules from the cell wall but instead resulted from the disruption of non-covalent interactions. Fixing the cells prior to extraction prevented release of non-covalently attached P1 fragments, providing an explanation why fixation preserved the adherence capability of mechanically agitated S. mutans cells.
Intermolecular Interactions of P1 Molecules and Fragments in Vitro
Given our speculation that an additional layer of non-covalently attached P1 molecules decorates the cell surface (presumably bound to a scaffolding layer of covalently attached P1), we sought to determine whether segments of the molecule containing the adhesive globular head and the C-terminal regions (6, 22, 44, 45) were capable of interacting with one another in vitro. We utilized a Biacore surface plasmon resonance assay in which a polypeptide fragment spanning the third A-repeat through the first P-repeat (A3VP1) was immobilized onto the gold chip surface (73). Binding of full-length rP1, recombinant A3VP1, and the C-terminal polypeptide to immobilized A3VP1 fragment was tested. The resulting sensorgrams are shown in Fig. 4A. MBP was included as a negative control protein as was an N-terminal fragment of P1 (NA1) that does not contain an adhesive domain. The C-terminal region of P1 was clearly interactive with A3VP1, which contains the globular head and one-third of the extended helical stalk. The globular head alone was also capable of interaction with the isolated C-terminal region by ELISA (data not shown). In addition, A3VP1 interacted with itself as well as with full-length P1. Additional experiments demonstrated the concentration-dependent nature of the interactions (Fig. 4B). The notably increased dissociation of the A3VP1 and C-terminal fragments compared with rP1 likely represents the presence of both adhesive domains within the full-length molecule compared with the single adhesive domain contained in each of the other polypeptides. Taken together, these results indicate that P1 is capable of molecular interactions between portions of the protein that are widely separated in the tertiary structure and not involved in intramolecular interactions.
FIGURE 4.
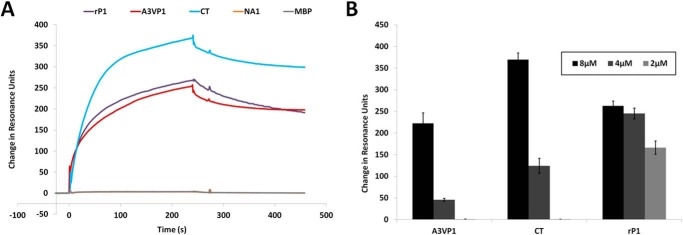
Intermolecular interactions between P1 fragments. A, Biacore surface plasmon resonance sensorgrams demonstrating the binding of each indicated polypeptide (8 μm) to immobilized A3VP1. The P1 NA1 fragment and MBP were used as negative controls. B, bar graph showing the maximum ΔRU of Biacore experiments illustrating the concentration dependence of binding of each of the recombinant P1 fragments (tested at 8, 4, and 2 μm) to immobilized A3VP1. Bars represent the average ± S.E. (error bars) of three separate experiments.
Regeneration of Adherence of Mechanically Extracted Cells by rP1 and P1 Fragments
Incubation of mechanically extracted S. mutans cells with rP1, A3VP1, or the C terminus regenerated the adherence capability of S. mutans (Fig. 5A). A3VP1 and the C terminus each conferred a significant increase in adherence (p value <0.005) compared with postextracted cells (Fig. 5B). In contrast, the modest increase in adherence observed with full-length rP1 is not statistically significant (p value >0.05). The negative control proteins NA1, which alone does not bind SAG (40), and MBP did not restore adherence of extracted cells. Incubation of the P1-deficient mutant PC3370 with rP1, A3VP1, or the C-terminal region of P1 did not increase adherence of this strain to immobilized SAG (Fig. 5C). This indicates that the regeneration of postextracted NG8 cells with P1 polypeptides involves their direct interaction with residual P1 on the surface of the cell rather than to other S. mutans surface proteins. Because A3VP1 and the C terminus each contain an adherent domain of P1, it is not surprising that either can independently restore adherence to postextracted cells. Adherence was not further improved by treating the postextracted cells with a mixture of A3VP1 and the C-terminal fragments (data not shown). Each of the fragments was effective at regenerating the adherent surface of S. mutans, whereas the full-length rP1 molecule was not. This suggests that the spatial context in which these particular P1 fragments assemble on the surface of S. mutans may be important to achieve the appropriate architecture necessary to mediate the functional interaction of P1 with immobilized SAG.
FIGURE 5.

Exogenously added P1 fragments can regenerate adherence of mechanically extracted S. mutans to immobilized SAG. A, Biacore surface plasmon resonance sensorgrams illustrating adherence to immobilized SAG of untreated, mechanically extracted, and mechanically extracted S. mutans strain NG8 following incubation with the indicated polypeptides (8 μm). The A3VP1 and the CT fragments display the greatest ability to regenerate adherence of S. mutans following mechanical extraction, whereas the full-length rP1 molecule is less effective. The P1 N-terminal fragment NA1, which does not contain a SAG binding domain, does not improve the adherence of mechanically extracted cells. MBP was also included as a negative control. PC3370 is an isogenic mutant of S. mutans strain NG8 that lacks P1. B, bar graph displaying the average ΔRU ± S.E. (error bars) of three independent experiments. A3VP1 and the C terminus both confer a significant increase in adherence to mechanically extracted S. mutans to SAG. Tukey's multiple comparison test determined differences among the groups: **, p ≤ 0.005. C, bar graph displaying the average ΔRU ± S.E. (error bars) of three independent experiments. Incubation with recombinant P1 polypeptides (8 μm) does not confer adherence to the non-adherent P1-deficient strain PC3370, indicating that regeneration of adherence of mechanically extracted wild-type S. mutans with recombinant P1 fragments involves their interaction with residual P1 on the cell surface.
Release of C-terminal P1 Fragments from the Surface of S. mutans following Treatment with Antibody or Trypsin
Incubation of S. mutans with a variety of anti-P1 mAbs caused the release of C-terminal fragments of P1 from the surface of unfixed but not fixed cells (Fig. 6A). Polypeptides reactive with a C terminus-specific rabbit polyclonal antiserum made against the most C-terminal 144 amino acids of P1 (58) (kindly provided by Dr. Song Lee, Dalhousie University) were detected following treatment of S. mutans whole cells with anti-P1 murine mAbs. In particular, prominent ∼50-kDa C-terminal fragments of P1 were released upon incubation of the cells with either mAb 1-6F or 6-8C. A protease-resistant C-terminal fragment was also detected by Western blotting following digestion of S. mutans whole cells with trypsin (Fig. 6B). The stable C-terminal fragment released into the supernatant following trypsin treatment likely represents the previously described ∼50-kDa protease-resistant antigen II component of P1 (29). This result confirms that the C terminus of P1 is present in two locations, both accessible on the cell surface and buried within the cell wall as demonstrated previously (58).
FIGURE 6.

C-terminal fragments of P1 are released from the surface of S. mutans following incubation with anti-P1 murine monoclonal antibodies (50) or treatment with trypsin. A, untreated or glutaraldehyde-fixed S. mutans cells were incubated with each indicated anti-P1 mAb, and the supernatant fluids were analyzed by Western blotting, probing with a specific rabbit antiserum made against the C-terminal 144 amino acids of P1 (58). Incubation with mAb 1-6F or 6-8C in particular results in the release of a predominant ∼50-kDa C-terminal fragment that corresponds in size to the previously described protease-resistant antigen II component of the adhesin (29). B, a similar ∼50-kDa C-terminal fragment is also released into the supernatant fluid following treatment of S. mutans cells with trypsin. This indicates that the C-terminal 144 amino acids of P1 contained within this fragment and recognized by the rabbit antiserum are also accessible on the surface of the cell and not exclusively buried within the cell wall peptidoglycan as reported previously (58).
AFM Imaging of S. mutans
We also used atomic force microscopy to characterize the cell surface properties of live S. mutans cells. To begin, we visualized the surface morphology of S. mutans NG8 wild-type cells and the PC3370 mutant strain devoid of P1 (ΔP1) using topographic imaging in air and under physiological conditions (PBS buffer, pH 7.4). Fig. 7, A and B, are low resolution AFM deflection images in air of wild-type and mutant cells, respectively. We report deflection images because given the large curvature of the cells this mode is more sensitive to the fine surface topography than height images. Consistent with earlier AFM (78) and electron microscopy (75, 81, 82) studies, the cells were elongated, had a smooth surface, and exhibited a well defined septum (arrow 1). In addition, annular structures were clearly visible at a certain distance from the poles (arrow 2). More physiological views of the cell surface are shown in Fig. 7, C and D, which present deflection images in buffer of isolated live wild-type and mutant cells immobilized in a porous membrane. Applying a low imaging force, images of the same area were obtained repeatedly without detaching the cell or significantly altering the surface morphology. For both the wild type and P1-deficient mutant, equatorial structures were notable and at a certain lateral distance from each other (arrow 2). Depending on how the cells were immobilized in the membrane, either the equatorial regions or the poles were exposed. Apart from the equatorial structures, the cell surface appeared relatively smooth; the root mean square roughness on height images was <1 nm (on 500 × 500-nm2 areas).
FIGURE 7.

AFM topographic imaging of S. mutans wild-type and ΔP1 mutant cells. A and B, deflection images recorded in air for wild type and mutant, respectively. C and D, deflection images of mechanically trapped live individual cells in PBS buffer for wild type and mutant, respectively. A well defined septum (arrow 1) and equatorial rings (arrow 2) are visible in both strains.
Detection and Stretching of Purified P1
We next used tips functionalized with specific antibodies to establish the ability of single molecule force spectroscopy to detect isolated, purified P1 proteins. Recombinant P1 bearing a His tag at its C-terminal end was attached to gold-coated substrates bearing Ni2+-NTA groups (Fig. 8A). A mAb reactive with the globular head (mAb 1-6F; Fig. 1C) was covalently attached to the AFM tip through a PEG linker, which offers firm attachment as well as flexibility (77). Fig. 8B shows the adhesion force histogram with representative retraction force curves obtained from the interaction between purified P1 and the anti-P1 antibody. About ∼20% of the curves showed binding events with the most probable rupture forces occurring around 40 ± 8 pN. We note that the profiles of the curves did not substantially change when recording consecutive force curves on different spots of the substrate. We attribute the ∼40-pN force to the rupture of a single P1-anti-P1 bond because we used a well established PEG-based chemistry that guarantees attachment of the antibodies on the tip at low density and because the ∼40-pN force is in the range of values reported for other antibody/antigen interactions at similar loading rates (83–88). Fig. 8C shows a scatter plot of adhesion force versus rupture distance obtained for the P1/anti-P1 interaction. Although the plot shows that most adhesion forces were concentrated around ∼40–50 pN, some larger adhesion forces up to 250 pN were also observed (see Fig. 8B, inset, two lower force curves), suggesting that multiple adhesins were probed. In addition, most rupture lengths varied between 50 and 150 nm with a few longer ruptures at 150–250 nm. The primary structure of the P1 molecule as shown in Fig. 1A consists of 1561 amino acids, which would correspond to a length of ∼562 nm if fully unfolded (1 amino acid = 0.36 nm (89)). Our results clearly show that a ∼40-pN pulling force was not capable of unfolding the P1 adhesin, consistent with the common observation that unraveling a β-sheeted structure such as that of the globular head or the C-terminal region would require much larger forces (89). Many of our rupture distances are consistent with the estimated ∼65-nm length of the molecule (Fig. 1B), suggesting that pulling single P1 molecules through their globular heads leads to the full elongation and extension of the rods until the antigen-antibody bond ruptures (see schematic in Fig. 8A). In addition, many ruptures were longer than 50 nm, a result that we attribute to two plausible scenarios. First, it is highly possible that there is a multilayer of P1 on the Ni2+-NTA surface because the adhesin could interact with itself to form multimers, meaning multiple adhesins would be probed together, thus leading to larger adhesions and longer extensions. Second, because the His-Ni2+-NTA bond is rather weak and because we may have a multilayer of proteins, we cannot exclude that during the course of the experiment P1 molecules (or multimers) were transferred to the AFM tip. This would imply that at some point P1-P1 homophilic bonds are being measured rather than P1-anti-P1, thus leading to greater adhesions and longer extensions particularly if P1 exists as a multimer. In support of this view, injection of free P1 molecules into the solution did not lead to an inhibition of the P1/anti-P1 interaction (data not shown) as would be expected for such a blocking experiment. This strongly suggests that upon blocking of the tip P1/P1 homophilic interactions were being measured.
FIGURE 8.

Single molecule force spectroscopy of purified P1 proteins. A, schematic of the surface chemistry used to functionalize model substrates with P1 and AFM tips with anti-P1 targeting the apical head (mAb 1-6F). V, variable. B, adhesion force histograms (n = 1024) and retraction force curves (inset) recorded between the P1 substrate and the anti-P1 tip. C, scatter plot of the specific adhesion forces as a function of the rupture length (21% of a total of n = 1024 force curves). Similar results were obtained in several independent experiments using different tip and sample preparations.
Molecular Mapping of the P1 Apical Head on Living Bacteria
Monoclonal antibodies that bind to different domains of P1 (Fig. 1C) have provided valuable insights into the antigenic regions of the protein as well as into the mechanism of protection observed with anti-P1 antibodies (49, 56, 75, 90, 91). We used single molecule force spectroscopy utilizing several different anti-P1 mAbs (Fig. 1C) as specific molecular probes to detect, localize, and stretch the different structural motifs of P1 on live S. mutans bacteria. Collectively, as detailed below, these studies demonstrated that the architecture of P1 on the cell surface is more complex than previously appreciated. We first probed the globular head of the adhesin using AFM tips bearing the mAb 1-6F (Fig. 9, A–D). Fig. 9, A–D, show the deflection image of a single S. mutans cell and the corresponding adhesion force map, representative retraction force curves, and adhesion versus extension plot obtained for the interaction between the cell in Fig. 9A and mAb 1-6F, respectively. A substantial fraction of the force curves, 18 ± 3% (from a total of n = 7000 curves from eight different maps obtained on both polar and side wall regions) showed single, well defined adhesion peaks of 43 ± 7 pN along with rupture lengths ranging from 50 to 125 nm. These force signatures were not observed in the bacterial mutant devoid of P1 (Fig. 9, E–H). This finding together with our prior experiments on a model surface (Fig. 8) leads us to believe that the ∼40-pN forces reflect the detection of single P1 adhesins. Sometimes larger forces (75 ± 8 pN) were seen, suggesting the detection of additional P1 moieties, which as described above are not likely full-length molecules. As the length of the intact P1 rod is estimated at ∼65 nm (44, 45), we attribute our 50–100-nm rupture lengths to represent the full extension of the rodlike structure of P1 together with stretching of additional smaller pieces of P1 arranged in a multilayer complex. In view of the small single adhesion peaks and short extensions, it is unlikely that secondary structures of the protein are being unfolded, consistent with the common observation that larger forces (∼200 pN) are needed to unravel β-sheeted structures (89). In contrast to experiments with purified P1, higher forces (>100 pN) and extended rupture lengths (>120 nm) were not as readily observed on live cells (see scatter plot in Fig. 9D). Adhesion force maps (Fig. 9B) revealed that adhesins were extensively exposed, corresponding to a maximum surface density of ∼700 sites/μm2. There was no clear evidence for clustering, which differs from the behavior of some adhesins shown to form nanoscale domains (65, 84). Finally, we note that the distribution, adhesion, and extension of P1 were similar on the poles and on the side walls of the cells, suggesting a relatively uniform distribution of P1 across the whole cells. This is in contrast to a previous report in which P1 (PAc) and other cell surface proteins were found to co-localize with the SecA component of the protein secretion machinery and to cluster in a discrete microdomain (92).
FIGURE 9.

Molecular mapping of the apical head of P1 proteins on live S. mutans cells. A and E, deflection images recorded in PBS buffer with a bare silicon nitride tip on the polar region of wild-type and ΔP1 mutant cells, respectively. V, variable. B and F, adhesion force maps recorded on the poles of wild-type and mutant cells (in the dashed square regions shown in A and E) using tips functionalized with anti-P1 antibodies targeting the apical head (mAb 1-6F). C and G, typical retraction force curves acquired in PBS buffer between anti-P1 tips and the poles of wild-type and ΔP1 mutant cells. D and H, scatter plot of adhesion versus rupture length for the adhesion events recorded on the poles of wild-type (force map in B) and ΔP1 mutant (force map in F) cells. The reported percentage values were from a total of n = 1024 force curves. Similar data were obtained in independent experiments using four tips and eight cells for wild-type cells and three tips and five cells for ΔP1 mutant cells.
Accessibility of the C- and N-terminal Regions of P1
We next used single molecule force spectroscopy to detect other regions of P1. To this end, we used anti-P1 antibodies directed against the C-terminal region (mAb 6-8C) as well as against a complex epitope that includes the N terminus and that is localized at the base of the A-P stalk (mAb 5-5D) (Fig. 10, A–H). The force data for both of these antibodies were qualitatively similar to the globular head region data (see the profiles of the retraction force curves (Fig. 10, C and G) as well as the binding forces (Fig. 10, D and H)), consistent with the detection of single antibody-antigen bonds. Similar to what was observed with mAb 1-6F, the adhesion maps did not suggest a clustered distribution of P1 (Fig. 10, B and F). In addition, rupture lengths (50–100 nm) were consistent with a rodlike structure that was not unfolded with moderate forces. However, there were notable differences in the number of adhesion events; the detection frequency was 8 ± 3% (n = 5000 curves from five different maps obtained on both polar and side wall regions) and 3 ± 2% (n = 5000 curves from five different maps) for mAbs 6-8C and 5-5D, respectively (versus 18% for mAb 1-6F). This behavior likely reflects differences in the accessibility of the epitopes to the antibodies and/or the probe tip. A previous model had suggested that the globular head is projected from the cell surface by the extended stalk emanating from the globular C-terminal domain (44). Our single molecule data support this model by showing that the globular head is most accessible on the cell surface and that a discontinuous epitope on the opposite end of the stalk is least accessible. Additionally, however, the detection of the C-terminal region by single molecule force spectroscopy is consistent with new data presented above that demonstrate that this segment is also exposed and accessible on the cell surface. If a single uniform monolayer of identically oriented covalently attached P1 molecules were correct, then the 6-8C epitope would be less accessible than the 5-5D epitope.
FIGURE 10.

Molecular mapping of the C terminus, stalk base, and stalk regions of P1 proteins on live S. mutans cells. A, E, and I, deflection images of wild-type cells recorded in PBS buffer with an unmodified silicon nitride tip. B, F, and J, adhesion force maps recorded on wild-type cells (in the dashed square regions shown in A, E, and I) using tips functionalized with antibodies targeting the C-terminal (mAb 6-8C), stalk base (mAb 5-5D), and stalk (mAb 4-10A) regions. C, G, and K, typical retraction force curves acquired on wild-type cells in PBS buffer using anti-P1 tips targeting the C-terminal, stalk base, and stalk regions. D, H, and L, scatter plots of adhesion versus rupture length corresponding to the adhesion events in the force maps in B, F, and J, respectively. The reported percentage values were from a total of n = 1024 force curves. Similar data were obtained in independent experiments using three tips and six cells for 6-8C, two tips and five cells for 5-5D, and two tips and five cells for 4-10A.
P1 Appears to Associate into a Multilayer Complex
To further evaluate the cell surface structure of P1, we also used mAb 4-10A that recognizes an epitope contained within the hybrid helical stalk (Fig. 10, I–L). Both A- and P-region sequences contribute to this discontinuous epitope (Fig. 1C) (49, 75). The detection frequency of 19% (n = 3000 curves from three different maps obtained on both polar and side wall regions) was as high as that of the globular head (Fig. 9D). This suggests that the stalk region may be more accessible than the C-terminal region or the base of the stalk but more likely reflects that this particular mAb binds to multiple sites because its epitope is repeated three times within each full-length molecule (see Fig. 1, B and C). The 4-10A force maps also suggested the absence of clustering (Fig. 10J). The majority of the adhesion values were in the range of those for typical single antibody-antigen bonds (Fig. 10L); however, the scatter plot also showed the presence of extended rupture lengths (120–200 nm; see Fig. 10K, last two retraction curves). These extended force profiles are not likely due to the dissociation of the A- and P-regions. Because the interaction of the α- and polyproline helices comprising the stalk is of high affinity (45), we believe that these extended force profiles instead reflect dissociation of additional P1 moieties associated with covalently anchored P1 proteins. Again, the variations in rupture distances likely reflect the fact that mAb 4-10A binds to a repeating epitope. As stated earlier, the initial discovery of P1 was as two separate extracellular antigens named AgI and AgII (29, 93). Given the propensity of P1 molecules and fragments thereof to interact with one another as well as the ability of P1 fragments to restore adhesion to mechanically extracted cells, our AFM results support a model whereby full-length Ag I/II is covalently coupled to the cell wall and serves as a scaffold for additional non-covalently linked fragments such as AgI and AgII to further extend the adhesive layer. Hence, the rupture lengths observed for whole cells are not identical to those observed on the P1 model surface in which only highly purified homogeneous full-length molecules were evaluated in the absence of the naturally occurring extracellular fragments that are routinely found to be associated with whole bacterial cells.
Immunogold Transmission Electron Microscopy
To confirm the results of the studies described above, the reactivity of anti-P1 mAbs 1-6F and 6-8C on the surface of the cell was visualized using immunogold electron microscopy. Untreated non-extracted cells, mechanically extracted cells, and cells subjected to glutaraldehyde fixation prior to mechanical extraction were evaluated (Fig. 11). Images of whole cells (Fig. 11A) as well as thin sections (Fig. 11B) were taken. The results are consistent with the dot blot and AFM results and the view that P1 is contained within a more complex architecture on the S. mutans cell surface rather than present as a single monolayer of identically oriented full-length molecules. In contrast to earlier EM studies (50, 94), it is now known where various anti-P1 mAbs map on the primary (49) as well as the tertiary structure of P1 (Fig. 1C). This new information allows a more informed perspective of P1 contained within the previously identified fuzzy coat layer on the cell surface (50, 95). The C-terminal epitope recognized by mAb 6-8C was clearly present and accessible on the surface of S. mutans when whole bacterial cells were viewed by immunogold transmission EM as compared with the seemingly different results when thin sections were viewed or dot blots of unfixed cells were performed (Fig. 3). This disparity can be explained by the extensive washing steps necessary during the thin section preparation and dot blot analysis compared with the brief soaking in PBS of a grid containing antibody-stained S. mutans cells that is used for transmission EM analysis. The reactivity observed by transmission EM for mAbs 1-6F and 6-8C, which map to opposite ends of the P1 ∼50-nm helical stalk, supports experimental evidence described above that a higher order ultrastructure of P1 exists on the cell surface. The results observed by immunogold EM could not occur if all P1 molecules were covalently attached to the cell surface and oriented such that the C terminus is only in close proximity with the cell wall and only the apex of the adhesin is projected outward. When cells were subjected to mechanical extraction, all reactivity with 6-8C was lost, whereas reactivity with 1-6F was retained. In contrast, when cells were fixed with glutaraldehyde prior to mechanical extraction and imaging, reactivity with 6-8C was retained, whereas that of 1-6F was masked. These results support the conclusions of our prior experiments, that is that a C-terminal polypeptide and possibly other fragments of P1 such as AgI are non-covalently associated with the cell surface. These fragments are removed by extraction procedures such as mechanical agitation or boiling in SDS. Glutaraldehyde treatment of the cells prior to the extraction fixes the fragments in place such that immunoreactivity with the C terminus-specific mAb 6-8C becomes evident, whereas that of mAb 1-6F is obscured. A P1-deficient mutant (68) was included as a negative control and was not reactive with the anti-P1 antibodies.
FIGURE 11.

Immunogold transmission electron microscopy demonstrating the reactivity of anti-P1 mAbs with S. mutans prior to and following mechanical extraction. mAbs 1-6F and 6-8C, which bind to opposite ends of the full-length intact P1 molecule (Fig. 1C), both react with epitopes exposed on the bacterial cell surface. Glutaraldehyde fixation of S. mutans prevents extraction of the C-terminal segment of P1 recognized by mAb 6-8C from the cell surface and masks the epitope recognized by mAb 1-6F. A, whole cell mount of untreated, mechanically extracted, and glutaraldehyde-fixed and extracted S. mutans incubated with mAbs 1-6F and 6-8C. B, thin sections (80–100 nm thick) of untreated, mechanically extracted, and glutaraldehyde-fixed S. mutans incubated with mAbs 1-6F and 6-8C.
DISCUSSION
Despite the recent availability of a complete tertiary model of P1 based upon a composite of several high resolution x-ray crystal structures in combination with velocity ultracentrifugation experiments (44–46, 48), little is known about how the molecule is configured on the cell surface or whether a specific architecture is required to mediate the functional property of adhesion to immobilized human salivary agglutinin. When bacterial thin sections were viewed in prior studies by electron microscopy, P1 appeared as a fuzzy fibrillar network along the surface of S. mutans (50, 95). P1 mutants lack this fibrillar layer and do not adhere to SAG-coated hydroxyapatite, nor do they aggregate in the presence of fluid-phase SAG (95). P1 has an LPXTG sortase A recognition motif characteristic of cell wall-anchored proteins (41, 42, 96–98); however, it has been recognized for many years that not all P1 is covalently linked to the cell surface and that the protein (mainly in the form of defined breakdown products) can be extracted by a variety of means (57, 59–63). To date, no experiments have evaluated the functional consequences of removal of non-covalently linked fragments of P1 from the cell surface or characterized the way in which the adhesin and its naturally occurring breakdown products may assemble to confer function. Our work demonstrates that such an assembly is necessary and culminates in a specialized architecture to mediate the interaction between S. mutans and immobilized SAG.
The reason for the processing of P1 into a stable protease-resistant C-terminal fragment (AgII) and a less stable N-terminal fragment (AgI) has never been fully understood. In light of our current data, a likely explanation for the protein being inherently unstable is that the two known adherence domains physically separate from the parent protein and then utilize the covalently linked P1 layer to build a functionally active supramolecular organization on the surface of the cell. Alternatively and/or additionally, non-covalently linked P1 may serve to neutralize SAG or immune mediators. Perturbing the quaternary structure of P1 from S. mutans whole cells by mechanical extraction results in a pronounced loss of adherent capability without interfering with the ability of these cells, which harbor readily detectable residual P1, to be aggregated by fluid-phase SAG. Fixing the extractable fragments in place by treatment with glutaraldehyde not only preserved the adherence of S. mutans subjected to mechanical agitation but also resulted in a markedly different reactivity profile with a panel of anti-P1 mAbs. Most notably, the cells gained reactivity with mAbs that map to the C-terminal AgII moiety, whereas epitopes recognized by other mAbs were no longer accessible. In further support of the assembly of a functional supramolecular organization on the cell surface, reconstitution experiments demonstrated that exogenously added P1 fragments containing the two known adherence domains, namely A3VP1 and the C terminus, restored adherence to postextracted cells to a significant degree, whereas the full-length molecule did not. AFM experiments were also performed in which the distribution and orientation of P1 on the surface of live cells were evaluated directly using mAbs that map to defined regions within the tertiary structure. These results are consistent with a model in which the globular head of P1 is projected away from the cell via its extended stalk. However, a C-terminal segment was also detectable and exposed on the cell surface similar to the globular head. The highly variable rupture lengths observed, particularly when using the 4-10A mAb, which maps to the P1 helical stalk, further suggest a model in which additional P1 moieties, possibly AgI, are arranged in a multilayer to decorate a scaffolding layer of ∼65-nm full-length P1 that is covalently anchored to the cell wall.
The ability of antibodies that map to the C terminus of P1 to interfere with adherence of S. mutans to immobilized SAG was initially puzzling because of their seeming lack of reactivity with whole cells of S. mutans (57). Our current work now provides an explanation. A previous report clearly showed that C-terminal epitopes were buried within the cell wall and made accessible following mutanolysin digestion of cell wall peptidoglycan (58), but this study did not evaluate the supernatant of the P1-stripped trypsin-treated cells for the presence of the C terminus of the protein. Our experiments identified a protease-resistant C-terminal fragment of P1 that was liberated from the cell surface by trypsin digestion. Release of cell surface-localized C-terminal fragments now offers an explanation why antibodies that appear non-reactive with the cell surface are so effective at inhibiting adherence. Antibody-triggered release of P1 fragments from the cell surface has been reported previously and hypothesized to represent a potential evasion mechanism by S. mutans when anti-P1 secretory IgA antibodies present in the oral cavity are encountered (59). In light of our current findings, we speculate that adherence-inhibiting anti-P1 antibodies act by two potential mechanisms. First, they may inhibit adherence directly by binding to and blocking a site that specifically mediates binding of P1 to SAG, and second, they may act indirectly by triggering the release of adherent fragments from the bacterial cell surface. A ∼50-kDa protein fragment was identified in the prior study (59) as part of an immune complex released following incubation with anti-P1 rabbit IgG or human colostral IgA, which contains natural anti-P1 activity. Interestingly, mAbs 1-6F and 6-8C are both highly inhibitory of S. mutans adherence. Unlike mAb 6-8C, the cognate epitope of mAb 1-6F is not contained within the C-terminal region of P1. The release of C-terminal fragments during incubation with these and other antibodies helps explain why, as observed in dot blot experiments, a pronounced shift in antigenicity of P1 occurs on the surface of the cell following fixation. Despite binding to opposite ends of the P1 molecule, mAbs 1-6F and 6-8C each appear to destabilize the quaternary structure of P1 on the cell surface in such a way as to cause the release of adherence-mediating P1 fragments, particularly the C terminus. Our immunogold electron microscopy experiments clearly demonstrate the presence of epitopes on the cell surface recognized by mAbs that map either to the apical head or the C terminus of P1, the latter of which is removed by mechanical extraction and preserved by chemical fixation.
Taken as a whole, our results suggest that a previously unrecognized functional quaternary structure exists in which C-terminal polypeptides of P1 and potentially other fragments decorate the outer layer of S. mutans by interacting with a covalently attached P1 scaffold. The intricate details of how P1 is assembled on the cell surface to confer its full range of capabilities need to be a focus of future studies if a complete understanding of the adhesive behavior of P1 and protective immunity against it is to be achieved. In addition, recent evidence of the propensity of P1 to form amyloid fibrils and hence contribute to functional amyloid formation by S. mutans (70) adds another dimension of complexity to the nature and functional properties of P1 on the surface of the cell.
Acknowledgments
We thank Karen Kelly and the University of Florida Interdisciplinary Center for Biotechnology Research Electron Microscopy core facility for valuable training and assistance with immunogold transmission electron microscopy and thin sectioning.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DE08007 and R01DE21789 from the NIDCR and Predoctoral Fellowship T90 DE021990-03. This work was also supported by a University of Florida Alumni fellowship; the National Foundation for Scientific Research (FNRS); the Université catholique de Louvain (Fondation Louvain-Prix De Merre); the Federal Office for Scientific, Technical and Cultural Affairs (Interuniversity Poles of Attraction Program); and the Research Department of the Communauté française de Belgique (Concerted Research Action).
- Ag
- antigen
- SAG
- salivary agglutinin complex
- AFM
- atomic force microscopy
- RU
- resonance units
- MBP
- maltose-binding protein
- rP1
- recombinant P1
- NTA
- nitrilotriacetic acid
- pN
- piconewtons
- CT
- C-terminal region.
REFERENCES
- 1. Hamada S., Slade H. D. (1980) Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol. Rev. 44, 331–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gauduchon V., Benito Y., Célard M., Mouren C., Delorme V., Philippe-Bert J., Etienne J., Vandenesch F. (2001) Molecular diagnosis of recurrent Streptococcus mutans endocarditis by PCR amplification and sequencing. Clin. Microbiol. Infect. 7, 36–37 [PubMed] [Google Scholar]
- 3. Ullman R. F., Miller S. J., Strampfer M. J., Cunha B. A. (1988) Streptococcus mutans endocarditis: report of three cases and review of the literature. Heart Lung 17, 209–212 [PubMed] [Google Scholar]
- 4. Kesavalu L., Lucas A. R., Verma R. K., Liu L., Dai E., Sampson E., Progulske-Fox A. (2012) Increased atherogenesis during Streptococcus mutans infection in apoE-null mice. J. Dent. Res. 91, 255–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abranches J., Zeng L., Bélanger M., Rodrigues P. H., Simpson-Haidaris P. J., Akin D., Dunn W. A., Jr., Progulske-Fox A., Burne R. A. (2009) Invasion of human coronary artery endothelial cells by Streptococcus mutans OMZ175. Oral Microbiol. Immunol. 24, 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brady L. J., Maddocks S. E., Larson M. R., Forsgren N., Persson K., Deivanayagam C. C., Jenkinson H. F. (2010) The changing faces of Streptococcus antigen I/II polypeptide family adhesins. Mol. Microbiol. 77, 276–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang S., Green N. M., Sitkiewicz I., Lefebvre R. B., Musser J. M. (2006) Identification and characterization of an antigen I/II family protein produced by group A Streptococcus. Infect. Immun. 74, 4200–4213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tettelin H., Masignani V., Cieslewicz M. J., Donati C., Medini D., Ward N. L., Angiuoli S. V., Crabtree J., Jones A. L., Durkin A. S., Deboy R. T., Davidsen T. M., Mora M., Scarselli M., Margarit y Ros I., Peterson J. D., Hauser C. R., Sundaram J. P., Nelson W. C., Madupu R., Brinkac L. M., Dodson R. J., Rosovitz M. J., Sullivan S. A., Daugherty S. C., Haft D. H., Selengut J., Gwinn M. L., Zhou L., Zafar N., Khouri H., Radune D., Dimitrov G., Watkins K., O'Connor K. J., Smith S., Utterback T. R., White O., Rubens C. E., Grandi G., Madoff L. C., Kasper D. L., Telford J. L., Wessels M. R., Rappuoli R., Fraser C. M. (2005) Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ray C. A., Gfell L. E., Buller T. L., Gregory R. L. (1999) Interactions of Streptococcus mutans fimbria-associated surface proteins with salivary components. Clin. Diagn. Lab. Immunol. 6, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prakobphol A., Xu F., Hoang V. M., Larsson T., Bergstrom J., Johansson I., Frängsmyr L., Holmskov U., Leffler H., Nilsson C., Borén T., Wright J. R., Strömberg N., Fisher S. J. (2000) Salivary agglutinin, which binds Streptococcus mutans and Helicobacter pylori, is the lung scavenger receptor cysteine-rich protein gp-340. J. Biol. Chem. 275, 39860–39866 [DOI] [PubMed] [Google Scholar]
- 11. Oho T., Yu H., Yamashita Y., Koga T. (1998) Binding of salivary glycoprotein-secretory immunoglobulin A complex to the surface protein antigen of Streptococcus mutans. Infect. Immun. 66, 115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brady L. J., Piacentini D. A., Crowley P. J., Oyston P. C., Bleiweis A. S. (1992) Differentiation of salivary agglutinin-mediated adherence and aggregation of mutans streptococci by use of monoclonal antibodies against the major surface adhesin P1. Infect. Immun. 60, 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bikker F. J., Ligtenberg A. J., Nazmi K., Veerman E. C., van't Hof W., Bolscher J. G., Poustka A., Nieuw Amerongen A. V., Mollenhauer J. (2002) Identification of the bacteria-binding peptide domain on salivary agglutinin (gp-340/DMBT1), a member of the scavenger receptor cysteine-rich superfamily. J. Biol. Chem. 277, 32109–32115 [DOI] [PubMed] [Google Scholar]
- 14. Beg A. M., Jones M. N., Miller-Torbert T., Holt R. G. (2002) Binding of Streptococcus mutans to extracellular matrix molecules and fibrinogen. Biochem. Biophys. Res. Commun. 298, 75–79 [DOI] [PubMed] [Google Scholar]
- 15. Handley P. S., Harty D. W., Wyatt J. E., Brown C. R., Doran J. P., Gibbs A. C. (1987) A comparison of the adhesion, coaggregation and cell-surface hydrophobicity properties of fibrillar and fimbriate strains of Streptococcus salivarius. J. Gen. Microbiol. 133, 3207–3217 [DOI] [PubMed] [Google Scholar]
- 16. Jenkinson H. F., Demuth D. R. (1997) Structure, function and immunogenicity of streptococcal antigen I/II polypeptides. Mol. Microbiol. 23, 183–190 [DOI] [PubMed] [Google Scholar]
- 17. Petersen F. C., Assev S., van der Mei H. C., Busscher H. J., Scheie A. A. (2002) Functional variation of the antigen I/II surface protein in Streptococcus mutans and Streptococcus intermedius. Infect. Immun. 70, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jakubovics N. S., Strömberg N., van Dolleweerd C. J., Kelly C. G., Jenkinson H. F. (2005) Differential binding specificities of oral streptococcal antigen I/II family adhesins for human or bacterial ligands. Mol. Microbiol. 55, 1591–1605 [DOI] [PubMed] [Google Scholar]
- 19. Lamont R. J., El-Sabaeny A., Park Y., Cook G. S., Costerton J. W., Demuth D. R. (2002) Role of the Streptococcus gordonii SspB protein in the development of Porphyromonas gingivalis biofilms on streptococcal substrates. Microbiology 148, 1627–1636 [DOI] [PubMed] [Google Scholar]
- 20. Lamont R. J., Hersey S. G., Rosan B. (1992) Characterization of the adherence of Porphyromonas gingivalis to oral streptococci. Oral Microbiol. Immunol. 7, 193–197 [DOI] [PubMed] [Google Scholar]
- 21. Nobbs A. H., Lamont R. J., Jenkinson H. F. (2009) Streptococcus adherence and colonization. Microbiol. Mol. Biol. Rev. 73, 407–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hajishengallis G., Koga T., Russell M. W. (1994) Affinity and specificity of the interactions between Streptococcus mutans antigen I/II and salivary components. J. Dent. Res. 73, 1493–1502 [DOI] [PubMed] [Google Scholar]
- 23. Shanmugam K. T., Masthan K. M. K., Balachander N., Jimson S., Sarangarajan R. (2013) Dental caries vaccine—a possible option? J. Clin. Diagn. Res. 7, 1250–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu Q., Katz J., Zhang P., Ashtekar A. R., Gaddis D. E., Fan M., Michalek S. M. (2011) Contribution of a Streptococcus mutans antigen expressed by a Salmonella vector vaccine in dendritic cell activation. Infect. Immun. 79, 3792–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang S. (2013) Dental caries and vaccination strategy against the major cariogenic pathogen, Streptococcus mutans. Curr. Pharm. Biotechnol. 14, 960–966 [DOI] [PubMed] [Google Scholar]
- 26. Zhao W., Minderman H., Russell M. W. (2014) Identification and characterization of intestinal antigen-presenting cells involved in uptake and processing of a nontoxic recombinant chimeric mucosal immunogen based on cholera toxin using imaging flow cytometry. Clin. Vaccine Immunol. 21, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Purushotham S., Deivanayagam C. (2014) The calcium-induced conformation and glycosylation of scavenger-rich cysteine repeat (SRCR) domains of glycoprotein 340 influence the high affinity interaction with antigen I/II homologs. J. Biol. Chem. 289, 21877–21887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hajishengallis G., Nikolova E., Russell M. W. (1992) Inhibition of Streptococcus mutans adherence to saliva-coated hydroxyapatite by human secretory immunoglobulin A (S-IgA) antibodies to cell surface protein antigen I/II: reversal by IgA1 protease cleavage. Infect. Immun. 60, 5057–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russell M. W., Bergmeier L. A., Zanders E. D., Lehner T. (1980) Protein antigens of Streptococcus mutans: purification and properties of a double antigen and its protease-resistant component. Infect. Immun. 28, 486–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Russell M. W., Mansson-Rahemtulla B. (1989) Interaction between surface protein antigens of Streptococcus mutans and human salivary components. Oral Microbiol. Immunol. 4, 106–111 [DOI] [PubMed] [Google Scholar]
- 31. Bleiweis A. S., Oyston P. C., Brady L. J. (1992) Molecular, immunological and functional characterization of the major surface adhesin of Streptococcus mutans. Adv. Exp. Med. Biol. 327, 229–241 [DOI] [PubMed] [Google Scholar]
- 32. Crowley P. J., Brady L. J., Piacentini D. A., Bleiweis A. S. (1993) Identification of a salivary agglutinin-binding domain within cell surface adhesin P1 of Streptococcus mutans. Infect. Immun. 61, 1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kishimoto E., Hay D. I., Gibbons R. J. (1989) A human salivary protein which promotes adhesion of Streptococcus mutans serotype c strains to hydroxyapatite. Infect. Immun. 57, 3702–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ericson T., Rundegren J. (1983) Characterization of a salivary agglutinin reacting with a serotype c strain of Streptococcus mutans. Eur. J. Biochem. 133, 255–261 [DOI] [PubMed] [Google Scholar]
- 35. Munro G. H., Evans P., Todryk S., Buckett P., Kelly C. G., Lehner T. (1993) A protein fragment of streptococcal cell surface antigen I/II which prevents adhesion of Streptococcus mutans. Infect. Immun. 61, 4590–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Dolleweerd C. J., Kelly C. G., Chargelegue D., Ma J. K. (2004) Peptide mapping of a novel discontinuous epitope of the major surface adhesin from Streptococcus mutans. J. Biol. Chem. 279, 22198–22203 [DOI] [PubMed] [Google Scholar]
- 37. Bikker F. J., Ligtenberg A. J., End C., Renner M., Blaich S., Lyer S., Wittig R., van't Hof W., Veerman E. C., Nazmi K., de Blieck-Hogervorst J. M., Kioschis P., Nieuw Amerongen A. V., Poustka A., Mollenhauer J. (2004) Bacteria binding by DMBT1/SAG/gp-340 is confined to the VEVLXXXXW motif in its scavenger receptor cysteine-rich domains. J. Biol. Chem. 279, 47699–47703 [DOI] [PubMed] [Google Scholar]
- 38. Madsen J., Mollenhauer J., Holmskov U. (2010) Review: Gp-340/DMBT1 in mucosal innate immunity. Innate Immun. 16, 160–167 [DOI] [PubMed] [Google Scholar]
- 39. Brady L. J., Crowley P. J., Ma J. K., Kelly C., Lee S. F., Lehner T., Bleiweis A. S. (1991) Restriction fragment length polymorphisms and sequence variation within the spaP gene of Streptococcus mutans serotype c isolates. Infect. Immun. 59, 1803–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heim K. P., Crowley P. J., Brady L. J. (2013) An intramolecular interaction involving the N terminus of a streptococcal adhesin affects its conformation and adhesive function. J. Biol. Chem. 288, 13762–13774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee S. F., Boran T. L. (2003) Roles of sortase in surface expression of the major protein adhesin P1, saliva-induced aggregation and adherence, and cariogenicity of Streptococcus mutans. Infect. Immun. 71, 676–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Navarre W. W., Schneewind O. (1994) Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. Mol. Microbiol. 14, 115–121 [DOI] [PubMed] [Google Scholar]
- 43. Lee S. G., Pancholi V., Fischetti V. A. (2002) Characterization of a unique glycosylated anchor endopeptidase that cleaves the LPXTG sequence motif of cell surface proteins of Gram-positive bacteria. J. Biol. Chem. 277, 46912–46922 [DOI] [PubMed] [Google Scholar]
- 44. Larson M. R., Rajashankar K. R., Crowley P. J., Kelly C., Mitchell T. J., Brady L. J., Deivanayagam C. (2011) Crystal structure of the C-terminal region of Streptococcus mutans antigen I/II and characterization of salivary agglutinin adherence domains. J. Biol. Chem. 286, 21657–21666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Larson M. R., Rajashankar K. R., Patel M. H., Robinette R. A., Crowley P. J., Michalek S., Brady L. J., Deivanayagam C. (2010) Elongated fibrillar structure of a streptococcal adhesin assembled by the high-affinity association of α- and PPII-helices. Proc. Natl. Acad. Sci. U.S.A. 107, 5983–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Troffer-Charlier N., Ogier J., Moras D., Cavarelli J. (2002) Crystal structure of the V-region of Streptococcus mutans antigen I/II at 2.4 Å resolution suggests a sugar preformed binding site. J. Mol. Biol. 318, 179–188 [DOI] [PubMed] [Google Scholar]
- 47. Nylander A., Forsgren N., Persson K. (2011) Structure of the C-terminal domain of the surface antigen SpaP from the caries pathogen Streptococcus mutans. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 23–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Heim K. P., Crowley P. J., Long J. R., Kailasan S., McKenna R., Brady L. J. (2014) An intramolecular lock facilitates folding and stabilizes the tertiary structure of Streptococcus mutans adhesin P1. Proc. Natl. Acad. Sci. U.S.A. 111, 15746–15751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McArthur W. P., Rhodin N. R., Seifert T. B., Oli M. W., Robinette R. A., Demuth D. R., Brady L. J. (2007) Characterization of epitopes recognized by anti-Streptococcus mutans P1 monoclonal antibodies. FEMS Immunol. Med. Microbiol. 50, 342–353 [DOI] [PubMed] [Google Scholar]
- 50. Ayakawa G. Y., Boushell L. W., Crowley P. J., Erdos G. W., McArthur W. P., Bleiweis A. S. (1987) Isolation and characterization of monoclonal antibodies specific for antigen P1, a major surface protein of mutans streptococci. Infect. Immun. 55, 2759–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Okahashi N., Takahashi I., Nakai M., Senpuku H., Nisizawa T., Koga T. (1993) Identification of antigenic epitopes in an alanine-rich repeating region of a surface protein antigen of Streptococcus mutans. Infect. Immun. 61, 1301–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Russell M. W., Lehner T. (1978) Characterisation of antigens extracted from cells and culture fluids of Streptococcus mutans serotype c. Arch. Oral Biol. 23, 7–15 [DOI] [PubMed] [Google Scholar]
- 53. Russell M. W., Zanders E. D., Bergmeier L. A., Lehner T. (1980) Affinity purification and characterization of protease-susceptible antigen I of Streptococcus mutans. Infect. Immun. 29, 999–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kelly C., Evans P., Bergmeier L., Lee S. F., Progulske-Fox A., Harris A. C., Aitken A., Bleiweis A. S., Lehner T. (1989) Sequence analysis of the cloned streptococcal cell surface antigen I/II. FEBS Lett. 258, 127–132 [DOI] [PubMed] [Google Scholar]
- 55. Okahashi N., Sasakawa C., Yoshikawa M., Hamada S., Koga T. (1989) Molecular characterization of a surface protein antigen gene from serotype c Streptococcus mutans, implicated in dental caries. Mol. Microbiol. 3, 673–678 [DOI] [PubMed] [Google Scholar]
- 56. Robinette R. A., Oli M. W., McArthur W. P., Brady L. J. (2009) Beneficial immunomodulation by Streptococcus mutans anti-P1 monoclonal antibodies is Fc independent and correlates with increased exposure of a relevant target epitope. J. Immunol. 183, 4628–4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brady L. J., Piacentini D. A., Crowley P. J., Bleiweis A. S. (1991) Identification of monoclonal antibody-binding domains within antigen P1 of Streptococcus mutans and cross-reactivity with related surface antigens of oral streptococci. Infect. Immun. 59, 4425–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Homonylo-McGavin M. K., Lee S. F., Bowden G. H. (1999) Subcellular localization of the Streptococcus mutans P1 protein C terminus. Can. J. Microbiol. 45, 536–539 [PubMed] [Google Scholar]
- 59. Lee S. F. (1995) Active release of bound antibody by Streptococcus mutans. Infect. Immun. 63, 1940–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Homonylo-McGavin M. K., Lee S. F. (1996) Role of the C terminus in antigen P1 surface localization in Streptococcus mutans and two related cocci. J. Bacteriol. 178, 801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Russell R. R. (1979) Wall-associated protein antigens of Streptococcus mutans. J. Gen Microbiol. 114, 109–115 [DOI] [PubMed] [Google Scholar]
- 62. Schöller M., Klein J. P., Frank R. M. (1981) Common antigens of streptococcal and non-streptococcal oral bacteria: immunochemical studies of extracellular and cell-wall-associated antigens from Streptococcus sanguis, Streptococcus mutans, Lactobacillus salivarius, and Actinomyces viscosus. Infect. Immun. 31, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Harrington D. J., Russell R. R. (1993) Multiple changes in cell wall antigens of isogenic mutants of Streptococcus mutans. J. Bacteriol. 175, 5925–5933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andre G., Leenhouts K., Hols P., Dufrêne Y. F. (2008) Detection and localization of single LysM-peptidoglycan interactions. J. Bacteriol. 190, 7079–7086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dupres V., Menozzi F. D., Locht C., Clare B. H., Abbott N. L., Cuenot S., Bompard C., Raze D., Dufrêne Y. F. (2005) Nanoscale mapping and functional analysis of individual adhesins on living bacteria. Nat. Methods 2, 515–520 [DOI] [PubMed] [Google Scholar]
- 66. Gilbert Y., Deghorain M., Wang L., Xu B., Pollheimer P. D., Gruber H. J., Errington J., Hallet B., Haulot X., Verbelen C., Hols P., Dufrêne Y. F. (2007) Single-molecule force spectroscopy and imaging of the vancomycin/D-Ala-D-Ala interaction. Nano Lett. 7, 796–801 [DOI] [PubMed] [Google Scholar]
- 67. Knox K. W., Wicken A. J. (1978) Effect of growth conditions on the antigenic components of Streptococcus mutans and lactobacilli. Adv. Exp. Med. Biol. 107, 629–637 [DOI] [PubMed] [Google Scholar]
- 68. Crowley P. J., Brady L. J., Michalek S. M., Bleiweis A. S. (1999) Virulence of a spaP mutant of Streptococcus mutans in a gnotobiotic rat model. Infect. Immun. 67, 1201–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rhodin N. R., Van Tilburg M. L., Oli M. W., McArthur W. P., Brady L. J. (2004) Further characterization of immunomodulation by a monoclonal antibody against Streptococcus mutans antigen P1. Infect. Immun. 72, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Oli M. W., Otoo H. N., Crowley P. J., Heim K. P., Nascimento M. M., Ramsook C. B., Lipke P. N., Brady L. J. (2012) Functional amyloid formation by Streptococcus mutans. Microbiology 158, 2903–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sivanathan V., Hochschild A. (2012) Generating extracellular amyloid aggregates using E. coli cells. Genes Dev. 26, 2659–2667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brady L. J., Cvitkovitch D. G., Geric C. M., Addison M. N., Joyce J. C., Crowley P. J., Bleiweis A. S. (1998) Deletion of the central proline-rich repeat domain results in altered antigenicity and lack of surface expression of the Streptococcus mutans P1 adhesin molecule. Infect. Immun. 66, 4274–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oli M. W., McArthur W. P., Brady L. J. (2006) A whole cell BIAcore assay to evaluate P1-mediated adherence of Streptococcus mutans to human salivary agglutinin and inhibition by specific antibodies. J. Microbiol. Methods 65, 503–511 [DOI] [PubMed] [Google Scholar]
- 74. Seifert T. B., Bleiweis A. S., Brady L. J. (2004) Contribution of the alanine-rich region of Streptococcus mutans P1 to antigenicity, surface expression, and interaction with the proline-rich repeat domain. Infect. Immun. 72, 4699–4706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Crowley P. J., Seifert T. B., Isoda R., van Tilburg M., Oli M. W., Robinette R. A., McArthur W. P., Bleiweis A. S., Brady L. J. (2008) Requirements for surface expression and function of adhesin P1 from Streptococcus mutans. Infect. Immun. 76, 2456–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Reynolds E. S. (1963) The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 17, 208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ebner A., Wildling L., Kamruzzahan A. S., Rankl C., Wruss J., Hahn C. D., Hölzl M., Zhu R., Kienberger F., Blaas D., Hinterdorfer P., Gruber H. J. (2007) A new, simple method for linking of antibodies to atomic force microscopy tips. Bioconjug. Chem. 18, 1176–1184 [DOI] [PubMed] [Google Scholar]
- 78. Cross S. E., Kreth J., Zhu L., Qi F., Pelling A. E., Shi W., Gimzewski J. K. (2006) Atomic force microscopy study of the structure-function relationships of the biofilm-forming bacterium Streptococcus mutans. Nanotechnology 17, S1–S7 [DOI] [PubMed] [Google Scholar]
- 79. Francius G., Alsteens D., Dupres V., Lebeer S., De Keersmaecker S., Vanderleyden J., Gruber H. J., Dufrêne Y. F. (2009) Stretching polysaccharides on live cells using single molecule force spectroscopy. Nat. Protoc. 4, 939–946 [DOI] [PubMed] [Google Scholar]
- 80. Hutter J. L., Bechhoefer J. (1993) Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 64, 1868–1873 [Google Scholar]
- 81. Liu B. H., Li K.-L., Kang K.-L., Huang W.-K., Liao J.-D. (2013) In situ biosensing of the nanomechanical property and electrochemical spectroscopy of Streptococcus mutans-containing biofilms. J. Phys. D Appl. Phys. 46, 275401 [Google Scholar]
- 82. Craig R. A., Riege D. H., Bleiweis A. S. (1979) Antigens of Streptococcus mutans: cellular localization of the serotype-specific polysaccharide of strain AHT and release during exponential growth. Infect. Immun. 26, 1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Allen S., Chen X., Davies J., Davies M. C., Dawkes A. C., Edwards J. C., Roberts C. J., Sefton J., Tendler S. J., Williams P. M. (1997) Detection of antigen-antibody binding events with the atomic force microscope. Biochemistry 36, 7457–7463 [DOI] [PubMed] [Google Scholar]
- 84. Alsteens D., Garcia M. C., Lipke P. N., Dufrêne Y. F. (2010) Force-induced formation and propagation of adhesion nanodomains in living fungal cells. Proc. Natl. Acad. Sci. U.S.A. 107, 20744–20749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Berquand A., Xia N., Castner D. G., Clare B. H., Abbott N. L., Dupres V., Adriaensen Y., Dufrêne Y. F. (2005) Antigen binding forces of single antilysozyme Fv fragments explored by atomic force microscopy. Langmuir 21, 5517–5523 [DOI] [PubMed] [Google Scholar]
- 86. Ros R., Schwesinger F., Anselmetti D., Kubon M., Schäfer R., Plückthun A., Tiefenauer L. (1998) Antigen binding forces of individually addressed single-chain Fv antibody molecules. Proc. Natl. Acad. Sci. U.S.A. 95, 7402–7405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schwesinger F., Ros R., Strunz T., Anselmetti D., Güntherodt H. J., Honegger A., Jermutus L., Tiefenauer L., Pluckthun A. (2000) Unbinding forces of single antibody-antigen complexes correlate with their thermal dissociation rates. Proc. Natl. Acad. Sci. U.S.A. 97, 9972–9977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Verbelen C., Christiaens N., Alsteens D., Dupres V., Baulard A. R., Dufrêne Y. F. (2009) Molecular mapping of lipoarabinomannans on mycobacteria. Langmuir 25, 4324–4327 [DOI] [PubMed] [Google Scholar]
- 89. Rief M., Gautel M., Oesterhelt F., Fernandez J. M., Gaub H. E. (1997) Reversible unfolding of individual titin immunoglobulin domains by AFM. Science 276, 1109–1112 [DOI] [PubMed] [Google Scholar]
- 90. Isoda R., Robinette R. A., Pinder T. L., McArthur W. P., Brady L. J. (2007) Basis of beneficial immunomodulation by monoclonal antibodies against Streptococcus mutans adhesin P1. FEMS Immunol. Med. Microbiol. 51, 102–111 [DOI] [PubMed] [Google Scholar]
- 91. Robinette R. A., Heim K. P., Oli M. W., Crowley P. J., McArthur W. P., Brady L. J. (2014) Alterations in immunodominance of Streptococcus mutans AgI/II: lessons learned from immunomodulatory antibodies. Vaccine 32, 375–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hu P., Bian Z., Fan M., Huang M., Zhang P. (2008) Sec translocase and sortase A are colocalised in a locus in the cytoplasmic membrane of Streptococcus mutans. Arch. Oral Biol. 53, 150–154 [DOI] [PubMed] [Google Scholar]
- 93. Lehner T., Russell M. W., Caldwell J. (1980) Immunisation with a purified protein from Streptococcus mutans against dental caries in rhesus monkeys. Lancet 1, 995–996 [DOI] [PubMed] [Google Scholar]
- 94. Moro I., Russell M. W. (1983) Ultrastructural localization of protein antigens I/II and III in Streptococcus mutans. Infect. Immun. 41, 410–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee S. F., Progulske-Fox A., Erdos G. W., Piacentini D. A., Ayakawa G. Y., Crowley P. J., Bleiweis A. S. (1989) Construction and characterization of isogenic mutants of Streptococcus mutans deficient in major surface protein antigen P1 (I/II). Infect. Immun. 57, 3306–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lee S. F., Gao L. (2000) Mutational analysis of the C-terminal anchoring domains of Streptococcus mutans P1 antigen: role of the LPXTGX motif in P1 association with the cell wall. Can. J. Microbiol. 46, 584–592 [PubMed] [Google Scholar]
- 97. Murakami Y., Nakano Y., Yamashita Y., Koga T. (1997) Identification of a frameshift mutation resulting in premature termination and loss of cell wall anchoring of the PAc antigen of Streptococcus mutans GS-5. Infect. Immun. 65, 794–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Murakami Y., Yamashita Y., Nakano Y., Ito H. O., Yu H., Koga T. (1997) Role of the charged tail in localization of a surface protein antigen of Streptococcus mutans. Infect. Immun. 65, 1531–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]