

Background: Rac 1 GTPase mediates glycogen phosphorylase activation and controls IL-2-stimulated T cell proliferation.
Results: PKCθ activates αPIX by serine phosphorylation and now this Rho-GEF activates Rac 1.
Conclusion: IL-2-stimulated T cells migration and proliferation require the involvement of the PKCθ/αPIX/Rac 1/PYGM pathway.
Significance: This new signaling cascade may be a viable therapeutic target to block the inflammatory response mediated by T cells.
Keywords: Guanine Nucleotide Exchange Factor (GEF), Phosphorylase, Protein Kinase C (PKC), Protein Phosphorylation, Ras-related C3 Botulinum Toxin Substrate 1 (Rac1)
Abstract
Recently, we have reported that the active form of Rac 1 GTPase binds to the glycogen phosphorylase muscle isoform (PYGM) and modulates its enzymatic activity leading to T cell proliferation. In the lymphoid system, Rac 1 and in general other small GTPases of the Rho family participate in the signaling cascades that are activated after engagement of the T cell antigen receptor. However, little is known about the IL-2-dependent Rac 1 activator molecules. For the first time, a signaling pathway leading to the activation of Rac 1/PYGM in response to IL-2-stimulated T cell proliferation is described. More specifically, αPIX, a known guanine nucleotide exchange factor for the small GTPases of the Rho family, preferentially Rac 1, mediates PYGM activation in Kit 225 T cells stimulated with IL-2. Using directed mutagenesis, phosphorylation of αPIX Rho-GEF serines 225 and 488 is required for activation of the Rac 1/PYGM pathway. IL-2-stimulated serine phosphorylation was corroborated in Kit 225 T cells cultures. A parallel pharmacological and genetic approach identified PKCθ as the serine/threonine kinase responsible for αPIX serine phosphorylation. The phosphorylated state of αPIX was required to activate first Rac 1 and subsequently PYGM. These results demonstrate that the IL-2 receptor activation, among other early events, leads to activation of PKCθ. To activate Rac 1 and consequently PYGM, PKCθ phosphorylates αPIX in T cells. The biological significance of this PKCθ/αPIX/Rac 1 GTPase/PYGM signaling pathway seems to be the control of different cellular responses such as migration and proliferation.
Introduction
Co-stimulation of T cell receptor and CD28 T cell receptors lead to IL-2 expression and secretion. An autocrine effect is necessary for expressing the IL-2 receptor (IL-2R) α chain (1, 2) and ultimately to culminate in T cell clonal expansion (3, 4). The IL-2 effect on T cells is not only restricted to the induction of lymphocyte proliferation only but, in the inflammatory response it is also necessary for T lymphocytes differentiation into effector T lymphocytes as well as regulatory T lymphocytes (5).
Binding of IL-2 to its high affinity receptor (IL-2R) drives the activation of a signaling network giving rise to many cellular responses, among them the three major signaling cascades best characterized are the Janus kinase (Jak)/STAT and MAPK pathways, which modulate gene expression and PI3K-mediated cell survival (4). To accomplish these cellular responses, IL-2-dependent T cells possibly require not only activation of these pathways but also a complex cooperation with other signaling networks mediated by tyrosine kinases such as lck, BTK (6), PLCγ (7, 8), and serine/threonine kinases such as protein kinase C family members (9, 10), and some GTPases of the Rho family. In fact, it has been reported that RhoA cooperates with ERK-dependent signaling pathways to transcribe c-fos in response to IL-2 (11). Moreover, Rac 1 has also been found to participate in IL-2-induced actin cytoskeleton rearrangement in a murine T cell line (12). More recently, our group reported that Rac 1 binds and activates the glycogen phosphorylase muscle isoform (PYGM)3 and thus established a novel metabolic pathway that participates actively in IL-2-stimulated cell proliferation in human T cells (13).
Signals emanating from a large variety of membrane receptors: growth factor receptors (14, 15), G protein-coupled receptors (16, 17), and tyrosine kinases-linked receptors such as TCR (5, 18), BCR (19, 20), and IL2-R (13), actively regulate Rho GTPase effects. Like other small GTPases, Rho GTPases function as molecular switches that cycle between the inactive GDP-bound and the active GTP-bound state. In the active state, GTPases interact with downstream effector molecules to promote a variety of biological responses, such as control of the appropriate actin cytoskeleton reorganization in response to extracellular signals, and their significant implications in additional biological processes, where gene expression regulation, cell polarity, and cell migration have also been reported (21–23).
The transition between the inactive to the active state is regulated by guanine nucleotide exchange factors (GEFs) (21–23). A key factor in the functioning of small GTPases lies in their selection and regulation of these GEFs. It is well established that upon IL-2/IL-2R ligation, Ras GEF, Son of Seven (Sos), associates to Grb2 and it is recruited through the adapter protein Shc, to the tyrosine-phosphorylated IL-2R β chain. In this configuration, Sos activates Ras and consequently the MAPK pathway (24, 25). Therefore, Sos exchange activity is indirectly regulated by tyrosine phosphorylation. However, the exchange activity of some GEFs of the Dbl family that activates Rac 1 GTPase are directly regulated by phosphorylation. In fact, in the immune system, Vav (Rac 1-specific GEF) must be tyrosine phosphorylated at residue 174 to turn on its GTPase activity (26, 27). Nevertheless, Tiam-1 and STEF, both members of the Tiam GEF family where the former is mainly expressed in the brain and in the immune system and the latter in the brain, are two additional GEFs with higher specificity for Rac 1 (28, 29) that are activated by threonine (30) and serine/threonine phosphorylation (31), respectively. Like Tiam-1, αPIX (also known as ARHGEF6 or Cool-2) (32–34), a Rho-GEF primarily expressed in neurons and hematopoietic cells (34), had its exchange activity predicted to be regulated by serine/threonine kinases phosphorylation by phosphoproteomic analysis (35–38). In the last few years, GTPases of the Rac subfamily gained increasing relevance in T cell biology (39, 40). In contrast to its well established Sos-mediated Ras activation mechanism in IL-2-stimulated T cells, the identity of the Rac GEF responsible for Rac activation in IL-2-stimulated signaling has not been determined.
Here we show that subsequent to IL-2 stimulation αPIX-Rho-GEF mediates PYGM activation in Kit 225 T cells; an IL-2-dependent human T cell line. Serines 225 and 488 of αPIX are critical to active Rac 1 and mediate PYGM activation in IL-2-stimulated cells. By combining pharmacological and genetic approaches, we identified PKCθ as the serine/threonine kinase that controls the phosphorylation of these serines and consequently the Rac 1/PYGM axis.
These results reveal that Rac 1/PYGM pathway activation stimulated by IL-2 is achieved through αPIX. Furthermore, our results identify PKCθ as the intermediary between the activated IL-2·IL-2R complex and αPIX. This novel intracellular signaling pathway actively participates in the regulation of the IL-2-stimulated T cell migration and proliferation.
EXPERIMENTAL PROCEDURES
Reagents
PKA inhibitor H-89 dihydrochloride, PKC inhibitors Gö6976 and Rottlerin, and PI 3-kinase inhibitor LY29004 hydrochloride were obtained from Sigma. Mouse monoclonal anti-HA antibody was obtained from Covance, mouse monoclonal anti-phosphoserine clone PSR-45 and rabbit monoclonal anti-glutathione S-transferase (GST) antibodies were from Sigma, mouse monoclonal anti-Rac 1 clone 23A8 antibody was obtained from Millipore, rabbit monoclonal anti-PKCα was obtained from Cell Signaling, and enhanced chemiluminescence (ECL) reagent was obtained from GE Healthcare. Phorbol 12-myristate 13-acetate (PMA) was obtained from Sigma. MISSION® esiRNA human PRKCQ (gene synonym PKCθ, reference EHU093601), MISSION esiRNA human ARHGEF6 (gene synonym αPIX, reference EHU133681), and MISSION esiRNA targeting EGFP (reference EHUEGFP) were from Sigma. IL-2 cytokine was provided by the “AIDS Research and Reference Reagent Program,” Division of AIDS (NIAD, National Institutes of Health).
Cell Culture and DNA/esiRNA Transfections
Kit 225 T cells were cultured as described by Hori et al. (41) in the presence of 16 units/ml of recombinant human IL-2. For transient transfections, cells were cultured in complete RPMI 1640 medium in the absence of IL-2 for 24 h. Thereafter, cells were washed, resuspended in 200 μl of serum-free medium, and placed in an electroporation cuvette (0.4 mm, Sigma) containing 10–20 μg of different plasmids, or 15 ng of esiRNAs. Electroporation was carried out in a Gene Pulser Xcell Electroporator (Bio-Rad) at 260 V and 950 microfarads (13). The cuvette content was collected into 10 ml of complete RPMI 1640 medium and cultured in the absence of IL-2 for an additional 24 h.
Agonists and Inhibitors
Kit 225 T cells were maintained in the absence of IL-2 for 48 h and subsequently stimulated with 500 units/ml of IL-2 at 37 °C (13). In some experiments, Kit 225 T cells were pretreated with 10 μm H-89 (PKA inhibitor) or 20 μm LY29004 (PI3K inhibitor) or 100 nm Gö6976 and different concentrations of Rottlerin (PKC inhibitors) for 1 h prior to IL-2 or PMA stimulation (13, 42).
Plasmid Construct and Site-directed Mutagenesis
The αPIX comprising amino acids 204–532 (αPIX204–532) was generated by PCR amplification using pMT2-HA-αPIX wt as template (forward oligonucleotide, 5′-CGG GAT CCA GAA CAG GCT GG-3′, and reverse oligonucleotide 5′-GCG GAT CCT GTG CAG TCA TTC C-3′, each harboring BamHI restriction sites (underlined). The BamHI αPIX204–532 fragment was subcloned into pGEX-4T3 (GE Healthcare) to generate the GST-αPIX204–532 fusion protein. pMT2-HA-αPIXS225A, pMT2-HA-αPIXS488A, and pMT2-HA-αPIXS225A/S488A single and double mutated constructs were generated according to manufacturer's instructions (QuikChange Lightning Site-directed Mutagenesis Kit, Stratagene). Oligonucleotides used for S225A mutation were: 5′-GAG AGA CCT CTC GCC CCA AAA GCC GTC-3′ (forward) and 5′-GAC GGC TTT TGG GGC GAG AGG TCT CTC-3′ (reverse) and for S488A mutation were: 5′-AGT CCT CGG ATG GCT GGC TTT ATC TAT-3′ (forward) and 5′-ATA GAT AAA GCC AGC CAT CCG AGG ACT-3′ (reverse).
Activity Assay for Glycogen Phosphorylase
The glycogen phosphorylase activity assay was performed as previously described (43, 44) with some modifications. Briefly, cells were washed twice with cold PBS and resuspended in 500 μl of TES buffer (20 mm Tris, pH 7.4, 1 mm EDTA, 225 mm sucrose, 2.5 mm DTT, 0.1 mm PMSF, 1 μg/ml of leupeptin, 1 μg/ml of aprotinin). Samples were sonicated and centrifuged at 12,300 × g for 10 min at 4 °C. Total protein (100 μg) was used to measure PYGM activity in assay buffer (50 mm KH2PO4, pH 7.5, 10 mm MgCl2, 5 mm EDTA, pH 8, 0.5 mm NADP+, 1.5 units/ml of glucose-6-phosphate dehydrogenase, 1 unit/ml of phosphoglucomutase, 0.1 mg/ml of glycogen (all from Sigma). Assay buffer containing 300 μl of TES without NADP+, glycogen, phosphoglucomutase, and glucose-6-phosphate dehydrogenase was added to 100 μg of total protein as a blank control. To carry out the metabolic activity assay the mixture was incubated at 37 °C for 20 min. By placing samples on ice the reaction was stopped. Sample absorbances were detected at 340 nm in a spectrophotometer (Ultrospec 3100 pro, Amersham Biosciences). The amount of NADPH formed was determined using a standard curve of known NADPH concentrations (Sigma).
Rac 1 Activation Assay
Rac 1 pulldown assay was performed using a GST fusion protein containing the Rac 1 binding domain of PAK1 (GST-RBD-PAK1). Transfected and untransfected cells kept in the absence of IL-2 for 48 h were stimulated with IL-2 for 10 min and lysed as described in Ref. 45. Cell lysates were centrifuged at 12,300 × g for 10 min at 4 °C and incubated for 1 h at 4 °C with 50 μg of GST-RBD-PAK1 fusion protein coupled to glutathione-Sepharose beads. Precipitated proteins were eluted from beads using 2× loading buffer (12 mm Tris, pH 6.8, 5% glycerol, 0.4% SDS, 140 mm 2-mercaptoethanol, 0.02% bromphenol blue), separated by SDS-PAGE, and analyzed by immunoblot with specific monoclonal antibodies. Immunoreactive bands were visualized using ECL.
Immunoprecipitation Assay
Transfected Kit 225 T cells with cDNA encoding for pMT2-HA-αPIX or empty vector (pMT2-HA) or esiRNAs to knock down PKCθ as indicated were treated or not with 500 units/ml of IL-2 for 10 min. Cells were washed three times in ice-cold PBS and lysed in RIPA buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% IGEPAL, 0.25% Na-deoxycolate, 1 mm EDTA, 1 mm PMSF, 1 mm Na3VO4, 1 mm NaF, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin). Ectopic HA-αPIX and endogenous αPIX were immunoprecipitated for 2 h at 4 °C using anti-HA or anti-αPIX antibodies. Immune complexes were recovered using Gamma Bind Plus-Sepharose beads (GE Healthcare, Pittsburgh, PA), washed, and eluted from beads and resolved electrophoretically by SDS-PAGE and analyzed by Western blot with anti-phosphoserine, anti-HA, or anti-αPIX antibodies. Immunoreactive bands were visualized using ECL.
In Vitro Kinase Assay
Kit 225 T cells transfected with PKCθ (wild type), PKCθK409R (dominant-negative), and/or the constitutively active forms of PKCα (PKCαA25E), PKCϵ (PKCϵA159E), and PKCθ (PKCθA148E) were incubated in the presence or absence of 500 units/ml of IL-2 for 10 min at 37 °C and washed twice with cold PBS. Thereafter, cells were lysed with lysis buffer (20 mm Tris, pH 7.4, 137 mm NaCl, 5 mm EDTA, 1 mm EGTA, 10 mm NaF, 1 mm sodium pyrophosphate, 100 mm β-glycerophosphate, 10 μg/ml of aprotinin, 1 mm PMSF, 10% glycerol, and 1% (v/v) Triton X-100) and lysates were clarified by centrifugation for 10 min at 12,300 × g at 4 °C. PKCs were immunoprecipitated with specific antibodies and immunocomplexes were recovered using Gamma Bind Plus-Sepharose beads (GE Healthcare). The immunocomplexes were washed twice with cold lysis buffer, twice with cold washing buffer (10 mm HEPES, pH 7.4, 100 mm NaCl, 20 μg/ml of aprotinin, and 0.5% IGEPAL-360) and twice with reaction buffer (20 mm Tris, pH 7.4, 20 mm NaCl, 1 mm DTT, 10 mm MgCl2, and 1 mm MnCl2). 500 ng of purified recombinant GST-αPIX204–532, which encompasses the two potential serine phosphorylation sites (Ser225 and Ser488) of αPIX (33), and 20 μm ATP was then added to the reaction mixture. The in vitro kinase reaction was carried out for 30 min at 30 °C after which it was stopped by adding 30 μl of 2× loading buffer. Proteins were separated by SDS-PAGE, followed by Western blot. Immunoreactive bands were visualized with anti-phosphoserine antibody and ECL.
Cell Migration Assay
esiRNA-transfected cell suspensions (2.5 × 105 cells in a 100-μl volume) were placed into the upper chamber, whereas 600 μl of medium with or without IL-2 (500 units/ml) was introduced into the lower chamber. Both chambers were incubated overnight at 37 °C in 5% CO2 and 95% air. Cells in the upper and bottom chamber were recovered separately into equal volumes for cell counting. The percentage of migrating cells was determined as follows: [number of cells migrating (lower chamber)/total number of cells (cells in the lower chamber + remaining cells in the upper chamber)]. Assay was performed using pore filters (8 μm, Corning® Costar Transwell and cell culture inserts were from Sigma) and cell counts were done in triplicate.
Cell Proliferation Measurement
esiRNA-transfected cells were seeded in 24-well plates in complete RPMI (106 cells/ml), and maintained in the absence of IL-2 for 48 h. Subsequently, cells (106) were incubated with 4 μm PKH26 following the manufacturer's instructions (Sigma). A sample (104 cells) was taken as the start control, another sample (104 cells) was left untreated, and the remaining cells were incubated in the presence of IL-2 (16 units/ml). Fluorescence was measured every 24 h for 3 days to the monitor cell division rate on a FACSCalibur (BD Biosciences) flow cytometer. Data obtained were analyzed using ModFit LT 3.0.
Statistical Analysis
Student's t test for the mean of two-paired samples was used to determine the significance between data means (**, p < 0.05; ***, p < 0.001).
RESULTS
αPIX-Rho-GEF Leads to PYGM Activation
To test if αPIX could be a link between IL-2 receptor and Rac 1 leading to the activation of PYGM, Kit 225 T cells were transfected with pMT2-HA or pMT2-HA-αPIX and stimulated or not with 500 units/ml of IL-2 for 10 min, lysed, and PYGM activity was determined as described under “Experimental Procedures.” As shown in Fig. 1A (first closed bar), IL-2 stimulated robust PYGM activity of empty vector-transfected Kit 225 T cells. This IL-2 stimulation of PYGM activity was already maximal and it was not further increased in αPIX-overexpressing cells (Fig. 1A, second closed bar compared with first closed bar). However, αPIX overexpression in the absence of IL-2 resulted in a significant increase of the PYGM activity when compared with control cells (Fig. 1A, second empty bar compared with first empty bar). Western blot shows αPIX expression levels (Fig. 1A, upper panel).
FIGURE 1.

IL-2 stimulates glycogen phosphorylase activity in Kit 225 T cells through αPIX Rho-GEF. Kit 225 T cells deprived of IL-2 for 24 h were transfected with plasmids coding for: A, pMT2-HA (empty vector) and pMT2-HA-αPIX; B, pcDNA3-HA (empty vector), pEF-Vav 1, pcDNA3-HA-Tiam-1, and pcDNA3-HA-STEF; and C, αpix(esiRNA) and egfp(esiRNA). As a control, 24 h post-transfection cells were stimulated (+) or not (−) with 500 units/ml of IL-2 for 10 min and lysed. Cell extracts from unstimulated or stimulated cells were used to measure glycogen phosphorylase activity, as described under “Experimental Procedures.” Western blot (W.B.) analysis of protein expression levels was carried out using specific antibodies, as indicated. Results show the mean of three independent experiments ± S.D. and statistical analysis shows a significant difference (***, p < 0.001).
Next, we aimed to determine whether other GEFs such as Vav, Tiam-1, and/or STEF/Tiam-2 that preferentially activate Rac 1 could also modulate the activity of the PYGM in our cellular system. For that purpose, following the same procedure described above, Kit 225 T cells were transfected with pcDNA3-HA (empty vector), pEF-Vav, pcDNA3-HA-Tiam-1, and pcDNA3-HA-STEF, and 24 h post-transfection PYGM activity was determined in Kit 225 T cells unstimulated and stimulated with IL-2 for 10 min. As expected, IL-2 significantly increased PYGM activity compared with unstimulated cells (Fig. 1B, first and second bars). This maximal increase in the PYGM activity induced by IL-2 also occurred in the presence of Vav and STEF (Fig. 1B, fourth and eighth bars compared with the second bar). However, in contrast to the αPIX outcome, Vav or STEF overexpression per se did not modify PYGM activity (Fig. 1B, third and seventh bars). In sharp contrast, Tiam-1 overexpression dramatically blocked PYGM activity (Fig. 1B, sixth bar). Western blots show Vav, STEF, and Tiam-1 expression levels (Fig. 1B, upper panel).
Finally, to corroborate that αPIX was specifically regulating the glycogen phosphorylase activity in IL-2-stimulated T cells, αpix was knocked down in Kit 225 T cells. To this end, Kit 225 T cells were transfected with esiRNA human ARHGEF6 (αPIX) or esiRNA targeting EGFP, as negative control. 24 h post-transfection PYGM activity was determined in Kit 225 T cells unstimulated and stimulated with IL-2 for 10 min. As shown in Fig. 1C, IL-2 stimulated robust PYGM activity in egfp(esiRNA)-transfected Kit 225 T cells. αpix knockdown (αpix(esiRNA)) cells stimulated by IL-2 for 10 min did not show any PYGM activity (Fig. 1C, second closed bar compared with first closed bar). Furthermore, in the absence of IL-2 αpix knockdown, cells also displayed no PYGM activity, in contrast to αPIX overexpressing cells (Fig. 1, C, second empty bar compared with A, second empty bar). Small aliquots of cell lysate from each condition were stored, electrophoretically resolved by SDS-PAGE, and followed by Western blot. Immunoreactive bands were visualized with specific antibodies as indicated. Fig. 1C, first and second panels show αPIX expression levels after esiRNA transfection and γtubulin, respectively. The γ-tubulin Western blot result shows that an equivalent amount of protein was used in each of the conditions analyzed.
Rac 1/PYGM Pathway Activation Depends on the Integrity of αPIX Serine 225 and 488 Residues
To demonstrate that αPIX functions as a Rac-activating molecule in Kit 225 T cells, αPIX was knocked-down with αpix(esiRNA), as we described above and the endogenous active Rac 1 was measured by the pulldown assay. As shown in Fig. 2A, in the absence of αPIX (knockdown), IL-2 was unable to stimulate Rac 1 activation (fourth lane compared with second lane). αPIX expression levels in the presence of esiRNA control (egfp) or αpix(esiRNA) were determined by Western blot (Fig. 2A, upper panel). Rac 1 detected in whole cell lysates shows that the total loaded proteins are equivalent in all lanes (Fig. 2A, third panel).
FIGURE 2.
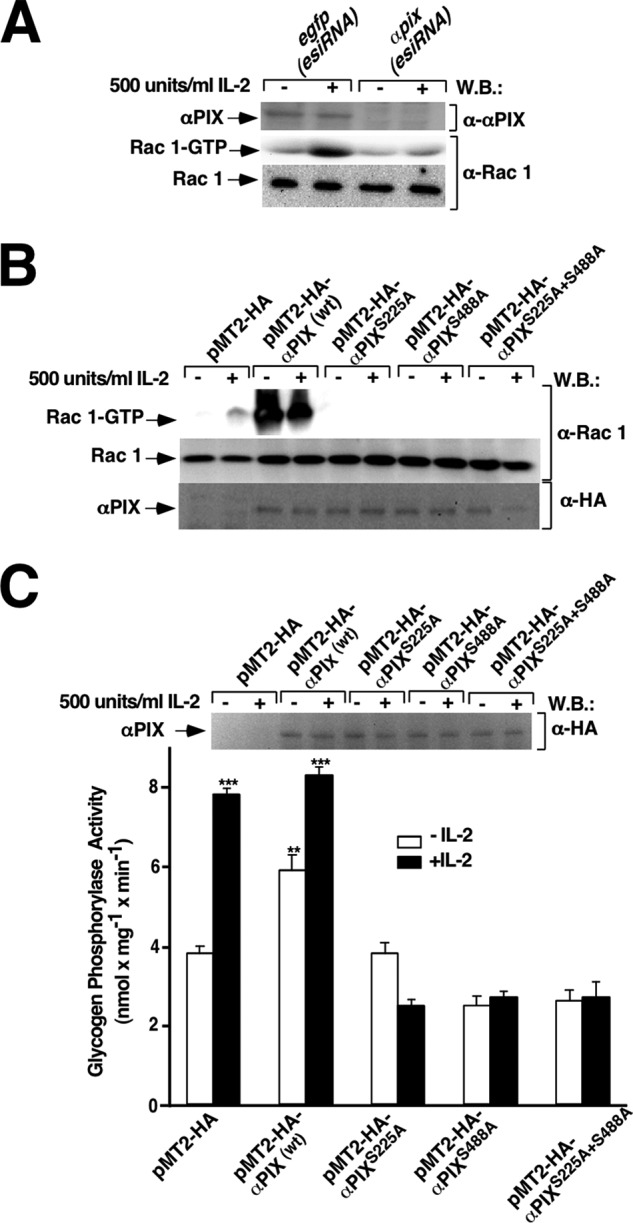
IL-2-stimulated Rac 1/PYGM pathway activation requires intact serine residues 225 and 488 of αPIX-Rho-GEF. Kit 225 T cells deprived of IL-2 for 24 h were transfected with egfp(esiRNA) and αpix(esiRNA), pMT2-HA (empty vector), pMT2-HA-αPIX wt, pMT2-HA-αPIXS225A, pMT2-HA-αPIXS488A, and αPIXS225A/S488A. 24 h post-transfection cells were stimulated (+) or not (−) with 500 units/ml of IL-2 for 10 min and lysed. A and B, cell lysates were used to measure Rac 1 activation by affinity precipitation assay. Precipitated active Rac1 (Rac1-GTP), total Rac1, and αPIX expression levels of the wild type and the mutant forms were analyzed by Western blot using anti-Rac1 and anti-HA antibodies, respectively. Results are representative of four independent experiments. C, cell extracts from unstimulated and stimulated cells were used to measure glycogen phosphorylase activity, as described under “Experimental Procedures.” Western blot (W.B.) analysis of protein expression levels was carried out using specific anti-HA antibody. Results show the mean of three independent experiments ±S.D. and statistical analysis shows a significant difference (***, p < 0.001).
Directed mutagenesis was used to investigate a putative role of serine 225 and 488 residues in activation of the Rac 1/PYGM pathway. To this end, the following single αPIX mutants were generated: αPIXS225A, αPIXS488A, and the αPIX double mutant, αPIXS225A/S488A. The effects of these αPIX mutants on IL-2-stimulated Rac 1 activation were examined in Kit 225 T cells previously transfected with αPIX wt, αPIXS225A, αPIX488A, αPIXS225A/S488A, or the empty vector (pMT2-HA) and stimulated or not with 500 units/ml of IL-2 for 10 min. The active form of Rac 1 present in whole cell lysates was pulled down using the fusion protein GST-RBD of PAK 1 and visualized as described under “Experimental Procedures.” As shown in Fig. 2B (first panel), IL-2 stimulated Rac 1 activation (lane 2) in control transfected cells. αPIX wt overexpression induced massive Rac 1 activation, which was not further increased by IL-2 stimulation (Fig. 2B, upper panel, lanes 3 and 4). In contrast, both αPIX single and double mutants dramatically blocked Rac 1 activation stimulated by IL-2 (Fig. 2B, upper panel, sixth, eighth, and tenth lanes). Furthermore, overexpression of these αPIX mutants in unstimulated cells did not induce any Rac 1 activation as it was observed with the αPIX wild type form (Fig. 2B, upper panel, fifth, seventh, and ninth lanes compared with third lane).
Next, the effect of these αPIX mutants on PYGM activity was also examined. As shown in Fig. 2C, IL-2 stimulated PYGM maximal activity of both empty vector and αPIX wt transfected Kit 225 T cells (Fig. 2C, second and fourth bars, respectively). αPIX overexpression without IL-2 stimulation also induced a significant increase in PYGM activity in comparison to control cells (Fig. 2C, third bar compared with first bar). In contrast, PYGM activity was completely blocked by both the single serine and double serine mutants with or without IL-2 stimulation (Fig. 2C, five right bars).
nPKCs Regulate Rac 1/PYGM Pathway Activation
To search for additional kinases also involved in IL-2 early signaling leading to PYGM activation, the possible involvement of PKA, PI3K, and/or PKCs was examined. To this end, Kit 225 T cells deprived of IL-2 for 48 h were pretreated for 1 h with vehicle, 10 μm H-89 (a PKA inhibitor), 20 μm LY294002 (a specific inhibitor of PI3K), 100 nm Gö6976 (an inhibitor of classic PKCs, mainly α and β), or 5 μm Rottlerin (which was initially described as a selective inhibitor of the novel PKC isoform δ (46)) and was subsequently described to inhibit also PKCθ (47, 48), followed by stimulation or not with 500 units/ml of IL-2 for 10 min. PYGM activity was determined as described above. Inhibition of PKA, PI3K, or classic PKCs did not affect PYGM activity stimulated by IL-2 (Fig. 3A, fourth, sixth, and eighth bars compared with second bar); notwithstanding, Rottlerin efficiently blocked PYGM activity stimulated by IL-2 (Fig. 3A, tenth bar) in a concentration-dependent manner (Fig. 3B). 2.5 μm Rottlerin was the minimal concentration leading to the maximal blockage (Fig. 3B).
FIGURE 3.

nPKCs regulate Rac 1/PYGM activation in IL-2-stimulated T cells. A–C, Kit 225 T cells were treated with inhibitors or vehicle (dimethyl sulfoxide) for 1 h and subsequently unstimulated (empty bars) or stimulated (closed bars) with 500 units/ml of IL-2 for 10 min (A and B) or 1 μm PMA for 15 min (C) as indicated. Cells were lysed and extracts were used to measure glycogen phosphorylase activity. Results show the mean of three independent experiments ± S.D., and statistical analysis shows a significant difference (***, p < 0.001). D and E, Kit 225 T cells were treated with PKC inhibitors or vehicle (dimethyl sulfoxide) for 1 h and stimulated or not with 500 units/ml of IL-2 for 10 min (D) or 1 μm PMA for 15 min (E) as indicated and lysed. Cell extracts were used to measure Rac1 activation by affinity precipitation assays. Precipitated active Rac 1 (Rac 1-GTP) and total Rac 1 from cell lysates were analyzed by Western blot (W.B.) using anti-Rac 1 specific antibody. Results are representative of three independent experiments.
To verify a putative connection between nPKC and PYGM in Kit 225 T cells, PMA, a natural DAG analog, was used to directly activate PKC. Kit 225 T cells deprived of IL-2 for 48 h were stimulated with 500 units/ml of IL-2 for 10 min or 1 μm PMA for 15 min. PMA was able to stimulate maximal PYGM activity; a response equivalent to the one produced by IL-2 (Fig. 3C, second and third bars). Accordingly, IL-2- and PMA-stimulated PYGM activity was blocked in Rottlerin (2.5 μm, for 1 h) pretreated cells (Fig. 3C, fifth and sixth bars). Next, the impact of PKCs inhibition on Rac 1 activation stimulated by IL-2 was examined. Results presented here confirmed that IL-2 stimulation correlated with an increase of the Rac 1 active form (Fig. 3D, first panel, second lane). IL-2 stimulated Rac 1 activation was unaffected when classical PKCs were inhibited by 100 nm Gö6975 (Fig. 3D, first panel, fourth lane). However, 2.5 μm Rottlerin almost completely blocked IL-2-stimulated Rac 1 activation (Fig. 3D, first panel, sixth lane). Finally, to determine whether PMA could stimulate Rac 1 activation, Kit 225 T cells deprived of IL-2 for 48 h were stimulated with 500 units/ml of IL-2 for 10 min or with 1 μm PMA for 15 min. As shown in Fig. 3E, both or IL-2-stimulated Rac 1 activation (first panel, second and third lanes) where completely blocked by 2.5 μm Rottlerin (Fig. 3E, first panel, fifth and sixth lanes).
To further characterize the involvement of nPKCs in signaling pathways stimulated by IL-2 leading to Rac 1/PYGM activation, Kit 225 T cells were transiently transfected with pcDNA3 (empty vector) or with cDNAs encoding the α, ϵ, and θ constitutively active isoforms of PKCs for 24 h, and Rac 1 activation was analyzed. As shown in Fig. 4A, transfection of the constitutively active form of PKCθ activated Rac 1 to a level comparable with those obtained by IL-2 stimulation. The potent stimulating effect of PKCθ was specific given that neither activated PKCα or PKCϵ had any effect on Rac 1 activation. In agreement with this neither PKCα nor PKCϵ increased the basal level of PYGM activity, whereas constitutively active PKCθ induced PYGM activity to levels comparable with that stimulated by IL-2 (Fig. 4B).
FIGURE 4.

PKCθ mediates Rac-1 and PYGM activation in Kit 225 T cells. Kit 225 T cells deprived of IL-2 for 24 h were transfected (A and B) with empty vector or with constitutively active mutants of PKCα, PKCϵ, and PKCθ (C and D) with empty vector or cDNAs encoding PKCθ (wt), PKCθA148E (constitutively active form), and PKCθK409R (dominant-negative form) and (E and F), with egfp(esiRNA) and pkcθ (esiRNA). 24 h post-transfection cells were stimulated or not with 500 units/ml of IL-2 for 10 min and lysed. A, C, and E, cell extracts were used to measure Rac 1 activation by affinity precipitation assays. Precipitated active Rac 1 (Rac 1-GTP) and total Rac 1 from cell lysates were analyzed by Western blot (W.B.) using anti-Rac 1 specific antibody. Results are representative of three independent experiments. The expression levels of PKCαA25E, PKCϵA159E, and PKCθA148E were visualized using anti-PKCα antibody to detect PKCαA25E, and anti-HA antibody to detect both PKCϵA159E and PKCθA148E, and endogenous PKCθ (E) was visualized using anti-PKCθ antibody. B, D, and F, cell lysates were used to examine glycogen phosphorylase activity. Results show the mean of three independent experiments ± S.D., and statistical analysis shows a significant difference (***, p < 0.001) in histograms B and D and (**, p < 0.05) in histogram E. Immunoreactive bands in B and D corresponding to the expression levels of PKCαA25E, PKCϵA159E, and PKCθA148E were visualized using anti-PKCα antibody to detect PKCαA25E, and anti-HA antibody to detect both PKCϵA159E and PKCθA148E. Endogenous PKCθ (E) and γ-tubulin (E) were visualized using anti-PKCθ and anti-γ-tubulin antibodies
To confirm the involvement of PKCθ in the Rac 1/PYGM pathway, Rac 1 and PYGM activation was determined in Kit 225 T cells overexpressing PKCθ (wt), PKCθA148E (constitutively active form of PKCθ), and PKCθK409R (dominant-negative form of PKCθ) with or without IL-2 stimulation. First, immunoblotting showed that all forms of PKCθ were equally expressed (Fig. 4, C, third panel, and D, upper panel). As shown in Fig. 4C, transfection of the PKCθ constitutively active form was found to activate Rac 1 to levels comparable with those stimulated by IL-2. In fact, IL-2 stimulation of cells overexpressing PKCθ (wt) or PKCθA148E did not increase Rac 1 activation any further (Fig. 4 C, fourth and sixth lanes compared with second). In contrast, transfection with PKCθK409R completely abolished Rac 1 activation either with or without IL-2 stimulation (Fig. 4C, seventh and eighth lanes). Similar results were obtained when PYGM activity was measured. PKCθA148E overexpression alone was enough to stimulate maximal PYGM activity; equivalent to PYGM activity stimulated by IL-2 (Fig. 4D, fifth bar compared with second, fourth, and sixth bars). Finally, PKCθK409R overexpression completely blocked PYGM activity either with or without IL-2 stimulation (Fig. 4D, seventh and eighth bars).
To confirm results obtained with PKCθ demonstrating that this serine-threonine kinase functions as a Rac 1/PYGM-activating molecule in Kit 225 T cells, PKCθ was knocked down with pkcθ (esiRNA), as described above. The endogenous Rac 1 active state was measured by the pulldown assay. As shown in Fig. 4E, in the absence of PKCθ (knockdown) IL-2 stimulation was unable to induce Rac 1 activation (fourth lane compared with second lane). The PKCθ expression level in cells transfected with esiRNA control (egfp) or pkcθ(esiRNA) was determined by Western blot (Fig. 4E, upper panel). Rac 1 detected in whole cell lysates shows that the loaded proteins were equivalent in all lanes (Fig. 4E, third panel). When PYGM activity in PKCθ knockdown Kit 225 T cells stimulated with IL-2 was examined, it was observed that in the absence of PKCθ IL-2 was unable to stimulate PYGM activation (Fig. 4F). Small aliquots of whole cell lysate from each condition were stored, electrophorectically separated on SDS-PAGE, and followed by Western blot. Immunoreactive bands were visualized with specific antibodies as indicated. Fig. 4F (first and second panels) shows PKCθ expression levels after esiRNA transfection and γ-tubulin, respectively. γ-Tubulin blot analysis indicates that equivalent amounts of protein were used in SDS-PAGE analysis.
PKCθ Controls αPIX Phosphorylation in Vivo and in Vitro
To test whether or not nPKCs could phosphorylate αPIX serine residues when Kit 225 T cells were stimulated by IL-2, HA-αPIX overexpressing cells were pretreated or not with 2.5 μm Rottlerin for 1 h, and stimulated or not with 500 units/ml of IL-2 for 10 min. HA-αPIX present in cell lysates was immunoprecipitated using anti-HA antibody, as described under “Experimental Procedures.” As shown in Fig. 5A (first left panel, fourth lane), IL-2 stimulated αPIX serine phosphorylation, which was blocked by Rottlerin (Fig. 5A, first right panel, fourth lane). Given that these experiments were performed in αPIX overexpressing cells, endogenous αPIX phosphorylation was also examined. Kit 225 T cells were transfected with pkcθ (esiRNA) to knockdown PKCθ and cell lysates were immunoprecipitated using anti-αPIX antibody, as described under “Experimental Procedures.” As shown in Fig. 5B (fourth lane 4 compared with second lane), in the absence of PKCθ expression, IL-2 was unable to induce αPIX serine phosphorylation. To determine the amount of immunoprecipitated αPIX, membranes that were used to examine the αPIX serine phosphorylation were stripped and reblotted with anti-αPIX antibody (Fig. 5B, second panel). PKCθ expression levels in the presence of esiRNA control (egfp) or pkcθ (esiRNA) present in whole cell lysates were also examined by SDS-PAGE and followed by Western blot. Immunoreactive bands were visualized using anti-PKCθ antibody (Fig. 5B, third panel).γTubulin shows that total protein was equivalent in all assay conditions (Fig. 5B, fourth panel). These results suggest that αPIX serine phosphorylation stimulated by IL-2 requires PKCθ protein expression.
FIGURE 5.
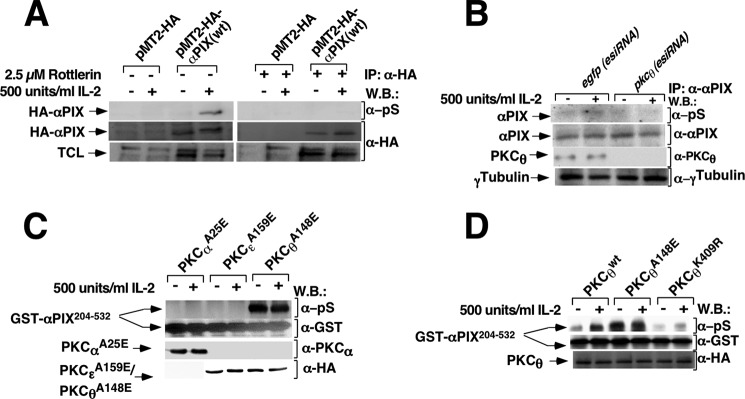
αPIX serine phosphorylation depends on PKCθ in intact Kit 225 T cells. Kit 225 T cells deprived of IL-2 for 24 h were transfected with (A), empty vector (pMT2-HA) or pMT2-HA-αPIX (wt), and (B) with egfp(esiRNA) and pkcθ(esiRNA). A, 24 h post-transfection an aliquot of cells were pretreated with 2.5 μm Rottlerin or vehicle (dimethyl sulfoxide) for 1 h. Subsequently, cells were stimulated or not with 500 units/ml of IL-2 for 10 min and lysed. B, 24 h post-transfection cells were stimulated or not with 500 units/ml of IL-2 for 10 min and lysed. Cell lysates were subjected to immunoprecipitation with anti-HA (A) or anti-αPIX (B) antibodies and immunoreactive bands were visualized using anti-phosphoserine, anti-HA, anti-αPIX, anti-PKCθ, and anti-γ-tubulin antibodies. Results are representative of three independent experiments. C, Kit 225 T cells deprived of IL-2 for 24 h were transfected with empty vector or with the constitutively active mutants of PKCα, PKCϵ, and PKCθ. D, Kit 225 T cells deprived of IL-2 for 24 h were transfected with cDNAs encoding PKCθ (wt), PKCθA148E (constitutively active form), and PKCθK409R (dominant-negative form). 24 h post-transfection cells were stimulated or not with 500 units/ml of IL-2 for 10 min and lysed. Cell extracts were immunoprecipitated with anti-PKCα and anti-HA antibodies (C) and anti-HA antibody (D). Immunocomplex activities were analyzed by an in vitro kinase assay followed by SDS-PAGE and Western blot. Immunoreactive bands were visualized using anti-phosphoserine (pS) antibodies. In addition, to determine the amount of GST-αPIX204–532, aliquots of lysates were analyzed by Western blot (W.B.) using anti-GST antibody. The expression levels of PKCαA25E, PKCϵA159E, PKCθ (wt), PKCθA148E, and PKCθK409R were visualized using anti-PKCα to detect PKCαA25E and anti-HA antibody to detect PKCϵA159E and PKCθ (wild type and mutants). Results are representative of three independent experiments.
As shown in Fig. 2, αPIX serine residues 225 and 488 are required for Rac 1 and PYGM activation in IL-2-stimulated Kit 225 T cells. Therefore, to determine whether or not PKCθ could phosphorylate the αPIX region comprising both serines, an in vitro kinase assay was performed using a GST-αPIX204–532 fusion protein as an exogenous substrate for PKCθ. The GST moiety was fused to the αPIX region spanning from 204 to 532 residues, as described under “Experimental Procedures.” Briefly, to carry out the in vitro kinase assay PKCαA25E, PKCϵA159E, and PKCθA148E were immunoprecipitated from cell lysates derived from stimulated or unstimulated Kit 225 T cells and immunocomplexes were incubated with ATP and GST-αPIX204–532. Subsequently, proteins were resolved by SDS-PAGE followed by Western blot and GST-αPIX204–532 serine phosphorylation was visualized using an anti-phosphoserine antibody. As illustrated in Fig. 5C, PKCθA148E induced robust GST-αPIX204–532 serine phosphorylation and it was not modified by IL-2 stimulation (fifth and sixth lanes). However, serine phosphorylation was not detectable either in the presence of PKCαA25E or PKCϵA159E (Fig. 5C, from first to fourth lanes).
To confirm PKCθ involvement in the αPIX204–532 region serine phosphorylation, Kit 225 T cells were transiently transfected with pcDNA3 (empty vector) or with cDNAs encoding PKCθ (wt), PKCθA148E (constitutively active form), and PKCθK409R (dominant-negative form). Equal expression levels of these PKCθ forms in Kit 225 T cells were confirmed by immunoblotting (Fig. 5D). As shown in Fig. 5D (second lane), IL-2 stimulates GST-αPIX204–532 serine phosphorylation in Kit 225 T cells overexpressing PKCθ (wt). When the effect of PKCθA148E overexpression on GST-αPIX204–532 phosphorylation was examined, a stronger level of exogenous substrate phosphorylation was observed than that found in cells overexpressing PKCθ (wt) and stimulated by IL-2 (Fig. 5D, third lane compared with second lane). IL-2 stimulation of PKCθA148E overexpressing cells did not increase the phophorylation level further than that of unstimulated PKCθA148E overexpressing cells (Fig. 5D, third and fourth lanes). In contrast, transfection with PKCθK409R (PKCθ, dominant-negative) completely blocked GST-αPIX204–532 phosphorylation independently of IL-2 stimulation (Fig. 5D, fifth and sixth lanes).
PKCθ and αPIX Are Needed for IL-2-induced Chemotaxis and Proliferation of Kit 225 T Cells
To investigate the role of PKCθ and αPIX in IL-2-stimulated Kit 225 T cell migration, cells were transfected with egfp (as control), pkcθ, or αpix (esiRNAs) and their effects on the IL-2-induced T cell chemotaxis through polyethylene terephthalate membranes were examined. As shown in Fig. 6A, IL-2 stimulated robust migration of Kit 225 T cells (first solid bar). On the other hand, lack of either PKCθ or αPIX expression abolished Kit 225 T cells migration stimulated by IL-2 (Fig. 6A, second and third solid bars). At the end of the experiment, cells in the upper and lower chambers from each transfected condition were mixed. Cells were lysed and whole cell lysates were separated by SDS-PAGE followed by Western blot. The expression levels of αPIX and PKCθ were visualized using specific antibodies as indicated in Fig. 6A (first and second panels). γ-Tubulin Western blot analysis shows that equal amounts of protein were used (Fig. 6A, third panel).
FIGURE 6.
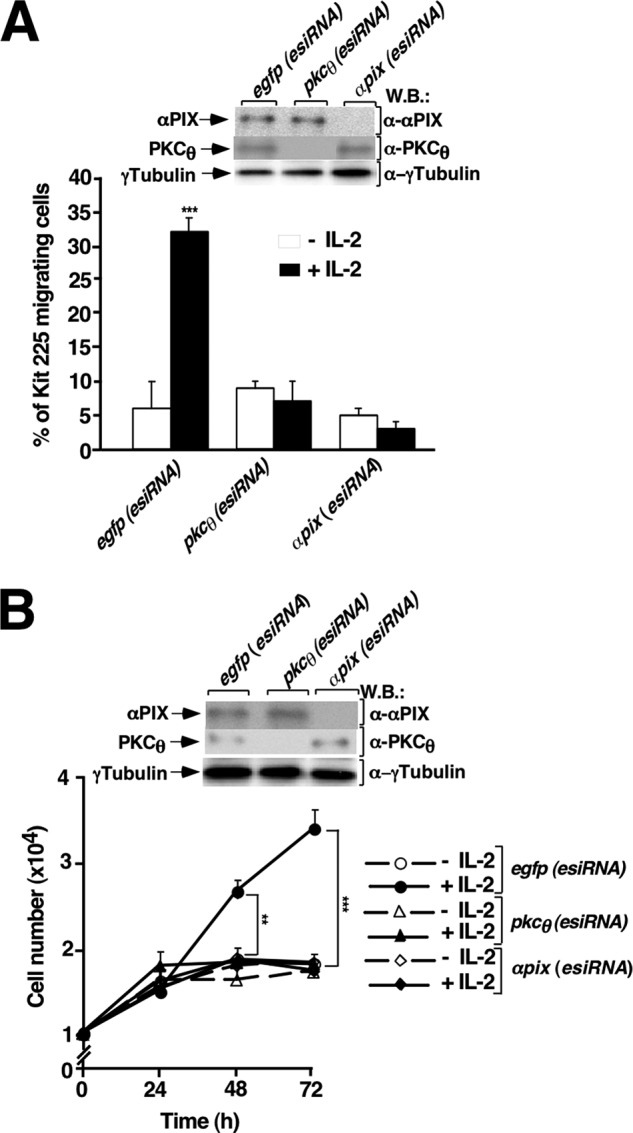
PKCθ/αPIX pathway mediates IL-2-stimulated Kit 225 chemotaxis and proliferation. A, esiRNA (egfp, pkcθ, αpix) transfected Kit 225 T cells migration was studied in a Transwell assay. Data representing the percentage of migrating cells are expressed as the mean of three independent experiments ± S.D. and statistical analysis shows a significant difference (***, p < 0.001). At the end of the assay, cells were recovered, lysed, and cell lysates were analyzed by Western blot (W.B.) using specific antibodies as indicated. B, esiRNA (egfp, pkcθ, αpix)-transfected Kit 225 T cells were stained with PKH26. Fluorescence was analyzed before IL-2 stimulation (−IL-2 (0 h) and after every 24-h incubation with IL-2 (+IL-2) for 3 consecutive days. Results represent the mean of three independent experiments ± S.D. and the statistical analysis showed a significant difference (**, p < 0.05 and ***, p < 0.001). 3 × 105 Kit 225 T cells were taken for each transfection condition, lysed, and cell lysates were analyzed by Western blot using specific antibodies as indicated.
To evaluate the role of PKCθ and αPIX in IL-2-stimulated cell proliferation, esiRNAs for egfp, pkcθ, and αpix were used. Cell proliferation was analyzed after flow cytometry by monitoring the decrease in fluorescence of dye PKH6 incorporated in cell membrane, which is diluted approximately 2-fold with each cell division. PKH26-labeled cells were treated with 16 units/ml of IL-2 every 24 h for 3 days. In control cells (egfp (esiRNA) transfected cells) cultured for 3 days, IL-2 stimulation resulted in approximately a 2-fold increase in cell number (Fig. 6B, closed circles) compared with IL-2-unstimulated cells (Fig. 6B, open circles). Remarkably, IL-2-stimulated cell proliferation was significantly reduced with either pkcθ (esiRNA) (Fig. 6B, closed triangles) or with αpix(esiRNA) (Fig. 6B, closed diamonds) knockdown. At the beginning of the experiment, 3 × 105 cells from each transfected condition were taken and lysed. Total protein in lysates was separated by SDS-PAGE followed by Western blot. The expression level of αPIX and PKCθ was visualized using specific antibodies as indicated in Fig. 6B (first and second panels). γ-Tubulin Western blot analysis indicates that equal amounts of protein were used in the analysis (Fig. 6B, third panel).
DISCUSSION
Small GTPases of the Rho family actively participate in the immune response after antigenic stimulation, allowing for appropriate actin cytoskeleton reorganization and regulation of transcription factors activity (53). Cooperation between transcription factors such as NFAT, NF-κB, and JNK is key to guarantee an adequate T cell clonal proliferation; in part by regulating transcription of IL-2 and the α chain of the IL-2Rα (49). Recently, we reported that upon IL-2R activation in Kit 225 T cells, the Rac 1 GTPase active form binds to and activates the metabolic enzyme PYGM, leading to cell proliferation (13). In the present study, we show that the IL-2/IL2-R engagement signals to the Rac 1/PYGM pathway through PKCθ and the GEF αPIX. More importantly, we identify αPIX as a Rac 1-specific GEF in IL-2-stimulated T cells and provide novel evidence demonstrating that αPIX requires serine phosphorylation by PKCθ to control the Rac 1/PYGM pathway, and thereby regulate T cell migration and proliferation.
Signals emanating from membrane receptors such as TCR, BCR, and IL-2R leading not only to the activation of protein-tyrosine kinases, but also of serine/threonine kinases, can positively regulate downstream effector molecules, including small GTPases of the Ras superfamily (18, 50). However, little is known about which Rho-GEF activates Rho GTPases after IL-2 receptor activation. Kit 225 T cells express IL-2R constitutively and depend exclusively on IL-2 for cellular proliferation (41). This feature represents an important advantage for IL-2-stimulated signaling studies and this cellular system has emphasized the importance of RhoA (51) and Rac 1 (12, 13) in T cell biology regulated by IL-2.
Given that tyrosine phosphorylation of cellular proteins is one of the most important and characteristic events in early cell signaling stimulated by IL-2, Osinalde et al. (35) used high resolution mass spectrometry, combined with phosphotyrosine immunoprecipitation and stable isotope labeling by amino acids in cell culture (SILAC) to identify 172 tyrosine-phosphorylated target proteins; among which is αPIX-RhoGEF. This result prompted us to examine the potential participation of αPIX in PYGM activation. Overexpression experiments with αPIX demonstrated a significant increase in PYGM activity in the absence of any stimuli. αPIX overexpression promotes its own spontaneous dimerization in the absence of any stimuli, and in this configuration αPIX activates Rac 1 (52). Furthermore, in the presence of IL-2, αPIX-transfected cells reached a maximum level of PYGM activity. However, these results do not exclude the possibility that other GEFs capable of undergoing tyrosine phosphorylation may be involved in this signaling pathway stimulated by IL-2. In fact, following this hypothesis we proposed that Rac 1 might also require Vav Rho-GEF to bind to and activate PYGM. Vav is the main GEF for Rac 1 in the hematopoietic system and its GEF function targeting the Rho family of GTPases is modulated by tyrosine phosphorylation at residue 174 (26, 27). Evans et al. (53) also reported than in peripheral blood lymphocytes, Vav is tyrosine phosphorylated after IL-2 stimulation. However, in Vav overexpressing Kit 225 T cells there is no increase of PYGM activity. In our view, this was quite an expected result. This is because specifically Vav Tyr174 must be phosphorylated (26, 27) to release its DH domain from its inhibitory configuration and thus gain access to and activate Rac 1 (27). When the effect of IL-2 on Tyr174 phosphorylation was examined, it was observed that this cytokine was not able to stimulate Tyr174 phosphorylation (data not shown). These results do not contradict the observations reported by Evans et al. (53), given that this group described general tyrosine phosphorylation of Vav occurring mostly in its SH2 domain and located at Vav carboxyl-terminal region; whereas, Tyr174 is located between Vav CH and DH domains at the amino-terminal region (26, 27).
From the start our results showed that αPIX, and not Vav, mediated PYGM activation in IL-2-stimulated Kit 225 T cells. Although, additional GEFs from the Dbl family of exchange factors such as Tiam-1 and STEF are capable of activating Rac 1 (28, 54), these GEFs have not been reported as being activated by tyrosine phosphorylation, but rather Tiam-1 was reported being activated by threonine phosphorylation (30), whereas STEF was reported being activated by serine/threonine phosphorylation (31). Even so, their effects on PYGM activity were evaluated. When these GEFs were individually overexpressed in Kit 225 T cells, it was observed that neither of them increased PYGM activity. Moreover, overexpression of Tiam-1 seems to have a dominant-negative effect on PYGM activity. Furthermore, when we examined the effects of αPIX knockdown with αpix(esiRNA) on either PYGM activity or Rac 1 activation, it was observed that the absence of the αPIX-Rho-GEF expression was enough to block activation of the Rac 1/PYGM pathway. Collectively, these results strongly suggest that αPIX is the only GEF responsible for activation of the Rac 1/PYGM pathway in Kit 225 T cells stimulated by IL-2.
Even if the αPIX predicted phosphorylation sites were the serines phosphorylated at residues 225 and 488 rather tyrosine residues (35), this prediction does not contradict experimental evidence obtained from SILAC-based quantitative phosphoproteomics data (35), because a protein enriched by immunoprecipitation with antiphosphotyrosine antibody upon IL-2 stimulation does not necessarily mean that it has been tyrosine phosphorylated in response to that cytokine. Any non-tyrosine-phosphorylated proteins may also be enriched if it is bound to a tyrosine-phosphorylated protein. The prediction that αPIX serine residues 225 and 488 may be susceptible to being phosphorylated in Kit 225 T cells (35) is consistent with the phosphoproteomic data obtained from ES and iPS cells (36), KG1 AML cells (37), and colorectal cancer cells (38).
From a functional point of view, phosphorylation/dephosphorylation cycles are major early events in intracellular signaling cascades. Therefore, the importance of these two αPIX-RhoGEF residues (Ser225 and Ser488) on Rac 1 and the subsequent PYGM activation was examined. The results obtained are compelling; both residues are essential to control Rac 1 activation and PYGM enzymatic activity. In addition, by pharmacological and genetic (either by PKCθ loss or gain of function) approaches we were able to find out that PKCθ was in control of αPIX phosphorylation and therefore activation of the Rac 1/PYGM pathway in Kit 225 T cells. The involvement of PKCθ in this cellular model is a novel finding although not a surprising one; given that its expression is restricted to certain tissues and cell types, including T cells (55). Since the relevance of this serine/threonine kinase in regulating the dynamics of the immunological synapse was described (56–58), many additional functions have been discovered, such as the control of NF-κB, AP-1, and NFAT transcription factors activation, which regulate the expression of proinflammatory cytokines and anti-proapoptotic molecules Bcl-XL (59). Moreover, the adhesive capacities of T lymphocytes (60) is the mechanism through which stable adhesion between T cells and antigen presenting cells is achieved (55, 61, 62). More recently, a new role for PKCθ in T cell physiology was described; i.e. PKCθ participation in the CCR7 downstream signaling driving T cell migration (63). Therefore, the present findings are in agreement with those reported by Cannon et al. (63) that not only stimulated IL-2 T cell proliferation but also T cell migration is regulated by the PKCθ/αPIX axis upstream of Rac 1/PYGM.
In conclusion, our findings reveal the mechanism through which IL-2 stimulates Rac 1 activation in Kit 225 T cells. We identified αPIX as the GEF that specifically activates Rac 1 and consequently regulates PYGM activation, as well as the molecular mechanism of αPIX activation of the Rac1/PYGM pathway. Regulation of this novel metabolic pathway requires that PKCθ phosphorylates αPIX serine residues participating in the control of T cell migration and/or proliferation. Mechanistically, the specific molecular players connecting the activated IL-2R and the PKCθ/αPIX/Rac 1/PYGM pathway, and also PYGM downstream signaling targets are still unknown. Future studies will allow us to characterize the signaling molecules upstream of PKCθ and effector molecules that participate in this signal transduction pathway.
Acknowledgments
We thank the AIDS Research and Reference Program, Division of AIDS (NIAD, National Institutes of Health) for the generous gift of recombinant IL-2, Dr. Kerstin Kutsche (Universitätsklinikum Hamburg-Eppendorf, Germany) for the generous gift of plasmid encoding for αPIX, and Dr. Gottfried Baier (Medical University of Innsbruck, Austria) for the generous gift of plasmids encoding for the constitutively active and dominant-negative forms of PKCθ.
Footnotes
- PYGM
- glycogen phosphorylase muscle isoform
- RBD
- Rho/Rac-binding domain
- GEF
- guanine nucleotide exchange factor
- BCR
- B cell receptor
- PMA
- phorbol 12-myristate 13-acetate
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Thomas R. M., Gao L., Wells A. D. (2005) Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J. Immunol. 174, 4639–4646 [DOI] [PubMed] [Google Scholar]
- 2. Smith K. A. (1988) The bimolecular structure of the interleukin 2 receptor. Immunol. Today 9, 36–37 [DOI] [PubMed] [Google Scholar]
- 3. Pei Y., Zhu P., Dang Y., Wu J., Yang X., Wan B., Liu J. O., Yi Q., Yu L. (2008) Nuclear export of NF90 to stabilize IL-2 mRNA is mediated by AKT-dependent phosphorylation at Ser647 in response to CD28 costimulation. J. Immunol. 180, 222–229 [DOI] [PubMed] [Google Scholar]
- 4. Malek T. R., Castro I. (2010) Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity 33, 153–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lan R. Y., Selmi C., Gershwin M. E. (2008) The regulatory, inflammatory, and T cell programming roles of interleukin-2 (IL-2). J. Autoimmun. 31, 7–12 [DOI] [PubMed] [Google Scholar]
- 6. Brockdorff J., Nielsen M., Kaltoft K., Mustelin T., Röpke C., Svejaard A., Geisler C., Odum N. (2000) Lck is involved in interleukin-2 induced proliferation but not cell survival in human T cells through a MAP kinase-independent pathway. Eur. Cytokine Netw. 11, 225–231 [PubMed] [Google Scholar]
- 7. Yang Y. R., Follo M. Y., Cocco L., Suh P. G. (2013) The physiological roles of primary phospholipase C. Adv. Biol. Regul. 53, 232–241 [DOI] [PubMed] [Google Scholar]
- 8. Kadamur G., Ross E. M. (2013) Mammalian phospholipase C. Annu. Rev. Physiol. 75, 127–154 [DOI] [PubMed] [Google Scholar]
- 9. Nishizuka Y. (1995) Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 9, 484–496 [PubMed] [Google Scholar]
- 10. Nishizuka Y. (2001) The protein kinase C family and lipid mediators for transmembrane signaling and cell regulation. Alcohol Clin. Exp. Res. 25, 3S–7S [DOI] [PubMed] [Google Scholar]
- 11. Arnaud M., Mzali R., Gesbert F., Crouin C., Guenzi C., Vermot-Desroches C., Wijdenes J., Courtois G., Bernard O., Bertoglio J. (2004) Interaction of the tyrosine phosphatase SHP-2 with Gab2 regulates Rho-dependent activation of the c-fos serum response element by interleukin-2. Biochem. J. 382, 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arrieumerlou C., Donnadieu E., Brennan P., Keryer G., Bismuth G., Cantrell D., Trautmann A. (1998) Involvement of phosphoinositide 3-kinase and Rac in membrane ruffling induced by IL-2 in T cells. Eur. J. Immunol. 28, 1877–1885 [DOI] [PubMed] [Google Scholar]
- 13. Arrizabalaga O., Lacerda H. M., Zubiaga A. M., Zugaza J. L. (2012) Rac1 protein regulates glycogen phosphorylase activation and controls interleukin (IL)-2-dependent T cell proliferation. J. Biol. Chem. 287, 11878–11890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beier I., Düsing R., Vetter H., Schmitz U. (2008) Epidermal growth factor stimulates Rac1 and p21-activated kinase in vascular smooth muscle cells. Atherosclerosis 196, 92–97 [DOI] [PubMed] [Google Scholar]
- 15. Bokoch G. M. (2003) Biology of the p21-activated kinases. Annu. Rev. Biochem. 72, 743–781 [DOI] [PubMed] [Google Scholar]
- 16. Chiariello M., Vaqué J. P., Crespo P., Gutkind J. S. (2010) Activation of Ras and Rho GTPases and MAP kinases by G-protein-coupled receptors. Methods Mol. Biol. 661, 137–150 [DOI] [PubMed] [Google Scholar]
- 17. Pelletier S., Duhamel F., Coulombe P., Popoff M. R., Meloche S. (2003) Rho family GTPases are required for activation of Jak/STAT signaling by G protein-coupled receptors. Mol. Cell. Biol. 23, 1316–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zugaza J. L., Caloca M. J., Bustelo X. R. (2004) Inverted signaling hierarchy between RAS and RAC in T-lymphocytes. Oncogene 23, 5823–5833 [DOI] [PubMed] [Google Scholar]
- 19. Caloca M. J., Zugaza J. L., Matallanas D., Crespo P., Bustelo X. R. (2003) Vav mediates Ras stimulation by direct activation of the GDP/GTP exchange factor Ras GRP1. EMBO J. 22, 3326–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caloca M. J., Zugaza J. L., Bustelo X. R. (2008) Mechanistic analysis of the amplification and diversification events induced by Vav proteins in B-lymphocytes. J. Biol. Chem. 283, 36454–36464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parri M., Chiarugi P. (2010) Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bustelo X. R., Sauzeau V., Berenjeno I. M. (2007) GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29, 356–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cherfils J., Zeghouf M. (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 24. Ravichandran K. S., Lorenz U., Shoelson S. E., Burakoff S. J. (1995) Interaction of Shc with Grb2 regulates the Grb2 association with mSOS. Ann. N.Y. Acad. Sci. 766, 202–203 [DOI] [PubMed] [Google Scholar]
- 25. Ravichandran K. S., Igras V., Shoelson S. E., Fesik S. W., Burakoff S. J. (1996) Evidence for a role for the phosphotyrosine-binding domain of Shc in interleukin 2 signaling. Proc. Natl. Acad. Sci. U.S.A. 93, 5275–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crespo P., Schuebel K. E., Ostrom A. A., Gutkind J. S., Bustelo X. R. (1997) Phosphotyrosine-dependent activation of Rac-1 GDP/GTP exchange by the vav proto-oncogene product. Nature 385, 169–172 [DOI] [PubMed] [Google Scholar]
- 27. Llorca O., Arias-Palomo E., Zugaza J. L., Bustelo X. R. (2005) Global conformational rearrangements during the activation of the GDP/GTP exchange factor Vav3. EMBO J. 24, 1330–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Collard J. G., Habets G. G., Michiels F., Stam J., van der Kammen R. A., van Leeuwen F. (1996) Role of Tiam 1 in Rac-mediated signal transduction pathways. Curr. Top. Microbiol. Immunol. 213, 253–265 [DOI] [PubMed] [Google Scholar]
- 29. Matsuo N., Hoshino M., Yoshizawa M., Nabeshima Y. (2002) Characterization of STEF, a guanine nucleotide exchange factor for Rac1, required for neurite growth. J. Biol. Chem. 277, 2860–2868 [DOI] [PubMed] [Google Scholar]
- 30. Fleming I. N., Elliott C. M., Collard J. G., Exton J. H. (1997) Lysophosphatidic acid induces threonine phosphorylation of Tiam1 in Swiss 3T3 fibroblasts via activation of protein kinase C. J. Biol. Chem. 272, 33105–33110 [DOI] [PubMed] [Google Scholar]
- 31. Goto A., Hoshino M., Matsuda M., Nakamura T. (2011) Phosphorylation of STEF/Tiam2 by protein kinase A is critical for Rac1 activation and neurite outgrowth in dibutyryl cAMP-treated PC12D cells. Mol. Biol. Cell. 22, 1780–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manser E., Loo T. H., Koh C. G., Zhao Z. S., Chen X. Q., Tan L., Tan I., Leung T., Lim L. (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1, 183–192 [DOI] [PubMed] [Google Scholar]
- 33. Obermeier A., Ahmed S., Manser E., Yen S. C., Hall C., Lim L. (1998) PAK promotes morphological changes by acting upstream of Rac. EMBO J. 17, 4328–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoshii S., Tanaka M., Otsuki Y., Wang D. Y., Guo R. J., Zhu Y., Takeda R., Hanai H., Kaneko E., Sugimura H. (1999) αPIX nucleotide exchange factor is activated by interaction with phosphatidylinositol 3-kinase. Oncogene 18, 5680–5690 [DOI] [PubMed] [Google Scholar]
- 35. Osinalde N., Moss H., Arrizabalaga O., Omaetxebarria M. J., Blagoev B., Zubiaga A. M., Fullaondo A., Arizmendi J. M., Kratchmarova I. (2011) Interleukin-2 signaling pathway analysis by quantitative phosphoproteomics. J. Proteomics 75, 177–191 [DOI] [PubMed] [Google Scholar]
- 36. Phanstiel D. H., Brumbaugh J., Wenger C. D., Tian S., Probasco M. D., Bailey D. J., Swaney D. L., Tervo M. A., Bolin J. M., Ruotti V., Stewart R., Thomson J. A., Coon J. J. (2011) Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods 8, 821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weber C., Schreiber T. B., Daub H. (2012) Dual phosphoproteomics and chemical proteomics analysis of erlotinib and gefitinib interference in acute myeloid leukemia cells. J. Proteomics 75, 1343–1356 [DOI] [PubMed] [Google Scholar]
- 38. Shiromizu T., Adachi J., Watanabe S., Murakami T., Kuga T., Muraoka S., Tomonaga T. (2013) Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J. Proteome Res. 12, 2414–2421 [DOI] [PubMed] [Google Scholar]
- 39. Poppe D., Tiede I., Fritz G., Becker C., Bartsch B., Wirtz S., Strand D., Tanaka S., Galle P. R., Bustelo X. R., Neurath M. F. (2006) Azathioprine suppresses ezrin-radixin-moesin-dependent T cell-APC conjugation through inhibition of Vav guanosine exchange activity on Rac proteins. J. Immunol. 176, 640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gérard A., van der Kammen R. A., Janssen H., Ellenbroek S. I., Collard J. G. (2009) The Rac activator Tiam1 controls efficient T-cell trafficking and route of transendothelial migration. Blood 113, 6138-6147 [DOI] [PubMed] [Google Scholar]
- 41. Hori T., Uchiyama T., Tsudo M., Umadome H., Ohno H., Fukuhara S., Kita K., Uchino H. (1987) Establishment of an interleukin 2-dependent human T cell line from a patient with T cell chronic lymphocytic leukemia who is not infected with human T cell leukemia/lymphoma virus. Blood 70, 1069–1072 [PubMed] [Google Scholar]
- 42. Manterola L., Hernando-Rodríguez M., Ruiz A., Apraiz A., Arrizabalaga O., Vellón L., Alberdi E., Cavaliere F., Lacerda H. M., Jimenez S., Parada L. A., Matute C., Zugaza J. L. (2013) 1–42 β-amyloid peptide requires PDK1/nPKC/Rac 1 pathway to induce neuronal death. Transl. Psychiatry 3, e219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersen B., Westergaard N. (2002) The effect of glucose on the potency of two distinct glycogen phosphorylase inhibitors. Biochem. J. 367, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McInerney M., Serrano Rodriguez G., Pawlina W., Hurt C. B., Fletcher B. S., Laipis P. J., Frost S. C. (2002) Glycogen phosphorylase is activated in response to glucose deprivation but is not responsible for enhanced glucose transport activity in 3T3-L1 adipocytes. Biochim. Biophys. Acta 1570, 53–62 [DOI] [PubMed] [Google Scholar]
- 45. Wery-Zennaro S., Zugaza J. L., Letourneur M., Bertoglio J., Pierre J. (2000) IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene 19, 1596–1604 [DOI] [PubMed] [Google Scholar]
- 46. Gschwendt M., Müller H. J., Kielbassa K., Zang R., Kittstein W., Rincke G., Marks F. (1994) Rottlerin, a novel protein kinase inhibitor. Biochem. Biophys. Res. Commun. 199, 93–98 [DOI] [PubMed] [Google Scholar]
- 47. Villalba M., Kasibhatla S., Genestier L., Mahboubi A., Green D. R., Altman A. (1999) Protein kinase Cθ cooperates with calcineurin to induce Fas ligand expression during activation-induced T cell death. J. Immunol. 163, 5813–5819 [PubMed] [Google Scholar]
- 48. Cordey M., Pike C. J. (2006) Conventional protein kinase C isoforms mediate neuroprotection induced by phorbol ester and estrogen. J. Neurochem. 96, 204–217 [DOI] [PubMed] [Google Scholar]
- 49. Billadeau D. D., Nolz J. C., Gomez T. S. (2007) Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 7, 131–143 [DOI] [PubMed] [Google Scholar]
- 50. Caloca M. J., Zugaza J. L., Bustelo X. R. (2003) Exchange factors of the RasGRP family mediate Ras activation in the Golgi. J. Biol. Chem. 278, 33465–33473 [DOI] [PubMed] [Google Scholar]
- 51. Gómez J., García A., R-Borlado L., Bonay P., Martínez-A C., Silva A., Fresno M., Carrera A. C., Eicher-Streiber C., Rebollo A. (1997) IL-2 signaling controls actin organization through Rho-like protein family, phosphatidylinositol 3-kinase, and protein kinase C-ζ. J. Immunol. 158, 1516–1522 [PubMed] [Google Scholar]
- 52. Feng Q., Baird D., Cerione R. A. (2004) Novel regulatory mechanisms for the Dbl family guanine nucleotide exchange factor Cool-2/α-Pix. EMBO J. 23, 3492–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Evans G. A., Howard O. M., Erwin R., Farrar W. L. (1993) Interleukin-2 induces tyrosine phosphorylation of the vav proto-oncogene product in human T cells: lack of requirement for the tyrosine kinase lck. Biochem. J. 294, 339–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zaldua N., Gastineau M., Hoshino M., Lezoualc'h F., Zugaza J. L. (2007) Epac signaling pathway involves STEF, a guanine nucleotide exchange factor for Rac, to regulate APP processing. FEBS Lett. 581, 5814–5818 [DOI] [PubMed] [Google Scholar]
- 55. Wang X., Chuang H. C., Li J. P., Tan T. H. (2012) Regulation of PKC-θ function by phosphorylation in T cell receptor signaling. Front. Immunol. 3, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arendt C. W., Albrecht B., Soos T. J., Littman D. R. (2002) Protein kinase C-θ: signaling from the center of the T-cell synapse. Curr. Opin. Immunol. 14, 323–330 [DOI] [PubMed] [Google Scholar]
- 57. Manicassamy S., Gupta S., Sun Z. (2006) Selective function of PKC-θ in T cells. Cell Mol. Immunol. 3, 263–270 [PubMed] [Google Scholar]
- 58. Isakov N., Altman A. (2012) PKC-theta-mediated signal delivery from the TCR/CD28 surface receptors. Front. Immunol. 3, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baier-Bitterlich G., Uberall F., Bauer B., Fresser F., Wachter H., Grunicke H., Utermann G., Altman A., Baier G. (1996) Protein kinase C-θ isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell. Biol. 16, 1842–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Letschka T., Kollmann V., Pfeifhofer-Obermair C., Lutz-Nicoladoni C., Obermair G. J., Fresser F., Leitges M., Hermann-Kleiter N., Kaminski S., Baier G. (2008) PKC-θ selectively controls the adhesion-stimulating molecule Rap1. Blood 112, 4617–4627 [DOI] [PubMed] [Google Scholar]
- 61. Freeley M., Long A. (2012) Regulating the regulator: phosphorylation of PKC θ in T cells. Front. Immunol. 3, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. von Essen M. R., Kongsbak M., Levring T. B., Hansen A. K., Boding L., Lauritsen J. P., Woetmann A., Baier G., Ødum N., Bonefeld C. M., Geisler C. (2013) PKC-θ exists in an oxidized inactive form in naive human T cells. Eur. J. Immunol. 43, 1659–1666 [DOI] [PubMed] [Google Scholar]
- 63. Cannon J. L., Asperti-Boursin F., Letendre K. A., Brown I. K., Korzekwa K. E., Blaine K. M., Oruganti S. R., Sperling A. I., Moses M. E. (2013) PKCθ regulates T cell motility via ezrin-radixin-moesin localization to the uropod. PLoS One 8, e78940. [DOI] [PMC free article] [PubMed] [Google Scholar]