

Summary
PTEN is proposed to function at the plasma membrane, where receptor tyrosine kinases are activated. However, the majority of PTEN is located throughout the cytoplasm. Here, we show that cytoplasmic PTEN is distributed along microtubules, tethered to vesicles via phosphatidylinositol 3-phosphate (PI(3)P), the signature lipid of endosomes. We demonstrate that the non-catalytic C2 domain of PTEN specifically binds PI(3)P through the CBR3 loop. Mutations render this loop incapable of PI(3)P binding and abrogate PTEN-mediated inhibition of PI 3-kinase/AKT signaling. This loss of function is rescued by fusion of the loop mutant PTEN to FYVE, the canonical PI(3)P binding domain, demonstrating the functional importance of targeting PTEN to endosomal membranes. Beyond revealing an upstream activation mechanism of PTEN, our data introduce the concept of PI 3-kinase signal activation on the vast plasma membrane that is contrasted by PTEN-mediated signal termination on the small, discrete surfaces of internalized vesicles.
Graphical abstract
Introduction
The PTEN tumor suppressor is among the most frequently altered genes in cancer (Li et al., 1997; Steck et al., 1997). The understanding that this phosphatase controls phospholipid second messengers (Maehama and Dixon, 1998) has profoundly changed our understanding of these signaling pathways in disease (Vanhaesebroeck et al., 2012). PTEN antagonizes the class I phosphatidylinositol (PI) 3-kinases (PI 3-Ks), which produce the lipid second messenger PI (3,4,5) trisphosphate (PI(3,4,5)P3/PIP3) in response to activation of receptor tyrosine kinases (RTKs), G-protein-coupled receptors, or membrane-bound oncogenes (Engelman et al., 2006). PIP3 then serves as a membrane platform for the coordinate activation of downstream signaling kinases, adaptor proteins, and phosphatases. Central among those kinases is the AKT proto-oncogene, which controls downstream targets to promote cell growth, proliferation, survival, metabolism, and motility pathways (Fayard et al., 2010).
PTEN antagonizes PI 3-K/AKT-mediated signaling (Vanhaesebroeck et al., 2012). PTEN is frequently deleted and mutated in sporadic cancer, and germline mutations cause a spectrum of familial cancer syndromes, referred to collectively as PTEN hamartoma syndrome (Liaw et al., 1997; Mester and Eng, 2013). In addition to proliferative disorders, PTEN malfunction is associated with neurological phenotypes, including autism spectrum disorders (ASD), which are diagnosed in ∼10% of patients with inherited heterozygous PTEN mutation (Butler et al., 2005; Varga et al., 2009). Human pathologies can be recapitulated in murine models of disease. Pten mutant mice demonstrate multiple facets of ASD (Kwon et al., 2006; Xiong et al., 2012), and in mouse models of cancer, partial loss of Pten suffices to recapitulate multiple tumor types, including those of breast and prostate (Alimonti et al., 2010; Trotman et al., 2003). Complete loss of PTEN, in synergy with other engineered genetic alterations, facilitates the formation of multiple other malignancies (Di Cristofano et al., 2001; Kwabi-Addo et al., 2001; Trotman et al., 2006; Hollander et al., 2011). In addition, complete PTEN gene loss can trigger growth arrest via p53 induction (Chen et al., 2005; Kim et al., 2006). Hence, PTEN has emerged as a prototypic haploinsufficient tumor suppressor (Berger et al., 2011).
PTEN activity and localization has received intense scrutiny (Leslie and Foti, 2011; Song et al., 2012). A major emerging concept from these studies has been the role of nuclear-cytoplasmic compartmentalization of PTEN. This distribution has been found to balance PTEN stability and PI 3-K-antagonizing cytoplasmic function (Trotman et al., 2007; Wang et al., 2007) with potential novel nuclear functions (Song et al., 2012). The importance of PTEN regulation extends beyond the disease context; failure to import PTEN into the nuclei of ischemic neurons results in cell death and increased infarct size (Howitt et al., 2012). Similar observations in brain development and wound healing underscore the necessity of curbing PTEN function to ensure cell viability and survival in a variety of developmental and post-insult scenarios (Naguib and Trotman, 2013). The importance of PTEN localization in terms of its partitioning between the cell nucleus and cytoplasm has been well established.
To date, a major paradox in our understanding of PTEN localization and activity has remained unsolved: classic PTEN function is ascribed to the plasma membrane, where RTKs are activated. However, plasma membrane localization of PTEN is rarely detectable in mammalian cells and tissues. This observation would, indeed, suggest that only a minor fraction of PTEN is engaged in pathway suppression at any given time. To better understand how PTEN is controlled, we focused on examining its predominant cytoplasmic localization. Our data reveal that cytoplasmic PTEN resides on PI(3)P-positive endosomes, introducing a novel site for hydrolysis of its PI(3,4,5)P3 substrate.
Results
Cytoplasmic PTEN Is Arranged along Microtubules
We observed prominent cytoplasmic and nuclear localization of Pten using light microscopy in primary mouse embryonic fibroblasts (MEFs) and various tissue culture cell lines (Figure 1A; Figure S1A). Upon inspection, we noticed that cytoplasmic Pten was found in discrete punctae, suggesting organization into distinct entities. Specificity of staining was confirmed with PTEN-deficient cell lines, two independent antibodies, and three independent methods of fixation (Figures 1A and S1A). We compared the distribution of PTEN to that of fluorescent transferrin (a vesicle marker) and observed highly similar staining patterns in primary MEFs (Figure 1B). As microtubules (MTs) guide most internal membrane transport, we also stained MT filaments and found Pten signals generally on or near MTs in a pattern that was very similar to the co-staining of transferrin and MTs (Figure 1B).
Figure 1. PTEN Is Organized along MTs.
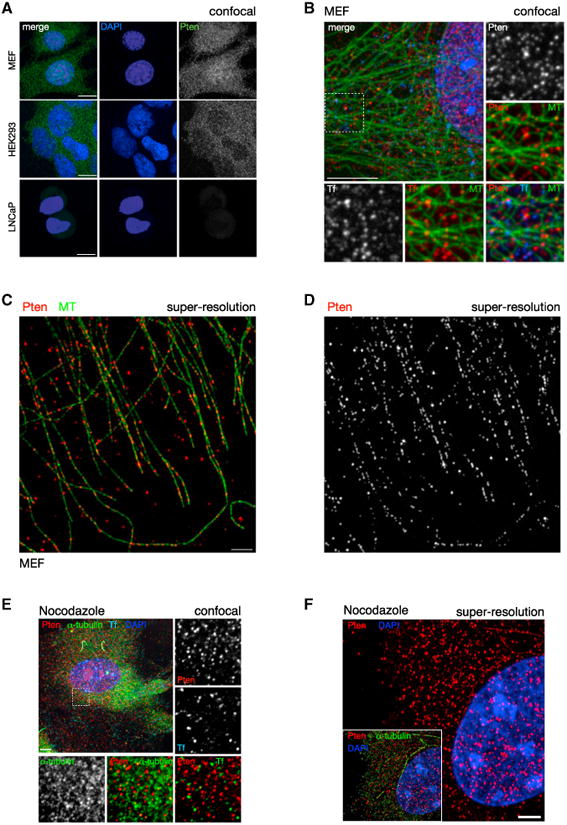
(A) Confocal immunofluorescence (IF) microscopy reveals that PTEN distribution is punctate in the cytoplasm. Punctate distribution is conserved in both primary WT MEFs (top panels) and HEK293 cell lines (middle panels). PTEN null cells (human prostate cancer-derived LNCaP) display minimal background staining, confirming antibody specificity (bottom panels). Images show total projections of all Z sections (extended focus). Scale bars, 10 μm.
(B) Cytoplasmic PTEN distribution is indistinguishable from that of labeled transferrin (Tf). Pten and Tf punctate stains show a similar close association with MTs. Images of WT MEFs, single section (Z = 1). Scale bar, 10 μm.
(C and D) Super-resolution light microscopy (OMX) reveals that PTEN is organized along MTs. Scale bar, 2 μm.
(E) Nocodazole treatment of WT MEFs abrogates MT assembly but does not dissolve the punctae of Pten or Transferrin, compared to alpha-tubulin. Z = 1. Scale bar, 10 μm.
(F) Nocodazole treatment abrogates the linear arrangement of PTEN along MTs. Z = 1. Scale bar, 2 μm.
See also Figure S1.
To better define and to quantify the relationship between Pten and MTs, we used super-resolution microscopy, which surpasses the optical diffraction limit of light microscopes (120 nm in XY planes and 300 nm in Z planes, compared to our confocal system: 214 nm in XY and 910 nm in Z). This approach revealed strong co-localization of cytoplasmic Pten and MTs (Figure 1C). Furthermore, the Pten signal pattern revealed that the majority of Pten is arranged along tracks that follow the local organization of the MT network (Figures 1D and S1B). Quantification of these data showed that 63% of the Pten punctae were directly on MTs and that 70% were within 0.25 μm of an MT, an association that was significantly different from the MT distance distribution of random locations inside the cell (p = 3.5E–36; Figure S1C) and comparable to the distance distribution between the endosomal protein Eea1 and MTs (Figure S1D). In contrast to endogenous PTEN, GFP-tagged exogenous PTEN was diffusely distributed in the cytoplasm of PTEN null cells (Figure S1E, top panels) and so did not recapitulate the endogenous PTEN pattern as visualized by GFP fluorescence. However, further investigation using antibody detection also revealed that exogenous EGFP-PTEN was distributed in a punctate pattern similar to that of endogenous PTEN but only in the lowest expressing cells (Figure S1E). These results showed that, when expressed at physiological levels, necessitating antibody-based detection for visualization, exogenous PTEN distribution is similar to that of endogenous PTEN. Identical results were obtained with anti-GFP or anti-PTEN antibodies or with otherwise tagged PTEN constructs (data not shown). In addition, we also developed a live-cell-compatible approach for imaging GFP-tagged PTEN (Figure S5). In wild-type (WT) MEFs, we observed that the pattern of endogenous Pten was not noticeably redistributed to the plasma membrane upon activation of PI 3-K pathway signaling by serum stimulation (Figure S1F). To summarize, we found that the majority of endogenous PTEN in the cytoplasm is organized along MTs.
To probe whether the MT association was direct or indirect via vesicles, we studied Pten after depolymerization of MTs. Using normal resolution confocal analysis, we found that nocodazole treatment efficiently depolymerized MTs, resulting in dispersed alpha-tubulin staining, but did not affect the size of Pten and Transferrin punctae (Figure 1E). Notably, nocodazole treatment completely abrogated any discernible arrangement of Pten along tracks, as revealed by super-resolution microscopy (Figure 1F). These data showed that, while the organization of Pten throughout the cytoplasm is dictated by MT filaments, the recruitment of Pten into punctae is not, which is consistent with MT binding via vesicles.
PTEN Directly Binds PI(3)P via Its C2 Domain
Many organelles and vesicles in eukaryotic cells are determined by their PI signature (Di Paolo and De Camilli, 2006; Liu and Bankaitis, 2010). To explore whether PTEN binds to a specific anchoring PIP that controls its cytoplasmic localization, we used protein-lipid overlay methodology, coupled with antibody detection (Dippold et al., 2009; Dowler et al., 2002; Yang et al., 2008). As shown in Figure 2A, we consistently detected direct binding of purified PTEN with PI mono-phosphates (PIPs). Notably, PTEN consistently bound with the highest affinity to PI(3)P (Figures 2A, S2A, and S2B), while weaker binding to PI(4)P and PI(5)P was intermittently detected. Recombinant PTEN purified from both bacterial and insect expression systems readily showed binding to PI(3)P, and binding specificity of this assay was confirmed using the known PI(3)P-binding PX domain of the p40 (PHOX) protein (Bravo et al., 2001) and the PH domain of PLCδ1, recognizing PI(4,5)P2 (Hodgkin et al., 2000) (Figure 2B).
Figure 2. PTEN Directly Binds to the Endosomal Signature Lipid PI(3)P.
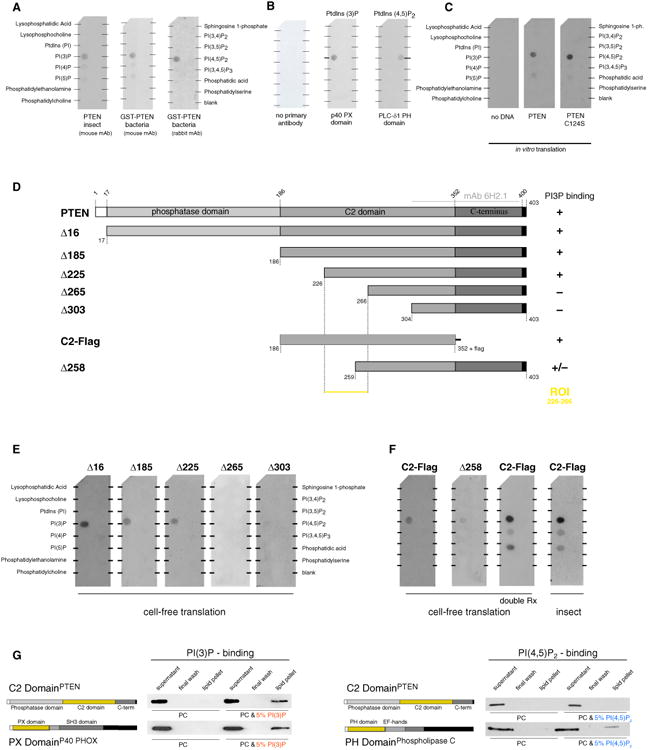
(A) Recombinant human PTEN protein directly binds PI(3)P as shown by incubation of GST-PTEN and untagged PTEN (generated using the indicated protein expression systems) with immobilized lipids. Note that binding to PI(4)P and PI(5)P is weak and variable. mAb, monoclonal antibody.
(B) Recombinant PX domain (from p40 PHOX) and PH domain (from phospholipase C-δ1) specifically recognize their respective lipid targets PI(3)P and PI(4,5)P2, while secondary antibody alone shows no staining. PtdIns, PI.
(C) Cell-free translation of PTEN using bacterial extract reproduces PI(3)P binding of WT PTEN (middle) and the C124S catalytically inactive mutant (right). ph, phosphate.
(D and E) Truncation maps (D) and overlay assays (E) performed with the indicated PTEN mutants identifies the ROI containing the minimal PI(3)P-binding element.
(F) The C2 domain of PTEN in isolation (with a C-terminal flag epitope for antibody detection) is necessary and sufficient for binding to PI(3)P. Doubling of reaction volumes for cell-free translation yields comparable results to reactions performed with insect-cell-purified C2 domain of PTEN.
(G) The C2 domain of PTEN binds synthetic liposomes containing 5% PI(3)P in a phosphatidylcholine (PC) membrane similar to the known PI(3)P-binding PX domain of the p40 PHOX protein. The C2 domain of PTEN does not interact with PI(4,5)P2 as shown using PC liposomes containing 5% PI(4,5)P2 and the known PI(4,5)P2-binding PH domain of phospholipase C as a positive control.
See also Figure S2.
Next, we proceeded to map the PI(3)P-binding domain on PTEN. First, we reproduced PI(3)P binding of PTEN using a cell-free bacterial-derived protein transcription/translation method (Figure 2C). Notably, this result confirmed that no post-translational modification of PTEN is needed for PI(3)P binding (Figure 2A, “GST-PTEN bacteria”). Furthermore, the catalytic-dead PTEN C124S mutant bound PI(3)P like WT PTEN. Note that the PTEN C124S mutant is not expected to bind the PI(3,4,5)P3 substrate (Flint et al., 1997) because of its wide catalytic pocket (Lee et al., 1999). Then, we tested whether PI(3)P binding could be mapped to a specific domain of PTEN. As shown using D16 in Figures 2D and 2E, deletion of the N-terminal 16 amino acids did not abolish PI(3)P binding, suggesting that this region—which has been implicated in binding to anionic lipids such as PI mono- and bisphosphates, as well as phosphatidyl serine (Campbell et al., 2003; McConnachie et al., 2003; Walker et al., 2004)—is not essential for PI(3)P interaction. Next, we saw that the PTEN catalytic domain was not needed for PI(3)P binding (Figures 2D and 2E, “Δ185”) and, therefore, constructed a series of truncations in the C2 domain (Δ225, Δ265, and Δ303), revealing a region of interest (ROI) between residues 226 and 266 of PTEN. We confirmed that the Flagepitope-tagged C2 domain on its own still bound PI(3)P (Figure 2F), as did the deletions of the COOH-terminal (C-terminal) tail and of the PDZ-binding site (data not shown). In contrast, PTEN Δ258 lacking most of the ROI showed only weak binding (Figure 2F, “Δ258”). Purification from insect cells confirmed that the C2 domain was sufficient for PI(3)P binding. Increased reaction material (in vitro) or higher PTEN purification yields (insect) also increased the relatively weak signal from PI(4)P and PI(5)P (Figure 2F; see “C2-Flag” double in vitro reaction volume and “insect”). Finally, we confirmed these results using a complementary, more physiological presentation of PIs on synthetic liposomes (Patki et al., 1998). As shown, the C2 domain of PTEN bound to PI(3)P-containing liposomes, similar to the PX domain of the p40 PHOX protein-positive control (Figure 2G, left panels). In contrast, the C2 domain did not bind to PI(4,5)P2, consistent with the lipid overlay results (Figure 2G, right panels). Taken together, our results demonstrated that the C2 domain of PTEN mediates binding to PI(3)P in vitro.
The discovery of PI lipid-binding domains and their development as cellular probes has greatly facilitated quantitative analysis of protein-lipid interactions in live and fixed cells (Lemmon, 2008; Schink et al., 2013). To assess the in vivo affinity of the PTEN C2 domain for PI(3)P, we used the prototypic FYVE domain as specific probe for the PI(3)P vesicle compartment (Gaullier et al., 1998). As shown in Figure 3A (left panels), fixed-cell imaging revealed extensive colocalization of the fluorescent-tagged C2PTEN domain fusion (CherryFP-C2PTEN, ChFP-C2PTEN) with the 23FYVE domain of HRS protein (GFP-2×FYVEHRS). Three-dimensional (3D) colocalization was confirmed by 3D reconstruction of these confocal images (Figure 3A, right panels). Note that fusion of the C2 domain with GFP produced identical results to tagging with ChFP (see Figure S2C; also, compare quantifications in Figures 3B and 3C with Figure 5D, right panels). Live imaging and tracking of ChFP-C2PTEN and GFP-2×FYVEHRS demonstrated extensive comigration of the two domains (Figure S3A; Movie S1 [note that color channel separation during motion bursts is due to sequential channel recording]). These results revealed a strong in vivo preference for PI(3)P binding of the PTEN C2 domain.
Figure 3. The C2 Domain of PTEN Is a Novel PI(3)P-Binding Domain.
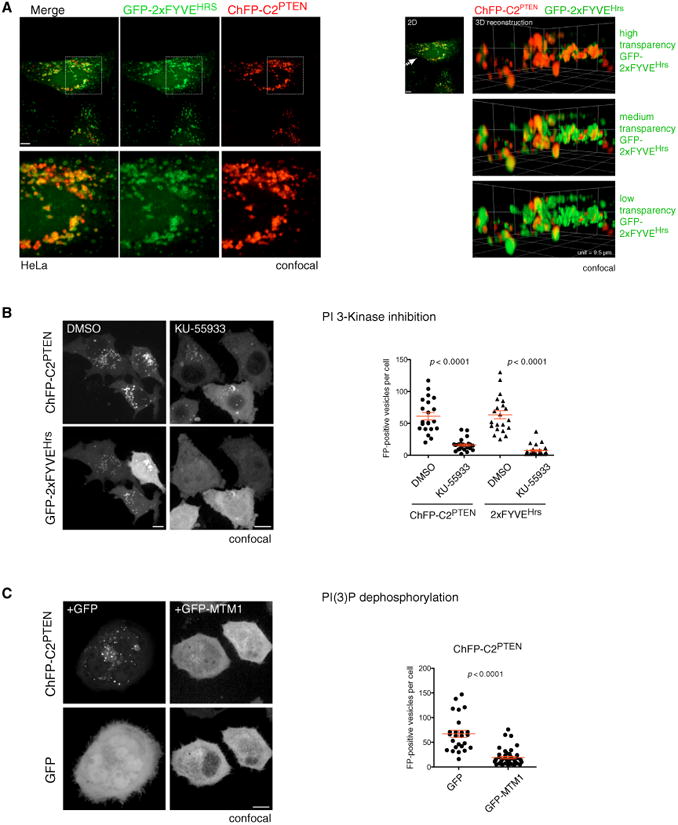
(A) Left panels: mCherry fluorescent protein (ChFP)-tagged PTEN C2 domain extensively co-localizes with the GFP-tagged FYVE domain of HRS protein, a prototypical PI(3)P probe in cells. Z = 1. Scale bar, 10 μm. Note that identical results are obtained with a GFP-tagged C2 domain (see Figures 5D and S2C). Right panels: 3D rendering of colocalization between PTEN C2 domain and 2×FYVEHRS. Arrow represents the angle of view. Three density settings are displayed for 2×FYVEHRS to allow complete visualization of the signal overlap with ChFP-C2PTEN domain. Unit length of the 3D floor is 9.5 μm.
(B) The number of PTEN C2-domain-positive and 2×FYVEHRS-domain-positive vesicles is diminished in HeLa cells subsequent to treatment with the PI 3-K inhibitor KU-55933. Scale bars, 10 μm, extended focus. Graph, red bars represent the mean value, and the whiskers represent SEM. p < 0.0001, two-tailed Student's t test.
(C) Overexpression of the PI(3)P phosphatase MTM1 (GFP-MTM1) suppresses C2PTEN-vesicle association. Scale bar, 10 μm, extended focus. Graph, red bars show mean with SEM, p < 0.0001, two-tailed Student's t test.
See also Figure S3.
Figure 5. PI3P-Dependent Localization and Activation Is Mediated by the CBR3 Loop of PTEN.
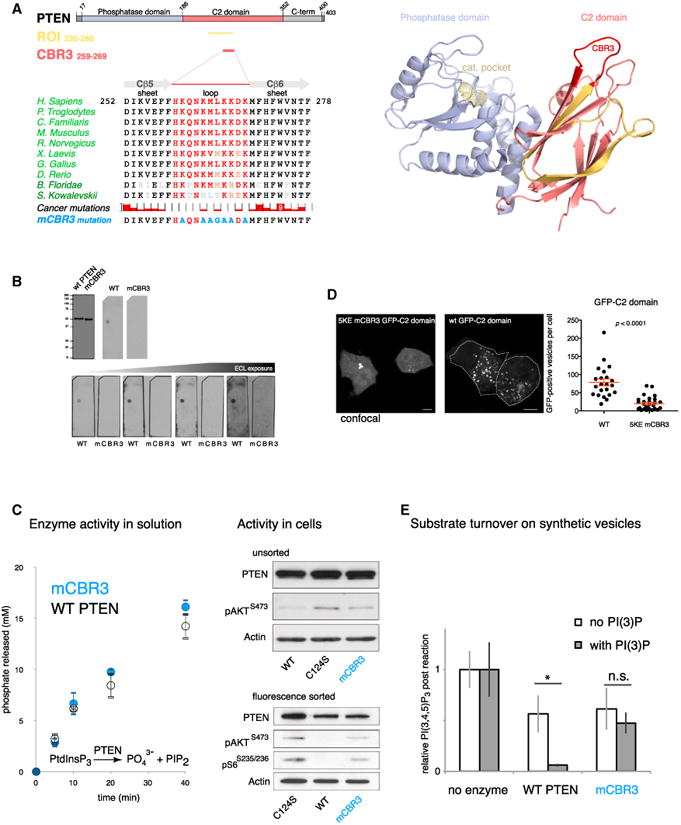
(A) Left: domain structure of PTEN with the identified ROI from Figure 2 (gold) and the CBR3 loop (red). Middle: conservation of the CBR3 loop (red) in the C2 domain among vertebrates and cancer-associated missense mutations. The top reported cancer mutation count (9) is found for the Trp-274 residue. Three cancer-associated missense mutations have been reported inside the CBR3 (Cerami et al., 2012). The last row shows mutations engineered to neutralize CBR3 loop function. Right: PTEN structure highlighting the CBR3 loop (red) in the ROI (gold). Note that the CBR3 is positioned to face the target membrane in concert with the catalytic pocket (Georgescu et al., 2000; Lee et al., 1999) (active-site cysteine is shown in yellow as ball-and-stick residue).
(B) Mutation of the CBR3 loop (mCBR3 PTEN), as indicated in (A), abolishes PTEN-PI(3)P binding.
(C) The mCBR3 PTEN mutant retains normal lipid phosphatase activity against soluble substrate (diC 8 PI(3,4,5)P3) (graph; error bars indicate SD, n = 2). Top: immunoblot analysis of overexpressed mCBR3 mutant GFP-PTEN reveals strongly impaired pathway antagonizing function compared to WT GFP-PTEN in PTEN-deficient PC3 cells. Bottom: immunoblot analysis revels that FACS-sorted mCBR3 mutant GFP-PTEN cells have higher AKT phosphorylation than cells transfected with WT GFP-PTEN.
(D) GFP-tagged 5KE mutant C2 domain of PTEN is less efficiently recruited to PI(3)P-positive membranes than the WT(wt) GFP-tagged C2PTEN domain. Student's t test, two tailed. Z = 1. Scale bar, 10 μm.
(E) Mass-spectrometry-based quantification of PI(3,4,5)P3 abundance shows that both WT and mCBR3 proteins are active on liposomes. More productive catalysis of WT enzyme is achieved by the addition of PI(3)P to liposome membranes, but, in contrast, no enhanced activity is observed with the mCBR3 enzyme. Error bars indicate SEM. *p < 0.05, Student's t test, two-tailed.
PI(3)P Controls Endogenous PTEN Localization
We then confirmed the specificity of this interaction. To this end, we first used pharmacological interference of the PI(3)P-producing Vps34 kinase. As shown and quantified in Figure 3B, the small molecule inhibitor KU-55933 (Hickson et al., 2004) strongly suppressed association of the C2PTEN domain with vesicles, similar to its effect on the 2×FYVEHRS probe as described elsewhere (Farkas et al., 2011) and recently reviewed (Schink et al., 2013). In corroboration, we found that overexpression of the PI(3)P-dephosphorylating MTM1 phosphatase (Taylor et al., 2000) strongly interfered with vesicle association of the C2PTEN domain (Figure 3C).
Using super-resolution microscopy (Figures 4A and 4B), we next confirmed that endogenous PTEN also co-localizes with the 2×FYVE domain into the PI(3)P vesicle compartment, similar to endogenous EEA1. These results suggested that the C2 domain can dictate steady-state localization of full-length endogenous PTEN. PI(3)P is mainly generated from PI through phosphorylation at the 3-position of inositol by the class III PI 3-K, VPS34 (Schu et al., 1993; Schink et al., 2013). Thus, we used Cre/loxP-mediated recombination to generate primary Vps34-knockout MEFs (Pik3c3Δ/Δ MEFs; Figure S3B). Pik3c3Δ/Δ MEFs showed more non-punctate Pten staining, while, in contrast, WT control MEFs displayed the typical punctate Pten distribution (Figure 4C). Quantification by profiling of normalized Pten fluorescence signal intensity along five cytoplasmic lines showed intense signal spikes that represent Pten punctae in WT cells (Figures 4D and S3B, graphs, green lines). In contrast, the same analysis revealed that intermediate signal intensity was more prominent in the Vps34-deficient MEFs, which stemmed from non-punctate signal between spikes along the lines (Figures 4D and S3B, graphs, red lines). Notably, the knockout MEFs presented previously described cytoplasmic vacuoles (Jaber et al., 2012; Johnson et al., 2006) (Figure 4C, arrowheads), functionally confirming loss of Vps34 activity. Ranking of the intensity distribution of Pten staining and plotting normalized intensity versus its normalized ranking confirmed the significant differences between Pten distribution in WT and knockout cells (Figure 4E). We used small interfering RNA (siRNA) to knock down Vps34 (Figure 4F). This knockdown approach and its quantification as discussed earlier (Figures S3C–S3E) also revealed a significant, non-punctate redistribution of endogenous PTEN throughout the cytoplasm, demarcating the vacuoles generated by loss of Vps34 function (Figure 4F, arrowheads).
Figure 4. PTEN Localization Is Dictated by PI(3)P.
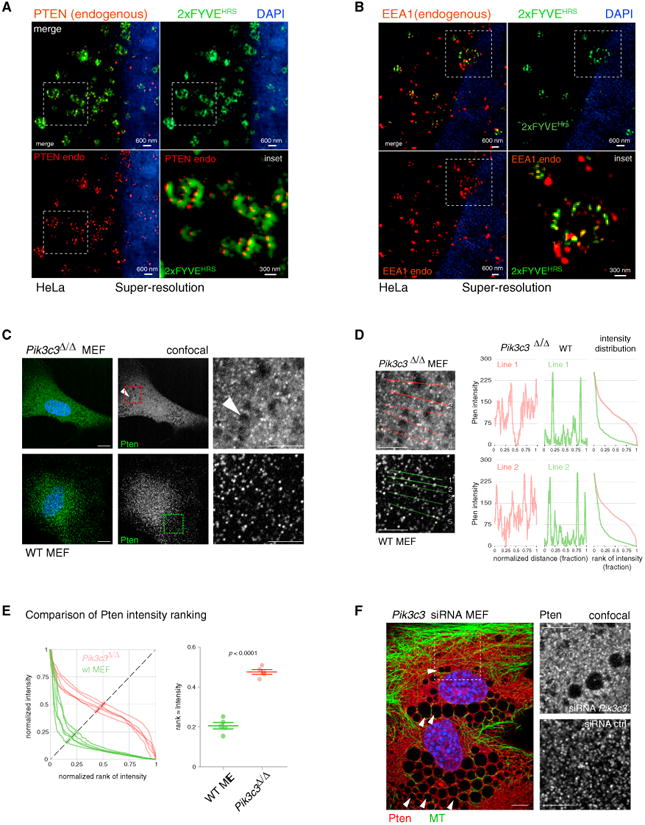
(A) Super-resolution microscopy demonstrates colocalization of endogenous (endo) PTEN with overexpressed GFP-2×FYVEHRS, Z = 1. 2D, two-dimensional.
(B) Similar results are obtained with endogenous EEA1 and overexpressed GFP-2×FYVEHRS, Z = 1.
(C) Pten shows non-punctate cytoplasmic staining in Vps34 (Pik3c3 gene) knockout MEFs (see Experimental Procedures). Arrowheads indicate large vacuole formation, which confirms loss of Vps34 function. Z = 1. Scale bar, 10 μm.
(D) Line intensity profiles of Pten in Pik3c3 null and WT MEFs from (C), Z =1. Scale bar, 10 μm. Right panels: absolute intensity is plotted versus normalized rank of absolute intensity.
(E) Quantification of Pten intensity plots. Normalized intensity is plotted versus normalized rank of intensity on five lines shown. Comparison of a measure for rank: intensity distribution (see Experimental Procedures) shows significant difference between WT and knockout cells. Bars represent the mean value, and the whiskers represent SEM. p < 0.0001, two-tailed Student's t test.
(F) siRNA against Vps34 (Pik3c3 gene) confirms loss of discrete Pten punctae in Vps34-knockdown MEFs. See Figures S3C and S3E for quantification. Arrowheads indicate vacuoles, confirming suppression of Vps34 function. Z = 1. Scale bars, 10 μm. Ctrl, control.
See also Figure S3.
Taken together, our results identified the C2 domain of PTEN as a novel targeting domain for PI(3)P-positive vesicles in cells. While we cannot exclude additional roles for other PIPs in regulating PTEN localization, our results suggest that PI(3)P is a major determinant in directing localization of endogenous PTEN in the cytoplasm.
PI(3)P Directs PTEN Function and Binding to Vesicles via the PTEN CBR3 Loop
The PI(3)P-binding ROI that we identified in the C2 domain (Figures 2D–2F) contains the membrane-interacting CBR3 loop of PTEN, as defined in the crystal structure (Georgescu et al., 2000; Lee et al., 1999) (Figure 5A). The CBR3 emanates from the C2 domain toward the membrane and lies on the same face as the catalytic pocket (Figure 5A, bottom). This feature is conserved among vertebrates (Figure 5A, sequence alignments) and represents a unique part of the ROI that does not contribute to PTEN structural integrity (Lee et al., 1999). The CBR3 loop has an expected +5 positive net charge from five lysine residues that flank two hydrophobic residues (Met264 and Leu265) at its tip— note that the His259-Asp268 pair is partially buried (Lee et al., 1999). Thus, we mutated the exposed features of the CBR3 loop as done previously (Lee et al., 1999) (Figure 5A, “mCBR3 mutation”) to produce PTEN mCBR3 protein from insect cells. As shown in Figure 5B, we could not detect PTEN mCBR3 binding to PI(3)P on lipid overlay strips, even at long exposures and in contrast to WT PTEN. Notably, the mutant protein retained full in vitro enzymatic activity, as shown by measurement of phosphate release using malachite green as a readout of soluble substrate turnover (Figure 5C, graph). These data demonstrated that the mCBR3 PTEN mutant was not structurally compromised as expected based on previous functional analysis (Lee et al., 1999). In cells, however, PTEN mCBR3 was significantly impaired in its ability to antagonize AKT signaling, consistent with previous reports (Georgescu et al., 2000; Orchiston et al., 2004) (Figure 5C, top right panels). This discrepancy was even more evident after fluorescence-activated cell sorting (FACS) and analysis of equal cell numbers transfected with GFP-tagged WT and mCBR3 mutant PTEN, confirming the low efficiency of the mCBR3 mutant PTEN in blocking AKT phosphorylation when present in cells (Figure 5C, bottom right). Next, we validated that CBR3 mutation affected PTEN localization. As shown in Figure 5D (right panels), the C2 domain with a charge converting the 5KE mutant CBR3 loop (see Experimental Procedures) showed significantly reduced vesicle association in cells.
Thus, collectively, our data demonstrated that the CBR3 loop controls PTEN in vivo function through binding to PI(3)P. The CBR3 loop is also mutated in cancer, albeit less than the immediately adjacent beta-strand structures, where point mutations are expected to interfere effectively with C2-domain folding (Lee et al., 1999) (see Figure 5A, “Cancer mutations”). Based on previous studies (Georgescu et al., 2000; Lee et al., 1999) and on our results, disruption of in vivo CBR3 function, indeed, requires multiple amino acid changes that translate to at least 8 nucleotide (nt) substitutions in the DNA sequence to be effective. This accumulation of events is extremely unlikely to occur spontaneously in tumors.
Next, we studied the CBR3-dependent PTEN function on synthetic liposomes. To this end, we incubated PTEN with (phosphatidylcholine) liposomes that contain the PTEN substrate PI(3,4,5)P3 alone or with 5% PI(3)P. To determine PTEN phosphatase activity, we directly measured PI(3,4,5)P3 levels by mass spectrometry (Figure 5E). This assay revealed a sharp increase in PI(3,4,5)P3 substrate turnover when the liposomes contained 5% PI(3)P in addition to substrate (see Figure 5E, “WT PTEN”), consistent with increased affinity and productive PTEN positioning on the vesicle surface, mediated by PI(3)P. The mCBR3 mutant of PTEN showed activity on substrate-only liposomes similar to that of WT PTEN (mCBR3-PTEN, no PI(3)P), fully consistent with its WT-like activity on soluble substrate (Figure 5E). In contrast to WT PTEN, however, the mCBR3 mutant showed no statistically significant increase in substrate turnover when PI(3)P was present on the substrate containing liposomes (mCBR3-PTEN, with PI(3)P). These data demonstrated that the WT CBR3 is essential for proper PTEN targeting and positioning (Georgescu et al., 2000) onto PI(3)P-containing vesicles and that this interaction greatly enhances PTEN function on its PI(3,4,5)P3 substrate. The results also suggested that the PI(3)P-CBR3 loop interaction is critical for PTEN activity in cells because it recruits and productively positions the enzyme onto PI(3)P-positive vesicles.
PTEN Acts as a Lipid Phosphatase on PI(3)P-Containing Membranes
We reasoned that, if mutation of the CBR3 loop of PTEN served to abolish its ability to bind PI(3)P and, thus, diminish its ability to antagonize PI3-K/AKT-pathway-mediated signaling, then restoration of the correct localization of PTEN to PI(3)P-containing endosomes would restore this function. Therefore, we fused the PI(3)P-targeting FYVE domain to inactive, mCBR3 PTEN and tested whether this would rescue PTEN function. As shown in Figure 6A, addition of the FYVE domain localized a significant proportion of GFP-PTEN to endosomal structures in a FYVE-copy-dependent manner, as expected for this canonical PI(3)P-binding domain (Gaullier et al., 1998; Schink et al., 2013) (compare GFP-PTEN, -PTEN-mCBR3-2×FYVEHRS, and -PTEN-mCBR3-3×FYVEHRS). Immunoblot analysis (Figure 6B) revealed that expression of WT PTEN diminished phospho-AKT levels, while similar levels of either catalytically inactive PTEN-C124S or PTEN-mCBR3 could not inhibit AKT activation, confirming our data shown in Figure 5C. Notably, the endosomal targeting observed with the FYVE-fusion constructs was able to completely restore the activity of the PTEN-mCBR3 mutant as seen by the ability of PTEN-mCBR3-2×FYVEHRS and -3×FYVEHRS to efficiently suppress Akt activation. The rescue constructs also suppressed cell proliferation by 70%, as quantified by measurement of proliferating cell nuclear antigen (PCNA) abundance (data not shown). In addition, we noted that these constructs suppressed AKT activation, even though they were present at far lower levels than those of WT or mCBR3 mutant PTEN.
Figure 6. Targeting of PTEN to PI(3)P Is Necessary for Function in Cells.
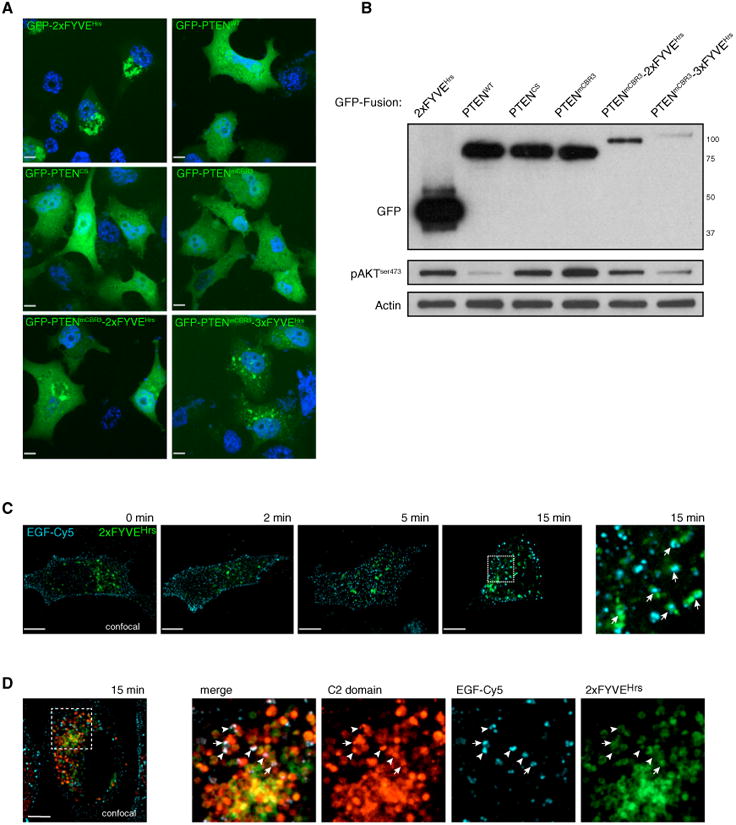
(A) Overexpression of multiple GFP-PTEN fusion proteins and their sub-cellular localization. Z = 1. Scale bar, 10 μm.
(B) Overexpression of PTEN constructs followed by FACS and interrogation of AKT phosphorylation. Fusion of mCBR3 PTEN to FYVE domains restores the ability of PTEN to efficiently antagonize PI3-K/AKT-mediated signaling.
(C) EGF-Cy5 internalization leads to colocalization with GFP-2×FYVEHRS after 15 min (arrows). Z = 1. Scale bar, 10 μm.
(D) Colocalization of ChFP-C2PTEN domain with EGF-Cy5 (arrowheads) and of ChFP-C2PTEN domain, EGF-Cy5, and GFP-2×FYVEHRS (arrows) is seen at 15 min post-EGF-Cy5 internalization. Z = 1. Scale bar, 10 μm.
See also Figure S4.
Thus, we concluded (1) that it is the degree of PTEN localization to endosomes that dictates its efficiency rather than overall cytoplasmic PTEN expression levels and (2) that PI(3)P-marked intracellular vesicles display a significant pool of PI(3,4,5)P3 at steady state, which the FYVE-fused or endogenous-vesicle-associated PTEN can inactivate to prevent AKT activation.
To further explore the notion that a major site of PTEN activity is on endocytic vesicles, we validated the intersection of the PI(3)P-positive vesicle compartment with PI(3,4,5)P3-containing membranes in cells. To this end, we first determined the time point for the intersection of incoming epidermal growth factor (EGF) receptor signals with the 2×FYVEHRS probe for PI(3)P. Using cold-synchronized pulse-chase experiments for EGF internalization, we found strong colocalization of Cy5-labeled EGF ligand with GFP-2×FYVEHRS at 15 min into the chase (Figure 6C), consistent with previous reports (Petiot et al., 2003). As shown in Figure 6D, this time point also revealed strong colocalization between the PTEN C2 domain, Cy5-labeled EGF ligand, and the 2×FYVEHRS probe for PI(3)P, consistent with the notion that activated, plasma-membrane-generated signal is internalized and that PI(3,4,5)P3-containing membranes intersect with the endosomal compartment.
To confirm that, once internalized, membranes harboring activated receptors not only intersected with the PI(3)P-rich, endosomal compartment but also possessed high PI(3,4,5)P3 content, we performed live imaging with the PH domain of AKT fused to EGFP as a probe for PTEN substrate. In PTEN null cells, coexpression of this probe with WT PTEN prevented enrichment of PHAKT in any cellular compartment (Figure S4A). In the absence of PTEN, a fraction of GFP-PHAKT localizes to structures in the cell periphery (Figure S4B, “0 min”). Subsequent to serum stimulation of cells, the plasma-membrane (ruffle)-associated PHAKT fraction is increased, and this is accompanied by significant contraction of the cell (Figure S4B, “contraction”; see also Movie S2), consistent with membrane uptake. As shown in Figure S4C, the time points of contraction coincide with redistribution of AKTPH domains that are found colocalizing (Figure S4C) and co-migrating (Figure S4D; Movie S2) with the C2 domain of PTEN. These data are in agreement with the notion that the C2 domain of PTEN is present on vesicles that are double positive for both the PI(3)P-targeting lipid and the PI(3,4,5)P3 substrate lipid of PTEN.
Collectively, our experiments revealed that the CBR3 loop in the PTEN C2 domain is both necessary and sufficient for the productive targeting of PTEN to PI(3P)-positive vesicle membranes that also contain the PTEN lipid substrate, PI(3,4,5)P3.
Exogenous PTEN Localization
To explore whether vesicle association is also found upon PTEN overexpresion, we tested the existence of different pools of full-length fluorescent-tagged PTEN, using the fluorescence loss in photobleaching (FLIP) technique. By bleaching the mobile, freely diffusing pool of overexpressed fluorescent-tagged PTEN, the vesicle-associated, slower moving fraction was made visible (Figures S5A and S5B). Post-bleach live imaging of catalytic dead ChFP-PTEN (full length) with the GFP-2×FYVEHRS probe revealed their extensive colocalization and co-migration with motion bursts (Figure S5A, “postbleach” live imaging; Movie S3 [note that color channel separation during motion bursts is due to sequential channel recording]). Using a PI(4,5)P2-binding probe as a marker for the plasma membrane (the PH domain of PLCδ1 fused to mCherry), we also confirmed that, after bleaching, PTEN enrichment is not overt at the plasma membrane or other sites to which the PLCδ1PH domain is targeted (Figure S5B). Thus, our results revealed that, when overexpressed, a fraction of exogenous PTEN stably interacts with an endosomal, PI(3)P-marked location, at which PI(3,4,5)P3 dephosphorylation can occur.
Discussion
Our results suggest an unexpected link between PTEN function, its localization, and the endocytic compartment. Intriguingly, this association was first hinted at 17 years ago, when the PTEN/MMAC1 gene was originally identified and two research teams independently noted that PTEN has significant sequence similarity to auxilin (Li et al., 1997; Steck et al., 1997). Auxilin is essential for endocytosis: it is recruited to clathrin-coated vesicles precisely after they pinch off from the plasma membrane to mediate the disassembly of the clathrin coat (Fotin et al., 2004). This is essential for the incoming vesicle membranes to be recognized and sorted. The event is tightly timed to ensure that clathrin lattice uncoating only occurs after vesicles have separated from the plasma membrane.
There are two remarkable consistencies between auxilin's structure and function and our findings on PTEN-vesicle interaction. First, it has been shown that the PTEN-like domain of auxilin is essential for recruitment of the protein to the vesicle membrane (Massol et al., 2006). The crystal structure of this domain of auxilin has been determined (Guan et al., 2010). As shown in Figure 7A, it is nearly identical to the structure of PTEN itself. This finding is consistent with a conserved vesicle-binding function between the two homologous domains. Second, several analyses demonstrated that the C2 domain in auxilin mediates the essential vesicle interactions, while the catalytic domain is dispensable based on mutation analysis (Guan et al., 2010; Lee et al., 2006; Massol et al., 2006). Most notably, it was found that auxilin's equivalent of the PTEN CBR3 loop (termed “loop 3” in auxilin) is critical for binding to PI(4)P (and the other monophosphorylated PIPs: PI(3)P and PI(5)P) (Guan et al., 2010; Lee et al., 2006; Massol et al., 2006).
Figure 7. PTEN Vesicle Binding May Be an Evolutionarily Conserved Function in Endocytosis.
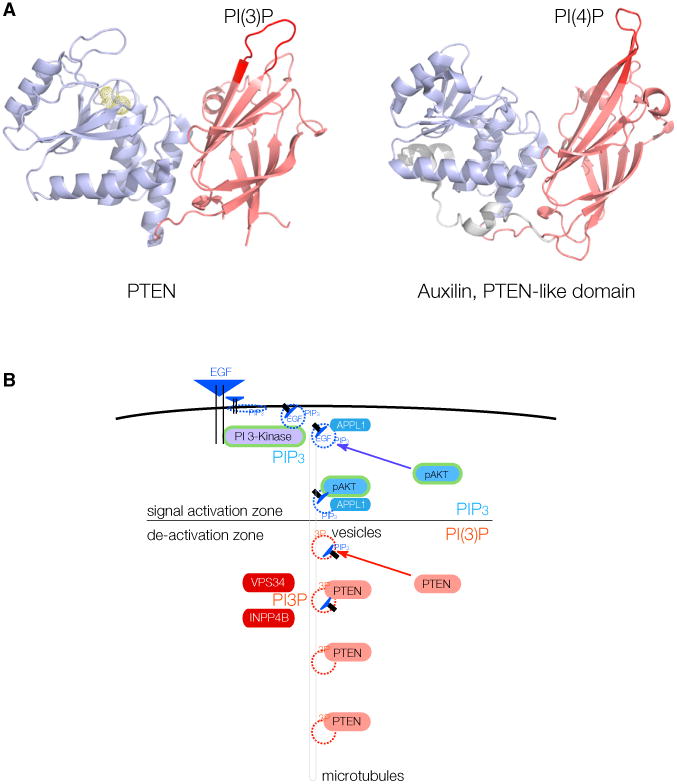
(A) Left: phosphatase (blue) and C2 domain (red) of PTEN, with the essential CBR3 PI(3)P-binding loop highlighted (left, crimson). Right: structure of the PTEN-like region of Auxilin, with the essential loop for PI(4)P binding highlighted (right, crimson).
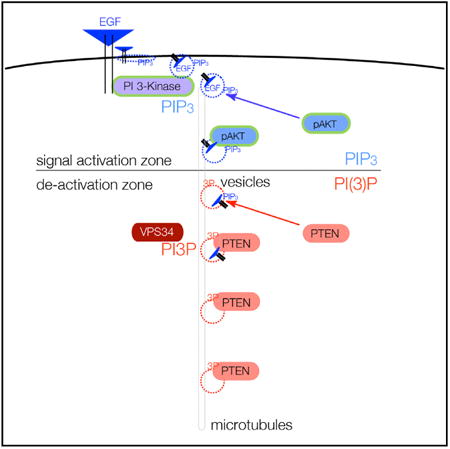
(B) Model for spatio-temporal maturation of vesicle-associated PIP3-lipid signaling through APPL1-positive vesicles (signal activation zone)and subsequent PIP3 signal termination by PTEN after generation of PI(3)P (also labeled 3P) onto vesicle membranes by, e.g., VPS34 or INPP4B (de-activation zone).
Collectively, these data are consistent with the intriguing possibility that the PTEN C2 domain may have evolved to recognize intracellular vesicle membranes based on their mono-phosphorylated PIP concentration. This concentration can be efficiently enriched and presented on the limited surfaces of vesicles after internalization from the cell periphery. This process can prevent massive dilution of the signaling lipid over the plasma membrane to facilitate protein recruitment. It will be interesting to see how this vesicle targeting of PTEN is affected by the many post-translational modifications of the extreme PTEN N-terminal 15 tail residues (reviewed by Leslie and Foti [2011]). Of note, this N-terminal tail is not conserved between PTEN and auxilin.
Our findings also suggest that enzyme-regulated control of PI(3)P may play a role in PTEN activation. While class I PI 3-Ks generate PI(3,4,5)P3, the class II (PIK3C2 α, β, γ) and class III (VPS34) enzymes generate PI(3)P (Vanhaesebroeck et al., 2012). We demonstrate that VPS34, in addition to its functions in endocytosis and autophagy (Backer, 2008; Funderburk et al., 2010), is needed for correct PTEN localization. Intriguingly, in spite of its pleiotropic roles, VPS34 is found deleted in up to 43% of human metastatic prostate cancers (Chen et al., 2011; Taylor et al., 2010). Downstream of these kinases, lipid phosphatases are critical to PI(3)P homeostasis. The INPP4b phosphatase, which produces PI(3)P from PI(3,4)P2, has been identified as a tumor suppressor (Chen et al., 2014; Fedele et al., 2010; Gewinner et al., 2009), consistent with a role in supporting PTEN via PI(3)P production. Finally, the Myotubularin phosphatase, which dephosphorylates PI(3)P (Taylor et al., 2000), is essential for cell survival through AKT signaling (Razidlo et al., 2011), consistent with suppression of the PI(3)P/PTEN axis. Intriguingly, cell death after MTM1 knockdown (high PI(3)P) can be specifically rescued by co-suppression of the class II PI 3-K PIK3C2B (Razidlo et al., 2011), which may point to a special role of PI 3-K C2-beta upstream of PTEN.
Conceptually, there are several interesting aspects to an alternative of PI 3-K signal termination on vesicle membranes, as opposed to stochastic termination on the plasma membrane (Figure 7B). First, a vesicle with a 100-nm diameter has a surface area that is five orders of magnitude smaller than the surface of a typical plasma membrane. It can be envisioned that this smaller surface and constitutive PTEN-vesicle binding makes PTEN-substrate interaction far more likely, processive, and controllable. Second, our findings may introduce a major missing link in the spatio-temporal control of PI 3-K signal termination (Platta and Stenmark, 2011). APPL1-positive endosomes have been shown to serve as a platform for RTK-mediated MAPK/AKT signaling (Miaczynska et al., 2004), and a recent study identified PI(3)P deposition as an essential switch for removal of APPL1 from vesicles and termination of vesicle signaling (Zoncu et al., 2009). Our findings could, therefore, link the termination of AKT signaling on APPL1-positive endosomes to recruitment of PTEN by PI(3)P deposition on endosomes. Beyond PI(3)P, it is highly likely that yet-unidentified proteins could control the endosomal sub-compartment specificity of the PTEN-vesicle interaction. It will, therefore, be interesting to see how PTEN function is impacted by perturbation of early steps of endocytosis and MT-mediated transport.
Experimental Procedures
Immunofluorescence Microscopy
Cells were fixed and permeabilized by one of three methods: methanol fixation, paraformaldehyde (PFA) fixation, or PHEMO fixation. Cells on coverslips were incubated at 4°C for 60 min in primary antibody diluted in 10% goat serum.
Structured Illumination for Super-Resolution Imaging
High-resolution images were acquired using an OMX 3D Structured Illumination microscope (Applied Precision). The z stacks were saved and processed using SoftWoRx 4.5.0 (Applied Precision).
Recombinant PTEN Protein Production
Untagged PTEN protein was expressed in insect cells using baculovirus and purified from Hi5 cell lysate by ion exchange chromatography on SP sepharose. For screening of PTEN mutants, PURExpress In Vitro Protein Synthesis kits (New England Biolabs) were used according to the manufacturer's instructions.
Pik3c3 Gene Knockdown and Knockout
Four independent siRNAs targeting Vps34 were pooled for use and compared to a scramble control (all siRNAs were from QIAGEN) and transfected into NIH 3T3 cells using Dharmafect1 reagent (Thermo Scientific) according to the manufacturer's instructions. Vps34fl/fl MEFs were treated with adenovirus-Cre-GFP or adenovirus-GFP control. Cell lysates were collected at the indicated times for immunoblot analysis with concurrent fixation of cells for immunofluorescence microscopy.
Confocal Microscopy and Image Analysis
All confocal microscopy was analyzed using Volocity software (v.6.3).
Extensive descriptions of methodology are included in the Supplemental Experimental Procedures.
Supplementary Material
In Brief.
PI 3-kinases can trigger cancerous growth of cells, which is efficiently curbed by the PTEN tumor suppressor. Here, Naguib et al. show that PTEN action is controlled by the cell's endocytosis machinery, which recruits PTEN to work on incoming signals that are presented as discretely parceled information on the surface of vesicles.
Highlights.
PTEN works efficiently on endocytic vesicles to inactivate PI 3-kinase/AKT signals
PI(3)P controls PTEN localization and function via the PTEN-C2 domain
PTEN-vesicle-binding faculty is used in essential endocytosis gene paralogs of PTEN
Acknowledgments
L.C.T. is a Research Scholar of the American Cancer Society and is supported by the Pershing Square Sohn Foundation and the U.S. Department of Defense (W81XWH-13-PCRP-IDA). We are grateful to have received generous funding of this work through grants to L.C.T. from the American Cancer Society (RSG-14-069-01-TBE); the NIH (CA137050); the Robertson Research Fund of Cold Spring Harbor Laboratory (CSHL); the NIH to the CSHL Cancer Center through Support Grant 5P30CA045508 for funding of microscopy, mass spectrometry, flow cytometry, and gene sequencing; and the generous donations from the Long Island Cruizin' For a Cure and the Glen Cove CARES Foundations. L.J.-T. is an investigator of the Howard Hughes Medical Institute.
We thank D. Spector, U. Greber, G.S. Taylor, Justin B. Kinney, and members of the L.C.T. and L.J.-T. laboratories for valuable discussion and help with analyses; Z. Lazar, S. Hearn, and D. Dai for help with confocal and super-resolution microscopy; P. Moody for help with FACS analysis; and S. Teplin for DNA sequencing.
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, five figures, one table, and three movies and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2015.03.011.
References
- Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476:163–169. doi: 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, Butler PJ, Lavenir I, Perisic O, Hawkins PT, et al. The crystal structure of the PX domain from p40(phox) bound to phosphatidylinositol 3-phosphate. Mol Cell. 2001;8:829–839. doi: 10.1016/s1097-2765(01)00372-0. [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RB, Liu F, Ross AH. Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2003;278:33617–33620. doi: 10.1074/jbc.C300296200. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O'Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell. 2011;20:173–186. doi: 10.1016/j.ccr.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Nowak DG, Trotman LC. Molecular pathways: PI3K pathway phosphatases as biomarkers for cancer prognosis and therapy. Clin Cancer Res. 2014;20:3057–3063. doi: 10.1158/1078-0432.CCR-12-3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–224. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber-Katz SE, Lee SK, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, et al. GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell. 2009;139:337–351. doi: 10.1016/j.cell.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowler S, Kular G, Alessi DR. Protein lipid overlay assay. Sci STKE. 2002;2002:l6. doi: 10.1126/stke.2002.129.pl6. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Farkas T, Daugaard M, Jäättelä M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem. 2011;286:38904–38912. doi: 10.1074/jbc.M111.269134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- Fedele CG, Ooms LM, Ho M, Vieusseux J, O'Toole SA, Millar EK, Lopez-Knowles E, Sriratana A, Gurung R, Baglietto L, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci USA. 2010;107:22231–22236. doi: 10.1073/pnas.1015245107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Proc Natl Acad Sci USA. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Grigorieff N, Walz T, Harrison SC, Kirchhausen T. Structure of an auxilin-bound clathrin coat and its implications for the mechanism of uncoating. Nature. 2004;432:649–653. doi: 10.1038/nature03078. [DOI] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaullier JM, Simonsen A, D'Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Kirsch KH, Kaloudis P, Yang H, Pavletich NP, Hanafusa H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000;60:7033–7038. [PubMed] [Google Scholar]
- Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Dai H, Harrison SC, Kirchhausen T. Structure of the PTEN-like region of auxilin, a detector of clathrin-coated vesicle budding. Structure. 2010;18:1191–1198. doi: 10.1016/j.str.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Hodgkin MN, Masson MR, Powner D, Saqib KM, Ponting CP, Wakelam MJ. Phospholipase D regulation and localisation is dependent upon a phosphatidylinositol 4,5-biphosphate-specific PH domain. Curr Biol. 2000;10:43–46. doi: 10.1016/s0960-9822(99)00264-x. [DOI] [PubMed] [Google Scholar]
- Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howitt J, Lackovic J, Low LH, Naguib A, Macintyre A, Goh CP, Callaway JK, Hammond V, Thomas T, Dixon M, et al. Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J Cell Biol. 2012;196:29–36. doi: 10.1083/jcb.201105009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, Zong WX. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci USA. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci. 2006;119:1219–1232. doi: 10.1242/jcs.02833. [DOI] [PubMed] [Google Scholar]
- Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2006;7:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwabi-Addo B, Giri D, Schmidt K, Podsypanina K, Parsons R, Greenberg N, Ittmann M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc Natl Acad Sci USA. 2001;98:11563–11568. doi: 10.1073/pnas.201167798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Lee DW, Wu X, Eisenberg E, Greene LE. Recruitment dynamics of GAK and auxilin to clathrin-coated pits during endocytosis. J Cell Sci. 2006;119:3502–3512. doi: 10.1242/jcs.03092. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Foti M. Non-genomic loss of PTEN function in cancer not in my genes. Trends Pharmacol Sci. 2011;32:131–140. doi: 10.1016/j.tips.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bankaitis VA. Phosphoinositide phosphatases in cell biology and disease. Prog Lipid Res. 2010;49:201–217. doi: 10.1016/j.plipres.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Massol RH, Boll W, Griffin AM, Kirchhausen T. A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc Natl Acad Sci USA. 2006;103:10265–10270. doi: 10.1073/pnas.0603369103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnachie G, Pass I, Walker SM, Downes CP. Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN evidence for activation by anionic phospholipids. Biochem J. 2003;371:947–955. doi: 10.1042/BJ20021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester J, Eng C. When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet. 2013;163C:114–121. doi: 10.1002/ajmg.c.31364. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Naguib A, Trotman LC. PTEN plasticity how the taming of a lethal gene can go too far. Trends Cell Biol. 2013;23:374–379. doi: 10.1016/j.tcb.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchiston EA, Bennett D, Leslie NR, Clarke RG, Winward L, Downes CP, Safrany ST. PTEN M-CBR3, a versatile and selective regulator of inositol 1,3,4,5,6-pentakisphosphate (Ins(1,3,4,5,6)P5). Evidence for Ins(1,3,4,5,6)P5 as a proliferative signal. J Biol Chem. 2004;279:1116–1122. doi: 10.1074/jbc.M310933200. [DOI] [PubMed] [Google Scholar]
- Patki V, Lawe DC, Corvera S, Virbasius JV, Chawla A. A functional PtdIns(3)P-binding motif. Nature. 1998;394:433–434. doi: 10.1038/28771. [DOI] [PubMed] [Google Scholar]
- Petiot A, Faure J, Stenmark H, Gruenberg J. PI3P signaling regulates receptor sorting but not transport in the endosomal pathway. J Cell Biol. 2003;162:971–979. doi: 10.1083/jcb.200303018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platta HW, Stenmark H. Endocytosis and signaling. Curr Opin Cell Biol. 2011;23:393–403. doi: 10.1016/j.ceb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Razidlo GL, Katafiasz D, Taylor GS. Myotubularin regulates Akt-dependent survival signaling via phosphatidylinositol 3-phosphate. J Biol Chem. 2011;286:20005–20019. doi: 10.1074/jbc.M110.197749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink KO, Raiborg C, Stenmark H. Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. Bioessays. 2013;35:900–912. doi: 10.1002/bies.201300064. [DOI] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Taylor GS, Maehama T, Dixon JE. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci USA. 2000;97:8910–8915. doi: 10.1073/pnas.160255697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay and macrocephaly. Genet Med. 2009;11:111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP. The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J. 2004;379:301–307. doi: 10.1042/BJ20031839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, Wang J, Erdjument-Bromage H, Tempst P, Cordon-Cardo C, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128:129–139. doi: 10.1016/j.cell.2006.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q, Oviedo HV, Trotman LC, Zador AM. PTEN regulation of local and long-range connections in mouse auditory cortex. J Neurosci. 2012;32:1643–1652. doi: 10.1523/JNEUROSCI.4480-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Kudlow JE, Michell RH, Olefsky JM, Field SJ, Evans RM. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451:964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P. A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell. 2009;136:1110–1121. doi: 10.1016/j.cell.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.