

Abstract
Objective:
To identify the genetic cause of pontocerebellar hypoplasia type III (PCH3).
Methods:
We studied the original reported pedigree of PCH3 and performed genetic analysis including genome-wide single nucleotide polymorphism genotyping, linkage analysis, whole-exome sequencing, and Sanger sequencing. Human fetal brain RNA sequencing data were then analyzed for the identified candidate gene.
Results:
The affected individuals presented with severe global developmental delay and seizures starting in the first year of life. Brain MRI of an affected individual showed diffuse atrophy of the cerebrum, cerebellum, and brainstem. Genome-wide single nucleotide polymorphism analysis confirmed the linkage to chromosome 7q we previously reported, and showed no other genomic areas of linkage. Whole-exome sequencing of 2 affected individuals identified a shared homozygous, nonsense variant in the PCLO (piccolo) gene. This variant segregated with the disease phenotype in the pedigree was rare in the population and was predicted to eliminate the PDZ and C2 domains in the C-terminus of the protein. RNA sequencing data of human fetal brain showed that PCLO was moderately expressed in the developing cerebral cortex.
Conclusions:
Here, we show that a homozygous, nonsense PCLO mutation underlies the autosomal recessive neurodegenerative disorder, PCH3. PCLO is a component of the presynaptic cytoskeletal matrix, and is thought to be involved in regulation of presynaptic proteins and synaptic vesicles. Our findings suggest that PCLO is crucial for the development and survival of a wide range of neuronal types in the human brain.
Pontocerebellar hypoplasia (PCH) is a group of disorders that share a common feature of an abnormally small cerebellum and pons. Involvement of the cerebrum, such as progressive microcephaly, is also common. It is highly heterogeneous, and 8 subtypes defined on a clinical and genetic basis are known.1,2 At least some subtypes are thought to be degenerative in nature and might not be true hypoplasia.1 PCH type III (PCH3) is one of the subtypes we previously reported in an Omani pedigree in 2003, and mapped to chromosome 7q11-21.3 In addition to a small cerebellar vermis, cerebellar hemispheres, and pons, imaging and clinical features of the affected children included reduced cerebral white matter volume, hypotonia, hyperreflexia, and seizures with onset in the first year of life.
The main objective of this study was to determine the genetic basis of PCH3. We extended the study of the previously reported pedigree, and identified a homozygous nonsense mutation in the PCLO gene to be the cause of PCH3.
METHODS
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained from participating individuals or their legal guardians, and research was performed according to institutional, national, and international human subject research guidelines.
Study subjects.
The clinical description and linkage analysis of part of the family was previously published.3
Genetic studies.
Genomic DNA samples were extracted from peripheral blood according to standard protocols. DNA samples were genotyped using Illumina HumanHap550-Duo BeadChip microarrays (Illumina, Inc., San Diego, CA). The data were visualized using the dChip software package.4,5 Multipoint logarithm of odds (LOD) scores were calculated using MERLIN,6 assuming a recessive mode of disease inheritance, full penetrance, and a disease allele frequency of 0.0001. Whole-exome sequencing in 2 affected individuals was performed using 2 different methods as follows:
Individual II:2: Coding regions were captured using Illumina TruSeq enrichment (62 Mb) with sequencing on a HiSeq (Illumina) with 101–base pair paired end reads. We obtained 9.1 G bases, of which 89.4% have a quality ≥Q30. Approximately 22.8% of the reads were marked as duplicates by Picard (version 1.101) and were excluded before mapping to the human genome reference sequence (GRCh37). The sequences were aligned to the reference sequence using BWA (Burrows-Wheeler Aligner) and variants called with mpileup (SAMtools). All variants were annotated using the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (September 2013), Complete Genomics 69 Genomes Data (February 2012), Single Nucleotide Polymorphism Database (dbSNP) (134–137), and the 1000 Genomes Project (May 2012) for minor allele frequency, using the Variant Explorer pipeline developed by the authors (K.S.-A. and K.M.).
Individual II:8: Coding regions were captured using the SureSelect Target Enrichment System (50 Mb; Agilent Technologies) with sequencing on a HiSeq (Illumina) with 76–base pair paired end reads. We obtained 9.2 G bases, of which 88% have a quality ≥Q30. Approximately 12.6% of the reads were marked as duplicates by Picard (version 1.46) and were excluded before mapping to the human genome reference sequence (GRCh37). Genome Analysis Toolkit (GATK; version 1.0.5777) was used to realign reads near potential insertion-deletion (indel) sites and to recalibrate base qualities, and single nucleotide variants were called using GATK and SAMtools (version 0.1.16) while indels were called using GATK and Dindel (version 1.01). All variants were annotated using dbSNP (134) and the 1000 Genomes Project pilot study (May 2011) for minor allele frequency. The variant consequences on protein structure were predicted by SNP Effect Predictor (VEP version 2.1) based on Ensembl (version 63). Variants were filtered out if the read depth was <4× or >1,200×, if the consensus quality was <20, or if the SNP quality was <25.
RNA sequencing.
Messenger RNA sequencing data from human fetal (9 weeks postconception) cerebral cortex was obtained as previously described.7
RESULTS
We studied a consanguineous pedigree with a total of 4 affected children from the Sultanate of Oman (figure 1A). I:1 and I:2 are first cousins once removed. I:3 is related to I:1 and I:2, although it is not known exactly how. Part of the pedigree (I:1, I:2, and their children) and detailed clinical information of 3 of the affected children (II:2, II:6, and II:8) were published previously.3 Briefly, the affected individuals presented with severe global developmental delay and seizures starting in the first year of life. Neurologic examination was remarkable for truncal hypotonia with increased deep tendon reflexes. II:2 had pale optic discs and II:6 had optic atrophy. Brain MRI of one of the affected individuals (II:2) showed diffuse atrophy of the cerebrum, cerebellum, and brainstem (figure 1B). The white matter volume was diminished, and the corpus callosum was thin. However, the state of myelination was age-appropriate. Subsequently, the father (I:2) remarried to I:3, and had a girl (II:9) who had the same neurologic presentation as her affected half-siblings. She is currently 11 years of age and started having generalized seizures starting at 9 months.
Figure 1. The pedigree studied and identification of a homozygous PCLO mutation.

(A) The pedigree studied with genotype of the identified PCLO mutation in each individual. Only the affected individuals show the homozygous C to T change. (B) Brain MRIs of individual II:2 at age 11 years, 10 months. Sagittal T1-weighted image shows a thin pons and small cerebellar vermis. The corpus callosum is thin. Coronal T2-weighted image shows generalized reduction of the cerebral white matter and atrophic cerebellar hemispheres. Scale bars = 2 cm. (C) Genome-wide linkage analysis. A region on chromosome 7 shows a maximum multipoint LOD score of 2.958. (D) Schematic representation of the PCLO protein. X = the location of the identified mutation. C2 = C2 domain; LOD = logarithm of odds; PDZ = PDZ domain; Zn = zinc finger domain.
We previously mapped the condition to chromosome 7q11-21 using genome-wide microsatellite marker analysis.3 Since then, we performed genome-wide SNP analysis to confirm the linkage. We performed SNP genotyping on 7 individuals in the pedigree (I:1, I:2, II:2, II:3, II:6, II:7, and II:8). Genome-wide linkage analysis yielded a maximum multipoint LOD score of 2.958 for the 7q11-21 region. The next highest LOD score in the genome was less than 1.5, thus confirming it as the candidate locus (figure 1C).
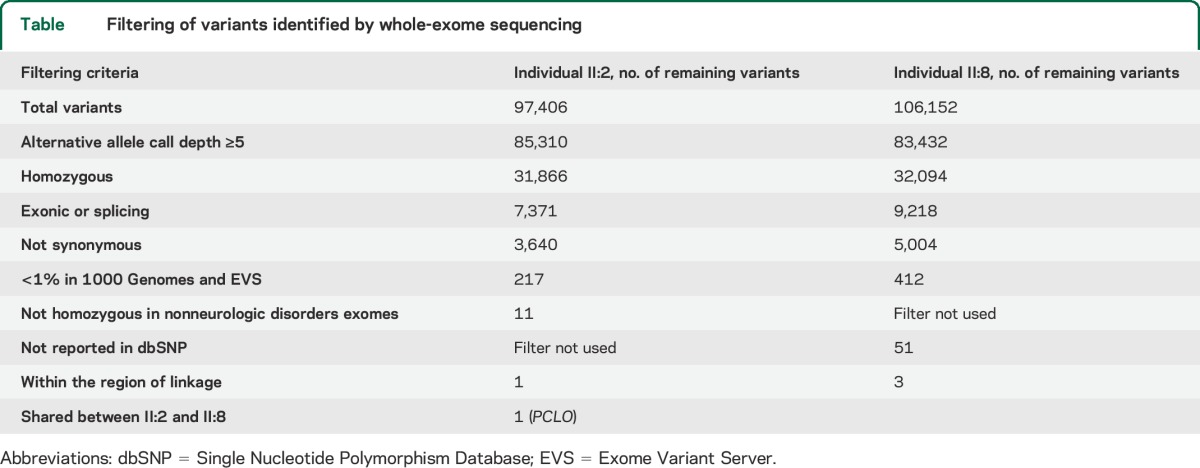
We then performed whole-exome sequencing on 2 affected individuals (II:2 and II:8). After filtering the identified variants for call quality, potential pathogenicity, population frequency, and localization within the candidate interval, only a single candidate variant shared between both individuals remained (table). The candidate variant, chr7:82579280 G>A (hg 19), is located within exon 6 of the PCLO (piccolo) gene and is predicted to cause a premature stop codon (NM_033026.5:c.10624C>T; p.R3542X). In individual II:8, 2 other variants persisted after filtering. They were chr7:72396170 GCGCCGCGCTCCCCAC>G within the POM121 gene and chr7:76062783 T>A within the ZP3 gene. The POM121 variant was found to be homozygous in 4 individuals in the Exome Aggregation Consortium (ExAC) Browser (November 2014), and the ZP3 variant was found to be homozygous in an individual without a neurologic condition in our in-house exome data. Thus, these 2 variants were ruled out. Sanger sequencing confirmed that the PCLO variant segregates with the disease in the family (figure 1A). There are 2 PCLO isoforms annotated (NM_014510.2, encoding 4,935 amino acids, and NM_033026.5, encoding 5,142 amino acids), and this variant affects both isoforms. This variant is not present in the 1000 Genomes (November 2014), the Exome Variant Server (November 2014), the ExAC Browser (November 2014), or in 122 Omani control chromosomes. In the Exome Variant Server, only 2 high-impact PCLO variants (nonsense, frameshift, or canonical splice site) are listed in one individual each, and both are heterozygous. In the ExAC Browser, there is no high-impact PCLO variant that is homozygous. This supports the notion that homozygous high-impact variants in PCLO could be deleterious. The PCLO protein has several identifiable domains. These include 2 piccolo zinc finger domains, a PDZ domain, and 2 C2 domains (figure 1D). The mutation localizes proximal to the PDZ and C2 domains, and is expected to eliminate these domains even if a truncated protein is produced.
Table.
Filtering of variants identified by whole-exome sequencing
Next, we investigated expression of PCLO in the developing human brain. Messenger RNA sequencing of the developing cerebral cortex from human fetal brain (9 weeks postconception) showed that PCLO is expressed at a moderate level (figure 2), approximately similar to that of a related gene, BSN (bassoon), and another synaptic protein gene, GRIK2 (glutamate receptor, ionotropic, kainate 2), which has been implicated in an intellectual disability syndrome.8 We also analyzed an additional RNA sequencing dataset of human fetal brain (13–16 weeks postconception).9 In this dataset, PCLO expression was very low in the ventricular zone (0.48 FPKM [fragments per kilobase of exon per million fragments mapped]), and moderately high in the cortical plate (12.10 FPKM). This dataset also shows that the level of expression and spatial pattern are similar to those of BSN (VZ = 1.45 FPKM; CP = 10.64 FPKM), and GRIK2 (VZ = 0.63 FPKM; CP = 19.03 FPKM). These data are consistent with known roles of these proteins in mature synapses.
Figure 2. Expression of PCLO in the developing human brain.

Expression of PCLO (top), a related presynaptic protein gene, BSN (middle), and another synaptic protein associated with intellectual disability, GRIK2 (bottom), seen in messenger RNA sequencing of the developing cerebral cortex of a 9-week human embryo. Y-axis units are in reads per million mapped reads (RPM). RNA isoforms as annotated in RefSeq are shown below each expression plot. The arrow indicates the location of the PCLO mutation.
We searched for additional deleterious PCLO alleles in our database of >500 exomes from individuals with various neurodevelopmental disorders, but did not find other convincing disease-causing mutations.
DISCUSSION
Our data show that a homozygous, nonsense mutation in the PCLO gene causes PCH3. Despite the term “pontocerebellar hypoplasia,” it is difficult to determine whether the condition in this family represents true hypoplasia of the pons and cerebellum, as no brain imaging of the affected individuals at an early age is available for comparison. The imaging findings are suggestive of atrophic changes, and so it may be more accurate to consider this entity as a degenerative disorder affecting both the cerebrum and cerebellum.
PCLO is a gene unique to vertebrates, which encodes a large protein component of the presynaptic active zone, a specialized area mediating neurotransmitter release.10–12 Piccolo has been found to interact with and control the assembly of presynaptic F-actin, which is important for the regulation of the synaptic vesicle cycle, suggesting that it may modulate neurotransmitter release through this interaction.13 It is interesting to note that the mutation in PCLO in humans leads not only to synaptic dysfunction but also to loss of cerebral and cerebellar volume, suggesting severe neuronal loss. Recently, piccolo and bassoon, another active zone component structurally related to piccolo, were shown to control ubiquitination of synapse proteins, and their loss led to aberrant degradation of multiple presynaptic proteins and eventually degeneration of synapses.14 Synaptic activity is essential for preventing apoptosis,15 so it is conceivable that degeneration of synapses due to absent PCLO triggers an apoptotic signal in neurons. For example, biallelic deletions of GRID2, which encodes a synaptic protein in Purkinje cells, also cause severe cerebellar atrophy in humans.16
Engineered mice that lack Pclo exon 14 have been reported.17 The homozygous mutant mice show a modest decrease in postnatal survival and weight, but interestingly do not show ultrastructural changes in synapses, defects in neurotransmission, or seizures.17 Although detailed neuroanatomical description of these mice has not been reported, the phenotype appears to be much milder compared to the individuals with the PCLO mutation described herein. Mouse isoforms of Pclo lacking exon 14 have been suggested.13 For example, the retinal photoreceptor and bipolar cell ribbon synapses express a unique Pclo isoform that is not affected by the absence of exon 14.18 This could, in part, explain the phenotypic difference between mice and humans, and raises the possibility that different PCLO isoforms have tissue-specific roles in the human CNS.
The affected individuals reported herein all had seizures starting during infancy. This could be explained by disruption of the equilibrium between excitatory and inhibitory neurotransmitter signaling due to neuronal loss, or by the effect of the PCLO mutation on synaptic vesicle recycling, which in turn might impair regulation of neuronal synchrony.
We did not find additional mutant alleles in our large cohort of neurodevelopmental disorders, but an individual from Turkey was reported to have a remarkably similar clinical presentation and displayed linkage to the chromosomal region encompassing PCLO, suggesting possible allelism.19 Since the brain MRI findings of PCH3 are rather nonspecific, with cerebral and cerebellar atrophy, it is possible that PCH3 is underdiagnosed. Molecular studies of additional cases of PCH3 will help delineate its full clinical spectrum and further elucidate the role of PCLO in the human brain.
ACKNOWLEDGMENT
The authors are grateful for the participation of the individuals and their family members reported herein. The authors thank Dr. A. James Barkovich for help with MRI interpretation, Dr. Almundher Al-Maawali for assistance with obtaining clinical information, and Dr. Tojo Nakayama for figure preparation. SNP genotyping was performed at the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University through the NIH Neuroscience Microarray Consortium. Human embryonic material was provided by the joint MRC/Wellcome Trust (grant 099175/Z/12/Z) Human Developmental Biology Resource (www.HDBR.org).
GLOSSARY
- dbSNP
Single Nucleotide Polymorphism Database
- ExAC
Exome Aggregation Consortium
- FPKM
fragments per kilobase of exon per million fragments mapped
- GATK
Genome Analysis Toolkit
- indel
insertion-deletion
- LOD
logarithm of odds
- NHLBI
National Heart, Lung, and Blood Institute
- PCH
pontocerebellar hypoplasia
- PCH3
pontocerebellar hypoplasia type III
AUTHOR CONTRIBUTIONS
A.A.-K., C.A.W., A.H.C., and G.H.M. designed the experiments. A.R. collected the clinical data. B.A.C., S.A.-T., and S.S. performed the experiments. M.A.P., A.Y.A.-M., and J.N.P. organized the clinical information and human samples. M.Y.A., B.A.C., K.S.-A., E.L.B., M.E.H., R.S.H., G.D.E., K.M., C.A.W., A.H.C., and G.H.M. analyzed the data. M.Y.A. and G.H.M. wrote the manuscript. A.H.C. and G.H.M. supervised and coordinated the project.
STUDY FUNDING
The study was supported by grants from the Manton Center for Orphan Disease Research, National Institute of Neurological Disorders and Stroke (R01NS035129), NIH/Fogarty International Center (R21TW008223), Dubai Harvard Foundation for Medical Research, Qatar National Research Fund, Boston Children's Hospital Faculty Career Development Award, Medical Research Council (G1002279), and Newlife Foundation for Disabled Children. C.A.W. is an investigator of the Howard Hughes Medical Institute. G.H.M. was also supported by grants from F. Hoffmann-La Roche Ltd., Qatar National Research Fund, and Boston Children's Hospital Faculty Career Development Award.
DISCLOSURE
M. Ahmed, B. Chioza, A. Rajab, K. Schmitz-Abe, A. Al-Khayat, S. Al-Turki, E. Baple, M. Patton, A. Al-Memar, M. Hurles, J. Partlow, R. Hill, G. Evrony, S. Servattalab, and K. Markianos report no disclosures relevant to the manuscript. C. Walsh is an investigator of the Howard Hughes Medical Institute, has received grants from the National Institute of Neurological Disorders and Stroke (NINDS) (R01NS035129; R01NS032457; R01NS079277), NIMH (R01MH083565), Fogarty International Center (R21TW008223), Paul G. Allen Family Foundation, Qatar National Research Fund, and Simons Foundation, and has a patent pending (PCT/US2011/63489). A. Crosby reports no disclosures relevant to the manuscript. G. Mochida has received research grants from F. Hoffmann-La Roche Ltd., received an honorarium from UCB Japan and Otsuka Pharmaceutical for a nonpromotional lecture, and has a patent pending (PCT/US2011/63489). Dr. Mochida has also been supported by grants from the NINDS (R01NS035129), Fogarty International Center (R21TW008223), Manton Center for Orphan Disease Research, Qatar National Research Fund, Simons Foundation, and Dubai Harvard Foundation for Medical Research. Go to Neurology.org for full disclosures.
ONLINE RESOURCES
MERLIN (http://www.sph.umich.edu/csg/abecasis/merlin/index.html)
SAMtools (http://samtools.sourceforge.net/)
NHLBI Exome Sequencing Project Exome Variant Server (http://evs.gs.washington.edu/EVS/)
Complete Genomics 69 Genomes Data (http://www.completegenomics.com/public-data/69-Genomes/)
dbSNP (http://www.ncbi.nlm.nih.gov/SNP/)
1000 Genomes Project (http://www.1000genomes.org/)
Dindel (https://www.sanger.ac.uk/resources/software/dindel/)
SNP Effect Predictor (http://www.ensembl.org/info/docs/tools/vep/index.html)
Exome Aggregation Consortium (ExAC) Browser (http://exac.broadinstitute.org/)
REFERENCES
- 1.Namavar Y, Barth PG, Poll-The BT, Baas F. Classification, diagnosis and potential mechanisms in pontocerebellar hypoplasia. Orphanet J Rare Dis 2011;6:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mochida GH, Ganesh VS, de Michelena MI, et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat Genet 2012;44:1260–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajab A, Mochida GH, Hill A, et al. A novel form of pontocerebellar hypoplasia maps to chromosome 7q11-21. Neurology 2003;60:1664–1667. [DOI] [PubMed] [Google Scholar]
- 4.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA 2001;98:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics 2004;20:1233–1240. [DOI] [PubMed] [Google Scholar]
- 6.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin: rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 7.Bae BI, Tietjen I, Atabay KD, et al. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 2014;343:764–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Motazacker MM, Rost BR, Hucho T, et al. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet 2007;81:792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fietz SA, Lachmann R, Brandl H, et al. Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc Natl Acad Sci USA 2012;109:11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cases-Langhoff C, Voss B, Garner AM, et al. Piccolo, a novel 420 kDa protein associated with the presynaptic cytomatrix. Eur J Cell Biol 1996;69:214–223. [PubMed] [Google Scholar]
- 11.Fenster SD, Chung WJ, Zhai R, et al. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron 2000;25:203–214. [DOI] [PubMed] [Google Scholar]
- 12.Rubin GM, Yandell MD, Wortman JR, et al. Comparative genomics of the eukaryotes. Science 2000;287:2204–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waites CL, Leal-Ortiz SA, Andlauer TF, Sigrist SJ, Garner CC. Piccolo regulates the dynamic assembly of presynaptic F-actin. J Neurosci 2011;31:14250–14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waites CL, Leal-Ortiz SA, Okerlund N, et al. Bassoon and Piccolo maintain synapse integrity by regulating protein ubiquitination and degradation. EMBO J 2013;32:954–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leveille F, Papadia S, Fricker M, et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J Neurosci 2010;30:2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hills LB, Masri A, Konno K, et al. Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology 2013;81:1378–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukherjee K, Yang X, Gerber SH, et al. Piccolo and bassoon maintain synaptic vesicle clustering without directly participating in vesicle exocytosis. Proc Natl Acad Sci USA 2010;107:6504–6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Regus-Leidig H, Ott C, Lohner M, et al. Identification and immunocytochemical characterization of Piccolino, a novel Piccolo splice variant selectively expressed at sensory ribbon synapses of the eye and ear. PLoS One 2013;8:e70373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Durmaz B, Wollnik B, Cogulu O, et al. Pontocerebellar hypoplasia type III (CLAM): extended phenotype and novel molecular findings. J Neurol 2009;256:416–419. [DOI] [PubMed] [Google Scholar]