

Background: Human age-related macular degeneration (AMD) retinas exhibit iron overload and localized C3 accumulation in the retinal pigment epithelium (RPE).
Results: Iron induces C3 expression via ERK1/2, SMAD3, and CCAAT/enhancer-binding protein-δ (C/EBP-δ) in a non-canonical TGF-β signaling pathway.
Conclusion: An iron-mediated pathway leads to complement dysregulation, an etiologic feature of AMD.
Significance: Regulating iron-induced C3 expression suggests a novel therapeutic approach for AMD.
Keywords: CCAAT/Enhancer-binding Protein (C/EBP), Extracellular Signal-regulated Kinase (ERK), Iron, Signaling, Transforming Growth Factor β (TGF-β), AMD, C3, RPE, SMAD3
Abstract
Dysregulation of iron homeostasis may be a pathogenic factor in age-related macular degeneration (AMD). Meanwhile, the formation of complement-containing deposits under the retinal pigment epithelial (RPE) cell layer is a pathognomonic feature of AMD. In this study, we investigated the molecular mechanisms by which complement component 3 (C3), a central protein in the complement cascade, is up-regulated by iron in RPE cells. Modulation of TGF-β signaling, involving ERK1/2, SMAD3, and CCAAT/enhancer-binding protein-δ, is responsible for iron-induced C3 expression. The differential effects of spatially distinct SMAD3 phosphorylation sites at the linker region and at the C terminus determined the up-regulation of C3. Pharmacologic inhibition of either ERK1/2 or SMAD3 phosphorylation decreased iron-induced C3 expression levels. Knockdown of SMAD3 blocked the iron-induced up-regulation and nuclear accumulation of CCAAT/enhancer-binding protein-δ, a transcription factor that has been shown previously to bind the basic leucine zipper 1 domain in the C3 promoter. We show herein that mutation of this domain reduced iron-induced C3 promoter activity. In vivo studies support our in vitro finding of iron-induced C3 up-regulation. Mice with a mosaic pattern of RPE-specific iron overload demonstrated co-localization of iron-induced ferritin and C3d deposits. Humans with aceruloplasminemia causing RPE iron overload had increased RPE C3d deposition. The molecular events in the iron-C3 pathway represent therapeutic targets for AMD or other diseases exacerbated by iron-induced local complement dysregulation.
Introduction
Dysregulation of iron homeostasis may be an etiologic factor in several neurodegenerative disorders such as age-related macular degeneration (AMD)4 (1), although the detailed mechanisms by which iron acts as a driver of AMD-like pathology have not been reported. AMD is considered a chronic, localized inflammatory disease (2, 3) with a genetic component (4). Several genetic factors that confer major risk for AMD are polymorphisms within complement component genes (5–10). Since complement dysregulation can lead to inflammatory reactions (11, 12), both genetic and environmental factors may work concurrently to determine the activity of the complement cascade in the outer retinal milieu. AMD is characterized by primary pathology in the retinal pigment epithelium (RPE) monolayer and the formation of sub-RPE deposits called drusen. In human drusen samples, both C3 and C3 activation fragments have been identified (13). C3, the central molecule of the complement cascade that includes the classical, alternative, and lectin pathways, is activated by numerous inciting factors that require a rapid inflammatory reaction (12, 14). In AMD, the aberrant activation of the alternative pathway and the resultant release of inflammatory mediators are involved in drusen formation (13). In the alternative pathway, C3 activation is under the control of Factor B, a key pathway component, and Factor H, a potent negative regulator, among others (15, 16). Both Factor B and Factor H are expressed by the RPE cell (15, 17, 18). Increased Factor B levels are accompanied by increased C3 expression and activation in aged mouse RPE (15). The interaction between C3 and Factor H is supported by the finding that C3b is a major physiological ligand of Factor H (19). Furthermore, both C3 and Factor H are genetically implicated as contributory factors to AMD etiology (7, 9).
Previous studies have shown elevated total iron levels in the RPE-Bruch's membrane complex as well as photoreceptors in the human macula of post-mortem dry and wet AMD eyes compared with those of healthy controls (20). The iron-containing species in these samples identified by histochemical and chelation tests comprise both loosely bound, chelatable iron and non-chelatable iron components. The non-chelatable iron components may be iron derivatives that are incorporated into and are essential for the function of reactive oxygen species-producing enzymes (21). The chelatable iron is capable of directly generating reactive oxygen species via Fenton chemistry. Evidence exists that reactive oxygen species can in turn damage iron-containing proteins to produce more labile iron (21, 22), suggesting the interchangeability of non-chelatable and chelatable iron. Thus, it is reasonable to study the biological impact of an increased intracellular labile iron pool on complement regulation in the RPE.
Our group has previously demonstrated increased RPE iron levels in mice with knock-out of the Cp gene (Cp−/−) and a naturally occurring Sla mutation in the Heph gene (HephSla/Sla or HephSla/Y), referred to as the double knock-out mouse model (23, 24). When compared with the wild-type mice, these double knock-out mice exhibited some pathological features of AMD, and the generalized high iron level in retina and RPE/choroid was associated with an accumulation of sub-RPE activated C3 fragments (24). Our group has also shown that systemic administration of the iron chelator deferiprone decreased murine retinal C3 mRNA levels in a light-induced degeneration model (25) and RPE C3 mRNA levels in a sodium iodate-induced degeneration model (26). These findings suggest that diminishing iron levels may reduce retinal complement expression. However, whether and how an increase in the labile iron pool within RPE cells is capable of triggering endogenous C3 activation are as yet unknown.
In cultured RPE cells, we found that iron exposure increases both C3 mRNA and protein levels. To understand the underlying molecular events, we first pursued a bioinformatics approach using a gene expression microarray, which identified the TGF-β signaling superfamily as the pathway(s) most strongly affected by iron. Interestingly, the TGF-β receptor type I gene, TGFBR1, is genetically associated with advanced AMD (27), and elevated urinary TGF-β1 protein levels are associated with early AMD (28). In canonical TGF-β signaling, TGF-β ligands bind to receptor serine/threonine kinases that phosphorylate transcription factors SMAD2 and SMAD3 at the C-terminal SSXS motif (29). Although non-phosphorylated SMAD proteins shuttle in and out of the nucleus dynamically, upon phosphorylation, SMAD2/3 docks with SMAD4 and undergoes nuclear translocation. Once in the nucleus, this complex recruits cofactors to form a larger complex and differentially regulates target gene expression. In this study, we identified in cultured human RPE cells an iron-induced, non-canonical TGF-β pathway leading to C3 up-regulation. Corresponding in vivo studies showed that deposition of the C3 activation product C3d is spatially associated with iron-overloaded RPE cells.
EXPERIMENTAL PROCEDURES
Cell Culture and Cell Treatment Reagents
ARPE-19 cells from the American Type Culture Collection (ATCC, Manassas, VA) were cultured in 1:1 DMEM/F-12 (Invitrogen) supplemented with 10% FBS (HyClone, Logan, UT). Once confluent, cells were maintained in medium with 1% FBS for 4 weeks prior to experiments to obtain mature monolayers (30). One day prior to experiments, cells were placed in serum-free medium to deplete residual serum complement components. Iron in the form of ferric ammonium citrate (FAC; MP Biomedicals, Santa Ana, CA) dissolved in serum-free medium was used to treat cells for the indicated times. Alamar Blue reagent for cell viability was from Invitrogen. Transition metals suitable for cell culture were from Sigma. Purified apo- and holo-transferrin were from Millipore (Billerica, MA). Expression plasmids pCS2 FLAG-SMAD3 (31), pCS2 FLAG-SMAD3 (EPSM) (31), and pCS2 FLAG SMAD3 EPSM A213S (32) were gifts from Joan Massagué (Addgene plasmids 14052, 14963, and 27113). Pharmacologic inhibitors, recombinant proteins, and neutralizing antibodies were obtained as follows: PD98059, U0126, SB202190, SP600125, and human recombinant TGF-β1, β2, β3 (Cell Signaling Technology, Danvers, MA); SIS3 (Millipore); SB431542 (Tocris, Minneapolis, MN); anti-TGF-β1/2/3 antibody and isotype control (R&D Systems, Minneapolis, MN).
RNA Extraction, Quantitative RT-PCR, Microarray Processing, and Data Analysis
Total RNA was isolated using QIAzol® reagent and the miRNeasy Mini kit from Qiagen (Valencia, CA). Quantitative reverse transcription-PCR (qRT-PCR) using the standard ΔΔCt method was performed using TaqMan® primers (Applied Biosystems, Waltham, MA) listed in Table 1 with 18S rRNA as the internal control. Microarray processing and data analysis services were provided by the Penn Molecular Profiling Facility using the Affymetrix GeneChip® Human Gene 2.0 ST Array (Affymetrix, Santa Clara, CA). For each group (untreated and FAC-treated), three independent arrays were performed, probing >40,000 transcript IDs from more than 24,800 genes. Processing steps were conducted as described in the Ambion WT Expression Manual and the Affymetrix GeneChip Expression Analysis Technical Manual. For data analysis, probe intensity (.cel) files were imported into Partek Genomics Suite (v6.6, Partek Inc., St. Louis, MO) where robust multiarray average normalization was applied yielding log2-transformed intensities. These values were tested for differential expression using Significance Analysis for Microarrays (SAM; samr v2.0, Stanford University) (33), yielding a fold change and q value (false discovery rate) for each transcript. To consider a transcript for input into DAVID Bioinformatics Resources 6.7 (accessed February 2015) for pathway analysis, the thresholds of fold change ≥1.5 (up or down) and q value ≤10% were applied. The KEGG and BioCarta pathway mapping databases as well as GOTERM_BP_FAT biological process database were included within the analysis. The top pathways/processes were determined by setting the corrected false discovery rate (Benjamini-Hochberg) to <10%. The complete array data set can be accessed in the supplemental table, and the .cel files have been deposited in NCBI's Gene Expression Omnibus, accessible through GEO series accession number GSE67603.
TABLE 1.
TaqMan qRT-PCR primers
| Gene name | Symbol | Catalog number |
|---|---|---|
| Complement component 3 (human) | C3 | Hs01100879_m1 |
| Transferrin receptor (human) | TFRC | Hs00174609_m1 |
| PAI-1 (human) | SERPINE1 (PAI-1) | Hs01126607_g1 |
| Transforming growth factor, β1 (human) | TGFB1 | Hs00998133_m1 |
| C/EBP-δ (human) | CEBPD | Hs00270931_s1 |
Whole Cell Protein Extraction, Cell Compartment Protein Extraction, and Western Blotting
Whole cell protein lysates were extracted using Laemmli SDS lysis buffer supplemented with protease/phosphatase inhibitor mixture and PMSF (Cell Signaling Technology) according to standard methods. Lysates were sonicated for 10 s prior to quantification with the 660 nm protein assay (Pierce). For each sample, 30 μg of protein were loaded onto a 4–12% Bis-Tris gel for Western blotting. Non-nuclear (cytoplasmic, membranous, and cytoskeletal) and nuclear protein lysates were extracted using the Qproteome® cell compartment kit (Qiagen) according to the manufacturer's instructions. Primary antibodies used are as follows: anti-p-ERK1/2, anti-ERK1/2, anti-p-SMAD2 (Ser-245/250/255), anti-GAPDH, and anti-α-tubulin clone 11H10 (Cell Signaling Technology); anti-SMAD3, anti-FLAG M2, and anti-actin AC-40 (Sigma); anti-p-SMAD3 (Ser-213) (Assay Biotech, Sunnyvale, CA); anti-transferrin receptor (TFRC; Invitrogen); and anti-p-SMAD3 (Ser-423/425) and anti-lamin B1 (Abcam, Cambridge, MA). Anti-C/EBP-δ (clone L46-743.92.69) was provided by BD Pharmingen as an outcome of an antibody co-development collaboration with the National Cancer Institute. Secondary antibodies IRDye® 680RD donkey anti-rabbit and 800CW donkey anti-mouse (LI-COR Biosciences, Lincoln, NE) were used in combination when possible for two-channel infrared detection with an Odyssey imager. ImageJ 1.46r software was used for band densitometry.
Knockdown of SMAD3 Using Lentiviral Vectors
This was performed according to the MISSION® shRNA instructions (Sigma). Briefly, ARPE-19 cells were seeded overnight to 70% confluence prior to treatment with lentiviral transduction particles (Sigma) that express shRNA targeting SMAD3 transcripts (sh-SMAD3). All particles used including controls are listed in Table 2. Cells were used for qRT-PCR 72 h post-transduction without selection, or puromycin was used to select for stably transduced cells for two to three passages prior to qRT-PCR and Western blotting.
TABLE 2.
Sigma MISSION lentiviral transduction particles
| Product name | Abbreviation used | Catalog number |
|---|---|---|
| pLKO.1-puro empty vector control | Vector | SHC001V |
| pLKO.1-puro non-mammalian shRNA control | sh-null | SHC002V |
| pLKO.1-puro-CMV-TurboGFPTM positive control | t-GFP (not shown) | SHC004V |
| pLKO.1-puro SMAD3 shRNA | sh-SMAD3 | SHCLNV-NM_005902 TRCN0000330127 |
Plasmid Construction, Transfection, and Dual-Luciferase Assays
DNA fragments consisting of four different sized, overlapping sequences of the C3 gene promoter flanked by 5′ and 3′ MluI and BglII sites, respectively, were amplified by the Q5® high fidelity DNA polymerase from New England Biolabs (Ipswich, MA). The fragments, which are ∼500 bp (−481 to +52), 1.0 kb (−1078 to +52), 1.5 kb (−1555 to +52), and 2.0 kb (−2047 to +52), were cloned into the pCR-Blunt II TOPO vector using the Zero Blunt TOPO kit (Life Technologies) according to the product instructions. Once sequence-verified, the fragments were subcloned into the pGL3-Basic vector (Promega, Madison, WI) for luciferase assays. The bZIP1 and bZIP2 mutant vectors with the same base substitutions as previously reported (34), were generated using the Q5 site-directed mutagenesis kit (New England Biolabs) from the pGL3 wild-type (WT) 500-bp vector template. All primers are listed in Table 3. ARPE-19 cells at 80% confluence grown in complete medium with no antibiotics were transfected with either empty or promoter-inserted pGL3 vector using the reagent Lipofectamine LTX (Life Technologies) at a 2:1 LTX:DNA ratio for 4 h before initiation of FAC treatment. Luciferase activity was then determined with the Dual-Luciferase reporter assay system kit (Promega) according to the manufacturer's protocols. Luminescence was read for 1.5 s by an Infinite M200 Pro plate reader (Tecan Systems, Morrisville, NC).
TABLE 3.
PCR primers used to generate C3 promoter fragments
| Fragment | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| 500 bp/WT | ACGTACGCGTAGTTTCTGTGCTGGGCTC | ACGTAGATCTGACAGTGCAGGGTCAGAG |
| 1 kb | ACGTACGCGTCTACTTGGGTGGTTGAGC | ACGTAGATCTGACAGTGCAGGGTCAGAG |
| 1.5 kb | ACGTACGCGTGAGGCAGGAGAATTGTTT | ACGTAGATCTGACAGTGCAGGGTCAGAG |
| 2 kb | ACGTACGCGTGCAGGGCTCATAAGTGTG | ACGTAGATCTGACAGTGCAGGGTCAGAG |
| 500 bp/bZIP1 | AATGGTATTGTGTCGACTGGGGCAGCC | TCCTAAGCTTTTCAGTCC |
| 500 bp/bZIP2 | GAAAAGCTTATCGCGAGGTATTGAGAAATCTGGGGCAGCCCCAAAAGG | AGTCCCTGGGGCCAACAT |
ELISA
Detections of secreted C3 precursor protein (Abcam), C3a (BD Biosciences), and Factor B fragment Ba (Quidel, San Diego, CA) in ARPE-19 cell culture conditioned medium were performed according to each manufacturer's instructions. The conditioned medium used for C3 assay did not require concentration, but those for C3a and Ba required an ∼70-fold concentration for detection in the standard range. Amicon Ultra-15 centrifugal filters with a 3-kDa-molecular mass cutoff membrane (Millipore) were used for concentration of media.
Immunofluorescence of Human Aceruloplasminemia Retina Paraffin Sections and Mouse Retinal Cryosections
Ocular tissue from a 60-year-old male donor with aceruloplasminemia was prepared as paraffin sections as described in a previous publication in adherence to the tenets of the Declaration of Helsinki (35). The paraffin sections were stained with anti-human C3d antibody (1:100; Abcam). Generation of the systemic ceruloplasmin-null, bestrophin promoter-driven Cre recombinase, hephaestin floxed/floxed mouse on a C57BL/6 background (BCre+, Cp−/−, HephF/F) has also been described in a previous publication (36). Background-matched C3-null (C3−/−) mice were provided by Wenchao Song (University of Pennsylvania). All mice were handled in accordance with the Institutional Animal Care and Use Committee of the University of Pennsylvania. Retina cryosections of 10-μm thickness derived from 12-month-old mice were used for immunolabeling studies. The primary antibodies used were rabbit anti-L-ferritin clone F-17 at a 1:100 dilution (a generous gift from Paolo Arosio, Università Degli Studi di Brescia, Brescia, Italy) and goat anti-mouse C3d at a 1:100 dilution (R&D Systems). Slides were examined on a Nikon Eclipse 80i microscope, and images were acquired using NIS-BR Elements v4.1 software (Nikon, Melville, NY).
Statistics
All experiments were performed in at least triplicate with mean ± S.E. reported for each comparison group. The means were analyzed using either a two-tailed Student's t test or a one-way analysis of variance followed by a Bonferroni post hoc test (Prism 5.0, GraphPad Software, San Diego, CA).
RESULTS
Iron Induced the Expression of Endogenous C3 in ARPE-19 Cells
In a previous publication, we have shown that photo-oxidative stress in mouse RPE cells can lead to elevated C3 mRNA levels (37). These changes are accompanied by altered expression of iron regulatory proteins. More directly, a mouse model of retinal iron overload, Cp−/−Hephsla, exhibited deposition of activated C3 fragments in the basolateral RPE and Bruch's membrane (24). Because C3 is a central player in the complement cascade, here we investigated whether iron can specifically induce C3 transcription and translation in the RPE cell. Using the human ARPE-19 cell line differentiated in low serum for 1 month and then cultured in serum-free medium, we determined that the viability of these cells decreased by 30% but remained stable at all iron (FAC) doses in the 25 μm to 1 mm range (Fig. 1A). In this same analysis, TFRC mRNA levels decreased by at least 50% in the same dose range (Fig. 1B), indicating an increase of the labile iron pool and an intact iron regulatory axis. Concurrently, the C3 mRNA levels increased by 3–4-fold relative to untreated cells (Fig. 1C). In a corresponding experiment, the C3 protein levels in the conditioned medium were found to increase 2–3-fold within the range of FAC used (Fig. 1D). Renewal of culture medium and continued treatment with FAC beyond 2 days (i.e. 4 and 8 days) resulted in further increases in C3 mRNA levels up to 20-fold (Fig. 1E). However, it was determined that only treatment with FAC, but not other transition metals (Mn2+, Ni2+, and Cu2+), induced increases in C3 mRNA levels relative to untreated (Fig. 1F). C3 mRNA up-regulation was also observed in cells treated with holo-transferrin, the extracellular iron carrier protein. The levels increased by ∼2.5-fold (Fig. 1G), whereas TFRC mRNA levels decreased by ∼40% (Fig. 1H). Overall, these data delineate a specific, iron-induced C3 gene response within the RPE cell.
FIGURE 1.

Iron up-regulates C3 expression in ARPE-19 cells. A, viability of cells grown as 1-month differentiated monolayers and treated with FAC at increasing doses for 2 days (d). Viability is decreased with 25 μm iron treatment compared with untreated but remains stable for higher doses examined. B and C, TFRC mRNA and C3 mRNA levels, respectively, with increasing FAC treatment doses for 2 days. TFRC mRNA levels decrease with 25 μm iron relative to untreated and are stable at higher doses; similarly, C3 mRNA levels are increased at the lowest iron dose relative to untreated but remain stable at the higher doses. D, C3 protein levels increase, relative to untreated, in the conditioned medium of cells treated with increasing doses of FAC for 2 days. E, C3 mRNA levels are increased relative to control cells at each of the 2-, 4-, and 8-day time points after the initial 250 μm FAC treatment. F, C3 mRNA levels of cells treated with Fe3+, Mn2+, Ni2+, or Cu2+ at 250 μm concentration for 2 days with only Fe3+/FAC showing an increase in C3 mRNA relative to untreated. G and H, C3 mRNA levels of cells treated with 9.75 mg/ml holo-transferrin (holo-Tf) are increased relative to those of apo-transferrin (apo-Tf)-treated cells with a corresponding decrease in TFRC mRNA. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001).
Pathway Enrichment Analysis Implicates TGF-β Signaling in Iron-induced C3 Expression
To characterize the potential molecular pathway(s) affected by iron treatment and identify the one(s) responsible for C3 induction, we performed a whole genome microarray on untreated ARPE-19 cells and cells treated with 250 μm FAC for 48 h/2 days (supplemental table). Iron loading resulted in 95 transcripts with up-regulated expression and 122 transcripts with down-regulated expression of magnitude ≥1.5-fold and q <10%. After eliminating non-annotated entries, we derived a list of 70 up-regulated and 87 down-regulated transcripts (total, 157) of which nine up-regulated transcripts and seven down-regulated transcripts were of a magnitude ≥2.0-fold (Table 4). Both C3 and TFRC are among these “top hits” (Table 4), validating our results above in Fig. 1. Using the list of 157 transcripts with expression change magnitude ≥1.5-fold as input into the program DAVID, we generated another list of “pathways” and “biological processes” that best represent these transcripts. The entry “TGF-β signaling” topped this list, fitting within the constraint of false discovery rate (Benjamini-Hochberg) <10%. Subsequent entries did not meet this statistical significance cutoff.
TABLE 4.
The most up-regulated and down-regulated annotated transcripts in ARPE-19 cells treated with FAC
| Transcript ID | RefSeq | Gene name | Gene symbol | -Fold change (log2)a |
|---|---|---|---|---|
| Transcripts with up-regulated expression | ||||
| 16955939 | NM_015541 | Leucine-rich repeats and immunoglobulin-like domains 1 | LRIG1 | 2.69089 |
| 16911261 | NM_001200 | Bone morphogenetic protein 2 | BMP2 | 2.44466 |
| 16996813 | NM_005582 | CD180 molecule | CD180 | 2.30087 |
| 17089003 | NM_182487 | Olfactomedin-like 2A | OLFML2A | 2.24716 |
| 16750996 | NM_020039 | Amiloride-sensitive cation channel 2, neuronal | ACCN2 | 2.18511 |
| 16867784 | NM_000064 | Complement component 3 | C3 | 2.18391 |
| 16829801 | NM_001124758 | Spinster homolog 2 (Drosophila) | SPNS2 | 2.18123 |
| 17096285 | NM_001333 | Cathepsin L2 | CTSL2 | 2.16549 |
| 16785938 | NM_015351 | Tetratricopeptide repeat domain 9 | TTC9 | 2.09855 |
| Transcripts with down-regulated expression | ||||
| 17056984 | NM_002192 | Inhibin, βA | INHBA | −2.02469 |
| 16907639 | NR_036227 | MicroRNA 2355 | MIR2355 | −2.07836 |
| 16976599 | NM_014465 | Sulfotransferase family, cytosolic, 1B, member 1 | SULT1B1 | −2.11595 |
| 16912362 | NM_181353 | Inhibitor of DNA binding 1, dominant negative helix-loop-helix protein | ID1 | −2.96384 |
| 16963241 | NM_003234 | Transferrin receptor (p90, CD71) | TFRC | −3.07206 |
| 16683377 | NM_002167 | Inhibitor of DNA binding 3, dominant negative helix-loop-helix protein | ID3 | −4.0267 |
| 17084723 | NM_001216 | Carbonic anhydrase IX | CA9 | −4.35892 |
a Data are relative to control, untreated cells.
Iron-induced C3 Up-regulation Is Independent of TGF-β Ligands
To determine whether TGF-β ligands drive C3 up-regulation following iron treatment, we incubated ARPE-19 cells with human recombinant TGF-β1, TGF-β2, or TGF-β3. By qRT-PCR analysis, TGF-β1 (Fig. 2A) and to a lesser degree TGF-β3 (data not shown) showed significant down-regulation of C3 mRNA levels, whereas TGF-β2 showed no significant difference when compared with untreated control (data not shown). These results suggest that the ligands, via canonical TGF-β pathway activation, do not account for the increase in C3 mRNA levels observed with iron loading. The significant up-regulation of specific downstream target genes of these ligands, such as plasminogen activator inhibitor type 1 (PAI-1) in the case of TGF-β1 (Fig. 2A), demonstrated that the canonical TGF-β pathway is intact and that TGF-β receptor blockade using SB431542 is efficacious. We then investigated the levels of TGFB1 mRNA and TGF-β1 protein under untreated and FAC-treated conditions and found that mRNA levels were increased slightly by ∼1.3-fold, but the secreted protein levels were moderately but significantly decreased by ∼20% (Fig. 2B). This led to the hypothesis that basal TGF-β levels, although suppressed by FAC, are important for regulating C3 mRNA levels. To test this hypothesis, we treated cells with anti-TGF-β1/2/3 neutralizing antibody and found that C3 mRNA levels were increased by ∼1.7-fold relative to control conditions, and PAI-1 mRNA levels were decreased by ∼60% in the same comparison (Fig. 2C). We further asked whether basal TGF-β activity affects the degree of FAC-induced C3 up-regulation. To determine this, we co-treated with anti-TGF-β1/2/3 antibody and FAC, which showed no significant difference in C3 mRNA levels compared with FAC only (∼2.5-fold over untreated) (Fig. 2D). PAI-1 mRNA levels were repressed by FAC but not further lowered by addition of neutralizing antibody (Fig. 2D).
FIGURE 2.

Iron-induced C3 expression is not dependent on TGF-β ligand/receptor-mediated canonical signaling. A, C3 mRNA levels in ARPE-19 cells are repressed by exogenous TGF-β1 (10 ng/ml; 2 days (d)), and this inhibition is partially relieved by TGF-β receptor inhibitor SB431542 (100 nm; 2 days). In contrast, PAI-1 mRNA levels are significantly increased by TGF-β1, and this increase is partially inhibited by SB431542. B, TGFB1 mRNA levels show an increase with FAC treatment; TGF-β1 protein levels in the conditioned medium of FAC-treated cells show a significant decrease relative to untreated. C, C3 mRNA levels are increased with addition of neutralizing antibody anti-TGF-β1/2/3 (10 μg/ml; 2 days) when compared with control or IgG treatment conditions. PAI-1 mRNA levels are decreased in the same comparison. D, C3 mRNA levels are not significantly changed with co-treatment with anti-TGF-β1/2/3 antibody and FAC when compared with FAC only. PAI-1 mRNA levels are decreased to similar levels with co-treatment with anti-TGF-β1/2/3 antibody and FAC compared with FAC only. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant).
Iron-induced C3 Up-regulation Involves ERK1/2 and SMAD3 Non-canonical TGF-β Signaling
Iron has been shown previously to induce ERK1/2 phosphorylation in rat hippocampal neurons and mouse neurosensory retina (38, 39). To examine whether ERK1/2 plays a role in iron induction of C3, we pretreated ARPE-19 cells with PD98059, a pharmacologic inhibitor of MEK1/2, the kinase upstream of ERK1/2, followed by FAC treatment. This inhibitor was able to block FAC-induced C3 up-regulation (Fig. 3A). Another inhibitor that functions by a similar mechanism, U0126, achieved the same effect (data not shown). In contrast, inhibitors of the two other MAPK classes, SB202190 against p38 and SP600125 against JNK1/2/3, did not suppress FAC-induced C3 up-regulation (Fig. 3A).
FIGURE 3.

Non-canonical TGF-β signaling involving ERK1/2 and SMAD3 mediates iron induction of C3. A, C3 mRNA levels in ARPE-19 cells treated with MEK1/2 inhibitor PD98059 (5 μm; 2 days (d)), p38 inhibitor SB202190 (20 μm; 2 days), or JNK1/2/3 inhibitor SP600125 (50 μm; 2 days) alone and plus FAC. Control contains vehicle only (DMSO). Relative to FAC alone, only PD98059 showed a significant decrease in C3 mRNA levels when co-treated with FAC. B, the specific inhibitor of SMAD3, SIS3 (2 μm; 2 days), inhibited FAC-induced C3 up-regulation. SIS3 decreased basal C3 mRNA levels. C, iron-induced increases in C3 protein levels in the conditioned media are diminished by SIS3 and PD98059. D, SIS3 inhibited PAI-1 mRNA levels to the same extent as FAC, whereas PD98059 had no significant effect. Co-treatment with SIS3 and FAC showed no additional suppression of PAI-1 mRNA levels, but co-treatment with PD98059 and FAC restored them to baseline. E, Western blot and densitometry analysis for p-ERK1/2 and ERK1/2 in lysates derived from cells at different time points (0, 1, 3, and 6 h) post-FAC treatment, with or without SIS3 co-treatment. SIS3 showed no effect on the FAC-induced increase of p-ERK1/2 at 1h. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant).
ERK1/2 can phosphorylate the linker region residues of SMAD2/3, central components in the TGF-β signaling pathway (40–44). In the present study, FAC did not induce linker region phosphorylation of SMAD2 (data not shown). Thus, to assess whether SMAD3 plays a direct role in C3 induction, we co-administered a specific inhibitor of SMAD3 function, SIS3 (45), with FAC and found that this compound suppressed the up-regulation (Fig. 3B). SIS3 alone also decreased C3 mRNA levels relative to the untreated baseline (Fig. 3B), suggesting that the inhibitor modulates basal C3 expression. Furthermore, ARPE-19 cells co-treated with SIS3 or PD98059 and FAC showed diminished levels of C3 protein in the conditioned medium compared with cells treated with FAC alone (Fig. 3C). Expression of the TGF-β1-responsive gene PAI-1 was inhibited by SIS3 but not by PD98059. The FAC-induced repression of PAI-1 mRNA levels was relieved by PD98059 (Fig. 3D). To determine the temporal sequence of ERK1/2 phosphorylation and SMAD3 activity, we treated cells with FAC for different lengths of time (0, 1, 3, and 6 h) in the absence and presence of SIS3 and found no difference in p-ERK1/2 levels by Western blotting at each of the time points examined (Fig. 3E). In contrast, PD98059 inhibited FAC-induced SMAD3 linker phosphorylation at Ser-213 (Fig. 4C). These findings place ERK1/2 upstream of SMAD3 in an FAC-induced signaling pathway.
FIGURE 4.

Iron-induced changes in SMAD3 phosphorylation are mediated by non-canonical TGF-β signaling. A, Western blot and densitometry analysis of ARPE-19 cell lysates for p-SMAD3 (Ser-213), p-SMAD3 (Ser-423/425), SMAD3, and TFRC in a FAC treatment time course experiment. The p-SMAD3 (Ser-213) 150-kDa band increased whereas the p-SMAD3 (Ser-423/425) and TFRC bands decreased in intensity in a time-dependent manner. The p-SMAD3 (Ser-213) antibody-detectable 52-kDa band showed no significant change in the same time course. The SMAD3 protein levels also remain unchanged. B, cells transfected with constructs expressing FLAG-tagged SMAD3, SMAD3 EPSM (linker mutations), and SMAD3 EPSM A213S (mutation reverted at residue 213) showed specificity of the antibody used to detect p-SMAD3 (Ser-213) in Western blots. C, Western blot and densitometry analysis of p-SMAD3 (Ser-213) shows that co-treatment with SIS3 or PD98059 and FAC reduced the intensity of this band relative to FAC alone. SIS3 decreased the basal p-SMAD3 (Ser-213) levels. Blotting and analysis of p-SMAD3 (Ser-423/425) demonstrated that SIS3 reduced band intensity to the same extent as FAC alone, whereas PD98059 had no significant effect. Co-treatment with either inhibitor with FAC showed no significant difference from FAC alone. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant).
Iron-induced Changes in SMAD3 Phosphorylation Are Distinct from Canonical TGF-β Effects
Since C3 up-regulation involves SMAD3 activity, we probed the phosphorylation status of SMAD3 following iron treatment. Using phospho-specific antibodies directed against a linker region residue (Ser-213) and the C-terminal residues (Ser-423/425), we found two directly opposite effects: p-SMAD3 (Ser-213), shown as a 150-kDa band, gradually increased at time points up to 48 h/2 days, whereas p-SMAD3 (Ser-423/425) decreased in the same time course (Fig. 4A). The p-SMAD3 (Ser-213) antibody-detectable 52-kDa band remained at baseline levels. TFRC protein levels diminished as a reflection of increasing labile iron pool (Fig. 4A). To test the specificity of the anti-p-SMAD3 (Ser-213) antibody in Western blotting (Fig. 4B), we transfected cells with a construct expressing FLAG-SMAD3 and detected a band at 54 kDa. We also transfected a construct expressing FLAG-SMAD3 EPSM (with mutated linker region phosphorylation sites) and detected virtually no band. Finally, we transfected a construct expressing FLAG-SMAD3 EPSM A213S (with residue 213 reverted back to serine) and detected the band once again. This, together with SMAD3 shRNA data shown below (see Fig. 6A), suggest that the 150-kDa band detected in untransfected cells contains p-SMAD3 (Ser-213). Western blots also showed that the SMAD3 inhibitor SIS3 and MEK1/2 inhibitor PD98059 can suppress iron-induced increases in p-SMAD3 (Ser-213) levels, supporting further the model that ERK1/2 and SMAD3 are in the same signaling pathway (Fig. 4C). Of note, p-SMAD3 (Ser-213) levels decreased with SIS3 treatment compared with the untreated baseline. This finding correlates with decreased C3 mRNA levels observed in the same comparison in Fig. 3B. In addition, SIS3 can suppress basal p-SMAD3 (Ser-423/425) levels to the same extent as FAC alone, whereas PD98059 showed no such effect on the phosphorylation of these C-terminal residues (Fig. 4C).
FIGURE 6.

SMAD3 gene silencing reduces the formation of p-SMAD3 (Ser-213)-containing complexes and suppresses iron-induced C3 up-regulation. A, Western blot and densitometry analysis for SMAD3, p-SMAD3 (Ser-213), and TFRC of ARPE-19 cell lysates derived from untransduced (control), vector, sh-null, and sh-SMAD3 lines, each in the absence or presence of FAC. Efficient knockdown of SMAD3 decreased the formation of p-SMAD3 (Ser-213)-containing complexes at 150 kDa. B, the FAC-induced increase in C3 mRNA levels is observed in the untransduced, vector, and sh-null cell lines but is absent in the sh-SMAD3 cell line. C, Western blot for non-nuclear (Non-Nucl) and nuclear (Nucl) lysates of untreated and FAC-treated cells. The p-SMAD3 (Ser-213)-containing complex at 150 kDa localizes only to the non-nuclear fractions. The p-SMAD3 (Ser-213) antibody-detectable band at 52 kDa is essentially unchanged by FAC treatment in both the non-nuclear and nuclear fractions. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant). d, days.
To gain insight into the role of endogenous TGF-β on the regulation of p-SMAD3 (Ser-213) and p-SMAD3 (Ser-423/425), we treated cells with BSA, IgG, or anti-TGF-β1/2/3 antibody with or without FAC. Anti-TGF-β1/2/3 antibody alone dramatically decreased basal, TGF-β-sustained p-SMAD3 (Ser-213) (Fig. 5A). In the presence of this antibody, iron retained its ability, albeit to a lesser extent, to induce formation of the p-SMAD3 (Ser-213) complex. Anti-TGF-β1/2/3 antibody alone also decreased p-SMAD3 (Ser-423/425) levels as expected, whereas iron treatment potentiated this suppressive effect. In contrast, cells treated with exogenous TGF-β1 for 1 h demonstrated no significant difference in p-SMAD3 (Ser-213) levels but exhibited up-regulated p-SMAD3 (Ser-423/425) levels, consistent with canonical signaling effects (Fig. 5B). This increase returned to basal level at 2 days (data not shown).
FIGURE 5.

Iron and TGF-β ligand differentially regulate spatially distinct SMAD3 phosphorylation sites. A, Western blot and densitometry analysis of p-SMAD3 (Ser-213) and p-SMAD3 (Ser-423/425) in cells treated with BSA, IgG, or anti-TGF-β1/2/3 antibody in the presence or absence of FAC show the combined effect of iron and neutralization of canonical TGF-β signaling. SMAD3 protein levels increased after anti-TGF-β1/2/3 antibody treatment relative to control condition, BSA, with no FAC. Only the antibody plus FAC condition is statistically significant. B, Western blot and densitometry analysis following treatment with exogenous TGF-β1 (10 ng/ml; 1 h) show no significant change in p-SMAD3 (Ser-213) and increased p-SMAD3 (Ser-423/425). Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; and ****, p ≤ 0.0001; ns, not significant).
To verify that the 150-kDa band does contain SMAD3 and more specifically p-SMAD3 (Ser-213), we transduced ARPE-19 cells with lentiviral constructs expressing shRNA to stably knock down SMAD3 protein (Fig. 6A). With SMAD3 knockdown (sh-SMAD3), the iron-induced increase in intensity of the 150-kDa band as detected by anti-p-SMAD3 (Ser-213) was diminished, unlike in the control cell lines (untransduced, vector, and sh-null), which are shown in the same panel (Fig. 6A). In addition, qRT-PCR measurement of C3 mRNA levels in sh-SMAD3 cells revealed a lack of FAC-induced up-regulation, which was present in the three control cell lines (Fig. 6B). To determine whether the p-SMAD3 (Ser-213)-containing protein complex (150 kDa) undergoes nuclear translocation, we performed cellular fractionation on untransduced cells with or without FAC treatment. This band, regardless of iron treatment, localized only to the non-nuclear fraction, suggesting that the complex does not translocate to the nucleus (Fig. 6C). In addition, the p-SMAD3 (Ser-213) antibody-detectable 52-kDa protein was not induced by FAC in either the non-nuclear or the nuclear compartment (Fig. 6C).
Iron-induced Increases in CEBPD mRNA Levels and C/EBP-δ Protein Levels in Both the Non-nuclear and Nuclear Compartments Are Dependent on SMAD3 Activity
It has been reported that SMAD3 physically interacts with C/EBP-δ and inhibits the transcription of C/EBP target genes important for adipocyte differentiation (46). To probe the regulatory relationship linking iron to SMAD3 and C/EBP-δ, we first measured CEBPD mRNA levels and found that FAC up-regulated CEBPD. Then co-treatment with SIS3 and FAC significantly decreased these levels (Fig. 7A). This finding posits a crucial role for SMAD3 in FAC-induced CEBPD gene regulation. In testing TGF-β-mediated basal CEBPD regulation, we found that anti-TGF-β1/2/3 antibody alone increased CEBPD mRNA levels by a modest ∼1.3-fold relative to control (Fig. 7B). FAC increased CEBPD mRNA levels by ∼2-fold relative to controls, although levels following co-treatment with FAC and anti-TGF-β1/2/3 antibody were not significantly different from those following FAC treatment alone (Fig. 7C). The trends in these neutralizing antibody experiments correlate with those found when measuring C3 mRNA levels (Fig. 2, C and D).
FIGURE 7.

Iron-induced CEBPD mRNA expression and C/EBP-δ protein nuclear translocation are dependent on SMAD3 activity. A, CEBPD mRNA levels in ARPE-19 cells are decreased by co-treatment with SIS3 and FAC compared with FAC alone, which is increased relative to control. B, CEBPD mRNA levels are increased with addition of neutralizing antibody anti-TGF-β1/2/3 (10 μg/ml; 2 days (d)) when compared with control or IgG treatment conditions. C, CEBPD mRNA levels are elevated with FAC treatment but not significantly changed with co-treatment with anti-TGF-β1/2/3 antibody and FAC. D, Western blot and densitometry analysis for C/EBP-δ in non-nuclear (Non-Nucl) and nuclear (Nucl) lysates of cells treated with FAC in a time course. C/EBP-δ protein levels in the non-nuclear fraction show a time-dependent increase. Nuclear accumulation of C/EBP-δ occurs with the duration of FAC treatment. E, knockdown of SMAD3 in sh-SMAD3 cells impaired FAC-induced increases in nuclear C/EBP-δ when compared with sh-null and untransduced cells. Basal C/EBP-δ levels increased in the sh-SMAD3 cell line relative to the controls. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant).
FAC also increased non-nuclear and nuclear C/EBP-δ protein levels in a time-dependent manner (Fig. 7D). Next, we aimed to understand the role of SMAD3 in iron-induced C/EBP-δ nuclear accumulation by comparing nuclear C/EBP-δ levels in the absence or presence of FAC for the sh-SMAD3 and control cell lines. We showed that SMAD3 knockdown blocked the iron-induced increase within the nuclear as well as the non-nuclear compartments (Fig. 7E). Interestingly, knockdown of SMAD3 elevated basal C/EBP-δ protein levels in both compartments in the absence of iron stimulus. In agreement with protein quantitation, CEBPD mRNA levels were increased in both the puromycin-selected (Fig. 7E) and unselected sh-SMAD3 cells relative to controls (data not shown). Taken together, these results suggest that SMAD3 mediates FAC-induced increases in CEBPD mRNA and C/EBP-δ protein in both the non-nuclear and nuclear compartments.
Iron Can Effect Transcriptional Up-regulation of C3 via the bZIP1 Domain of the C3 Promoter
It has been reported that C/EBP-δ binds to the bZIP1 domain of the C3 promoter following stimulation by IL-1 (34, 47, 48). To determine whether iron can similarly stimulate the C3 promoter, we generated a set of luciferase constructs harboring four overlapping but discretely sized C3 promoter fragments and transfected them into ARPE-19 cells (Fig. 8A). Cells transfected with each of the four fragments, ∼500 bp, 1 kb, 1.5 kb, or 2.0 kb in length relative to the C3 transcription start site, responded to iron treatment with increased firefly luciferase activity, whereas Renilla luciferase, which served as a transfection control, showed no significant difference between the with or without iron conditions (Fig. 8A). In a follow-up luciferase assay, cells transfected with a vector harboring the wild-type proximal 500-bp promoter fragment were compared with cells transfected to contain the same fragment with either the bZIP1 or bZIP2 domain mutations described previously (34). As shown, bZIP1, but not bZIP2, lost its iron-inducible luciferase activity (Fig. 8B). Therefore, bZIP1, the putative C/EBP-δ binding site, is likely to be important for iron-mediated C3 transcriptional up-regulation.
FIGURE 8.

The bZIP1 domain, a putative C/EBP-δ binding site in the C3 promoter, is critical for iron-induced transcriptional up-regulation of C3. A, ARPE-19 cells transfected with luciferase vectors containing a set of nested but different sized C3 promoter fragments as schematically indicated uniformly displayed responsiveness to FAC treatment. B, cells transfected with the smallest fragment (500 bp) containing a WT sequence, the same fragment with a mutant bZIP1 domain, or the same fragment with a mutant bZIP2 domain are treated with or without FAC. The bZIP1 mutant suppressed baseline and FAC-inducible luciferase activities. In A and B, results were corrected for transfection efficiency as measured by Renilla luciferase activity, which was not significantly changed by FAC. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant). TSS, transcription start site; Luc, luciferase; d, days.
Iron-induced C3 Protein Activation and Alternative Complement Pathway Activation Can Be Suppressed by Pharmacologic Inhibition of SMAD3 and ERK1/2
To examine the effect of iron on C3 protein activation, we used ELISA to measure the levels of the cleavage by-product of C3 activation, C3a, in ARPE-19 cell-conditioned media. There was an iron dose-dependent increase in C3a levels (Fig. 9A). We also found a significant decrease in C3a levels when comparing cells that were co-treated with SIS3 or PD98059 inhibitor and FAC with cells treated with FAC alone (Fig. 9B). Evidently, pharmacologic suppression of C3 mRNA (Fig. 3, A and B) and protein (Fig. 3C) levels also translated into decreased C3 protein activation. By approximation, iron induced a comparable 2-fold change in both C3 and C3a levels relative to untreated (Figs. 3C and 9B). Analogously, co-treatment with SIS3 or PD98059 and FAC compared with FAC alone resulted in an ∼25% decrease in both C3 and C3a levels (Figs. 3C and 9B). In addition, co-treatment with both inhibitors and FAC achieved commensurate levels of C3a protein as co-treatment of a single inhibitor and FAC (Fig. 9B). To study the alternative complement pathway activation status, we measured the levels of the Factor B activation product, Factor Ba, and determined that, relative to untreated, FAC induced Factor Ba production. This effect was inhibited by co-treatment with SIS3 or PD98059 inhibitor and FAC (Fig. 9C). Although this finding points to a possible co-regulation of Factor B and C3 expression, it is not further explored herein.
FIGURE 9.
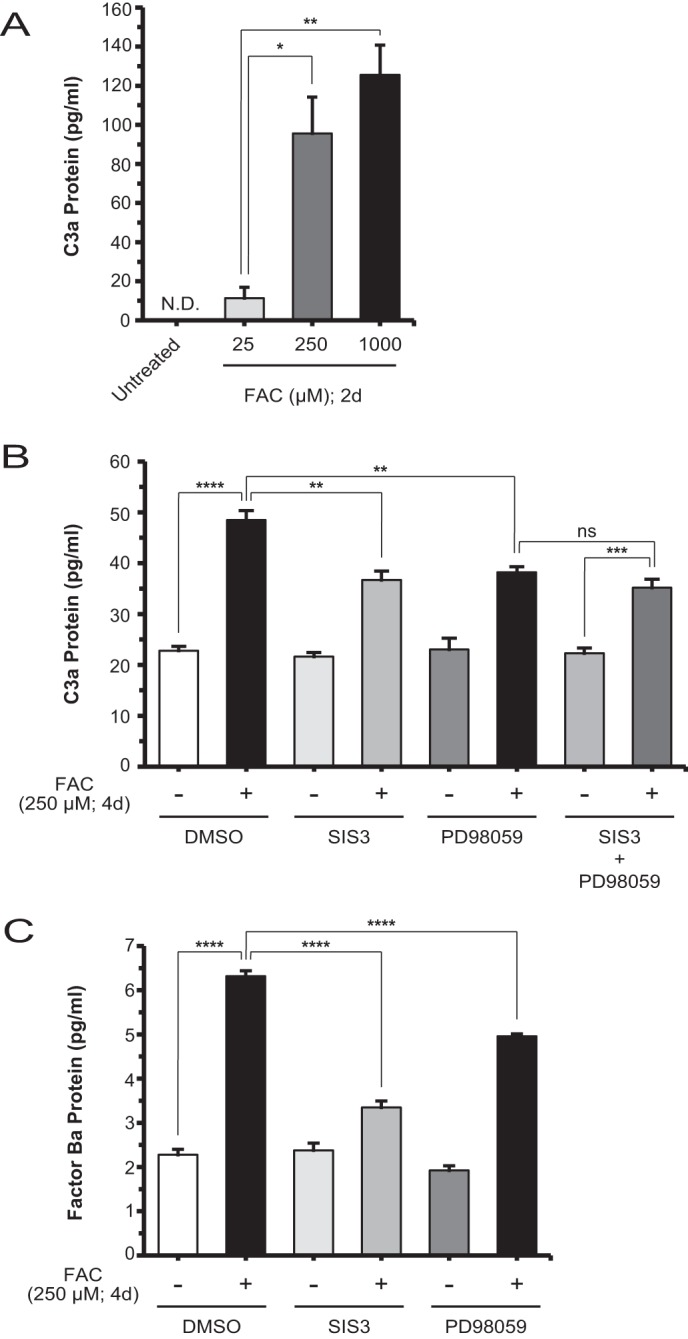
Iron-induced C3 processing and alternative pathway activation can be suppressed by inhibitors of SMAD3 and MEK1/2. A, ELISA of C3a from the conditioned medium of untreated ARPE-19 cells and those treated with different doses of FAC for 2 days (d). Compared with either untreated or 25 μm FAC-treated cells, the 250 μm FAC-treated cells showed a significant increase in C3a production. B, C3a production is significantly decreased by co-treatment with SIS3 or PD98059 and FAC compared with FAC alone at 4 days; co-treatment with both inhibitors and FAC did not further reduce C3a levels compared with co-treatment with a single inhibitor and FAC. C, ELISA using some of the same conditions as in B showed that production of Factor Ba, a marker of alternative complement pathway activation, is decreased by co-treatment with SIS3 or PD98059 and FAC compared with FAC alone. Data are expressed as mean ± S.E. (error bars) (n ≥ 3 with the following statistical notations: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, not significant).
Chronic Iron Overload in the RPE Is Associated with Increased C3 Expression and Activation as Demonstrated by Localized C3d Deposition in Vivo
Our group has published a case report of post-mortem retinal findings in a human patient with aceruloplasminemia showing iron overload in the RPE cells (35). In the present study, we used paraffin-embedded sections containing RPE cells of the macular region where iron deposition is the most concentrated and detected stronger signals for C3d, a C3 activation fragment, than in the same region of an eye from an age-matched patient without retinal disease (Fig. 10A). The fluorescence pattern is punctate and distributed throughout the cell, a pattern that is absent in the sections from the normal eye. Furthermore, a ferroxidase conditional knock-out in the RPE serves as a good ocular disease model for the human disease aceruloplasminemia. As such, these BCre+, Cp−/−, HephF/F mice have a mosaic pattern of gene ablation with select RPE cells exhibiting iron overload detected by L-ferritin antibody staining. Cells showing strong L-ferritin signal are highly correlated with cells showing increased C3d deposition (Fig. 10B).
FIGURE 10.
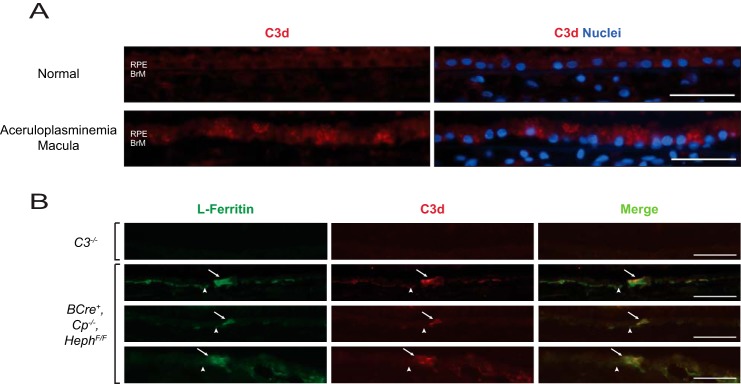
Iron overload in the RPE is associated with C3 activation as demonstrated by localized C3d deposition. A, paraffin-embedded sections of normal and aceruloplasminemia human macular retina with RPE and Bruch's membrane (BrM) labeled are stained with antibodies against C3d (red) and DAPI for nuclei (blue). B, representative images of RPE cells (arrow) and Bruch's membrane (arrowhead) in Optimal Cutting Temperature compound-embedded retina sections of 12-month-old C3−/− and BCre+, Cp−/−, HephF/F mice. Cells with increased L-ferritin levels (green; left panels) have an associated increase in C3d deposition (red; middle panels). The merged images demonstrating co-localization are shown in the right panels. Scale bars are equivalent to 50 μm.
DISCUSSION
The main findings of our study suggest a model, schematically diagrammed in Fig. 11, as follows. First, increased iron in RPE cells stimulated the phosphorylation of ERK1/2 followed by SMAD3 (Ser-213) phosphorylation in a pathway leading to C3 up-regulation. Second, iron induced increases in linker region phosphorylation, i.e. p-SMAD3 (Ser-213), and decreases in C-terminal phosphorylation, i.e. p-SMAD3 (Ser-423/425). Together these changes represent the effects of cross-talk between an iron-induced non-canonical TGF-β pathway and an intrinsic, canonical TGF-β signaling pathway. Third, SMAD3 mediated the iron-induced up-regulation and nuclear accumulation of C/EBP-δ, a transcriptional inducer of C3. Fourth, the bZIP1 domain, previously identified as a C/EBP-δ-binding region within the C3 promoter, was responsive to iron stimulation. Fifth, activation of C3 protein and Factor B, an activator in the alternative pathway, was induced by increased iron. Sixth, in vivo studies supported the molecular association between RPE intracellular iron overload and increased deposition of activated C3 fragments.
FIGURE 11.
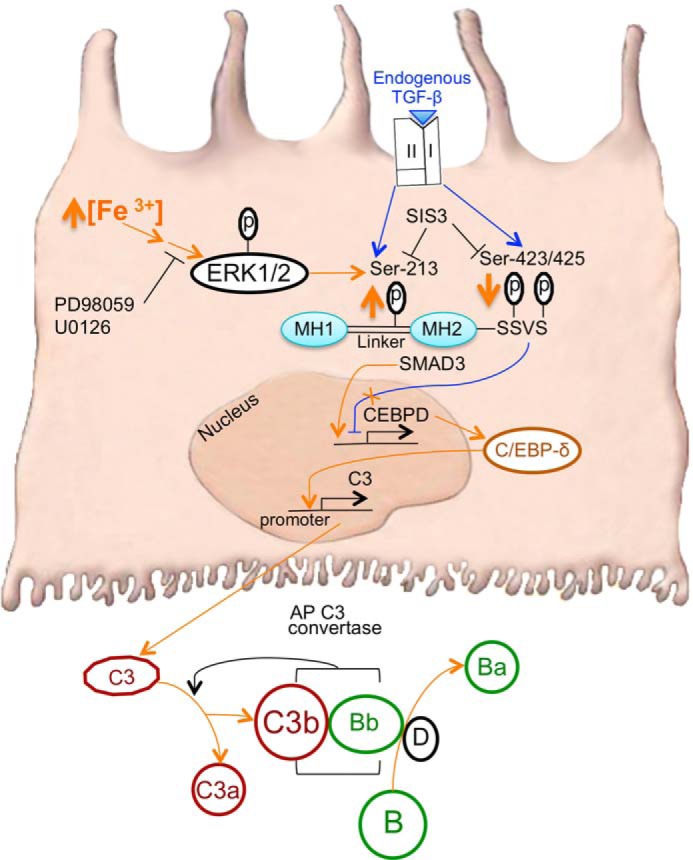
The molecular mechanism of iron-induced RPE C3 production involves ERK1/2, SMAD3, and C/EBP-δ signaling. Increased intracellular iron (Fe3+) in RPE cells treated with FAC stimulates the phosphorylation of ERK1/2 followed by SMAD3 linker (Ser-213) phosphorylation (orange up arrow) in a non-canonical TGF-β pathway leading to up-regulation of C3, the central molecule of the complement cascade (the FAC-induced pathway is delineated by orange arrows). MEK1/2 inhibitor PD98059 and SMAD3 inhibitor SIS3 can block FAC-induced C3 up-regulation. FAC also results in decreased phosphorylation of SMAD3 C-terminal residues (Ser-423/425) at the SSVS motif (orange down arrow). Through SMAD3, FAC increases CEBPD mRNA and C/EBP-δ protein levels possibly by inducing CEBPD expression and relieving SMAD3-mediated inhibition at baseline (orange X over blue inhibitory line). The p-SMAD3 (Ser-213) complex remains extranuclear. In addition, FAC promotes the nuclear accumulation of C/EBP-δ protein. C/EBP-δ likely binds to the bZIP1 domain of the C3 promoter, inducing C3 expression. Once translated into protein, C3 is secreted and cleaved into C3a and C3b. Concurrently, Factor D catalyzes the activation of Factor B to form Bb and the cleavage product Factor Ba. Factors C3b and Bb together form the alternative pathway (AP) C3 convertase to amplify the activation of C3 by forming more C3b and C3a. The iron-induced non-canonical TGF-β pathway (orange) may cross-talk with the basal, canonical TGF-β pathway (blue; see “Discussion”), which is initiated by the binding of TGF-β ligand to the TGF-β receptor complex (types I and II) at the plasma membrane. Endogenous TGF-β binding maintains the phosphorylation levels of SMAD3 at both Ser-213 and Ser-423/425, ultimately acting as a negative regulator of basal C3 expression. Stimulatory arrows and inhibitory lines represent a functional link between the subsequent entities, not necessarily a direct interaction.
The mechanistic findings in the present study are relevant to AMD as the disease is associated with elevated RPE iron levels (20). The increased iron leads to oxidative stress by Fenton chemistry in RPE cells, likely overwhelming their antioxidant defenses. As a general concept, oxidative stress is considered as an inciting factor of complement overactivation in the RPE (30, 49, 50). To determine whether iron can up-regulate RPE complement immune-mediated defenses, we treated ARPE-19 cells with a standard range of FAC doses (51) and measured C3 mRNA and protein levels. C3 activation indicates a mobilized arm of innate immunity and a possible buildup of an adaptive immune response (11, 12). We have for the first time provided evidence that iron triggers the transcriptional up-regulation, protein secretion, and proteolytic activation of C3. Several other transition metals did not lead to C3 up-regulation in the same experimental model. We have also identified components of the TGF-β signaling superfamily that mediate iron-induced C3 expression.
Exogenous TGF-β1 ligand stimulates the canonical pathway (42, 52), resulting in repression of C3 and induction of PAI-1 mRNA levels. These findings demonstrate that in ARPE-19 cells the machinery of the canonical TGF-β pathway is intact. When endogenous TGF-β ligands were neutralized in the absence of FAC, the resultant up-regulation of C3 and down-regulation of PAI-1 mRNA levels revealed the regulatory role of the canonical TGF-β pathway at baseline. Because addition of anti-TGF-β1/2/3 antibody alone up-regulated C3 expression, one could hypothesize that signaling events of the iron-induced non-canonical TGF-β pathway could cross-talk with those of the canonical pathway, leading to inhibition of the inhibitory effect of the latter on C3 expression. Consistent with this hypothesis, we found that iron treatment resulted in a modest but significant decrease in endogenous TGF-β1 levels, thereby attenuating canonical signaling. Apparently, iron-mediated effects on the non-canonical and canonical TGF-β pathways act together to up-regulate C3. We further explored the relative contributions of these two pathways to C3 up-regulation. The up-regulation of C3 as an outcome of activated ERK1/2 and SMAD3 in non-canonical signaling was ∼1.5-fold more robust than the effect of neutralizing endogenous TGF-β signaling (Fig. 2, C and D). Furthermore, co-treatment with anti-TGF-β1/2/3 antibody and iron did not show any additive effect on C3 up-regulation when compared with iron alone. Taken together, it is plausible that inhibition of the inhibitory canonical TGF-β signaling plays an adjunctive role in iron-induced C3 up-regulation.
In the context of iron stimulation, the regulatory cross-talk between these two pathways may be mediated in part by ERK1/2. ERK1/2 is thought to have a pathogenic role in both atrophic and neovascular AMD because p-ERK1/2 is increased in human geographic atrophy retinas (53), and RAS/ERK signaling is implicated in oxidative stress-mediated VEGF secretion by ARPE-19 cells (54). ERK1/2 has been shown previously to phosphorylate several SMAD3 linker region residues in a variety of cellular contexts (31, 41, 42, 55, 56). In the present study, we observed that iron-induced activation of ERK1/2, but not p38 or JNK1/2/3, up-regulated C3 in a non-canonical TGF-β pathway. Furthermore, the MEK1/2 inhibitor PD98059 blocked iron-induced increases in p-SMAD3 (Ser-213). Taken together, this suggests that iron activates ERK1/2 to phosphorylate SMAD3 at Ser-213, which likely contributes to C3 up-regulation. Meanwhile, phosphorylation of C-terminal residues (Ser-423/425) was not affected by ERK1/2 inhibition in the absence or presence of iron (Fig. 4C). Although induction of the TGF-β1 target gene PAI-1 is SMAD3 C-terminal phosphorylation-dependent (57, 58), phosphorylation of the linker region by activated ERK1/2 may be important for the inhibitory effect of iron on PAI-1 expression. We have shown that inhibition of ERK1/2 activity restored iron-suppressed PAI-1 mRNA levels back to baseline (Fig. 3D). This finding implies that phosphorylation of the SMAD3 linker region plays a regulatory role in canonical, C-terminal phosphorylation-dependent gene expression. Nevertheless, iron appears to utilize spatially distinct SMAD3 phospho-isoforms as a platform to convey different signals to effector genes as exemplified by the transcriptional up-regulation of C3 versus the down-regulation of PAI-1.
In ARPE-19 cells, the 150-kDa band detected by p-SMAD3 (Ser-213) antibody and up-regulated by iron contains SMAD3. Results from antibody specificity tests, SIS3 inhibition, and targeted knockdown of SMAD3 support this finding. Previously, it was determined that SMAD3-containing complexes are distributed over a wide range of molecular masses (59), and 150 kDa is within this range. However, this 150-kDa complex should have dissociated under SDS-PAGE conditions. The lack of dissociation then raises the possibility that either the complex is extraordinarily stable or it represents a covalently modified form of SMAD3 that cannot be effectively disrupted by SDS. In post-mortem human RPE/choroid tissues, the most significant band detected by the same antibody against p-SMAD3 (Ser-213) was higher than 52 kDa (∼70 kDa; data not shown). Taken together, these results suggest that the apparent molecular masses of p-SMAD3 (Ser-213)-containing species are context-dependent. The 150-kDa, p-SMAD3 (Ser-213)-containing species remained in the non-nuclear compartment after iron treatment. Phosphorylation at Ser-213 may affect the interaction of SMAD3 with other signaling mediators, one of which could be transcription factor C/EBP-δ, to effect the nuclear accumulation of the latter. SMAD3 has been shown to directly interact with C/EBP-δ in the context of adipocyte differentiation (46), although the details of how p-SMAD3 (Ser-213) in ARPE-19 cells may regulate this interaction remain unknown.
C/EBP-δ acts as the major protein responsible for acute phase C3 gene expression by binding to the bZIP1 domain in the promoter sequence (34, 47, 48). The bZIP1 domain is required for basal and cytokine-inducible C3 expression. Similarly, we found this domain to be important for iron-induced C3 transcription in ARPE-19 cells. Also, we showed that iron-induced C3 up-regulation was accompanied by increased CEBPD mRNA and C/EBP-δ protein levels. SMAD3 activity appears to mediate these increases because SIS3 blocked iron-induced increases in CEBPD mRNA, and there was no significant difference in nuclear C/EBP-δ levels between untreated and iron-treated sh-SMAD3 cells. It should be noted that C/EBP-δ protein levels were elevated in sh-SMAD3 cells compared with the control cell lines, suggesting that SMAD3 inhibits basal C/EBP-δ expression. In other words, the observed iron-induced increases in C/EBP-δ levels may result at least in part from the relief of SMAD3 inhibition of basal C/EBP-δ expression. This mechanistic model is consistent with that of vascular smooth muscle cells in an anti-inflammatory state, in which TGF-β1-stimulated SMAD3 represses C/EBP-δ expression (60). In a further study of basal repression of CEBPD, neutralization of endogenous TGF-β ligands in the absence of FAC resulted in a modest up-regulation of CEBPD that was comparable in magnitude with the iron-induced up-regulation of CEBPD (Fig. 7, B and C). Thus, similar to the regulation of C3 mRNA levels, CEBPD mRNA levels are likely induced by iron through a non-canonical TGF-β pathway and through the relief of SMAD3-mediated basal inhibition.
In AMD pathogenesis, it is thought that overactivation of the complement cascade, especially in the alternative pathway, contributes to the chronic inflammatory state. Our findings support the concept of a locally produced complement-mediated disease process as iron insult of the RPE cell resulted not only in increased C3 transcription but also in protein secretion and activation. With more activated C3 in the RPE milieu, the complement cascade is mobilized to initiate immune-mediated inflammation. Within the cascade, formation of the alternative pathway C3 convertase requires Factor B and generates by-product Factor Ba so that more C3 can be activated (14, 61). Although C3 activation and its downstream effects on RPE pathology are complex, our findings of increased C3a and Factor Ba levels are still suggestive of C3 and alternative pathway activation, respectively. Befitting this complexity, although pharmacologic blockade of ERK1/2 or SMAD3 incompletely suppressed iron-induced C3a formation, blockade of both targets provided no synergistic suppression (Fig. 9B). This supports our model of ERK1/2 and SMAD3 functioning in a single iron-mediated pathway. The incomplete suppression of C3a by the inhibitors singly or in combination points to the likely involvement of other iron-induced factors or processes in C3 activation. Additionally, we provide strong evidence that excess intra-RPE iron can lead to local complement dysregulation as reflected by C3d accumulation in the individual RPE cells of an aceruloplasminemia patient and an animal model of RPE-specific iron overload. In AMD, localized complement production and activation within the RPE layer may be more active in individuals with disease-associated complement gene polymorphisms. Since ERK1/2, SMAD3, C/EBP-δ, and endogenous TGF-β are now implicated in RPE iron-C3 dysregulation, these signaling molecules may serve as useful therapeutic targets for ameliorating RPE pathology in AMD.
Supplementary Material
Acknowledgments
We thank J. Samuel Zigler, Jr. for critical reading of the manuscript and Xiaomin Ma for preparing the schematic illustration.
This work was supported, in whole or in part, by National Institutes of Health Grants R01EY015240 (to J. L. D.), R01EY023709 (to W. S.), and T32EY007035, a vision science training grant (to the University of Pennsylvania). This work was also supported by Research to Prevent Blindness, The Beckman Institute for Macular Research, the Paul and Evanina Bell Mackall Foundation Trust, and the F. M. Kirby Foundation.

This article contains a supplemental table.
- AMD
- age-related macular degeneration
- C3
- complement component 3
- RPE
- retinal pigment epithelial/epithelium
- C/EBP-δ
- CCAAT/enhancer-binding protein-δ
- bZIP
- basic leucine zipper
- SIS3
- specific inhibitor of SMAD3
- FAC
- ferric ammonium citrate
- qRT-PCR
- quantitative RT-PCR
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- p-
- phospho-
- TFRC
- transferrin receptor
- PAI-1
- plasminogen activator inhibitor type 1.
REFERENCES
- 1. Dunaief J. L. (2006) Iron induced oxidative damage as a potential factor in age-related macular degeneration: The Cogan Lecture. Invest. Ophthalmol. Vis. Sci. 47, 4660–4664 [DOI] [PubMed] [Google Scholar]
- 2. Zipfel P., Lauer N., Skerka C. (2010) in Inflammation and Retinal Disease: Complement Biology and Pathology (Lambris J. D., Adamis A. P., eds) pp. 9–24, Springer, New York [Google Scholar]
- 3. Johnson L. V., Leitner W. P., Staples M. K., Anderson D. H. (2001) Complement activation and inflammatory processes in drusen formation and age-related macular degeneration. Exp. Eye Res. 73, 887–896 [DOI] [PubMed] [Google Scholar]
- 4. Grassmann F., Heid I. M., Weber B. H. (2014) Genetic risk models in age-related macular degeneration. Adv. Exp. Med. Biol. 801, 291–300 [DOI] [PubMed] [Google Scholar]
- 5. Edwards A. O., Ritter R., 3rd, Abel K. J., Manning A., Panhuysen C., Farrer L. A. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 [DOI] [PubMed] [Google Scholar]
- 6. Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., Spencer K. L., Kwan S. Y., Noureddine M., Gilbert J. R., Schnetz-Boutaud N., Agarwal A., Postel E. A., Pericak-Vance M. A. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 [DOI] [PubMed] [Google Scholar]
- 7. Klein R. J., Zeiss C., Chew E. Y., Tsai J.-Y., Sackler R. S., Haynes C., Henning A. K., SanGiovanni J. P., Mane S. M., Mayne S. T., Bracken M. B., Ferris F. L., Ott J., Barnstable C., Hoh J. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gold B., Merriam J. E., Zernant J., Hancox L. S., Taiber A. J., Gehrs K., Cramer K., Neel J., Bergeron J., Barile G. R., Smith R. T., AMD Genetics Clinical Study Group, Hageman G. S., Dean M., Allikmets R. (2006) Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 38, 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yates J. R., Sepp T., Matharu B. K., Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Hayward C., Morgan J., Wright A. F., Armbrecht A. M., Dhillon B., Deary I. J., Redmond E., Bird A. C., Moore A. T., and Genetic Factors in AMD Study Group (2007) Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 357, 553–561 [DOI] [PubMed] [Google Scholar]
- 10. Seddon J. M., Yu Y., Miller E. C., Reynolds R., Tan P. L., Gowrisankar S., Goldstein J. I., Triebwasser M., Anderson H. E., Zerbib J., Kavanagh D., Souied E., Katsanis N., Daly M. J., Atkinson J. P., Raychaudhuri S. (2013) Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 45, 1366–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan B. P., Marchbank K. J., Longhi M. P., Harris C. L., Gallimore A. M. (2005) Complement: central to innate immunity and bridging to adaptive responses. Immunol. Lett. 97, 171–179 [DOI] [PubMed] [Google Scholar]
- 12. Markiewski M. M., Lambris J. D. (2007) The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 171, 715–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anderson D. H., Mullins R. F., Hageman G. S., Johnson L. V. (2002) A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 134, 411–431 [DOI] [PubMed] [Google Scholar]
- 14. Sahu A., Lambris J. D. (2001) Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 180, 35–48 [DOI] [PubMed] [Google Scholar]
- 15. Chen M., Muckersie E., Robertson M., Forrester J. V., Xu H. (2008) Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp. Eye Res. 87, 543–550 [DOI] [PubMed] [Google Scholar]
- 16. Pickering M. C., Cook H. T., Warren J., Bygrave A. E., Moss J., Walport M. J., Botto M. (2002) Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat. Genet. 31, 424–428 [DOI] [PubMed] [Google Scholar]
- 17. Chen M., Forrester J. V., Xu H. (2007) Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 84, 635–645 [DOI] [PubMed] [Google Scholar]
- 18. Coffey P. J., Gias C., McDermott C. J., Lundh P., Pickering M. C., Sethi C., Bird A., Fitzke F. W., Maass A., Chen L. L., Holder G. E., Luthert P. J., Salt T. E., Moss S. E., Greenwood J. (2007) Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. U.S.A. 104, 16651–16656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perkins S. J., Nan R., Li K., Khan S., Miller A. (2012) Complement factor H-ligand interactions: self-association, multivalency and dissociation constants. Immunobiology 217, 281–297 [DOI] [PubMed] [Google Scholar]
- 20. Hahn P., Milam A. H., Dunaief J. L. (2003) Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch's membrane. Arch. Ophthalmol. 121, 1099–1105 [DOI] [PubMed] [Google Scholar]
- 21. Dixon S. J., Stockwell B. R. (2014) The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 [DOI] [PubMed] [Google Scholar]
- 22. Flint D. H., Tuminello J. F., Emptage M. H. (1993) The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 268, 22369–22376 [PubMed] [Google Scholar]
- 23. Hahn P., Qian Y., Dentchev T., Chen L., Beard J., Harris Z. L., Dunaief J. L. (2004) Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 101, 13850–13855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hadziahmetovic M., Dentchev T., Song Y., Haddad N., He X., Hahn P., Pratico D., Wen R., Harris Z. L., Lambris J. D., Beard J., Dunaief J. L. (2008) Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest. Ophthalmol. Vis. Sci. 49, 2728–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song D., Song Y., Hadziahmetovic M., Zhong Y., Dunaief J. L. (2012) Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic. Biol. Med. 53, 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hadziahmetovic M., Pajic M., Grieco S., Song Y., Song D., Li Y., Cwanger A., Iacovelli J., Chu S., Ying G., Connelly J., Spino M., Dunaief J. L. (2012) The oral iron chelator deferiprone protects against retinal degeneration induced through diverse mechanisms. Transl. Vis. Sci. Technol. 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fritsche L. G., Chen W., Schu M., Yaspan B. L., Yu Y., Thorleifsson G., Zack D. J., Arakawa S., Cipriani V., Ripke S., Igo R. P., Jr., Buitendijk G. H., Sim X., Weeks D. E., Guymer R. H., Merriam J. E., Francis P. J., Hannum G., Agarwal A., Armbrecht A. M., Audo I., Aung T., Barile G. R., Benchaboune M., Bird A. C., Bishop P. N., Branham K. E., Brooks M., Brucker A. J., Cade W. H., Cain M. S., Campochiaro P. A., Chan C. C., Cheng C. Y., Chew E. Y., Chin K. A., Chowers I., Clayton D. G., Cojocaru R., Conley Y. P., Cornes B. K., Daly M. J., Dhillon B., Edwards A. O., Evangelou E., Fagerness J., Ferreyra H. A., Friedman J. S., Geirsdottir A., George R. J., Gieger C., Gupta N., Hagstrom S. A., Harding S. P., Haritoglou C., Heckenlively J. R., Holz F. G., Hughes G., Ioannidis J. P., Ishibashi T., Joseph P., Jun G., Kamatani Y., Katsanis N. N., Keilhauer C., Khan J. C., Kim I. K., Kiyohara Y., Klein B. E., Klein R., Kovach J. L., Kozak I., Lee C. J., Lee K. E., Lichtner P., Lotery A. J., Meitinger T., Mitchell P., Mohand-Saïd S., Moore A. T., Morgan D. J., Morrison M. A., Myers C. E., Naj A. C., Nakamura Y., Okada Y., Orlin A., Ortube M. C., Othman M. I., Pappas C., Park K. H., Pauer G. J., Peachey N. S., Poch O., Priya R. R., Reynolds R., Richardson A. J., Ripp R., Rudolph G., Ryu E., Sahel J. A., Schaumberg D. A., Scholl H. P., Schwartz S. G., Scott W. K., Shahid H., Sigurdsson H., Silvestri G., Sivakumaran T. A., Smith R. T., Sobrin L., Souied E. H., Stambolian D. E., Stefansson H., Sturgill-Short G. M., Takahashi A., Tosakulwong N., Truitt B. J., Tsironi E. E., Uitterlinden A. G., van Duijn C. M., Vijaya L., Vingerling J. R., Vithana E. N., Webster A. R., Wichmann H. E., Winkler T. W., Wong T. Y., Wright A. F., Zelenika D., Zhang M., Zhao L., Zhang K., Klein M. L., Hageman G. S., Lathrop G. M., Stefansson K., Allikmets R., Baird P. N., Gorin M. B., Wang J. J., Klaver C. C., Seddon J. M., Pericak-Vance M. A., Iyengar S. K., Yates J. R., Swaroop A., Weber B. H., Kubo M., Deangelis M. M., Léveillard T., Thorsteinsdottir U., Haines J. L., Farrer L. A., Heid I. M., Abecasis G. R. (2013) Seven new loci associated with age-related macular degeneration. Nat. Genet. 45, 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guymer R. H., Tao L. W., Goh J. K., Liew D., Ischenko O., Robman L. D., Aung K., Cipriani T., Cain M., Richardson A. J., Baird P. N., Langham R. (2011) Identification of urinary biomarkers for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 52, 4639–4644 [DOI] [PubMed] [Google Scholar]
- 29. Feng X.-H., Derynck R. (2005) Specificity and versatility in TGF-β signaling through SMADS. Annu. Rev. Cell Dev. Biol. 21, 659–693 [DOI] [PubMed] [Google Scholar]
- 30. Kunchithapautham K., Atkinson C., Rohrer B. (2014) Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J. Biol. Chem. 289, 14534–14546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kretzschmar M., Doody J., Timokhina I., Massagué J. (1999) A mechanism of repression of TGFβ/Smad signaling by oncogenic Ras. Genes Dev. 13, 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gao S., Alarcón C., Sapkota G., Rahman S., Chen P.-Y., Goerner N., Macias M. J., Erdjument-Bromage H., Tempst P., Massagué J. (2009) Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-β signaling. Mol. Cell 36, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tusher V. G., Tibshirani R., Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. 98, 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Juan T. S., Wilson D. R., Wilde M. D., Darlington G. J. (1993) Participation of the transcription factor C/EBP delta in the acute-phase regulation of the human gene for complement component C3. Proc. Natl. Acad. Sci. 90, 2584–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wolkow N., Song Y., Wu T.-D., Qian J., Guerquin-Kern J.-L., Dunaief J. L. (2011) Aceruloplasminemia: retinal histopathology and iron-mediated melanosome degradation. Arch. Ophthalmol. 129, 1466–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolkow N., Song D., Song Y., Chu S., Hadziahmetovic M., Lee J. C., Iacovelli J., Grieco S., Dunaief J. L. (2012) Ferroxidase hephaestin's cell-autonomous role in the retinal pigment epithelium. Am. J. Pathol. 180, 1614–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hadziahmetovic M., Kumar U., Song Y., Grieco S., Song D., Li Y., Tobias J. W., Dunaief J. L. (2012) Microarray analysis of murine retinal light damage reveals changes in iron regulatory, complement, and antioxidant genes in the neurosensory retina and isolated RPE. Invest. Ophthalmol. Vis. Sci. 53, 5231–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muñoz P., Humeres A., Elgueta C., Kirkwood A., Hidalgo C., Núñez M. T. (2011) Iron mediates N-methyl-d-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 286, 13382–13392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hadziahmetovic M., Song Y., Ponnuru P., Iacovelli J., Hunter A., Haddad N., Beard J., Connor J. R., Vaulont S., Dunaief J. L. (2011) Age-dependent retinal iron accumulation and degeneration in hepcidin knockout mice. Invest. Ophthalmol. Vis. Sci. 52, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kamato D., Rostam M. A., Piva T. J., Babaahmadi Rezaei H., Getachew R., Thach L., Bernard R., Zheng W., Little P. J., Osman N. (2014) Transforming growth factor β-mediated site-specific Smad linker region phosphorylation in vascular endothelial cells. J. Pharm. Pharmacol. 66, 1722–1733 [DOI] [PubMed] [Google Scholar]
- 41. Matsuura I., Wang G., He D., Liu F. (2005) Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry 44, 12546–12553 [DOI] [PubMed] [Google Scholar]
- 42. Wang G., Matsuura I., He D., Liu F. (2009) Transforming growth factor-β-inducible phosphorylation of Smad3. J. Biol. Chem. 284, 9663–9673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bae E., Kim S.-J., Hong S., Liu F., Ooshima A. (2012) Smad3 linker phosphorylation attenuates Smad3 transcriptional activity and TGF-β1/Smad3-induced epithelial-mesenchymal transition in renal epithelial cells. Biochem. Biophys. Res. Commun. 427, 593–599 [DOI] [PubMed] [Google Scholar]
- 44. Hough C., Radu M., Doré J. J. (2012) TGF-β induced Erk phosphorylation of Smad linker region regulates Smad signaling. PLoS One 7, e42513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jinnin M., Ihn H., Tamaki K. (2006) Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-β1-induced extracellular matrix expression. Mol. Pharmacol. 69, 597–607 [DOI] [PubMed] [Google Scholar]
- 46. Choy L., Derynck R. (2003) Transforming growth factor-β inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J. Biol. Chem. 278, 9609–9619 [DOI] [PubMed] [Google Scholar]
- 47. Wilson D. R., Juan T. S., Wilde M. D., Fey G. H., Darlington G. J. (1990) A 58-base-pair region of the human C3 gene confers synergistic inducibility by interleukin-1 and interleukin-6. Mol. Cell. Biol. 10, 6181–6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maranto J., Rappaport J., Datta P. (2008) Regulation of complement component C3 in astrocytes by IL-1β and morphine. J. Neuroimmune Pharmacol. 3, 43–51 [DOI] [PubMed] [Google Scholar]
- 49. Handa J. T. (2012) How does the macula protect itself from oxidative stress? Mol. Aspects Med. 33, 418–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thurman J. M., Renner B., Kunchithapautham K., Ferreira V. P., Pangburn M. K., Ablonczy Z., Tomlinson S., Holers V. M., Rohrer B. (2009) Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 284, 16939–16947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Salvador G. A., Oteiza P. I. (2011) Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. NeuroToxicology 32, 75–82 [DOI] [PubMed] [Google Scholar]
- 52. Massagué J. (2012) TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dridi S., Hirano Y., Tarallo V., Kim Y., Fowler B. J., Ambati B. K., Bogdanovich S., Chiodo V. A., Hauswirth W. W., Kugel J. F., Goodrich J. A., Ponicsan S. L., Hinton D. R., Kleinman M. E., Baffi J. Z., Gelfand B. D., Ambati J. (2012) ERK1/2 activation is a therapeutic target in age-related macular degeneration. Proc. Natl. Acad. Sci. 109, 13781–13786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kunchithapautham K., Rohrer B. (2011) Sub-lytic membrane-attack-complex (MAC) activation alters regulated rather than constitutive vascular endothelial growth factor (VEGF) secretion in retinal pigment epithelium monolayers. J. Biol. Chem. 286, 23717–23724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tarasewicz E., Jeruss J. S. (2012) Phospho-specific Smad3 signaling: impact on breast oncogenesis. Cell Cycle. 11, 2443–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang G. (2008) The Smad3 Linker Region: Transcriptional Activity and Phosphorylation-mediated Regulation. Ph.D. thesis, Rutgers, The State University of New Jersey, New Brunswick, NJ [Google Scholar]
- 57. Liu X., Sun Y., Constantinescu S. N., Karam E., Weinberg R. A., Lodish H. F. (1997) Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10669–10674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hua X., Liu X., Ansari D. O., Lodish H. F. (1998) Synergistic cooperation of TFE3 and Smad proteins in TGF-β-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 12, 3084–3095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jayaraman L., Massague J. (2000) Distinct oligomeric states of SMAD proteins in the transforming growth factor-β pathway. J. Biol. Chem. 275, 40710–40717 [DOI] [PubMed] [Google Scholar]
- 60. Feinberg M. W., Watanabe M., Lebedeva M. A., Depina A. S., Hanai J., Mammoto T., Frederick J. P., Wang X.-F., Sukhatme V. P., Jain M. K. (2004) Transforming growth factor-β1 inhibition of vascular smooth muscle cell activation is mediated via Smad3. J. Biol. Chem. 279, 16388–16393 [DOI] [PubMed] [Google Scholar]
- 61. Kolb W. P., Morrow P. R., Tamerius J. D. (1989) Ba and Bb fragments of factor B activation: fragment production, biological activities, neoepitope expression and quantitation in clinical samples. Complement Inflamm. 6, 175–204 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.