

Background: In striatum, cortical glutamatergic and midbrain dopaminergic inputs are integrated via cAMP.
Results: PDE10A, the major cAMP-hydrolyzing enzyme in striatum, is targeted into a signaling complex containing the scaffolding proteins AKAP150, PSD95, and the NMDA receptor and released upon phosphorylation.
Conclusion: Targeting of PDE10 is under control of cAMP/PKA activity.
Significance: Phosphorylation-dependent release of PDE10 gives rise to a feed forward mechanism.
Keywords: A-kinase Anchoring Protein (AKAP), Cell Signaling, Cyclic AMP (cAMP), Phosphodiesterases, Protein Complex, Protein Kinase A (PKA)
Abstract
Phosphodiesterase 10A (PDE10A) is a dual substrate PDE that can hydrolyze both cGMP and cAMP. In brain, PDE10A is almost exclusively expressed in the striatum. In several studies, PDE10A has been implicated in regulation of striatal output using either specific inhibitors or PDE10A knock-out mice and has been suggested as a promising target for novel antipsychotic drugs. In striatal medium spiny neurons, PDE10A is localized at the plasma membrane and in dendritic spines close to postsynaptic densities. In the present study, we identify PDE10A as the major cAMP PDE in mouse striatum and monitor PKA-dependent PDE10A phosphorylation. With recombinantly expressed PDE10A we demonstrate that phosphorylation does not alter PDE10A activity. In striatum, PDE10A was found to be associated with the A kinase anchoring protein AKAP150 suggesting the existence of a multiprotein signaling complex localizing PDE10A to a specific functional context at synaptic membranes. Furthermore, the cAMP effector PKA, the NMDA receptor subunits NR2A and -B, as well as PSD95, were tethered to the complex. In agreement, PDE10A was almost exclusively found in multiprotein complexes as indicated by migration in high molecular weight fractions in size exclusion chromatography. Finally, affinity of PDE10A to the signaling complexes formed around AKAP150 was reduced by PDE10A phosphorylation. The data indicate that phosphorylation of PDE10 has an impact on the interaction with other signaling proteins and adds an additional line of complexity to the role of PDE10 in regulation of synaptic transmission.
Introduction
To date, 11 families of phosphodiesterases (PDE1 to PDE11)2 have been identified that are differentially expressed in mammalian tissues and cells (1). Phosphodiesterases tightly regulate cyclic nucleotide signaling by hydrolysis of the second messengers cGMP and cAMP. In the central nervous system, comparatively high cyclic nucleotide-degrading activity is found. Here, many neurons express multiple PDEs with different subcellular localization and/or substrate specificities.
PDE10 was initially discovered in brain and is capable of hydrolyzing both second messengers, cAMP and cGMP, but has higher affinity for cAMP (2–4). PDE10 contains a tandem of regulatory GAF domains. cAMP binds to the second of these GAF domains, GAF-B (5), and activates the enzyme at least 3-fold (6). PDE10 is encoded by one gene (PDE10A) that gives rise to several splice variants (7, 8) of which PDE10A2 appears to be the major neuronal form in various species. The highest expression of PDE10A has been detected in the striatum (6, 9) where it is restricted to the GABAergic medium spiny neurons (10–12). These neurons represent ∼95% of striatal neurons and integrate cortical glutamatergic input and midbrain dopaminergic signaling. They form the major input station for the basal ganglia that are involved in planning and modulation of movement pathways and a variety of other cognitive processes. Dysfunction of striatal circuitry is implicated in the pathophysiology of many brain disorders such as Parkinson disease, Huntington disease, schizophrenia, and substance abuse (13–15).
The high expression of PDE10A in medium spiny neurons suggests an important role in modulation of striatal function through cAMP degradation and termination of cAMP/PKA signaling. Consequently, enhancement of cAMP signaling through PDE10A knock-out or pharmacological inhibition of PDE10A has been shown to affect locomotor activity and acquisition of conditioned avoidance (16–19). These results led to the proposal of PDE10A as a promising target for novel antipsychotic therapies.
Here, we describe PDE10A as the major cAMP PDE in striatum. Interaction of PDE10A with a postsynaptic signaling complex is demonstrated by coprecipitation of PDE10 with the scaffold protein AKAP150, PKA as well as PSD95 and NMDA receptor subunits. Such a complex might put PDE10A into the position of a “gate keeper” that limits cAMP accumulation at postsynaptic sites, prevents spreading of synaptic signals into the cell body, and ensures precisely timed phosphorylation and thereby regulation of NMDA receptors. We demonstrate PKA-dependent phosphorylation of PDE10A in striatal slices and identify protein phosphatase 2A (PP2A) as the responsible phosphatase. Moreover, phosphorylation of PDE10A2 does not alter enzymatic properties but releases PDE10A from the identified signaling complex.
MATERIALS AND METHODS
Analysis of Subcellular Distribution of PDE10A
All experiments were performed using 3-month-old male mice of the C57BL/6 strain. After sacrificing the animals, the brain was quickly removed and the striata from the left and right hemisphere were isolated in less than 2 min. The striata of two mice were immediately homogenized in 10 volumes (w/v) of ice-cold lysis buffer (50 mm NaCl, 1 mm EDTA, 2 mm dl-dithiothreitol, 50 mm triethanolamine/hydrochloride, pH 7.4, containing the protease inhibitors phenylmethylsulfonyl fluoride (0.4 mm), benzamidine (0.2 mm), and pepstatin A (1 μm)) using a glass-glass Potter-Elvehjem homogenizer. The lysate was cleared from nuclei and cellular debris by centrifugation at 800 × g (10 min, 4 °C) and then recentrifuged at 125,000 × g (40 min, 4 °C) to obtain the cytosolic fraction. Membranes were resuspended in the original volume of lysis buffer. Equal volumes of all fractions (corresponding to 20 μg of protein in the homogenate) were applied to SDS-PAGE. Synaptosomal cytosol and membranes were prepared as described (20).
Cell Culture, Cloning, Transfection, and in Vitro Phosphorylation
HEK293 cells were grown in DMEM with 5% heat-inactivated fetal calf serum and 1% penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere. Mouse PDE10A2 was cloned into the pcDNA3.1 zeo+ vector via NheI/NotI sites and 8 μg of vector/75 cm2 bottle were transfected into HEK293 cells using the FuGENE6 protocol (Promega). Transfected cells were harvested 48–72 h post-transfection, washed twice with phosphate-buffered saline, resuspended in 0.5 ml of lysis buffer, and lysed by sonication (two 5-s pulses). Cellular debris was eliminated by centrifugation (800 × g, 10 min, 4 °C). The lysate was split for comparative analysis of phosphorylated versus non-phosphorylated PDE10A2 and preincubated for 10 min with 5 mm MgCl2, 0.5 mm ATP, 1 μm okadaic acid and PhosSTOP (Roche Applied Science) in lysis buffer. In vitro phosphorylation was performed by addition of the catalytic subunit of PKA (0.5 μg, Jena Bioscience) for 30 min at 37 °C. For non-phospho control, ATP and PKA were omitted.
Characterization of pPDE10 Antibody
PDE10A2-expressing HEK293 cells were homogenized in cell extraction buffer (50 mm NaCl, 50 mm triethanolamine/hydrochloride, pH 7.4, containing 0.4 mm phenylmethylsulfonyl fluoride, 0.2 mm benzamidine, 1 μm pepstatin A, and 1 μm okadaic acid) by 15 strokes with a glass-glass Potter-Elvehjem homogenizer. The cell extract was diluted with PKA buffer (0.5 mm ATP, 3 mm MgCl2, 0.5 mg/ml of BSA, 2 mm dl-dithiothreitol, 50 mm triethanolamine/hydrochloride, pH 7.4, final concentrations) and incubated with or without 0.5 μg (800 units) of PKA (Jena Biosciences, PR-318) for 30 min at 37 °C. An aliquot of the phosphorylated sample was saved for Western blot before purification of pPDE10 using the Pierce phosphoprotein enrichment kit (Thermo Scientific) following the suppliers instructions as follows. The sample was diluted 10-fold with the supplied lysis/binding/wash buffer containing CHAPS (0.25%) and then incubated with the column for 1 h at 4 °C with gentle agitation. Flow-through was discarded and the phosphorylated protein was eluted with 1 ml of supplied elution buffer without CHAPS. All samples were applied to a 9% SDS gel. For detection, PDE10 antibody (6) and pPDE10 antibody were used. The pPDE10 antibody was generated against phosphorylated amino acids 11–20 of PDE10A2 in rabbits and purified according to the manufacturer's protocol (PSL, Heidelberg, Germany).
Assay of PDE Activity
PDE activity was measured by the conversion of [32P]cyclic nucleotide monophosphate into [32P]nucleotide monophosphate as described previously (21). Reaction mixtures contained 0.1 μl of the striatal or HEK cell homogenates. Substrate concentrations were 0.03 to 1 μm cAMP or 1 μm cGMP, as indicated. Data are mean ± S.E. of at least three independent experiments performed in duplicates. PDE10A2 activation by the GAF domain ligand 7-CH-cAMP (Biolog) was determined at a substrate concentration of 0.03 μm cAMP. To determine the dissociation rate of cAMP from the PDE10A2 GAF domain, PDE10A2 was preincubated with 3 mm MgCl2 and 10 μm cAMP for 2 min at 37 °C. PDE10A2 was then diluted 100-fold and PDE activity was determined at various time points between 0 and 32 min.
Preparation and Incubation of Striatal Slices
Three-month-old male mice were killed, and brains were removed and placed in ice-cold Krebs-Henseleit (KH) buffer containing the following: 118 mm NaCl, 4.7 mm KCl, 2.55 mm CaCl2, 1.2 mm KH2PO4, 1.2 mm MgSO4, 25 mm NaHCO3, and 27.8 mm d-glucose equilibrated to pH 7.4 by aeration with 95% O2, 5% CO2. Brain slices (coronal, 300 μm thick) were cut using a Vibroslice (NVSLM1, World Precision Instruments). The striatum was isolated and immediately transferred to a nylon net submerged in KH buffer, kept at pH 7.4 by aeration as described above (room temperature). After 1 h of recovery, slices were transferred to incubation chambers containing 10 ml of KH, kept at 37 °C, and equilibrated for a further 30 min. Slices were preincubated with PDE and phosphatase inhibitors for 10 min as indicated, transferred to new incubation chambers containing NMDA with or without phosphatase inhibitors as indicated for an additional 5 min. Slices were instantly frozen in liquid nitrogen. For immunodetection, single slices were homogenized in 240 μl of lysis buffer as described above. SDS-PAGE sample buffer (60 μl, ×4) was immediately added and 20 μl of each sample was applied to electrophoresis.
Immunoprecipitation of PDE10A
The striata of two mice were homogenized in 1 ml of ice-cold Nonidet P-40 buffer (150 mm NaCl, 1.5 mm EDTA, 1% Nonidet P-40, 50 mm Tris, pH 7.4, containing protease inhibitors as above) using a glass-glass Potter-Elvehjem homogenizer. The lysate was cleared by centrifugation at 800 × g (10 min, 4 °C) and added to Dynabeads Protein A (Invitrogen) that were loaded with ∼50 μg of anti-PDE10A antibody (6) per 10 mg of beads. Incubation was for 1 h at 4 °C in the absence or presence of the purified antigen (500 μg). Unspecifically bound proteins were removed by three washes with PBS and precipitated proteins were eluted by boiling in SDS sample buffer (30 μl/mg of Dynabeads) and loaded to SDS gels (15–20 μl per lane). One-tenth of the striatal lysate was applied for input loading.
Immunoprecipitation of Phosphorylated PDE10A
For immunoprecipitation of phosphorylated PDE10, striatal slices from 2 mice were prepared. Phosphorylation was induced by incubation of the slices with papaverine (100 μm) and forskolin (10 μm) for 10 min in the presence of phosphatase inhibitors (0.5 μm okadaic acid, 5 μm cyclosporin A, 0.2 μm calyculin A). Slices were pooled and instantly homogenized in 1 ml of Nonidet P-40 buffer (as above, but additionally containing phosphatase inhibitors) before precipitation by PDE10 antibody coupled to Dynabeads (see above).
Determination of Precipitated PDE Activity
AKAP150 and the NMDA receptor subunit NR2A were precipitated from striata of 2 mice as described above using 10 μg of antibodies (AKAP150 sc-6445; NMDAϵ1 sc-1468, Santa Cruz) per 10 mg of beads. After washing, 55 μg of Dynabeads with bound proteins/sample were used in PDE assays. Measurements were performed in duplicates. Data represent mean ± S.E. of at least three independent experiments.
Western Blot
Protein samples were separated by SDS-PAGE and transferred to nitrocellulose membranes (Protran BA-85, Schleicher & Schuell) using standard procedures. After blocking (Roti-Block; Carl Roth), proteins were detected using the following antibodies: PDE10A, AKAP150, and NMDAϵ1 (see above), NMDAϵ2 (sc-1469, Santa Cruz), PP2B (sc-6123, Santa Cruz), and PSD95 (MA1–046, Thermo Scientific). Anti-pPDE10A was generated in rabbits against the phosphorylated N-terminal peptide (amino acids 11–20). Purification of the antiserum was performed according to the manufacturer's instructions (PSL, Heidelberg, Germany). Secondary peroxidase-labeled anti-goat, anti-rabbit, and anti-mouse IgG were obtained from Sigma. Detection was performed with SuperSignal West Dura chemiluminescent substrate (Pierce) and a CCD camera (GDS 8000; UVP). Optical densities of Western signals were measured using the LabWorks 4.0 software (UVP). For quantification of PDE10 phosphorylation, pPDE10A and PDE10A in the same sample were measured, normalized to mean intensity of specific bands on a membrane to account for intensity variation of different blots, and expressed as pPDE/PDE ratio.
Phosphorylation of Precipitated PDE10A
Dynabeads (5 mg) with precipitated proteins were washed three times with PBS and resuspended in PKA buffer (10 μm cAMP, 0.5 mm ATP, 3 mm MgCl2, 0.5 mg/ml BSA, 2 mm dl-dithiothreitol, 50 mm triethanolamine/hydrochloride, pH 7.4) containing 1 μm MP-10 and PhosSTOP. In the negative control, cAMP and ATP were omitted. Incubation was for 10 min at 37 °C. Dynabeads were washed and proteins were eluted with 100 μl of SDS sample buffer. Volumes corresponding to 0.75 mg of Dynabeads were applied per lane of a 9% SDS gel.
Size Exclusion Chromatography of PDE10-containing Complexes from Striatum
For gel filtration chromatography, the striata of 5 male mice were homogenized in Nonidet P-40 buffer (see above) and centrifuged for 30 min at 20,000 × g (4 °C). The resulting supernatant was applied to a HiLoad 26/60 Superdex 200pg column. The column was run with 0.68 column volumes of the Nonidet P-40 buffer containing 25 mm Tris, pH 7.4. Separation of proteins was followed by online detection of absorption at 280 nm. Fractions of 1.25 ml were collected and protein containing fractions were screened for PDE activity in the absence and presence of MP-10 (0.1 μm). For calibration, thyroglobulin (660 kDa), immunoglobulin G (150 kDa), bovine serum albumin (67 kDa), and ovalbumin (43 kDa) were separated on the same column and molecular weights were calculated using semi-logarithmic regression.
Statistical Analysis
Statistical testing was performed in Excel using Student's t test or Holm-Bonferroni's test (multiple comparisons of PDE activities with PDE inhibitors). For multiple comparisons of PDE10A phosphorylation, data were log transformed, homoscedasticity was checked by Levene's test, and data were further analyzed by one-way analysis of variance followed by Bonferroni's multiple comparisons post hoc tests using IBM SPSS 22. Differences are reported as significant at p < 0.05. Data are expressed as mean ± S.E. (error bars).
RESULTS
PDE10A Is the Major cAMP PDE in Mouse Striatum
Phosphodiesterases are critical regulators of cyclic nucleotide signaling in brain. In striatum, multiple PDEs with different substrate specificities and subcellular localization are expressed (11, 12, 22–24). Here, we set out to determine the relative contribution of the dual substrate PDE10A for cyclic nucleotide turnover in striatum. To this end, we measured cAMP and cGMP hydrolytic activity in striatal homogenates from mouse in the absence and presence of various PDE inhibitors at substrate concentrations of 0.1 μm cAMP (Fig. 1A) or 1 μm cGMP (Fig. 1B). Inhibition of PDEs 3, 4, 5, 9, or 11 did not affect cAMP or cGMP hydrolysis. Inhibition of PDE1 (50 μm vinpocetine) or PDE2 (0.2 μm Bay 60-7550) reduced the overall cAMP degradation by ∼10% each. The greatest effect on cAMP hydrolysis was observed in the presence of two different PDE10 inhibitors: papaverine (10 μm) inhibited ∼70%, and the more specific MP-10 (0.01 μm) inhibited ∼60% of cAMP hydrolysis (Fig. 1A). In the following, papaverine was used in some experiments because of the limited availability of MP-10.
FIGURE 1.

PDE10A is the major cAMP PDE in striatum. A, cAMP, and B, cGMP PDE activities were determined in lysates of mouse striatum in the absence (total activity) and presence of specific PDE inhibitors (PDE1, 50 μm vinpocetine; PDE2, 0.2 μm BAY 60-7550; PDE3, 10 μm cilostamide; PDE4, 5 μm rolipram; PDE5 and -11, 1 μm tadalafil; PDE9, 1 μm PF-04447943; PDE10A, 0.01 μm MP-10 and 10 μm papaverine) with 0.1 μm cAMP (A) or 1 μm cGMP (B) as substrate. Activities are displayed in percent of total cyclic nucleotide turnover. Shown are mean ± S.E. of n ≥ 3 independent experiments performed in duplicates. *, p < 0.0003 (A) or p < 0.007 (B) using Holm-Bonferroni's test. C, PDE10 content of cytosolic and membrane fractions derived from striatum (left) and striatal synaptosomes (right) was analyzed in Western blots (GAPDH, PSD-95: cytosolic and membrane markers, respectively). Shown are representative blots and statistical analysis (n = 9, *, p < 0.005 (total striatum); n = 3, *, p < 0.01 (synaptosomes) by Student's t test). Larger sections of Western blots are shown in supplemental Fig. S1.
For cGMP turnover, PDEs 1 and 2 are most important as inhibition of these PDEs resulted in a ∼35 and ∼20% reduction of cGMP degradation, respectively. Although PDE10A is considered a dual specific PDE, its inhibition by papaverine only resulted in a slight reduction of cGMP-degrading activity in striatum (Fig. 1B). Thus, we conclude that PDE10A acts as the major cAMP PDE in striatum at the substrate concentration tested and controls most of the cAMP/PKA signal transduction in medium spiny neurons.
PDE10A Is Primarily Associated to Synaptosomal Membranes in Mouse Striatum
Next we analyzed subcellular localization of PDE10A in mouse striatum. To assess compartmental localization, mice striata were separated into a cytosolic and a membrane fraction. Adequate fractionation was ensured by analysis of cytosolic and membrane marker proteins GAPDH and PSD95, respectively. As shown by Western immunodetection, PDE10A was enriched in the membrane fraction approximately ∼3-fold compared with the cytosolic fraction. These results are in accordance with several previous reports (10, 25). In addition, we prepared synaptosomes. As shown in Fig. 1C, PDE10A distribution in the synaptosomal cytosol and membranes mirrored the distribution in total striatum. We therefore conclude that PDE10A is present in striatal synapses.
cAMP-dependent Phosphorylation of PDE10A Occurs in Vivo and Is Reversed by PP2A/PP1
PDE10A has been shown to be phosphorylated at Thr-16 by PKA in vitro and in heterologous expression systems (26, 27). However, evidence for phosphorylation to occur in vivo is sparse. To examine PDE10A2 phosphorylation in mouse striatum, we generated rabbit polyclonal antibodies that specifically recognize phosphorylated PDE10A2. This antibody reacted strongly with heterologously expressed PDE10A2 only after in vitro phosphorylation by PKA catalytic subunit (Fig. 2).
FIGURE 2.
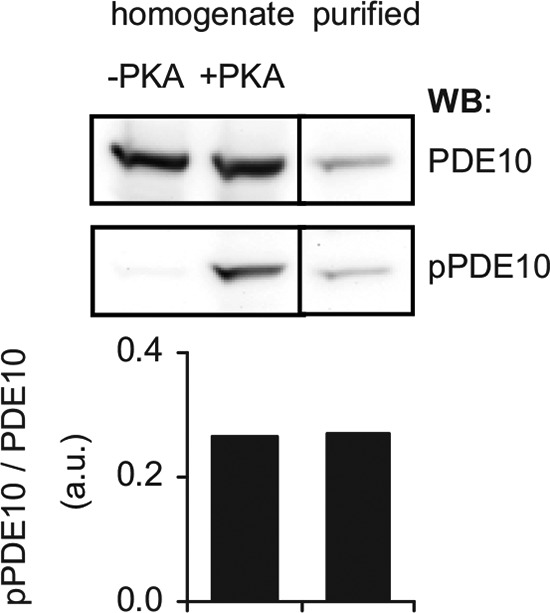
Characterization of pPDE10 antibody. Heterologously expressed PDE10A2 was incubated with ATP (0.5 mm) and MgCl2 (3 mm) in the absence or presence of PKA (800 units) for 30 min at 37 °C. An aliquot of the phosphorylated sample was saved for Western blot before purification of pPDE10 using the Pierce phosphoprotein enrichment kit. Samples were analyzed using PDE10 and pPDE10 antibodies. Graphs depict densitometric analysis of PDE10 phosphorylation. Shown is the ratio of phospho-PDE10 to PDE10 signal before and after purification of the phosphorylated protein.
In vivo phosphorylation of PDE10A2 was analyzed in mouse striatal slices. In lysate of untreated slices, phosphorylation of PDE10A2 was undetectable (Figs. 3A and 4, A and B, first lane). However, treatment with the adenylyl cyclase stimulator forskolin or the PDE10 inhibitors papaverine or MP-10 to raise intracellular cAMP and consecutively stimulate PKA led to a robust approximately 3–4-fold increase in phosphorylation (Figs. 3A and 4, A and B, respectively). This effect was enhanced by the simultaneous application of the PP2A/PP1 inhibitor okadaic acid (1 μm, Fig. 4A, third lane)3 but not by cyclosporin A, a protein phosphatase 2B (calcineurin) inhibitor (Fig. 4B, third lane).
FIGURE 3.
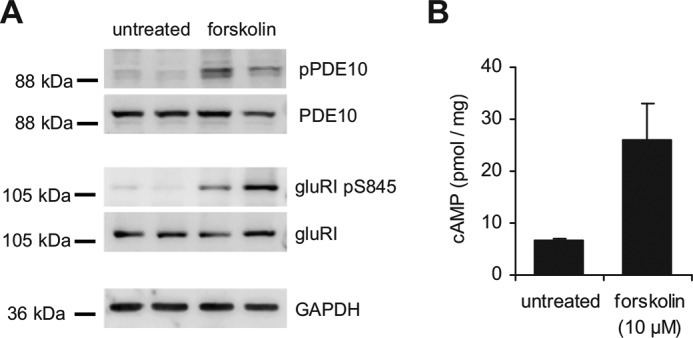
Phosphorylation of PDE10A and GluRI pS845 is mediated by PKA. A, mouse striatal slices were prepared as described under ”Materials and Methods“ and treated without (basal, first and second lanes) or with the adenylyl cyclase stimulator forskolin (10 μm, third and fourth lanes) for 5 min at 37 °C. Single slices were lysed and phosphorylation of PDE10A and GluRI pS845 was detected in Western blots using phospho-specific antibodies. Shown is a representative Western blot. B, cAMP levels in the lysates were determined by radioimmunoassay. Measurements were performed in duplicate. Shown are mean ± S.E. of n = 6 slices of three mice.
FIGURE 4.
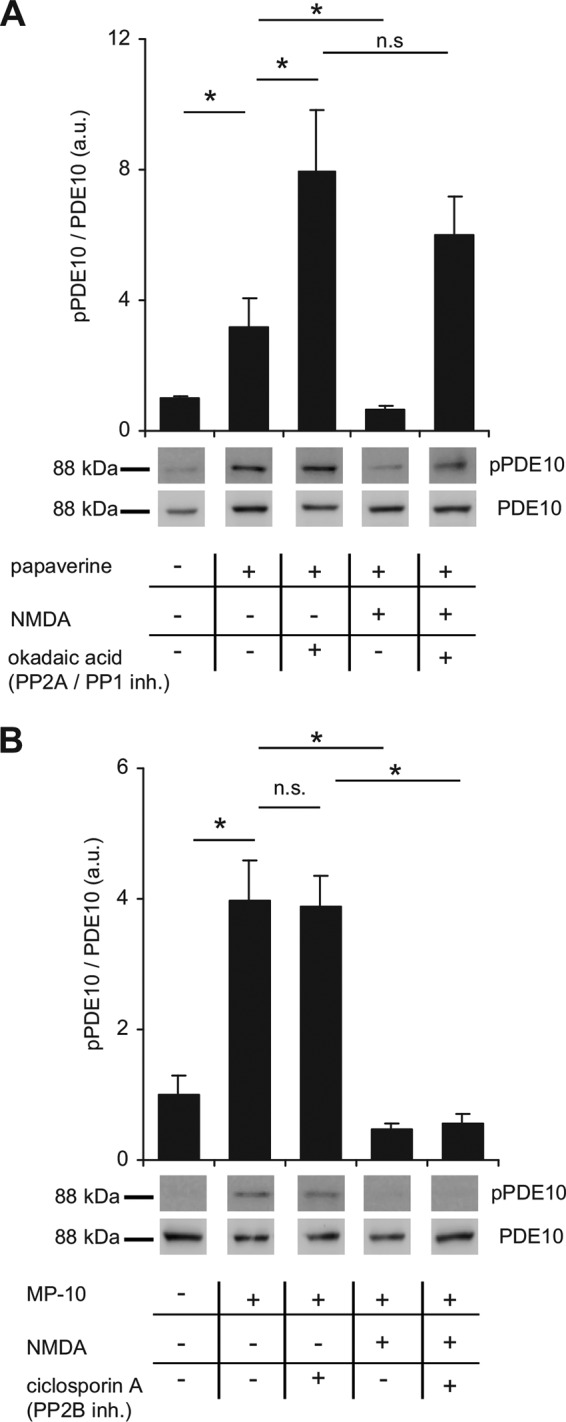
PDE10A phosphorylation is reversed by PP2A/PP1 inhibition. A and B, mouse striatal slices were preincubated with PDE10 inhibitors and (A) PP2A/PP1 or (B) PP2B phosphatase inhibitors as indicated (PDE10, 100 μm papaverine or 1 μm MP-10; PP2A/PP1, 1 μm okadaic acid; PP2B, 5 μm cyclosporin A). Subsequently, NMDA (300 μm) was added as indicated and slices were incubated for an additional 5 min. Phosphorylation of PDE10A2 was detected in lysates of single slices using pPDE10A antibodies and normalized to total PDE10 content. Shown are representative blots and graphs depicting mean ± S.E. values obtained by densitometric analysis (n = 8 slices of 4 animals for each condition). *, p < 0.05, one-way analysis of variance followed by Bonferroni's test. Larger sections of Western blots are shown in supplemental Fig. S2.
Next, we tested the effect of NMDA-mediated Ca2+ elevation on PDE10A phosphorylation as Ca2+ is known to directly or indirectly regulate the activity of multiple phosphatases. Indeed, activation of NMDA receptors (300 μm NMDA) resulted in dephosphorylation of PDE10A2 (Fig. 4, A and B, fourth lanes). The NMDA-dependent dephosphorylation was prevented by okadaic acid but not by cyclosporin A (Fig. 4, A and B, fifth lanes, respectively).
To ensure phosphatase inhibitors were used in concentrations allowing specific inhibition, each slice was additionally analyzed for phosphorylation of the GluRI subunit of the AMPA receptor. GluRI Ser-845 is known to be phosphorylated by PKA and dephosphorylated by PP2B (28, 29). As expected, and in contrast to PDE10, GluRI phosphorylation was slightly enhanced by treatment with cyclosporin A but not influenced by okadaic acid (Fig. 5). The results demonstrate that PDE10A2 is endogenously phosphorylated by PKA and identify PP2A and/or PP1, but not PP2B, as responsible phosphatases.
FIGURE 5.
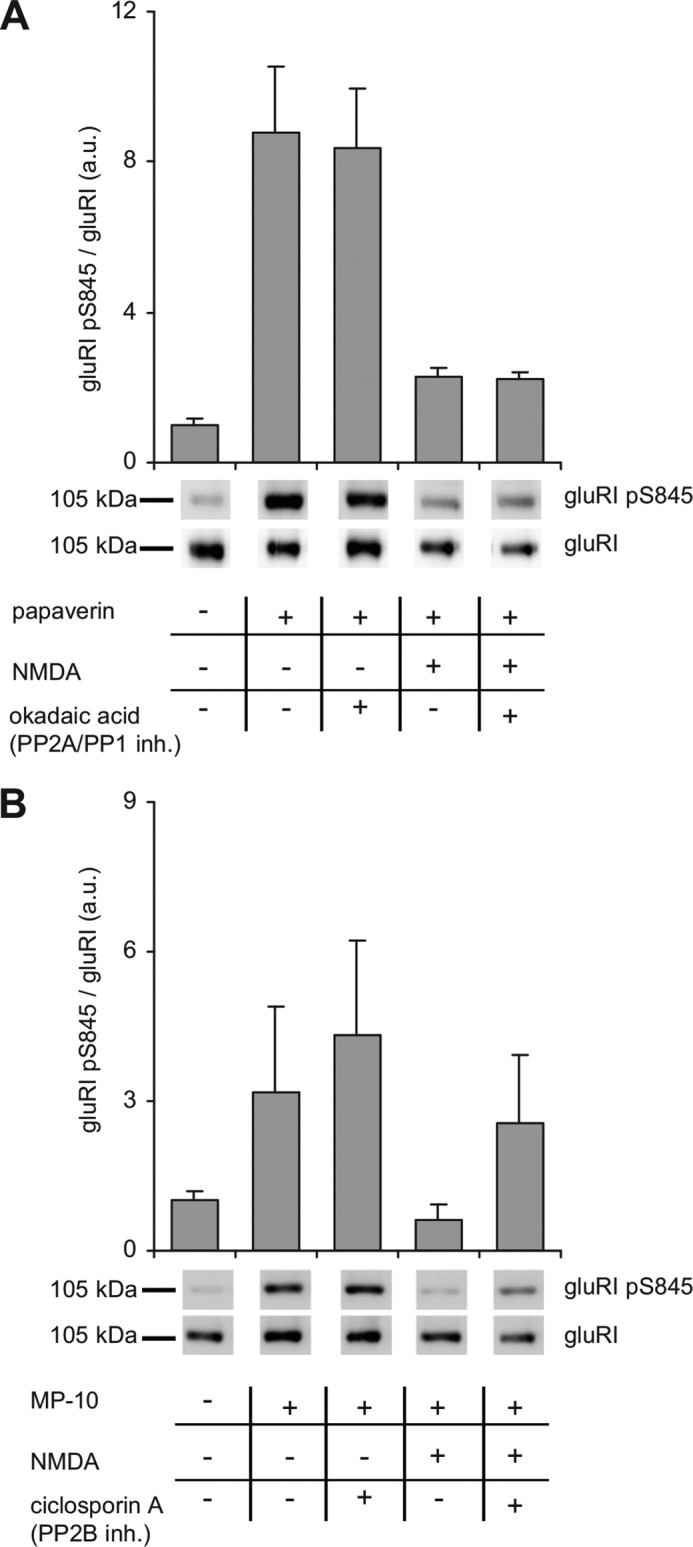
PKA-mediated phosphorylation of GluRI Ser(P)-845 and dephosphorylation by PP2B. A and B, mouse striatal slices were preincubated with PDE10 inhibitors and (A) PP2A/PP1 or (B) PP2B phosphatase inhibitors as indicated (PDE10, 100 μm papaverine or 1 μm MP-10; PP2A/PP1, 1 μm okadaic acid; PP2B, 5 μm cyclosporin A). Subsequently, NMDA (300 μm) was added as indicated and slices were incubated for an additional 5 min. Phosphorylation of GluRI was detected using GluRI Ser(P)-845-specific antibodies and normalized to total GluRI content. Shown are representative blots and graphs depicting mean ± S.E. values obtained by densitometric analysis (n = 8 slices of 4 animals for each condition). Larger sections of Western blots are shown in supplemental Fig. S3.
Phosphorylation Does Not Affect Enzymatic Properties of PDE10A2
We then asked whether phosphorylation impacts PDE10A2 enzymatic properties and analyzed phosphorylated versus non-phosphorylated PDE10A2 with an enzyme expressed in HEK293 cells and in vitro phosphorylated by PKA. Stoichiometric phosphorylation was ensured by Western blots before and after specific enrichment of the phosphorylated PDE10A. As the pPDE10/total PDE10 ratio was unchanged after enrichment of the phosphoprotein, we conclude that PDE10 was fully phosphorylated under our experimental conditions (see Fig. 2). As shown in Fig. 6A, cAMP breakdown of a substrate concentration of 1 μm cAMP was unaffected by phosphorylation. Also, the concentration-dependent activation by the GAF domain ligand 7-CH-cAMP (6) was unchanged (Fig. 6B). We wondered if phosphorylation might impact dissociation of cAMP from the GAF domains. Therefore, phosphorylated and non-phosphorylated PDE10A2 were incubated with a high concentration of cAMP (10 μm) to saturate the GAF domains and then quickly diluted 100-fold. The decline in PDE activity was then determined in samples drawn at various time points. As shown in Fig. 6C, there was no difference in cAMP dissociation from the GAF domains. In conclusion, phosphorylation does not appear to play a role in regulation of PDE10A2 activity.
FIGURE 6.
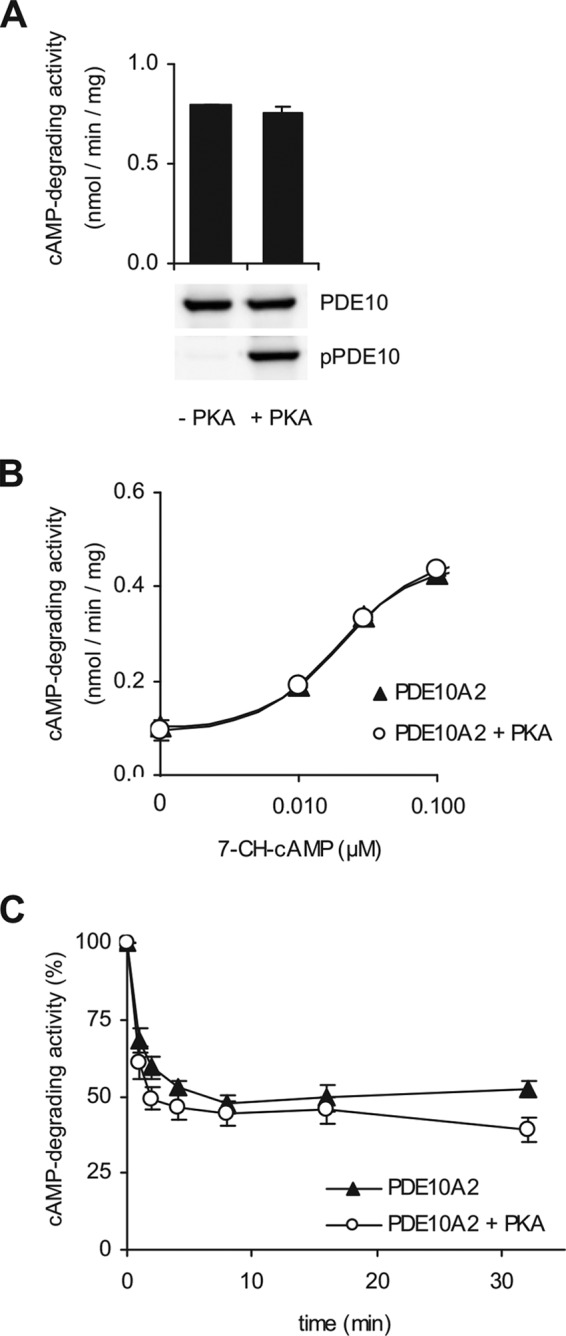
Enzymatic properties do not differ between phosphorylated and non-phosphorylated PDE10A. PDE10A2 expressed in HEK293 cells was phosphorylated by addition of the PKA catalytic subunit to the cell lysate. A, specific activity of phosphorylated versus non-phosphorylated PDE10A2 was determined at a substrate concentration of 1 μm cAMP. Phosphorylation of PDE10A2 was checked by immunodetection. B, cAMP-degrading activity of phosphorylated (open circles) and non-phosphorylated (closed triangles) PDE10A2 was determined in the presence of increasing concentrations of the stimulator 7-CH-cAMP (substrate 0.03 μm cAMP). C, deactivation caused by dissociation of cAMP from the GAF domains of phosphorylated (open circles) or non-phosphorylated (closed triangles) PDE10A2 was measured over a 30-min time course. Results represent mean ± S.E. of n = 3.
PDE10A Precipitates the cAMP-dependent Protein Kinase
For further analysis, PDE10A was enriched by precipitating the enzyme from mouse striatal extracts using specific antibodies coupled to Dynabeads. In an attempt to phosphorylate PDE10A in vitro, we found to our surprise that phosphorylation did not require addition of PKA but addition of cAMP and ATP was sufficient to enhance phosphorylation of the precipitated PDE10A 2-fold (Fig. 7A). This suggests co-precipitation of PKA with PDE10A. Indeed, using PKA RIIα-specific antibodies, we found that PKA co-precipitated with PDE10A (Fig. 7B). Precipitation of PKA was prevented when the experiment was carried out in the presence of the antigen used to generate the PDE10 antibodies.
FIGURE 7.
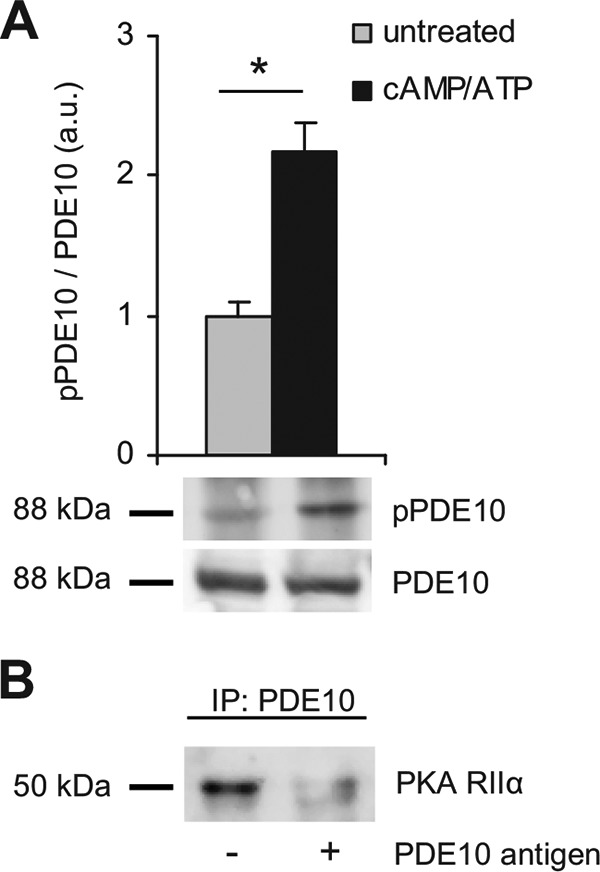
PDE10A interacts with PKA. PDE10A was precipitated from lysates of mouse striatum using specific antibodies coupled to Dynabeads. A, the precipitates were incubated with or without ATP (0.5 mm), MgCl2 (3 mm), and cAMP (10 μm) and PDE10A2 phosphorylation was detected using pPDE10A antibodies. Shown are representative blots and mean ± S.E. of pPDE10A/PDE10A ratio after densitometric analysis; n = 3; *, p < 0.005, Student's t test. B, immunoprecipitation (IP) of PDE10 was performed in the absence or presence of the PDE10A antigen used for antibody generation and PKA RIIα was detected by Western blot. Larger sections of Western blots are shown in supplemental Fig. S4.
PDE10A Interacts with AKAP150
As the phosphorylation did not affect enzymatic activity, we speculated about an effect of phosphorylation on subcellular distribution. As shown above, we found that PKA co-precipitates with PDE10A. Classically, PKA is tethered to its target proteins by members of the protein kinase A anchoring protein family (AKAPs) (for review, see Refs. 30 and 31). In mouse striatum, AKAP150 is highly abundant (32). Therefore it was tempting to speculate that PDE10A interacts with AKAP150 that in turn has bound PKA.
To confirm this hypothesis, we immunoprecipitated AKAP150 from solubilized striatal membranes (Fig. 8A). In the precipitate, we measured PDE activity at a substrate concentration of 0.1 μm cAMP and indeed could show that AKAP150 pulls down cAMP-hydrolyzing activity that was inhibited by MP-10 (Fig. 8B). No PDE activity was precipitated when the AKAP antibody was omitted.
FIGURE 8.

PDE10A interacts with AKAP150. A, AKAP150 was precipitated from mouse striatal lysate using specific antibodies. For control, the primary antibody was omitted. B, PDE activity co-precipitated with AKAP150 was determined at a substrate concentration of 0.1 μm cAMP in the absence and presence of the PDE10 inhibitor MP-10 (1 μm) (*, p < 0.005, Student's t test, n = 3, n.d., below detection limit). C, PDE10A was precipitated from mouse striatal lysate and co-precipitated AKAP150 was detected with specific antibodies. One-tenth of the total protein was run in the input lane. For control, the PDE10A antibody was omitted. D, PDE10A immunoprecipitation (IP) was performed in the absence or presence of purified PDE10A antigen added to the striatal lysate prior to precipitation. Data are representative of at least three independent experiments. Full Western blots are shown in supplemental Fig. S5.
The results suggest an interaction between PDE10A and AKAP150. This interaction was confirmed by reverse precipitation using PDE10A-specific antibodies. As shown in Fig. 8C, AKAP150 co-precipitated with PDE10A, whereas in the control (omission of first antibody) neither PDE10A nor AKAP150 were detected. Additionally, precipitation of AKAP150 was abrogated when the experiment was carried out in the presence of the PDE10A antigen (Fig. 8D). Thus, the results clearly demonstrate a specific interaction of PDE10A with the anchoring protein AKAP150.
Targeting of the PDE10A/AKAP150 Complex to Glutamate Receptors
Our findings suggest formation of a signaling complex around AKAP150 that ensures tight spatio-temporal control of cAMP signal transduction in medium spiny neurons of the striatum.
Physiologically, the anchoring PKA and PDE10A enables selective and timed phosphorylation of possible target proteins. Colledge et al. (33) showed that in rat brain extracts AKAP150 targets PKA to AMPA receptors via the scaffolding protein SAP97 and NMDA receptors via the scaffold PSD95. We wondered if in striatum PDE10A is bound to similar signaling complexes. To test this idea, we analyzed the PDE10A precipitate for the scaffolding protein PSD95 as well as AMPA receptor (GluRI) and NMDA receptor subunits (NR2A, NR2B) (Fig. 9). The GluRI subunit of the AMPA receptor previously shown to be targeted to AKAP150 via SAP97 was not bound to the complex (Fig. 9A). However, PSD95 and NMDA receptor subunits NR2A and NR2B were precipitated by PDE10A antibodies, but not in controls without first antibody (Fig. 9A). Again, the presence of the PDE10A antigen efficiently blocked the precipitation (Fig. 9B). Thus, PDE10A specifically interacts with a signaling complex that targets PKA to the NMDA receptor. In light of many publications showing that AKAP150 binds PP2B/calcineurin (34–37) it is interesting to note that we could not detect the phosphatase in the PDE10A precipitate. This might be explained by a general difficulty of maintaining this very dynamic, modest affinity interaction (38).
FIGURE 9.
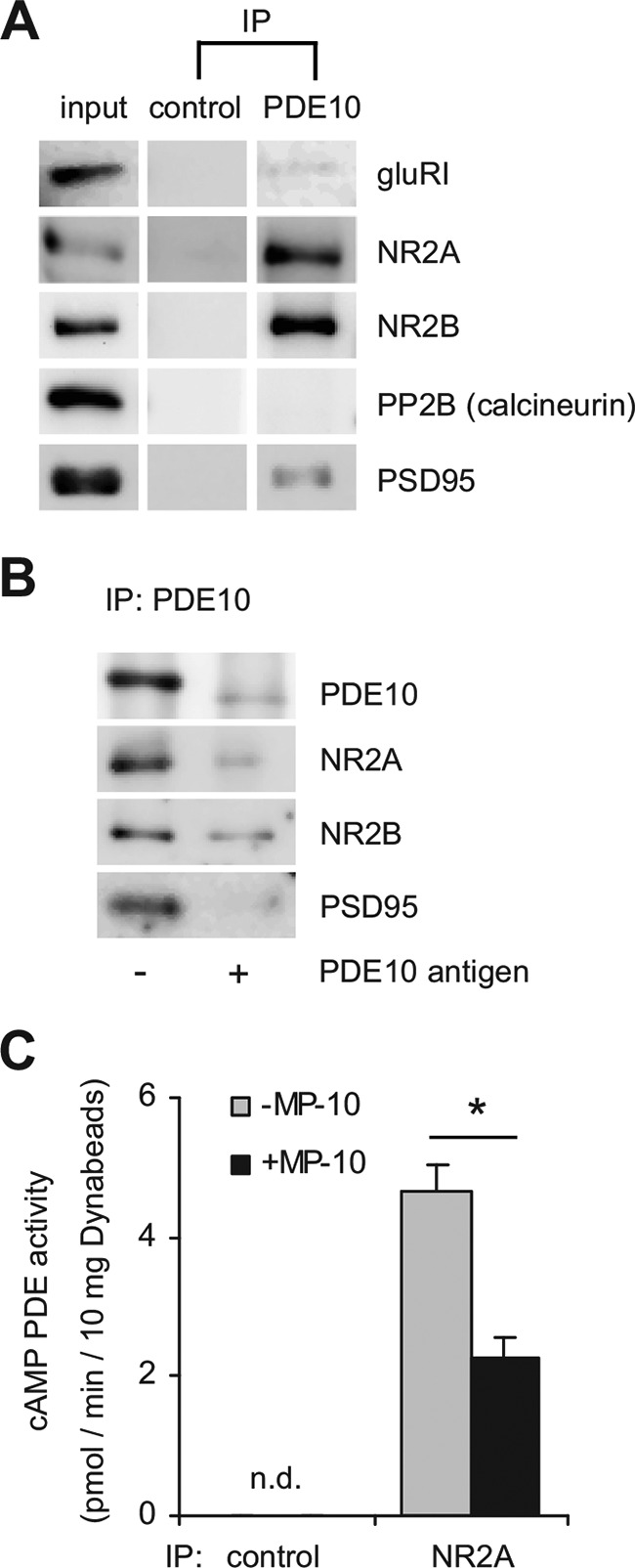
The PDE10A-precipitated protein complex contains PSD95 and the NMDA receptor. A, PDE10A was immunoprecipitated (IP) from striatal extracts and co-precipitated proteins were detected in Western blots. One-tenth of the total protein was run in the input lane. For control, the PDE10A antibody was omitted. B, PDE10A immunoprecipitation was performed in the absence or presence of purified PDE10A antigen added to the striatal lysate prior to precipitation and co-precipitated proteins were detected in Western blots. Larger sections of Western blots are shown in supplemental Fig. S6. C, NR2A subunit of the NMDA receptor was immunoprecipitated from mouse striatal lysates and cAMP PDE activity in the precipitate was determined in the absence (gray bar) or presence (black bar) of the PDE10A inhibitor MP-10 (1 μm). For control, the NR2A antibody was omitted. Data are representative of three independent experiments. *, p < 0.01, Student's t test; n.d., below detection limit.
Conversely, we examined if PDE10A co-precipitates with NMDA receptors. As depicted in Fig. 9C, antibodies against the NMDA receptor subunit NR2A precipitated cAMP PDE activity that can be attributed to PDE10A as the PDE10 inhibitor MP-10 reduced the activity by 50%. In control precipitations without NR2A antibody, cAMP-degrading activity was below the detection limit. Our results indicate that PDE10A exists in a highly ordered multimeric protein complex.
PDE10A Almost Completely Exists in High Molecular Weight Complexes
To estimate the fraction of PDE10A that is tied to the signaling complexes described above, we fractionated solubilized striatal membranes by size exclusion chromatography. Elution of PDE10A was followed by measuring cAMP PDE activity in the absence and presence of MP-10 (0.1 μm) (Fig. 10A) and in Western blots using PDE10-specific antibodies (Fig. 10B). As shown, virtually all PDE10A catalytic activity migrated in fractions that correspond to a molecular mass above 450 kDa indicating that very little of the enzyme exists as single protein (i.e. PDE10A homodimer). Additionally, catalytic activity is tracked by the intensity of the PDE10A bands in Western blots.
FIGURE 10.

PDE10A migrates in high molecular weight complexes. A, solubilized striatal membranes were subjected to size exclusion chromatography (Superdex 200pg HiLoad 26/60). Eluted fractions were tested for PDE10A by measurement of cAMP PDE activity (0.1 μm cAMP) in the absence (closed circles) and presence of MP-10 (0.1 μm, open circles). B, Western blots of eluted fractions using PDE10-specific antibodies. Input was diluted 50-fold; numbered fractions were 1.25 ml each starting with 121 ml. Shown is a representative experiment of n = 4.
Formation of PDE10A Protein Complexes Is Regulated by Phosphorylation
The PDE10A containing protein complexes described above were detected under basal conditions, i.e. low cAMP. To evaluate if phosphorylation of PDE10A affects its interaction with the signaling complex, we performed immunoprecipitation experiments under phosphorylating conditions (PDE10 inhibition, forskolin, and phosphatase inhibitors). Effective phosphorylation of PDE10A in the treated sample was verified and amounts of precipitated PDE10 were comparable with those under non-phosphorylating conditions (Fig. 11). Interestingly, precipitation of interacting proteins was clearly reduced by phosphorylation. Quantification of signal intensities normalized to PDE10 in the same lane revealed a ∼60% reduction of AKAP150 binding to phosphorylated PDE10. For the co-precipitated NMDA receptor and PSD95, a 75% reduction was determined. Thus, phosphorylation of PDE10 reduces the affinity of PDE10 to the signaling complex suggesting that phosphorylation of PDE10 releases the enzyme from the AKAP and NMDA receptor-containing signaling complex.
FIGURE 11.
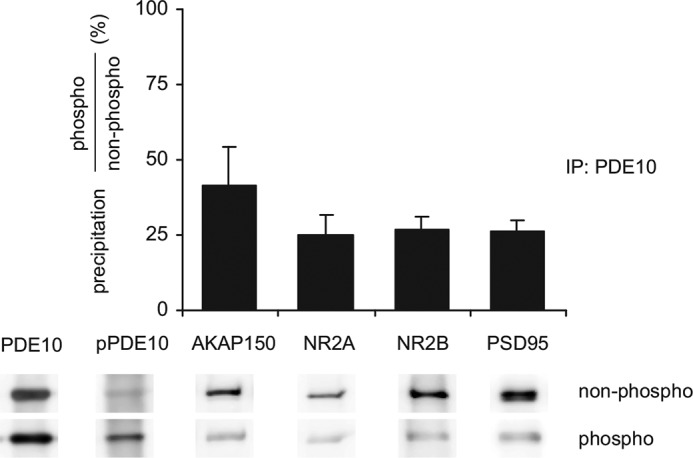
PDE10A phosphorylation reduces precipitation of the protein complex. PDE10A was precipitated from homogenates of mouse striatal slices either untreated (non-phospho) or preincubated (phospho) with papaverine (100 μm), forskolin (10 μm), and phosphatase inhibitors (0.5 μm okadaic acid, 5 μm cyclosporin A, 0.2 μm calyculin A). Precipitated PDE10 and pPDE10 were analyzed in Western blots. Co-precipitation of proteins under both conditions was analyzed in Western blots, densitometrically quantified, normalized to PDE10A content in the same lane, and expressed as fractional precipitation (phospho/non-phospho). Shown are representative Western blots and mean ± S.E. of n = 3 independent experiments. Larger sections of Western blots are shown in supplemental Fig. S7.
DISCUSSION
Striatal medium spiny neurons integrate glutamatergic input from the cortex and dopaminergic input from the substantia nigra. The second messenger cAMP is the key mediator of dopaminergic input in these neurons. PDE10A, a phosphodiesterase with clear preference for degradation of cAMP (6), has been shown to be primarily expressed in medium spiny neurons by in situ hybridization and immunological techniques (9, 10, 39). Furthermore, subcellular fractionation indicated that the protein was associated with synaptosomal membranes (10). Here, we confirmed the association of PDE10A with synaptosomal membranes of the striatum. By analyzing cAMP-degrading activity, PDE10A was found to be the major cAMP PDE in this tissue being responsible for more than 60% of overall cAMP-degrading activity at the substrate concentration tested (see Fig. 1). Accordingly, inhibition of PDE10A enhanced phosphorylation of GluR1, an established target of PKA, indicating a cAMP increase (see Figs. 4 and 5). Furthermore, inhibition of PDE10A led to phosphorylation of PDE10A itself, which was previously only detectable after enrichment of PDE10A (27). Because forskolin likewise induced PDE10A phosphorylation, the phosphorylation can be ascribed to PKA as already suggested by others (25, 27). Here, we answered two open questions: (i) which phosphatase is responsible for PDE10A dephosphorylation and (ii) what are the functional consequences of PDE10A phosphorylation.
In striatum, three phosphatases are relevant, PP1, PP2A, and the Ca2+-stimulated PP2B (calcineurin). Under PKA-activating conditions, a combined PP2A/PP1 inhibitor increased PDE10A phosphorylation. This suggests dephosphorylation of PDE10A by PP2A, because PKA activation has been shown to indirectly inhibit PP1 activity via phosphorylation of the striatum-specific DARPP32 at Ser-34 (40). However, dephosphorylation of PDE10A by PP1 cannot be excluded but would imply incomplete inhibition of PP1 by DARPP32. A second line of evidence for the involvement of PP2A in PDE10A phosphorylation emerges from our observation that NMDA-induced Ca2+ increases induced dephosphorylation of PDE10. In striatum, the PR72 subunit of PP2A is highly expressed and mediates Ca2+ activation of PP2A (41). Furthermore, the PP2A/PP1 inhibitor abolished Ca2+-induced dephosphorylation. Involvement of the second Ca2+-activated phosphatase PP2B (calcineurin) can be excluded, because the PP2B inhibitor did not affect Ca2+-induced dephosphorylation. Last, the observation that the PP2B inhibitor did not abolish NMDA-induced dephosphorylation of PDE10A also argues against the involvement of PP1, because the Ca2+-activated PP2B dephosphorylates DARPP32 at Ser-34 leading to PP1 activation (42, 43). In summary, the results provide evidence that PP2A is the phosphatase responsible for dephosphorylation of PDE10A in striatum.
Our second aim was to elucidate the functional consequences of PDE10A phosphorylation. In two previous reports the impact of phosphorylation on subcellular localization of the enzyme was analyzed. Kotera et al. (25) were the first to show that phosphorylation of the striatal PDE10A isoform, PDE10A2, regulates subcellular localization. The authors hypothesized that membrane association of the enzyme was caused by either N-glycosylation or association with AKAP proteins. In a later study, Charych et al. (27) identified palmitoylation as the underlying mechanism of plasma membrane association of PDE10A and suggested irreversible N-palmitoylation of the enzyme. Furthermore, PKA-mediated phosphorylation of the enzyme prevented palmitoylation and membrane trafficking but did not cause redistribution of the already palmitoylated enzyme from the membrane. Consequently, phosphorylation of PDE10A at the site of PDE10A synthesis was discussed as a switch irreversibly directing PDE10A to either the cytosolic or membrane compartments (27).
Because multiple PDE families are regulated by phosphorylation (PDEs 1A, 3, 5, and 4 (44–49), for review about PDE4 phosphorylation see Ref. 50) or have been reported to be phosphorylated (PDEs 7 and 11 (51, 52), for review, see Ref. 53) we studied the impact of phosphorylation on enzymatic properties with recombinantly expressed enzyme. However, the phosphorylated and the non-phosphorylated enzymes did not differ in catalytic activity, activation, or dissociation of cAMP from the GAF domain (see Fig. 6). Hence, phosphorylation does not regulate PDE10A2 activity.
Although in vitro phosphorylating the endogenous, immunoprecipitated striatal PDE10A, we found PKA to be present in the precipitate. Our hypothesis of PDE10A being part of a larger signaling complex was confirmed by co-precipitation of the multidomain scaffold protein AKAP150. AKAPs play pivotal roles by assembling cAMP generator and effector molecules (PKA, PDEs). In the striatum, AKAP150 is highly expressed and targeted to dendritic spines (54). In the rat brain, AKAP150 is linked to the NMDA receptor via PSD95 (33). In line, the PDE10A precipitate contained NMDA receptor subunits NR2A and NR2B and the postsynaptic protein PSD95 giving rise to the assumption of a large multiprotein complex. As co-precipitation of the interacting proteins was reduced under phosphorylating conditions, we conclude that affinity of PDE10A to the signaling complex is reduced by phosphorylation. Thus, a model emerges in which the unphosphorylated PDE10A is targeted to membranes in dendritic spines by palmitoylation and subsequently integrates into complexes containing AKAP150 and PKA. In case cAMP synthesis exceeds the cAMP-degrading activity of the enzyme, PKA is activated and phosphorylates PDE10. Thereupon, PDE10A is released from the complex but is still bound to the membrane by palmitoylation allowing lateral diffusion. Functionally, release of PDE10A should result in steep cAMP increases once a certain cAMP concentration is exceeded.
Supplementary Material
Acknowledgments
We gratefully acknowledge the technical assistance of Ulla Krabbe, Medah Özcan, Arkadius Pacha, and Caroline Vollmers.
This work was supported by Deutsche Forschungsgemeinschaft Grant KO 1157/4-1.

This article contains supplemental Figs. S1–S7.
The bands in the second and third lanes display similar density on the representative blot shown, which has been selected because it is representative for the other statistically significant differences.
- PDE
- phosphodiesterase
- AKAP
- A-kinase anchor protein
- MP-10
- 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline
- PP2A
- protein phosphatase 2A.
REFERENCES
- 1. Bender A. T., Beavo J. A. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 58, 488–520 [DOI] [PubMed] [Google Scholar]
- 2. Fujishige K., Kotera J., Michibata H., Yuasa K., Takebayashi S., Okumura K., Omori K. (1999) Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J. Biol. Chem. 274, 18438–18445 [DOI] [PubMed] [Google Scholar]
- 3. Loughney K., Snyder P. B., Uher L., Rosman G. J., Ferguson K., Florio V. A. (1999) Isolation and characterization of PDE10A, a novel human 3′,5′-cyclic nucleotide phosphodiesterase. Gene 234, 109–117 [DOI] [PubMed] [Google Scholar]
- 4. Soderling S. H., Bayuga S. J., Beavo J. A. (1999) Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl. Acad. Sci. U.S.A. 96, 7071–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gross-Langenhoff M., Hofbauer K., Weber J., Schultz A., Schultz J. E. (2006) cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem. 281, 2841–2846 [DOI] [PubMed] [Google Scholar]
- 6. Jäger R., Russwurm C., Schwede F., Genieser H.-G., Koesling D., Russwurm M. (2012) Activation of PDE10 and PDE11 phosphodiesterases. J. Biol. Chem. 287, 1210–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujishige K., Kotera J., Yuasa K., Omori K. (2000) The human phosphodiesterase PDE10A gene genomic organization and evolutionary relatedness with other PDEs containing GAF domains. Eur. J. Biochem. 267, 5943–5951 [DOI] [PubMed] [Google Scholar]
- 8. O'Connor V., Genin A., Davis S., Karishma K. K., Doyère V., De Zeeuw C. I., Sanger G., Hunt S. P., Richter-Levin G., Mallet J., Laroche S., Bliss T. V., French P. J. (2004) Differential amplification of intron-containing transcripts reveals long term potentiation-associated up-regulation of specific Pde10A phosphodiesterase splice variants. J. Biol. Chem. 279, 15841–15849 [DOI] [PubMed] [Google Scholar]
- 9. Fujishige K., Kotera J., Omori K. (1999) Striatum- and testis-specific phosphodiesterase PDE10A isolation and characterization of a rat PDE10A. Eur. J. Biochem. 266, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 10. Xie Z., Adamowicz W. O., Eldred W. D., Jakowski A. B., Kleiman R. J., Morton D. G., Stephenson D. T., Strick C. A., Williams R. D., Menniti F. S. (2006) Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience 139, 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishi A., Kuroiwa M., Miller D. B., O'Callaghan J. P., Bateup H. S., Shuto T., Sotogaku N., Fukuda T., Heintz N., Greengard P., Snyder G. L. (2008) Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. J. Neurosci. 28, 10460–10471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sano H., Nagai Y., Miyakawa T., Shigemoto R., Yokoi M. (2008) Increased social interaction in mice deficient of the striatal medium spiny neuron-specific phosphodiesterase 10A2. J. Neurochem. 105, 546–556 [DOI] [PubMed] [Google Scholar]
- 13. Kapur S. (2004) How antipsychotics become anti-“psychotic”: from dopamine to salience to psychosis. Trends Pharmacol. Sci. 25, 402–406 [DOI] [PubMed] [Google Scholar]
- 14. Everitt B. J., Robbins T. W. (2005) Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 8, 1481–1489 [DOI] [PubMed] [Google Scholar]
- 15. DeLong M. R., Wichmann T. (2007) Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 64, 20–24 [DOI] [PubMed] [Google Scholar]
- 16. Siuciak J. A., McCarthy S. A., Chapin D. S., Fujiwara R. A., James L. C., Williams R. D., Stock J. L., McNeish J. D., Strick C. A., Menniti F. S., Schmidt C. J. (2006) Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: evidence for altered striatal function. Neuropharmacology 51, 374–385 [DOI] [PubMed] [Google Scholar]
- 17. Siuciak J. A., Chapin D. S., Harms J. F., Lebel L. A., McCarthy S. A., Chambers L., Shrikhande A., Wong S., Menniti F. S., Schmidt C. J. (2006) Inhibition of the striatum-enriched phosphodiesterase PDE10A: a novel approach to the treatment of psychosis. Neuropharmacology 51, 386–396 [DOI] [PubMed] [Google Scholar]
- 18. Hebb A. L., Robertson H. A., Denovan-Wright E. M. (2008) Phosphodiesterase 10A inhibition is associated with locomotor and cognitive deficits and increased anxiety in mice. Eur. Neuropsychopharmacol. 18, 339–363 [DOI] [PubMed] [Google Scholar]
- 19. Schmidt C. J., Chapin D. S., Cianfrogna J., Corman M. L., Hajos M., Harms J. F., Hoffman W. E., Lebel L. A., McCarthy S. A., Nelson F. R., Proulx-LaFrance C., Majchrzak M. J., Ramirez A. D., Schmidt K., Seymour P. A., Siuciak J. A., Tingley F. D., 3rd, Williams R. D., Verhoest P. R., Menniti F. S. (2008) Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J. Pharmacol. Exp. Ther. 325, 681–690 [DOI] [PubMed] [Google Scholar]
- 20. Russwurm M., Wittau N., Koesling D. (2001) Guanylyl cyclase/PSD-95 interaction: targeting of the nitric oxide-sensitive α2β1 guanylyl cyclase to synaptic membranes. J. Biol. Chem. 276, 44647-44652 [DOI] [PubMed] [Google Scholar]
- 21. Jäger R., Schwede F., Genieser H.-G., Koesling D., Russwurm M. (2010) Activation of PDE2 and PDE5 by specific GAF ligands: delayed activation of PDE5. Br. J. Pharmacol. 161, 1645–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nishi A., Snyder G. L. (2010) Advanced research on dopamine signaling to develop drugs for the treatment of mental disorders: biochemical and behavioral profiles of phosphodiesterase inhibition in dopaminergic neurotransmission. J. Pharmacol. Sci. 114, 6–16 [DOI] [PubMed] [Google Scholar]
- 23. Russwurm C., Zoidl G., Koesling D., Russwurm M. (2009) Dual acylation of PDE2A splice variant 3: targeting to synaptic membranes. J. Biol. Chem. 284, 25782–25790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin D. T., Fretier P., Jiang C., Vincent S. R. (2010) Nitric oxide signaling via cGMP-stimulated phosphodiesterase in striatal neurons. Synapse 64, 460–466 [DOI] [PubMed] [Google Scholar]
- 25. Kotera J., Sasaki T., Kobayashi T., Fujishige K., Yamashita Y., Omori K. (2004) Subcellular localization of cyclic nucleotide phosphodiesterase type 10A variants, and alteration of the localization by cAMP-dependent protein kinase-dependent phosphorylation. J. Biol. Chem. 279, 4366–4375 [DOI] [PubMed] [Google Scholar]
- 26. Kotera J., Fujishige K., Yuasa K., Omori K. (1999) Characterization and phosphorylation of PDE10A2, a novel alternative splice variant of human phosphodiesterase that hydrolyzes cAMP and cGMP. Biochem. Biophys. Res. Commun. 261, 551–557 [DOI] [PubMed] [Google Scholar]
- 27. Charych E. I., Jiang L.-X., Lo F., Sullivan K., Brandon N. J. (2010) Interplay of palmitoylation and phosphorylation in the trafficking and localization of phosphodiesterase 10A: implications for the treatment of schizophrenia. J. Neurosci. 30, 9027–9037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roche K. W., O'Brien R. J., Mammen A. L., Bernhardt J., Huganir R. L. (1996) Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 29. Sanderson J. L., Gorski J. A., Gibson E. S., Lam P., Freund R. K., Chick W. S., Dell'Acqua M. L. (2012) AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J. Neurosci. 32, 15036–15052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colledge M., Scott J. D. (1999) AKAPs: from structure to function. Trends Cell Biol. 9, 216–221 [DOI] [PubMed] [Google Scholar]
- 31. Welch E. J., Jones B. W., Scott J. D. (2010) Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol. Interv. 10, 86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ostroveanu A., Van der Zee E. A., Dolga A. M., Luiten P. G., Eisel U. L., Nijholt I. M. (2007) A-kinase anchoring protein 150 in the mouse brain is concentrated in areas involved in learning and memory. Brain Res. 1145, 97–107 [DOI] [PubMed] [Google Scholar]
- 33. Colledge M., Dean R. A., Scott G. K., Langeberg L. K., Huganir R. L., Scott J. D. (2000) Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27, 107–119 [DOI] [PubMed] [Google Scholar]
- 34. Coghlan V. M., Perrino B. A., Howard M., Langeberg L. K., Hicks J. B., Gallatin W. M., Scott J. D. (1995) Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267, 108–111 [DOI] [PubMed] [Google Scholar]
- 35. Dell'Acqua M. L., Dodge K. L., Tavalin S. J., Scott J. D. (2002) Mapping the protein phosphatase-2B anchoring site on AKAP79: binding and inhibition of phosphatase activity are mediated by residues 315–360. J. Biol. Chem. 277, 48796–48802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oliveria S. F., Gomez L. L., Dell'Acqua M. L. (2003) Imaging kinase-AKAP79-phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J. Cell Biol. 160, 101-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oliveria S. F., Dell'Acqua M. L., Sather W. A. (2007) AKAP79/150 anchoring of calcineurin controls neuronal l-type Ca2+ channel activity and nuclear signaling. Neuron 55, 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li H., Pink M. D., Murphy J. G., Stein A., Dell'Acqua M. L., Hogan P. G. (2012) Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat. Struct. Mol. Biol. 19, 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seeger T. F., Bartlett B., Coskran T. M., Culp J. S., James L. C., Krull D. L., Lanfear J., Ryan A. M., Schmidt C. J., Strick C. A., Varghese A. H., Williams R. D., Wylie P. G., Menniti F. S. (2003) Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 985, 113–126 [DOI] [PubMed] [Google Scholar]
- 40. Svenningsson P., Nishi A., Fisone G., Girault J.-A., Nairn A. C., Greengard P. (2004) DARPP-32: an integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296 [DOI] [PubMed] [Google Scholar]
- 41. Ahn J.-H., Sung J. Y., McAvoy T., Nishi A., Janssens V., Goris J., Greengard P., Nairn A. C. (2007) The B"/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc. Natl. Acad. Sci. U.S.A. 104, 9876–9881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nishi A., Snyder G. L., Nairn A. C., Greengard P. (1999) Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J. Neurochem. 72, 2015–2021 [DOI] [PubMed] [Google Scholar]
- 43. Nishi A., Bibb J. A., Snyder G. L., Higashi H., Nairn A. C., Greengard P. (2000) Amplification of dopaminergic signaling by a positive feedback loop. Proc. Natl. Acad. Sci. U.S.A. 97, 12840–12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma R. K., Wang J. H. (1985) Differential regulation of bovine brain calmodulin-dependent cyclic nucleotide phosphodiesterase isoenzymes by cyclic AMP-dependent protein kinase and calmodulin-dependent phosphatase. Proc. Natl. Acad. Sci. U.S.A. 82, 2603–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grant P. G., Mannarino A. F., Colman R. W. (1988) cAMP-mediated phosphorylation of the low-Km cAMP phosphodiesterase markedly stimulates its catalytic activity. Proc. Natl. Acad. Sci. U.S.A. 85, 9071–9075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wyatt T. A., Naftilan A. J., Francis S. H., Corbin J. D. (1998) ANF elicits phosphorylation of the cGMP phosphodiesterase in vascular smooth muscle cells. Am. J. Physiol. 274, H448–H455 [DOI] [PubMed] [Google Scholar]
- 47. Liu H., Maurice D. H. (1999) Phosphorylation-mediated activation and translocation of the cyclic AMP-specific phosphodiesterase PDE4D3 by cyclic AMP-dependent protein kinase and mitogen-activated protein kinases: a potential mechanism allowing for the coordinated regulation of PDE4D activity and targeting. J. Biol. Chem. 274, 10557–10565 [DOI] [PubMed] [Google Scholar]
- 48. Sette C., Iona S., Conti M. (1994) The short-term activation of a rolipram-sensitive, cAMP-specific phosphodiesterase by thyroid-stimulating hormone in thyroid FRTL-5 cells is mediated by a cAMP-dependent phosphorylation. J. Biol. Chem. 269, 9245–9252 [PubMed] [Google Scholar]
- 49. Sette C., Conti M. (1996) Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase: involvement of serine 54 in the enzyme activation. J. Biol. Chem. 271, 16526–16534 [DOI] [PubMed] [Google Scholar]
- 50. Conti M., Beavo J. (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 76, 481–511 [DOI] [PubMed] [Google Scholar]
- 51. Han P., Zhu X., Michaeli T. (1997) Alternative splicing of the high affinity cAMP-specific phosphodiesterase (PDE7A) mRNA in human skeletal muscle and heart. J. Biol. Chem. 272, 16152–16157 [DOI] [PubMed] [Google Scholar]
- 52. Yuasa K., Kotera J., Fujishige K., Michibata H., Sasaki T., Omori K. (2000) Isolation and characterization of two novel phosphodiesterase PDE11A variants showing unique structure and tissue-specific expression. J. Biol. Chem. 275, 31469–31479 [DOI] [PubMed] [Google Scholar]
- 53. Omori K., Kotera J. (2007) Overview of PDEs and their regulation. Circ. Res. 100, 309–327 [DOI] [PubMed] [Google Scholar]
- 54. Dell'Acqua M. L., Faux M. C., Thorburn J., Thorburn A., Scott J. D. (1998) Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4,5-bisphosphate. EMBO J. 17, 2246–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.