

Abstract
Several cytoplasmic proteins that are involved in G protein-coupled receptor signaling cascades are known to translocate to the plasma membrane upon receptor activation, such as beta-arrestin2. Based on this example and in order to identify new cytoplasmic proteins implicated in the ON-and-OFF cycle of G protein-coupled receptor, a live-imaging screen of fluorescently labeled cytoplasmic proteins was performed using translocation criteria. The screening of 193 fluorescently tagged human proteins identified eight proteins that responded to activation of the tachykinin NK2 receptor by a change in their intracellular localization. Previously we have presented the functional characterization of one of these proteins, REDD1, that translocates to the plasma membrane. Here we report the results of the entire screening. The process of cell activation was recorded on videos at different time points and all the videos can be visualized on a dedicated website. The proteins BAIAP3 and BIN1, partially translocated to the plasma membrane upon activation of NK2 receptors. Proteins ARHGAP12 and PKM2 translocated toward membrane blebs. Three proteins that associate with the cytoskeleton were of particular interest : PLEKHH2 rearranged from individual dots located near the cell-substrate adhesion surface into lines of dots. The speriolin-like protein, SPATC1L, redistributed to cell-cell junctions. The Chloride intracellular Channel protein, CLIC2, translocated from actin-enriched plasma membrane bundles to cell-cell junctions upon activation of NK2 receptors. CLIC2, and one of its close paralogs, CLIC4, were further shown to respond with the same translocation pattern to muscarinic M3 and lysophosphatidic LPA receptors. This screen allowed us to identify potential actors in signaling pathways downstream of G protein-coupled receptors and could be scaled-up for high-content screening.
Eukaryotic cells have evolved to segregate cellular functions into specialized intracellular membrane compartments. Information regarding the precise intracellular localization of a poorly characterized protein is thus important when evaluating its function. However, localization per se, without additional information, is not sufficient to indicate the potential function of cytoplasmic, or cytoplasmic and nuclear proteins that account for almost half of the proteome diversity, when compared with proteins located in dedicated organelles, such as the Golgi apparatus or lysosomes. To tackle this need for additional information, we have implemented a live-imaging screening strategy that is based on changes in localization of fluorescently labeled cytoplasmic proteins studied upon the activation of cells. Indeed, cytoplasmic and nuclear proteins often respond to the activation of a given signaling cascade by translocating to another cell compartment, for instance by moving from the cytoplasm to the plasma membrane, or to the nucleus. Classical signaling proteins that translocate following receptor activation include AKT/PKB (1), raf1 (2), or NF-KB (3). Therefore, finding that a given cytoplasmic protein responds to the activation of a given signaling cascade provides further insight into its function.
Studies dealing with the deciphering of intracellular localization of the proteome have mainly used mass-spectrometry based approaches (4, 5). Microscopy techniques have also been successfully used and validated: the localization of 97% of the yeast Saccharomyces cerevisiae proteome was achieved by tagging each protein with the fluorescent GFP protein (6). In human cells, a similar approach with a fluorescently tagged full-length ORF plasmid collection was used for the GFP-cDNA localization project (7) and it was also completed in mice with the Alliance for Cellular Signaling (8). The use of fluorescently tagged overexpressed proteins to probe intracellular localization was validated by protein correlation profiling although less correlation was observed for cytosolic proteins (9). More recently, the ≪protein Atlas≫ project has found a good correlation between endogenous proteins localization using immunofluorescence techniques compared with intracellular localization of the same proteins as determined in the GFP-cDNA localization project (10). Therefore, the cDNA library of this GFP-cDNA localization project was selected for our live-imaging screen: from this library, we screened the proteins that had been identified residing in the cytoplasm or cytoplasm and nucleus.
The signaling pathway that we selected for our case study is governed by the Gq-coupled tachykinin NK2 receptor, a prototypical G protein-coupled receptor (GPCR)1, that triggers an elevation of intracellular calcium concentration upon activation with the neuropeptide, neurokinin A (11). GPCRs share a common overall structure with 7-transmembrane alpha-helices embedded in the lipid bilayer mostly at the cell surface, from where they receive ligand-mediated messages to transduce into metabolic intracellular responses. The main, but not exclusive, intracellular effectors of GPCRs are heterotrimeric G proteins that belong to several groups depending on their secondary messenger modulation (Gq/11 : calcium elevation, Gs : cAMP elevation, Gi/o : cAMP reduction, G12/13 : actin cytoskeleton rearrangements) (12). Sustained activation leads to desensitization of the receptor through phosphorylations by specific GPCR kinases or Protein Kinases A and C (PKCA) that modify serine, threonine, and tyrosine residues on the intracellular part of the receptor. These phosphorylations participate in the uncoupling of the receptor from the G-proteins and the recruitment of beta-arrestins, such as ARRB2, that serve as specific adaptors of clathrin-coated pit formation. The receptors then internalize (13).
These receptors have an extremely dynamic and flexible structure, and it has been shown in recent years that they can adopt multiple conformations that can be differentially stabilized depending on the extracellular ligands. This is at the origin of complex signaling cascades that depend on the conformation adopted by the receptor and on its capacity to stimulate intracellular effectors (now referred to as “biased agonism”) (14). Not all the signaling cascades following activation have been fully characterized, making these receptors good targets to search for undiscovered cytoplasmic proteins involved in their signaling pathway.
The present article is dedicated to the full description of the results obtained in this live-imaging screen. Previously, we reported the identification and characterization of one of these translocated proteins, REDD1, as a novel target of GPCR signaling (15). The data are compared with the results of phospho-proteome investigations of signaling pathways governed by GPCRs (16–22).
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293 cells and clonal cell lines stably expressing either human NK2Gly361Glu Receptor, or CLIC2-mYFP or CLIC4-mYFP were cultivated as previously described (23).
Live-imaging Screening by Microscopy as Described in (15)
On day 0, HEK293 cells expressing the non-fluorescent NK2-Gly361Glu receptor (NK2R-HEK cells) were plated into 8-well Labtek chambers with glass-slides (NUNC). On day 1, 150 ng of individual vectors were transiently transfected using the calcium phosphate technique. Each clone was transfected twice, in two separate eight-well chambers. Transfection efficiency was of at least 80% unless otherwise stated (column headed “localization during screening” of the supplemental Table S1 and on the web site http://www.gfpcdnalive-gpcr.cnrs.fr). Each fluorescent protein was observed after 24 h of expression under the spinning-disk ultraview confocal microscope (Perkin Elmer) equipped with an incubation box allowing maintenance at 37 °C using the 63 × objective. The majority of the screened fluorescent proteins presented a level of expression that was in the range of expression of ARRB2-YFP and PKCA-YFP, the two proteins used as positive controls for translocation. Weak expression of a given fluorescent protein is also indicated (supplemental Table S1 and on the web site). In addition, the field to be filmed (displaying around 5–10 cells) was chosen manually by assessing the fluorescent expression of each cDNA clone in all the cells of the well from the Labtek chamber. Cells selected displayed a localization of the fluorescent signal that was representative of the transfected cells in the well. Cells were also chosen for detectable but low and homogenous amount of emitted fluorescence. Images acquisition was recorded for 90 s in the case of the activation process : the focus was placed in the z axis middle of the cell; activation with 100 nm NKA (in synthetic buffer HEPES-BSA, in mm: 137.5 NaCl, 1.25 MgCl2, 1.25 CaCl2, 6 KCl, 5.6 glucose,10 HEPES, 0.4 NaH2PO4, 1% bovine serum albumin (w/v), pH 7.4) was performed after 20 s recording while acquisition was in progress, allowing the visualization of the fluorescent protein before activation and its eventual short-term translocation upon induction. Unless otherwise stated on the website: image acquisition was continuous with exposure-time between 399ms to 999ms with Binning 1 for YFP samples and Binning 4 for CFP samples. Videos were generated using ImageJ software. Videos are accelerated showing 4 frames/seconds. The activation time, at around 20 s, is detectable on the videos by a change in the focal plane, because of the tips of the pipette slightly touching the culture plate. Sometimes, the focal plane was manually moved during the recording. This is indicated on the website. After activation, subsequent short videos of around 15 s were recorded ∼1 h and 4h after activation, this information is indicated on the website. If some translocation was detected during the experiment, the second well with the same clone nonactivated was first analyzed to verify that there was no change in localization of this un-activated clone. In a second time, this un-activated clone was used to reproduce the experiment of NKA activation.
Creating Generic Expression Vector With the LIC System
The CLIC2 and CLIC4 fragments were generated by PCR using pd-humanCLIC-EYFP as a template (7). Expression vectors for fusion proteins containing the mYFP fluorophore were generated with the LIC method as described in (23). The CLIC protein are separated from the mYFP by a six-residue linker, LeuSerAsnGluAsnGlu.
Confocal Fluorescent Microscopy
Cells were plated in 24-well plates on 12-mm glass coverslips for 24 h. Cells were activated in synthetic HEPES-BSA buffer with agonists for different period of time. Then, cells were rinsed in warm PHEM buffer (60 mm Pipes, 25 mm Hepes, 10 mm EGTA, and 2 mm MgCl2) then fixed for 12 min. in paraformaldehyde 4%/glutaraldehyde 0,05%/TritonX-100 0,05%/1xPHEM. Coverslips were rinsed with PBS, then incubated 10 min. twice in NaBH4 1 mg/ml. Actin was detected by a 15 min. staining with 1 Unit Texas-Red-X-Phalloidin (Molecular Probes, Invitrogen, Illkirch, France). Coverslips were mounted onto microscope slides using Mowiol antifading agent (Calbiochem, Molsheim, France). Image acquisition was performed as described in (23) with an inverted microscope (Leica, Nanterre, France) and a laser scanning confocal imaging system (SP2-UV or AOBS SP2 RS) using a HCX PL APO CS 100.0 × 1.40 oil immersion UV objective. To obtain a good signal to noise ratio, the images were averages from eight consecutive acquisitions.
Live-confocal Microscopy
HEK293 cells stably expressing CLIC2-mYFP or CLIC4-mYFP were plated into four 8-well Labtek chambers with glass-slides (NUNC). Two days later, cells were incubated 3 h in 200 μl of synthetic HEPES-BSA buffer. Image acquisition was performed with an inverted microscope and a laser scanning confocal imaging system (Leica-SP8) using a HCX PL APO CS 63 × 1.20 oil immersion objective. Activation was performed with 22 μl agonists in HEPES-BSA buffer (final concentration ACh, 10 μm, LPA 1 μm) or with 22 μl vehicle as a negative control. Images were acquired in multiposition mode (4 positions) over 15 min with z-stacks. Each video was generated using FIJI software and represent all the z-stack images at a given time-frame.
Figure. Preparation
Figs. of microscopy images were prepared with FigureJ (24).
RESULTS
Setting-up the Translocation Screen of the Cytoplasmic Protein Collection
The aim of the screen was to find cytoplasmic proteins that would respond to activation of the cells with the agonist neurokinin A (NKA) by changing their intracellular localization as an indication that the hit protein might be a downstream target of the NK2 receptor signaling cascades. Fig. 1A presents examples of changes in intracellular localization expected to occur for a cytoplasmic protein fluorescently labeled upon activation of the cells with the neuropeptide agonist. Upon activation of the cell expressing NK2 receptors with NKA, a cytoplasmic protein could, for example, translocate into the nucleus, or to the plasma membrane, or could migrate toward intracellular structures.
Fig. 1.
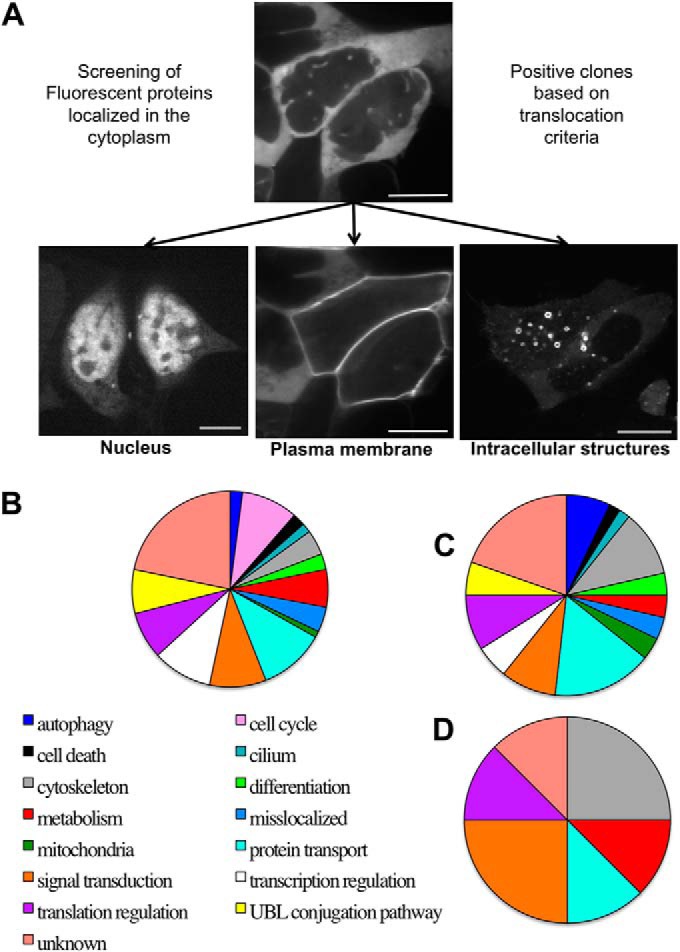
Global analysis of the results of the screening. A, Scheme of the translocation criteria upon NK2R-HEK cell activation with the neurokinin A agonist (100 nm). Examples of images extracted from the videos displaying fluorescent proteins in the cytoplasm (PKCA-YFP), in the nucleus (CFP-FAM58A), at the plasma membrane (PKCA-YFP upon NKA activation of the cells), in intracellular structures (ARRB2-YFP after NKA activation). B, Classification and statistics of the 193 proteins screened according to 15 categories based on known cellular processes. C, Classification of the 53 proteins displaying a specific localization before activation according to the 15 categories. D, Classification of the eight proteins that respond to NKA activation according to the 15 categories. Scale bar 5 μm.
The human tachykinin NK2 receptor that we used was mutated in its C-terminal tail at position Gly 361 (replaced by Glu). This mutation results in a predominant coupling with heterotrimeric Gq protein and triggers an intracellular calcium increase upon NKA stimulation, but no cAMP accumulation, contrary to the wild-type receptor (25). This mutant receptor was chosen to bias the assay toward Gq-coupling and calcium signaling. Human Embryonic kidney cells, that do not express endogenously tachykinin receptors (26) were stabilized to over-express human NK2-Gly361Glu receptor (NK2R-HEK cells).
In practice, NK2R-HEK cells were transiently transfected with individual vectors from the fluorescently tagged human full-length ORF plasmid collection encoding cytoplasmic proteins (7). This plasmid collection is composed of ORFs of poorly characterized function that are fused in-frame with the cDNA encoding a fluorescent protein (ECFP on the Nter of the ORF and alternatively EYFP Enhanced Yellow Fluorescent protein on the Cter of the ORF).
Optimizations were made to determine the amount of DNA to be transfected such that it would be suitable for screening the entire plasmid collection. This optimization was performed by testing four clones of the library, together with two positive control vectors: (1) PKCA, previously, we have shown that Protein Kinase C (PKCA) phosphorylates the NK2 receptor upon activation (27) and this implies PKCA translocation from the cytoplasm to the plasma membrane in a very transient manner within seconds (illustrated for PKCA-YFP in Fig. 1A). (2) beta-arrestin 2, we have also shown that beta-arrestin 2 (ARRB2) translocates at the plasma membrane, interacts with the activated receptor (23, 28) and triggers the internalization of NK2R into clathrin-coated vesicles (illustrated for ARRB2-YFP in Fig. 1A). Thus, ARRB2-YFP displays a fast, but sustained, translocation upon NKA activation. Of note, we observed that ARRB2-YFP translocation is best detected when the level of expression of the protein is low. Thus, the level of DNA to be transfected was adjusted in order to give a good transfection efficiency, whereas maintaining low expression of the encoded fluorescent protein.
In another control experiment setting-up the screen, we confirmed that HEK293 cells without exogenous expression of the NK2 receptor that were transiently transfected with PKCA-YFP or ARRB2-YFP, did not show translocation of these fluorescent proteins when treated with the NKA ligand. There was also no change in the actin cytoskeleton upon addition of NKA to HEK293 cells without exogenous expression of NK2 receptor. We have also never detected any binding of the radiolabeled selective NK2-antagonist, SR48989, or of the fluorescently labeled NKA to HEK293 cells without exogenous expression of NK2 receptor or any calcium responses to NKA (25, 27–29). Therefore, we are confident that changes in localization of a given fluorescent protein upon NKA stimulation occur because of the specific activation of the tachykinin NK2 receptor overexpressed in NK2R-HEK cells.
Twenty-four hours after transfection, the precise localization of the cytoplasmic fluorescent protein under study was analyzed in living NK2R-HEK cells using a spinning-disk confocal microscope. Fluorescence of the cells was recorded for ∼20 s before NKA activation, then followed for over a minute. Further videos were recorded depending on the behavior of the cells and/or of the fluorescent cytoplasmic protein under observation. Eight rounds of transfection were necessary to screen the vector collection (almost 400 individual vectors) and at each transfection, the positive control vectors expressing fluorescently labeled PKCA-YFP and ARRB2-YFP were tested in duplicate. ARRB2-YFP was found to translocate during the first 70 s of activation 13 times out of 16. Within minutes to hours after NKA activation, the majority of the cells in a well were found to display ARRB2-YFP around vesicles in all the experiments (16 out of 16). PKCA-YFP was found to translocate during the process that recorded activation 13 times out of 16. Of note, 2 videos display a very faint and transient translocation of PKCA-YFP (videos “T3pvideo 2” and “T5video 1” on the website).
Global Analysis of the Results of the Screening
Since the first description of the fluorescent cDNA library, most of the proteins encoded by this collection of expression vectors are now fully annotated and some functions have been attributed to them as well. However, the whole screening procedure, the analysis of the movies and the criteria for selecting the potential hit proteins were performed in blind by using the identification number associated with each vector without the knowledge of the protein encoded. The final analysis of the proteomic data was achieved with the knowledge of the proteins encoded.
The Name and Swissprot ID of each screened protein is indicated in Table I. We screened 140 fluorescent fusion proteins that had previously been determined as localizing in the cytoplasm of resting cells (7) and 53 that were localized both in the cytoplasm and the nucleus. Each of these proteins was screened once with the fluorophore ECFP fused to its amino-terminal part and once with the fluorophore EYFP fused to its carboxyl-terminal part (unless otherwise stated). Fig. 1B indicates the repartition of the screened proteins according to the known biological processes in which they are described in the uniprot database at http://www.uniprot.org/. The screened proteins belong to 13 categories of biological processes, with the addition of two categories for proteins that are still of unknown function (22% of the screened proteins) and for clearly mislocalized proteins (4% of the screened proteins), such as integral membrane proteins and those that should be secreted. Categories representing around 10% of the screened proteins are: cell cycle, protein transport, signal transduction, and transcription regulation.
Table I. List of all the screened proteins. Name and SwissprotID of the screened proteins are listed in alphabetical order: in green, fluorescent proteins with specific localization before activation with Neurokinin A, in yellow, fluorescent proteins that translocate upon tachykinin receptor activation.
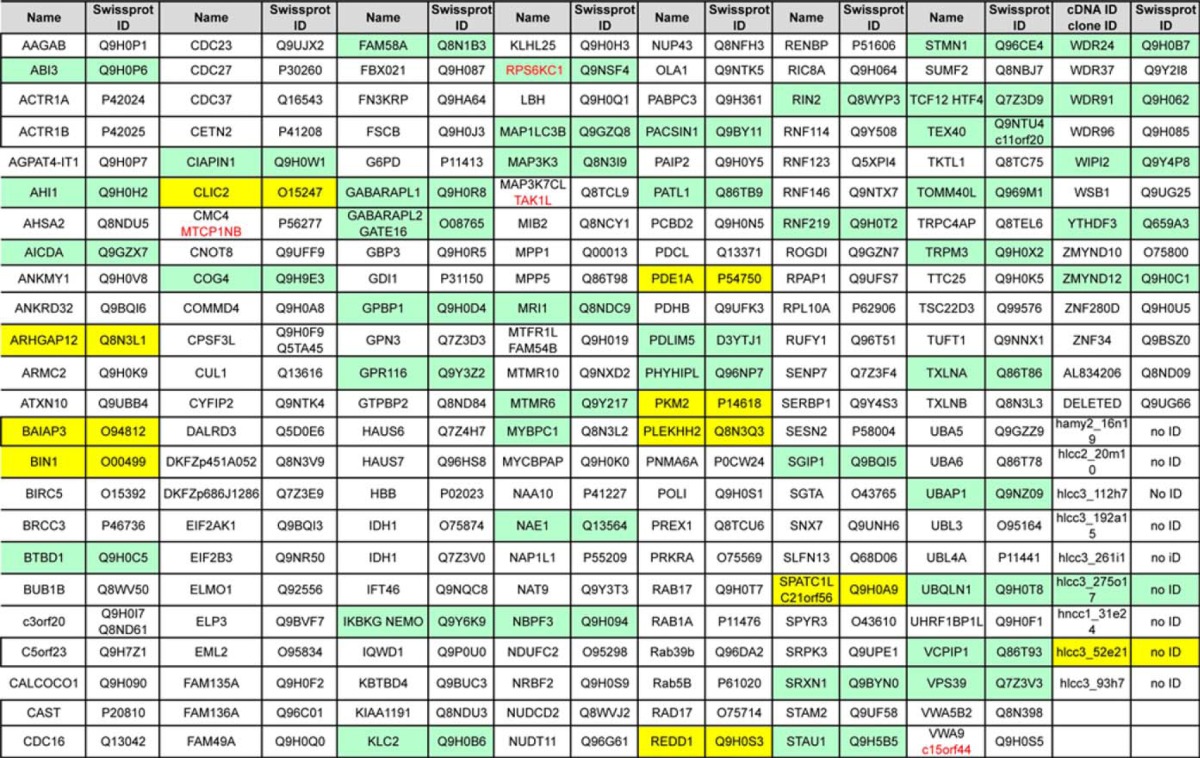
Fifty-six proteins were selected as potential hits and were classified into three different classes according to their localization depending on the activated state of the cells (1, special localization before activation; 2, fast translocation upon activation (usually recorded in the video of activation); 3, slow translocation observed several minutes or hours after NKA activation). Fifty-three fluorescent proteins (27% of the screened proteins) displayed a special localization before activation, in addition to their diffuse location in the cytoplasm or the cytoplasm + nucleus, that could be associated, for example, to fast moving vesicles or dots, centrosomes, cytoskeleton, mitochondria or undefined structures (green cases in Table I). Fig. 1C illustrates the proportion of these proteins with specific localizations according to their known biological function: categories representing around 10% or more of these 53 proteins are: unknown (20%), protein transport (16%), and cytoskeleton element (10%).
Eight fluorescent proteins were selected because they displayed translocation upon NKA activation of NK2R-HEK cells (yellow cases in Table I). Six of them already displayed a special location before activation (ARHGAP12, BAIAP3, CLIC2, PLEKHH2, SPATC1L), the others being BIN1, PKM2, and REDD1. The 8 proteins identified in the live-imaging screen belong to six out of the 14 categories of “cellular functions” (excluding the mislocalized proteins): signal transduction (BAIAP3, ARHGAP12), cytoskeleton (CLIC2, PLEKHH2), metabolism (PKM2), protein transport (BIN1), translation regulation (REDD1), or still unknown function (SPATC1L, Fig. 1D).
Additional Information Collected from Analysis of the Videos From the Screen
Additional information was collected from the careful analysis of all the videos that recorded cell activation (212 videos analyzed): Shrinkage and even blebbing of the cells was often observed.
Blebbing is a protrusion of the plasma membrane that looks like a bubble and that occurs because of the transient uncoupling of the lipid bilayer from the underlying cortico-cytoskeleton (30). It happens both physiologically (for example, during mitosis and cell migration) or upon cellular stresses (apoptosis, necrosis, cell-substrate detachment and even as a light-artifact observed during microscopic analysis of living cells). Among the videos recorded upon cell activation by NKA, 30% display membrane blebbing of the cells. Activation of GPCRs has been described as triggering a cellular blebbing that lasts several minutes and is continuously dynamic (31). On the other hand, the blebbing observed during our screening occurred just after activation and was short-lasting. In addition, although HEK293 cells are adherent, they can easily detach from the surface of the slide on which they grow. The blebbing observed during recordings of NKA activation appeared to be mainly because of cell detachment and shrinkage.
The results of the video analysis are listed in the supplemental Table S1 in which proteins are classified by their name (as well as their full-name, original clone ID, cDNA ID, Swissprot ID and fusion orientation). supplemental Table S1 also presents (1) the localization of the endogenously expressed protein as determined by immunofluorescence studies in (10) (2) the known biological processes, (3) the localization of the fluorescent clone as determined in the original study of intracellular localization (7) compared with the localization observed during the screening. Also noted are any noticeable features at the time of the screening, the classification of the protein dynamics categories (special location or moving vesicles or dots or translocation) and the cell dynamics categories (detachment, shrinkage or blebbing).
Most of the videos recorded during the live-imaging screen can be viewed on-line at http://www.GFPcDNAlive-GPCR.cnrs.fr, in which the proteins of interest are listed in a table similar to supplemental Table S1. The videos of the positive controls, ARRB2-YFP and PKCA-YFP, transfected at each round of transfection, are also displayed on the website.
Fluorescently Labeled Proteins Identified During the Screen that Display Specific Localizations Independent of Both the Expression and the Activation of NK2 Receptor
53 fluorescently labeled proteins were identified during the screen with an additional localization other than the cytoplasm and/or nucleus. 47 of them did not respond to NKA application. Interestingly, not all of these specific localizations were described in the original screening for localization performed in Vero cells (7). However, most of those identified correlate well with the known biological processes that have been attributed to the corresponding proteins (see supplemental Table S1). These results are an indication of the reliability of the screening procedure for detecting intracellular localization.
Figs. 2 and 3 show images extracted from the videos of NK2R-HEK cells expressing some of these fluorescently modified proteins with the expected specific localization. Several proteins implicated in the UBL-conjugation pathway were detected during the screen for their localization in vesicles, probably corresponding to different compartments of the endocytic pathway (UBAP1, UBQLN1, NAE1, VPS39 Fig. 2A, 2B, 2C, 2D respectively and corresponding videos). Likewise, proteins known to be implicated in autophagy (MAP1LC3B, GABARAPL1, WIPI2, and GABARAPL2) were all detected for their specific localizations, that possibly correspond to autophagic structures (Fig. 2E, 2F, 2G, 2H, respectively and corresponding videos).
Fig. 2.
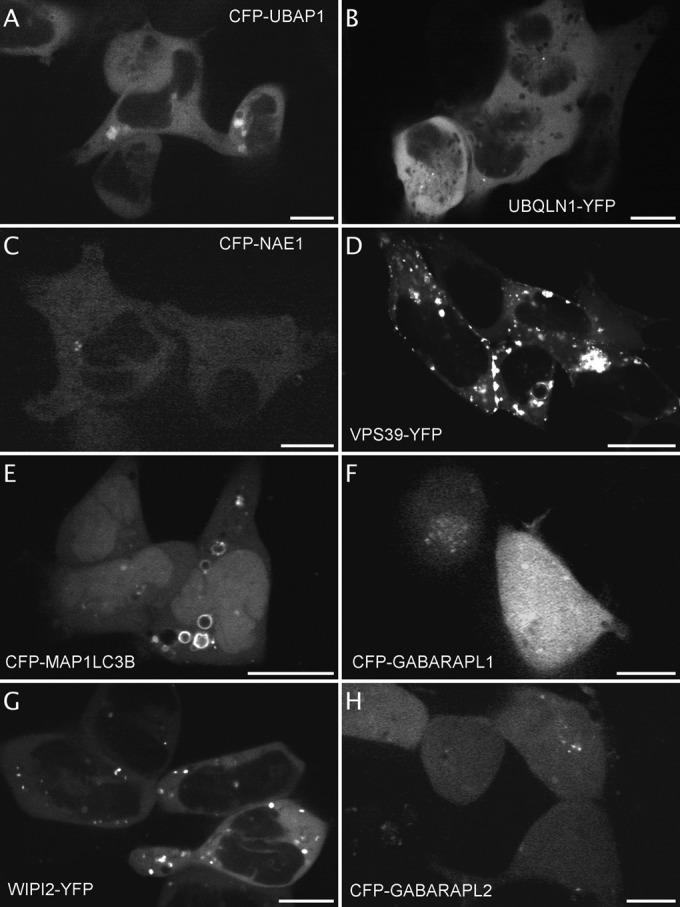
Correct detection in HEK-NK2R of fluorescent proteins implicated in internalization and in autophagy during the screen as blind internal controls. A, The image of cells expressing CFP-UBAP1 in the cytoplasm and around large vesicular structures is extracted from video1 (out of 2 videos on the website). Five cells out of eight are displayed. B, The image of cells expressing UBQLN1-YFP in the cytoplasm and few spots are extracted from video2. Four cells out of five are displayed. C, The image of cells expressing CFP-NAE1 in the cytoplasm and dots is extracted from video1 on the website. Two cells can be visualized out of six in the video. D, The image of cells expressing VPS39-YFP in the cytoplasm and aggregates is extracted from video1 on the website. Four cells out of eight are displayed. E, The image of cells expressing CFP-MAP1LC3B in the cytoplasm, around vacuole-type compartments and enriched in the nucleus is extracted from video 1 out of four videos. Three cells out of four are displayed. F, The image of cells expressing CFP-GABARAPL1 in the cytoplasm, dots and the nucleus is extracted from video 1 out of three videos. Two cells out of nine are displayed. G, The image of cells expressing WIPI2-YFP in the cytoplasm and vesicles is extracted from video1 out of four videos. Five cells out of eight are displayed. H, The image of cells expressing CFP-GABARAPL2 in the cytoplasm, dots and nucleus is extracted from video 1 out of four videos. Five cells out of seven are displayed. Scale bar 5 μm.
Fig. 3.
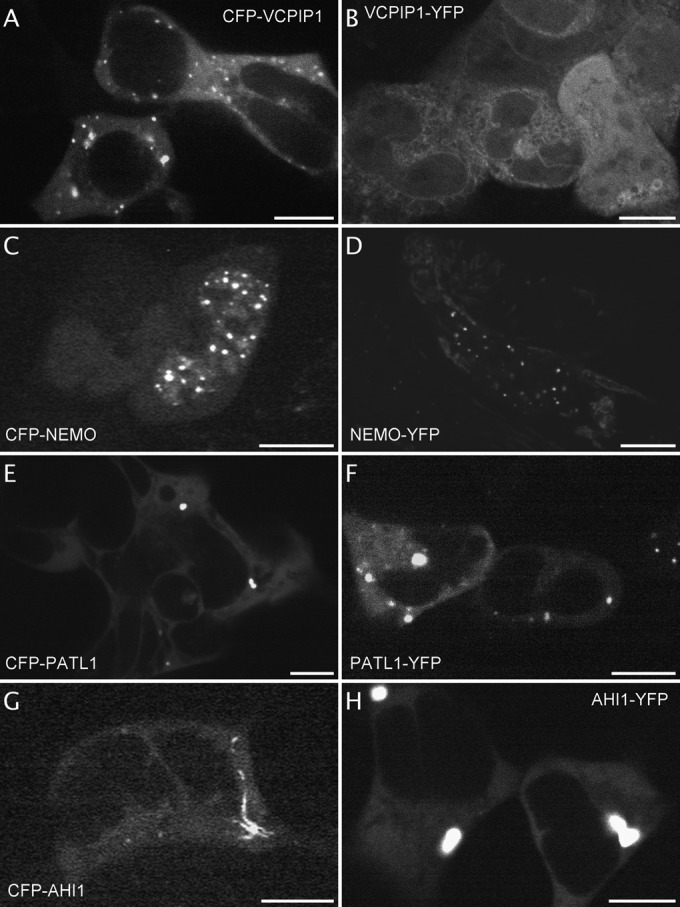
Identification of variation of localization depending on the fluorophore orientation in the fusion protein expressed in HEK-NK2R cells. Images extracted from the videos of VCPIP1 expressing cells with the fluorophore fused to its Nter (A, extracted from video 1 out of two, 3 cells displayed out of 10) or to its Cter (B, extracted from video 7 out of seven, 6 cells displayed out of 21). Images extracted from the videos of NEMO expressing cells with the fluorophore fused to its Nter (C, from video1 out of two, 3 cells displayed out of 6) or to its Cter (D, from video 1, 2 cells displayed out of 8). Images extracted from the videos of PATL1 expressing cells with the fluorophore fused to its Nter (E, from video 2, 6 cells displayed out of 8) or to its Cter (F, from video 3 out of five, 2 cells displayed out of 13). Images extracted from the videos of AHI1-expressing cells with the fluorophore fused to its Nter (G, from video 3, 1 cell displayed out of 14) or to its Cter (H, from video 1 out of 3, 2 cells displayed out of 13). Scale bar 5 μm.
Noticeable differences can be found in the pattern of the fluorescent signal according to the fusion orientation of the fluorophore with respect to the protein tested for proteins that carry signals on either their Nter parts (such as signal peptide) or their Cter parts (like autophagic proteins, such as GABARAPL1, that becomes conjugated with phosphatidylethanolamine on its cleaved C terminus) as was expected and observed previously (7). Fig. 3 shows images of NK2R-HEK cells expressing fluorescently labeled proteins with known localizations, but for which we have found that this localization depends upon the fusion orientation. For example, VCPIP1 is implicated in protein transport between the endoplasmic reticulum (ER) and the Golgi apparatus. Although the VCPIP1-YFP became excluded from the nucleus and associated with the ER membrane (Fig. 3B, and 7 videos on the website), the CFP-VCPIP1 localized in cytoplasm and is enriched in vesicles (Fig. 3A and 2 videos on the website). The transcription regulator, CFP-NEMO (Fig. 3C and 2 videos on the website), was more abundant in nucleus speckles than was NEMO-YFP (Fig. 3D and 1 video on the website) (32). The RNA-binding protein, CFP-PATL1, was less abundant than PATL1-YFP in PML bodies (Fig. 3E and 3F, 2 and 5 videos on the website respectively) (33, 34). Finally, although the cilium protein, CFP-AHI1 localized in the cytoplasm and concentrated in cilium-like structures, the AHI1-YFP localized in the cytoplasm and concentrated into one or two large cytoplasmic spots (Fig. 3G and 3H, respectively, 3 videos each on the website) (35).
For other fluorescently labeled proteins, the identification of a specific localization is a novel information including, most of the time, a different pattern depending on the fusion orientation. CFP-YTHDF3 was excluded from the nucleus whereas the protein with the fluorophore on its Cter was enriched in spots of the nucleus (Fig. 4A and 4B, 2 and 9 videos on the website respectively). Fluorescent YTHDF2 fusion is described in the literature (36) and it becomes phosphorylated upon Gq-AT1 receptor activation (17). Fluorescent PDLIM5 was enriched at microtubule-like cytoskeleton elements whether fused to the Cter (Fig. 4C, 4 videos on the website) or to the Nter (not shown). In the literature, homologs of PDLIM5, such as LMP1-GFP (37) or GFP-Enigma (= PDLIM7) have been found associated with the actin cytoskeleton (38, 39). CFP-CIAPIN accumulated at the ER membrane whereas CIAPIN-YFP did not (Fig. 4D). WDR91-YFP and PHYHIPL-YFP, both proteins of unknown function, concentrate in cytoplasmic dots (Fig. 4E and 4F, 3 and 2 videos on the website, respectively). Novel localizations are also illustrated in Figure 5 : CFP-COG4, implicated in protein transport at the Golgi apparatus, was enriched in vesicles (Fig. 5A) (40), CFP-FAM58A and CFP-GPBP1, implicated in transcription regulation, are enriched in dots and vesicles (Fig. 5B and 5C). Both the CFP-NBPF3 and the unknown testis-expressed sequence 40 protein CFP-TEX40 displayed perinuclear localization (Fig. 5D and 5E and 2 videos on the website). STMN1, which is implicated in microtubule dynamics and becomes phosphorylated upon AT1R and CXCR4 activation (17, 18, 20) displayed a vesicular localization when tagged on its Nter part that was not described previously for the same fusion protein (Fig. 5F) (41, 42).
Fig. 4.
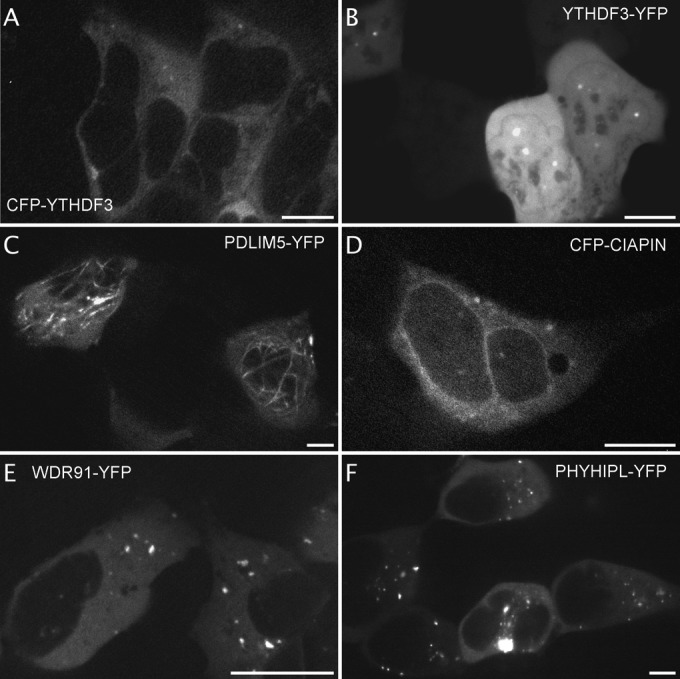
Identification of fluorescent proteins with special localization independent of NK2R-HEK cells activation. Examples of images extracted from the videos displaying fluorescent proteins in dots within the cytoplasm (A, CFP-YTHDF3, from video 2, 5 cells displayed out of 9) or dots within the nucleus (B, YTHDF3-YFP, from video 2 out of 9, 5 cells displayed out of 12), cytoskeleton localization (C, PDLIM5-YFP, from video 2 out of 4, 3 cells displayed out of 6), perinuclear localization (D, CFP-CIAPIN, from video 1, 1 cell displayed out of 4), intracellular vesicles (E and F, WDR91-YFP, from video 2 out of 3, 3 cells displayed out of 10 and PHYHIPL-YFP, from video 1 out of 2, 5 cells displayed out of 19). Scale bar 5 μm.
Fig. 5.
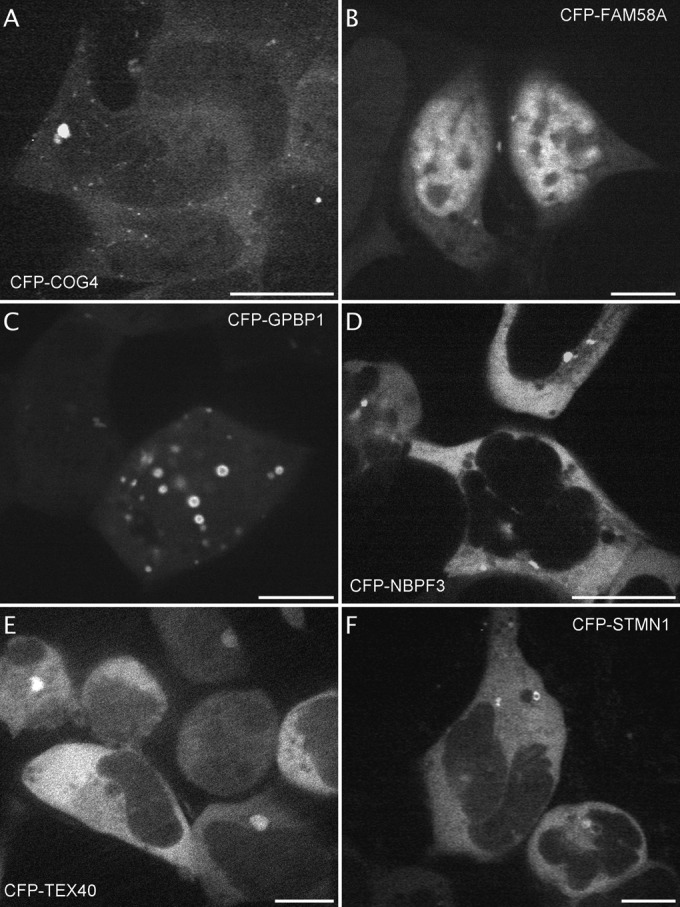
Identification of fluorescent proteins with special localization independent of NK2R-HEK cells activation. Examples of images extracted from the videos displaying fluorescent proteins moving vesicles within the cytoplasm (A, CFP-COG4, from video 4, 7 cells displayed out of 12) or dots near or within the nucleus (B, C, CFP-FAM58A, from video 1, 4 cells displayed out of 6 and CFP-GPBP1, from video 1 out of 2, 2 cells displayed out of 9), perinuclear localization (D, E, CFP-NBPF3, from video 2, 3 cells displayed out of 9 and CFP-TEX40, from video 1 out of 2, 7 cells displayed out of 13) and intracellular vesicles (F, CFP-STMN1, from video1, 3 cells displayed out of 5). Scale bar 5 μm.
Fluorescently Labeled Proteins Identified During the Screen that are Potentially Implicated in Blebbing
Although the blebbing of activated NK2R-HEK cells observed during the screening was mainly because of cell detachment and not to NK2R activation per se, it allowed us to detect two fluorescently labeled proteins displaying a peculiar translocation toward blebs, suggesting that these proteins might have a role in triggering bleb formation and/or resorption (Fig. 6).
Fig. 6.

Identification of fluorescent proteins implicated in blebbing and in internalization. Images extracted from video 7 out of 9 of ARHGAP12-YFP expressing NK2R-HEK cells before (A) and 1 min after formation of the blebb (B). The protein concentrates at the opening of blebbs and their sites of resorption. Images extracted from video1 of PKM2-YFP expressing NK2R-HEK cells before (C) and few seconds after cell shrinkage (D, from video 1, 1 cell displayed out of 10). Cytoplasmic BIN1-YFP (E) transiently accumulates at plasma membrane sites around 1 min after NK2 receptors activation (F, from video 1 Part 1 out of 6 videos, 5 cells displayed out of 10). Images extracted from video1 out of 12 videos, 2 SGIP1-YFP expressing cells out of 8 before (G) and 1 min after NKA activation (H). The fluorescent protein is enriched into plasma membrane spots (probably together with clathrin) and, although cells are shrinking, NK2 receptor activation does not modify the localization of SGIP1-YFP. Scale bar 5 μm.
ARHGAP12
Fluorescently labeled ARHGAP12 was found in a reproducible manner to accumulate at the plasma membrane at the circumference of the entrance of the blebs (see Fig. 6A–6B and 9 videos on the website). It mainly accumulates at the circumference when blebs are retracting and it stays enriched at the plasma membrane zone, like a scar, after the bleb has resorbed. ARHGAP12 belongs to the Rho-GAP family containing GTPase-activity toward the small G-protein Rho (43). Blebbing is triggered by RhoA/Rock signaling and thus it is interesting to find the precise localization of ARHGAP12 at the entrance of the bleb upon retraction, especially as it is difficult to determine the role of each one of the Rho-GAP family members (probably at least 70 proteins in human).
PKM2
Upon blebbing, PKM2-YFP was found to accumulate inside the bleb when compared with most of the other fluorescently labeled proteins (see Fig. 6C–6D and corresponding video). The pyruvate-kinase isoenzyme 2, PKM2, has been found in exosome extracts (44) but it has not yet been reported to be present in blebs. Interestingly, PKM2 interacts with DAPK that triggers blebbing upon necrosis (45). Tyrosine-phosphorylation of PKM2 was significantly increased in the cortical frontal region of the brain of morphine-dependent rats (16).
Fluorescently Labeled Proteins That Respond to NKA Activation by a Rapid Change in Intracellular Localization Identified During the Screen
During the screen, two fluorescently labeled proteins were rapidly translocated to the plasma membrane upon NK2 receptor activation : REDD1-YFP and BIN1-YFP. The mechanism of REDD1-YFP translocation has been confirmed and characterized in our previous study (15), demonstrating that REDD1 is a novel link between GPCR and the mammalian Target Of Rapamycin Complex 1 mTORC1 kinase signaling that regulates translation.
BIN1
BIN1-YFP was localized in the cytoplasm before activation of the cell. Several seconds after activation, part of the fluorescent signal appeared at the plasma membrane (Fig. 6E–6F and 6 videos on the website). BIN1 (also known as Amphiphysin2) participates in clathrin-coated pit formation (46) and therefore it is interesting to detect that it responds to the activation of the NK2 receptor, a receptor that internalizes via clathrin.
Of note, another protein tested during the screening, SGIP1, participates in specific cargo loading into clathrin-coated pits (47). In our screen, SGIP1 displayed a special localization before NKA activation. Indeed, SGIP1-YFP, and to a lesser extent, CFP-SGIP1, were concentrated in dots at the plasma membrane similar to those formed by GFP-clathrin (Fig. 6G–6H) (48). SGIP1 was not found to react upon NK2 receptor activation suggesting SGIP1 is unnecessary for NK2 receptor internalization (Fig. 6G–6H and 12 videos on the website). This might be explained by the fact that beta-arrestins are specialized in loading GPCRs into clathrin-coated pits and that no further clathrin-associated sorting proteins are required.
Identification of Fluorescently Labeled Proteins that Respond to NKA Activation by a Long-lasting Change in Intracellular Localization
BAIAP3
Upon NK2 receptor activation, CFP-BAIAP3 (that otherwise localizes within the cytoplasm and in vesicles) partially translocated to the plasma membrane where it stayed for several minutes (Fig. 7A–7B and 5 videos on the website). BAIAP3 was first isolated during a two-hybrid screen as a binding partner of the carboxyl-terminal part of the BAI1 adhesion GPCR and its function is still unknown (49). Our data show that BAIAP3 responds to Gq-coupled receptor stimulation.
Fig. 7.

Identification of fluorescent proteins of unknown function that respond to NK2 receptors activation by a change in localization. Images extracted from videos of CFP-BAIAP3 expressing NK2R-HEK cells before (A) and within seconds after NKA activation (B, from video 1 out of 5). Representative epifluorescence images acquired close to the slide of fixed PLEKHH2-YFP expressing NK2R-HEK cells before (C) and after 3 h NKA activation (D), n = 3 independent experiments. Images extracted from videos of CLIC2-YFP expressing NK2R-HEK cells before (E) and 2 min after NKA activation (F, from video 2 out of 7 videos, 4 cells displayed out of 8). Images extracted from video1 of HEK293 cells stably expressing CLIC2-mYFP before (G) and 20 min after activation of endogenous Gq-coupled muscarinic M3 receptors with 10 μm ACh (H) n = 3 independent experiments. Images extracted from video1 of HEK293 cells stably expressing CLIC2-mYFP before (I) and 25 min after activation of endogenous G12-coupled lysophosphatidic acid receptors with 1 μm LPA (J). n = 2 independent experiments. Scale bar 5 μm.
PLEKHH2
At steady state, PLEKHH2-YFP was present in two other specific structures in addition to a cytoplasmic localization: A few big aggregates inside the cytoplasm and small individual spots at the plasma membrane that were grouped (Fig. 7C and 7 videos on the website). These assemblies of spots are probably at the level of the interaction between the plasma membrane and the surface of the glass slide. After several hours of NKA activation, the small individual spots rearranged into several short linear succession of dots, usually in lamellipodia (Fig. 7D). Our data illustrate a very peculiar localization of PLEKHH2 and indicate that it responds to activation of a prototypical Gq-coupled receptor.
SPATC1L
This speriolin-like protein is of unknown function, whereas speriolin is a spermatogenic cell-specific centrosomal protein (50). Before activation, CFP-SPATC1L was distributed in the cytoplasm, the nucleus and in the perinuclear region. After one hour of activation, fluorescence was also detectable at the top of the cell-cell junction (see the 5 videos on the website).
CLIC2
At steady state, fluorescently labeled CLIC2 was found in the cytoplasm and in the nucleus but it was also enriched in plasma membrane bundles (Fig. 7E). Upon NKA activation, some CLIC2 expressing cells displayed rapid and transient translocation of the fluorescent signal, then the bundle structures spread and the protein became more diffuse at the plasma membrane (Fig. 7F). These specific localizations before activation and after NKA, the diffuse plasma membrane staining occurred with both orientations of the fusion protein with the fluorophore (CFP-CLIC2 and CLIC2-YFP, 10 and 6 videos on the website, respectively). This was a slow process that lasted over an hour after NK2 receptor activation. CLIC2, 247 residues, belongs to the CLIC family of Chloride Intracellular Channel proteins that include 6 cytoplasmic proteins of unclear function (51).
More importantly, CLIC4 has been previously described to transiently translocate to the plasma membrane upon activation of the G12-coupled receptor LPA via a RhoA mechanism (52). CLIC4 is present in the GFP-tagged plasmid collection but was not included in the original screen. We decided to further study the behavior of CLIC2 and to extend the analysis to CLIC4. Stable HEK293 cell lines were generated, expressing either CLIC2-mYFP or CLIC4-mYFP. The two fluorescent proteins display the same pattern of localization (Fig. 7G and 7I for CLIC2-mYFP and videos of CLIC2 and CLIC4 on the website) : cytoplasmic, nuclear and enriched in plasma membrane bundles. Latter, the bundles get numerous at the top of cells when the cells reach confluence. Stable cell lines were observed live under a confocal microscope at 37 °C near confluence. When endogenously expressed muscarinic M3 Gq-GPCR was activated with 10 μm of its acetylcholine agonist, the CLIC2-mYFP became slightly enriched at the plasma membrane and then, over a period of 60 min, some of the proteins underwent re-organization at the top of cell junctions and the bundles diminished in number (Fig. 7H and the 3 videos on the website). Several GPCRs responding to the LPA agonists are endogenously expressed in HEK293 cells. When these receptors were activated with 1 μm LPA, fast translocation of CLIC2-mYFP to the plasma membrane was triggered and physiological blebbing was observed (see on the website, the last videos taken over a period of 17 min activation with LPA following a section in the middle of the z axis of the cells). This was followed by a strong rearrangement within the cell and part of the CLIC2-mYFP accumulated over a period of 60 min at the top of the cell, where they form junctions although the bundles enriched in CLIC2-mYFFP did not diminish in numbers (Fig. 7J and the 8 videos on the website). We found that CLIC4-mYFP responded to LPA in the same manner as CLIC2-mYFP (2 videos on the website).
To get further insight into the change in localization of the CLIC2 protein upon activation of GPCRs, CLIC2-mYFP-expressing cells were treated with muscarinic M3 agonist, fixed and stained with Texas-Red phalloidin to detect the actin cytoskeleton. Before activation, the CLIC2-mYFP is localized in the cytoplasm and the nucleus (Fig. 8A, 8D) and is enriched in plasma membrane bundles at the top of the cells and the fluorescent protein co-localize with the actin cytoskeleton (Fig. 8G CLIC2-mYFP, 8H Actin, 8I overlay). One hour after activation of the cells, the CLIC2-mYFP signal at the top of the cell is re-organized at the cell–cell junctions where actin is also enriched (Fig. 8P CLIC2-mYFP, 8Q Actin, 8R overlay). Hence, we describe CLIC2 and CLIC4 as novel proteins that rearrange to cell–cell junctions upon stimulation of at least two GPCRs.
Fig. 8.

Co-localization of CLIC2-mYFP with actin. Confocal imaging of CLIC2-mYFP cells before (A to I) and 1 h after agonist challenge (10 μm carbachol, J to R). Cells were fixed and stained with Texas-Red phalloidin. Z-stack images of the cells were collected from the bottom (A, B, C, J, K, L), the middle (D, E, F, M, N, O) and the top of the cells (G, H, I, P, Q, R). The CLIC2-mYFP signal is in (A, D, G, J, M, P). The actin is visualized in (B, E, H, K, N, Q). Co-localized YFP (in green) and Texas-Red (in red) signals appear yellow in the overlay (C, F, I, L, O, R). n = 2 independent experiments. Scale bar 5 μm.
DISCUSSION
One hundred ninety-three human proteins or open-reading frames, fluorescently labeled at their N terminus and alternatively their C terminus, were independently transfected in cells stably expressing a GPCR, the tachykinin NK2 receptor. The fluorescence of each protein was then observed in the cytoplasm of the cells using confocal microscopy. The process of cell activation with the NK2 receptor agonist was recorded on videos at different time points and positive fluorescent clones were selected based upon a change in their intracellular localization. The screening reveals new specific intracellular localizations, specifies which end of the protein can be properly tagged and more importantly, identifies eight proteins that responded by translocating to the activation of the Gq-coupled NK2 receptor.
Four percent of the tested proteins were found to potentially respond to NK2 receptor activation. By comparison, the recent studies by mass spectrometry that have identified proteins that belong to GPCR signaling pathways by quantitative phosphoproteomics have usually found that 5% to 10% of the 2000 to 3000 phosphorylated proteins detected were modulated by GPCR activation. Hence, 5% of the detected phosphoproteome were found modulated by activation of the G12-coupled lysophosphatidic acid (LPA) receptors (19), 5–15% by activation of the Gi-coupled CXCR4 chemokine receptor (18, 20, 22), 10% were modulated by activation of the Gs-coupled V2 receptor (21) and up to 20% when the Gq-coupled angiotensin II type 1 AT1R was activated (17).
In our screen, no hit was detected for cellular processes like autophagy, cell cycle, cell death, cilium, differentiation, mitochondria, transcription regulation or ubiquitin-like (UBL) conjugation pathway. This might infer that proteins from these biological processes do not respond by changing their intracellular localization. Indeed, proteins of the UBL-conjugation pathway become phosphorylated upon activation of GPCRs, such as WIPI2, UBAP2L (17, 22) and UBA1 (17). WIPI2, UBAP1, UBA5, and UBA6 are present in the collection of fluorescent ORFs and were screened, but were negative for translocation. However, one would have expected to detect nuclear translocation of fluorescently labeled-proteins implicated in transcription because this function was well represented in proteins from our screen (Fig. 1B). Slow and sustained translocation to the nucleus would have been identified, although the set-up of the screen, which did not use multipositioning of the confocal platform at the time of activation, could account for missing some proteins that were translocating rapidly and transiently. It is probable that a live-imaging screen using the complete cytoplasmic proteome fluorescently labeled, instead of only a subset of 193 proteins, would result in the identification of proteins that respond to NK2 receptor activation by nuclear translocation.
Proteins Downstream of Calcium Secondary Messenger Identified in the Live-imaging Screen
Most phosphoproteome analyses of GPCR signaling have detected modulation of several components of the mTOR translation regulation pathway, but the mTORC1 inhibitor, REDD1, was not among the phosphoproteins detected in these studies (17, 18, 20–22). This is probably because of the low abundance of this ubiquitously expressed protein and also because our previous study on REDD1 has shown that its main phosphorylated residues (ser19, thr23, and thr25) were not necessary for REDD1 translocation in response to GPCR activation (15). Low abundant proteins and the absence of triggered phosphorylations are two obvious drawbacks of the phosphoproteomic approaches for analyzing new proteins of the GPCR signaling pathways. In our previous study, the translocation of REDD1 was further characterized using a quantitative assay based on Bioluminescence Resonance Energy Transfer performed in living cells that permits quantification of dynamic interactions of proteins with the plasma membrane (15). Using this technique, we could show that REDD1 is specifically translocated to the plasma membrane in response to activation of five out of six endogenously expressed GPCRs that were tested, namely, those that couple preferentially to Gq, Gs or Gi heterotrimeric G proteins, but not to G12. Calcium elevation was found to be necessary although not sufficient. It would be interesting to implement this plasma membrane BRET assay in the case of the BAIAP3 protein that we identified in the screen. Indeed, BAIAP3 contains a C2 domain that is a structural domain involved in targeting protein to cell membranes and that can bind calcium. Considering that PKCA contains a prototypical C2 domain implicated in its plasma-membrane targeting, the C2 domain of BAIAP3 might have triggered its calcium-dependent translocation to the plasma membrane.
NK2 Receptor Internalization after Activation
Upon stimulation of the NK2 receptor, the translocated ARRB2 binds the receptor and targets it to clathrin-coated pits. We have now found that the fluorescent BIN1-YFP rapidly translocates to the plasma membrane upon cell activation with NKA as well. The fluorescent signal of BIN1-YFP was different from the signal of ARRB2-YFP. After plasma membrane translocation, ARRB2-YFP concentrates with receptors in plasma membrane domains that become internalized (28) and the 15 videos recorded during the screening on the website). After translocation, the BIN1-YFP signal does not concentrate or internalize. BIN1 does not follow the receptors inside the cell, which is in agreement with its role, because BIN1-mcherry, has been found to be recruited early to the nascent clathrin-coated pit and to induce membrane curvature through its BAR domain (Bin-Amphiphysin-Rvs) before scission (53). Because BIN1 is implicated in several brain diseases (46), its specific recruitment by GPCR activation at the plasma membrane might have a significant functional role, because GPCRs are enriched in the central nervous system.
Of note, other proteins of the endocytic machinery are targeted by GPCR activation: dynamin 1, the AP2 subunit beta1 and AP2-associated protein kinase 1 are becoming phosphorylated upon LPA activation (19), PACSIN1 upon angiotensin II stimulation (17).
Cytoskeleton Rearrangement
It is well established that GPCRs modify cytoskeletal dynamics, acting on both tubulin and actin (54). NK2R-HEK cells activated with NKA also display actin rearrangements. The dynamics of the actin cytoskeleton are regulated by the small Rho-GTPases, and the regulators of their GTP cycle: the GTPase activating proteins GAP, the guanine nucleotide exchange factor (GEF) and the guanine nucleotide dissociation inhibitors (GDI). Using phophoproteomic approaches, the G12-coupled LPA receptor and the Gi-coupled CXCR4 chemokine receptor were the main receptors found to trigger phosphorylation of proteins implicated in motility and adhesion (19, 22). In particular, several ARHGAP members were identified : ARHGAP29 phosphorylated upon LPA activation (19), ARHGAP15 upon CXCR4 activation (20). Interestingly, ARHGAP5 and 21 became phosphorylated upon activation of the Gq-coupled AT1R (17). The only ARHGAP that was tested in our screen was ARHGAP12 that reacted upon blebbing of the cells. It might be of interest to test whether some ARHGAP proteins translocate upon GPCR activation or whether other members of this protein family display specific translocation during the blebbing process.
The following proteins, that are known or suspected to associate with cytoskeletal elements, were not previously identified as participating in GPCR signaling:
The intracellular localization of PLEKHH2-YFP that we have observed, is in agreement with the involvement of PLEKHH2 in cell adhesion and association with actin. PLEKHH2 is mainly described in the kidney, where it is considered to be a podocyte protein involved in matrix adhesion (interaction with the focal adhesion protein Hic-5 was detected by two-hybrid-screening) and actin dynamics (55). PLEKHH2 is a large protein of 1493 residues that contains two domains found in a number of cytoskeletal-associated proteins (FERM and MyTH4) and two Pleckstrin-Homology PH domains often present in intracellular signaling and/or cytoskeletal proteins. PLEKKH2 is homologous to the headless myosinX isoform (76.9% identity) (56, 57). MAX-1, the ortholog of headless myosinX in Caenorhabditis elegans plays a role in axon guidance by modulating the netrin receptor signaling pathway (58). A potential role of PLEKHH2 in GPCR signaling has never been reported before.
The chloride intracellular channel CLIC proteins are not well characterized at the functional level. These proteins are expressed in a wide variety of tissues in multicellular organisms and can be found associated to specific cellular membranes. CLIC proteins are capable in vitro of changing conformation from a globular, soluble state to a membrane-inserted state in which they provide chloride conductance. Several members of the family associate with the actin cytoskeleton, both in vitro and in vivo. Interestingly, several CLIC family members have been linked to GPCR signaling. Hence, CLIC6 interacts with the dopamine D2-like GPCR (59) and CLIC4 was shown to interact with the histamine H3 GPCR (60). In addition, CLIC4 was found to translocate to the plasma membrane upon G12-coupled LPA receptor simulation (52). However, the CLICs have not been identified by phosphoproteome analysis upon activation of GPCR. This could be because of the fact that the residues found to be necessary for translocation of CLIC4 are not phosphorylated but belong to the conserved redox-sensitive domain (52). Although, the mechanism of translocation was well characterized in this previous study, no function for CLIC4 was established in GPCR signaling.
We found that the fluorescent CLIC2 translocates from actin-enriched plasma membrane bundles to cell-cell junctions upon NK2 receptor activation, and also upon activation of endogenously expressed Gq-coupled muscarinic M3 and G12-coupled LPA receptors. We further show that CLIC4 displays the same intracellular localization and the same response to GPCR activation. Although CLIC2 is mainly described as interacting and regulating the Ryanodine receptor intracellular calcium releasing channel on the endoplasmic reticulum ER membrane (61), CLIC4, has already been found associated with cell-cell junctions (62). Our study thus proposes for the first time that CLIC2 plays a role in GPCR signaling and that CLIC2 and CLIC4 could participate in the organization of cellular junctions upon GPCR activation. Of note, we also observed the translocation of SPATC1L at cellular junctions after NK2 receptor activation, suggesting that GPCR signaling could regulate junction formation.
CONCLUSION
This screen selected positive clones based on a wider spectrum of criteria than screens that search for interacting partners, such as two-hybrid screens. On the other hand, it was less systematic than the phosphoproteomic approaches that have been applied to find the signaling pathways of a given GPCR. However, the results are complementary with the phosphoproteomic approaches and give additional insight by way of localization. The functions of ARHGAP12, SPATC1L, PLEKHH2, and the CLIC proteins remain unclear and the information concerning their localization, and their response by changes in intracellular localization are important for understanding their roles. With the development of high-content screening imaging apparatuses, this type of screening is becoming more easily applicable to decipher the function of cytoplasmic proteins and to indicate in which cascade of biological response they participate.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Hans WD Matthes for all we have learned from him. We thank Pierrick Bossert, CNRS, the excellent webmaster and conceptor of the GFP-cDNA Live and G-Protein Coupled Receptors website. We are grateful to all members of the imaging center at IGBMC particularly Pascal Kessler. We would like to thank members of the Galzi/Simonin lab, particularly Renaud Wagner and Brigitte Ilien for support, Valérie Utard for maintenance of the cell culture facility and Jeremy Garwood for critical reading of the manuscript.
Footnotes
Author contributions: H.W.D.M. did cloning experiments, designed and prepared polyclonal antibodies, R.P. participated in the design of the live-imaging screen, J.C.S. shared in the design and performance of the live-imaging screen, J.L.G. participated in the design and follow-up of the project and in the writing of the manuscript, SL designed and coordinated the project and experiments, performed research and interpreted the data, prepared the figures and wrote the manuscript.
* The work was supported by the French Ministry of Research, EMBO and HFSP short-term fellowships to SL.
 This article contains supplemental Table S1.
This article contains supplemental Table S1.
Conflict of Interest: The authors declare that they have no conflict of interest.
1 The abbreviations used are:
- GPCR
- G protein-coupled receptor
- NKA
- neurokinin A
- LPA
- lysophosphatidic acid
- ACh
- acéthylcholine
- GFP
- Green Fluorescent Protein
- CFP
- Cyan Fluorescent Protein
- YFP
- Yellow Fluorescent protein
- mYFP
- monomeric YFP.
REFERENCES
- 1. Franke T. F., Kaplan D. R., Cantley L. C., Toker A. (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275, 665–668 [DOI] [PubMed] [Google Scholar]
- 2. Rizzo M. A., Shome K., Vasudevan C., Stolz D. B., Sung T. C., Frohman M. A., Watkins S. C., Romero G. (1999) Phospholipase D and its product, phosphatidic acid, mediate agonist-dependent raf-1 translocation to the plasma membrane and the activation of the mitogen-activated protein kinase pathway. J. Biol. Chem. 274, 1131–1139 [DOI] [PubMed] [Google Scholar]
- 3. Edwards D. R. (1994) Cell signaling and the control of gene transcription. Trends Pharmacol. Sci. 15, 239–244 [DOI] [PubMed] [Google Scholar]
- 4. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 5. Yates J. R., 3rd, Gilchrist A., Howell K. E., Bergeron J. J. (2005) Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 6, 702–714 [DOI] [PubMed] [Google Scholar]
- 6. Huh W. K., Falvo J. V., Gerke L. C., Carroll A. S., Howson R. W., Weissman J. S., O'Shea E. K. (2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 7. Simpson J. C., Wellenreuther R., Poustka A., Pepperkok R., Wiemann S. (2000) Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO Rep. 1, 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zavzavadjian J. R., Couture S., Park W. S., Whalen J., Lyon S., Lee G., Fung E., Mi Q., Liu J., Wall E., Santat L., Dhandapani K., Kivork C., Driver A., Zhu X., Chang M. S., Randhawa B., Gehrig E., Bryan H., Verghese M., Maer A., Saunders B., Ning Y., Subramaniam S., Meyer T., Simon M. I., O'Rourke N., Chandy G., Fraser I. D. (2007) The alliance for cellular signaling plasmid collection: a flexible resource for protein localization studies and signaling pathway analysis. Mol. Cell. Proteomics 6, 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Foster L. J., de Hoog C. L., Zhang Y., Zhang Y., Xie X., Mootha V. K., Mann M. (2006) A mammalian organelle map by protein correlation profiling. Cell 125, 187–199 [DOI] [PubMed] [Google Scholar]
- 10. Stadler C., Rexhepaj E., Singan V. R., Murphy R. F., Pepperkok R., Uhlen M., Simpson J. C., Lundberg E. (2013) Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 10, 315–323 [DOI] [PubMed] [Google Scholar]
- 11. Steinhoff M. S., von Mentzer B., Geppetti P., Pothoulakis C., Bunnett N. W. (2014) Tachykinins and their receptors: contributions to physiological control and the mechanisms of disease. Physiol. Rev. 94, 265–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Denis C., Sauliere A., Galandrin S., Senard J. M., Gales C. (2012) Probing heterotrimeric G protein activation: applications to biased ligands. Curr. Pharmaceut. Des. 18, 128–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shenoy S. K., Lefkowitz R. J. (2011) beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galandrin S., Oligny-Longpre G., Bouvier M. (2007) The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol. Sci. 28, 423–430 [DOI] [PubMed] [Google Scholar]
- 15. Michel G., Matthes H. W., Hachet-Haas M., El Baghdadi K., de Mey J., Pepperkok R., Simpson J. C., Galzi J. L., Lecat S. (2014) Plasma membrane translocation of REDD1 governed by GPCRs contributes to mTORC1 activation. J. Cell Sci. 127, 773–787 [DOI] [PubMed] [Google Scholar]
- 16. Kim S. Y., Chudapongse N., Lee S. M., Levin M. C., Oh J. T., Park H. J., Ho I. K. (2005) Proteomic analysis of phosphotyrosyl proteins in morphine-dependent rat brains. Brain Res. Mol. Brain Res. 133, 58–70 [DOI] [PubMed] [Google Scholar]
- 17. Christensen G. L., Kelstrup C. D., Lyngso C., Sarwar U., Bogebo R., Sheikh S. P., Gammeltoft S., Olsen J. V., Hansen J. L. (2010) Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol. Cell. Proteomics 9, 1540–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Hayre M., Salanga C. L., Kipps T. J., Messmer D., Dorrestein P. C., Handel T. M. (2010) Elucidating the CXCL12/CXCR4 signaling network in chronic lymphocytic leukemia through phosphoproteomics analysis. PloS One 5, e11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schreiber T. B., Mausbacher N., Keri G., Cox J., Daub H. (2010) An integrated phosphoproteomics work flow reveals extensive network regulation in early lysophosphatidic acid signaling. Mol. Cell. Proteomics 9, 1047–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wojcechowskyj J. A., Lee J. Y., Seeholzer S. H., Doms R. W. (2011) Quantitative phosphoproteomics of CXCL12 (SDF-1) signaling. PloS One 6, e24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoffert J. D., Pisitkun T., Saeed F., Song J. H., Chou C. L., Knepper M. A. (2012) Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol. Cell. Proteomics 11, M111 014613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yi T., Zhai B., Yu Y., Kiyotsugu Y., Raschle T., Etzkorn M., Seo H. C., Nagiec M., Luna R. E., Reinherz E. L., Blenis J., Gygi S. P., Wagner G. (2014) Quantitative phosphoproteomic analysis reveals system-wide signaling pathways downstream of SDF-1/CXCR4 in breast cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, E2182–E2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hachet-Haas M., Converset N., Marchal O., Matthes H., Gioria S., Galzi J. L., Lecat S. (2006) FRET and colocalization analyzer – a method to validate measurements of sensitized emission FRET acquired by confocal microscopy and available as an ImageJ Plug-in. Microsc. Res. Tech. 69, 941–956 [DOI] [PubMed] [Google Scholar]
- 24. Mutterer J., Zinck E. (2013) Quick-and-clean article figures with FigureJ. J. Microsc. 252, 89–91 [DOI] [PubMed] [Google Scholar]
- 25. Lecat S., Bucher B., Mely Y., Galzi J. L. (2002) Mutations in the extracellular amino-terminal domain of the NK2 neurokinin receptor abolish cAMP signaling but preserve intracellular calcium responses. J. Biol. Chem. 277, 42034–42048 [DOI] [PubMed] [Google Scholar]
- 26. Atwood B. K., Lopez J., Wager-Miller J., Mackie K., Straiker A. (2011) Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vollmer J. Y., Alix P., Chollet A., Takeda K., Galzi J. L. (1999) Subcellular compartmentalization of activation and desensitization of responses mediated by NK2 neurokinin receptors. J. Biol. Chem. 274, 37915–37922 [DOI] [PubMed] [Google Scholar]
- 28. Cezanne L., Lecat S., Lagane B., Millot C., Vollmer J. Y., Matthes H., Galzi J. L., Lopez A. (2004) Dynamic confinement of NK2 receptors in the plasma membrane. Improved FRAP analysis and biological relevance. J. Biol. Chem. 279, 45057–45067 [DOI] [PubMed] [Google Scholar]
- 29. Palanche T., Ilien B., Zoffmann S., Reck M. P., Bucher B., Edelstein S. J., Galzi J. L. (2001) The neurokinin A receptor activates calcium and cAMP responses through distinct conformational states. J. Biol. Chem. 276, 34853–34861 [DOI] [PubMed] [Google Scholar]
- 30. Charras G. T. (2008) A short history of blebbing. J. Microsc, 231, 466–478 [DOI] [PubMed] [Google Scholar]
- 31. Godin C. M., Ferguson S. S. (2010) The angiotensin II type 1 receptor induces membrane blebbing by coupling to Rho A, Rho kinase, and myosin light chain kinase. Mol, Pharmacol. 77, 903–911 [DOI] [PubMed] [Google Scholar]
- 32. Nakamori Y., Emoto M., Fukuda N., Taguchi A., Okuya S., Tajiri M., Miyagishi M., Taira K., Wada Y., Tanizawa Y. (2006) Myosin motor Myo1c and its receptor NEMO/IKK-gamma promote TNF-alpha-induced serine307 phosphorylation of IRS-1. J, Cell Biol. 173, 665–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Braun J. E., Tritschler F., Haas G., Igreja C., Truffault V., Weichenrieder O., Izaurralde E. (2010) The C-terminal alpha–alpha superhelix of Pat is required for mRNA decapping in metazoa. EMBO J. 29, 2368–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scheller N., Resa-Infante P., de la Luna S., Galao R. P., Albrecht M., Kaestner L., Lipp P., Lengauer T., Meyerhans A., Diez J. (2007) Identification of PatL1, a human homolog to yeast P body component Pat1. Biochim. Biophys. Acta 1773, 1786–1792 [DOI] [PubMed] [Google Scholar]
- 35. Hsiao Y. C., Tong Z. J., Westfall J. E., Ault J. G., Page-McCaw P. S., Ferland R. J. (2009) Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis, and vesicle trafficking. Human Mol. Genet. 18, 3926–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X., Lu Z., Gomez A., Hon G. C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., Ren B., Pan T., He C. (2014) N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Strohbach C., Kleinman S., Linkhart T., Amaar Y., Chen S. T., Mohan S., Strong D. (2008) Potential involvement of the interaction between insulin-like growth factor binding protein (IGFBP)-6 and LIM mineralization protein (LMP)-1 in regulating osteoblast differentiation. J. Cell. Biochem. 104, 1890–1905 [DOI] [PubMed] [Google Scholar]
- 38. Kadrmas J. L., Beckerle M. C. (2004) The LIM domain: from the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 5, 920–931 [DOI] [PubMed] [Google Scholar]
- 39. Barres R., Gremeaux T., Gual P., Gonzalez T., Gugenheim J., Tran A., Le Marchand-Brustel Y., Tanti J. F. (2006) Enigma interacts with adaptor protein with PH and SH2 domains to control insulin-induced actin cytoskeleton remodeling and glucose transporter 4 translocation. Mol. Endocrinol. 20, 2864–2875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laufman O., Kedan A., Hong W., Lev S. (2009) Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 28, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamada K., Matsuzaki S., Hattori T., Kuwahara R., Taniguchi M., Hashimoto H., Shintani N., Baba A., Kumamoto N., Yamada K., Yoshikawa T., Katayama T., Tohyama M. (2010) Increased stathmin1 expression in the dentate gyrus of mice causes abnormal axonal arborizations. PloS One 5, e8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hsieh S. Y., Huang S. F., Yu M. C., Yeh T. S., Chen T. C., Lin Y. J., Chang C. J., Sung C. M., Lee Y. L., Hsu C. Y. (2010) Stathmin1 overexpression associated with polyploidy, tumor-cell invasion, early recurrence, and poor prognosis in human hepatoma. Mol. Carcinogen. 49, 476–487 [DOI] [PubMed] [Google Scholar]
- 43. Tcherkezian J., Lamarche-Vane N. (2007) Current knowledge of the large RhoGAP family of proteins, Biol. Cell 99, 67–86 [DOI] [PubMed] [Google Scholar]
- 44. Mathivanan S., Ji H., Simpson R. J. (2010) Exosomes: extracellular organelles important in intercellular communication. J. Proteomics 73, 1907–1920 [DOI] [PubMed] [Google Scholar]
- 45. Bialik S., Kimchi A. (2014) The DAP-kinase interactome. Apoptosis 19, 316–328 [DOI] [PubMed] [Google Scholar]
- 46. Tan M. S., Yu J. T., Tan L. (2013) Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol. Med. 19, 594–603 [DOI] [PubMed] [Google Scholar]
- 47. Conibear E. (2010) Converging views of endocytosis in yeast and mammals. Curr. Opin. Cell Biol. 22, 513–518 [DOI] [PubMed] [Google Scholar]
- 48. Gaidarov I., Santini F., Warren R. A., Keen J. H. (1999) Spatial control of coated-pit dynamics in living cells. Nat. Cell Biol. 1, 1–7 [DOI] [PubMed] [Google Scholar]
- 49. Stephenson J. R., Purcell R. H., Hall R. A. (2014) The BAI subfamily of adhesion GPCRs: synaptic regulation and beyond. Trends Pharmacol. Sci. 35, 208–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goto M., O'Brien D. A., Eddy E. M. (2010) Speriolin is a novel human and mouse sperm centrosome protein. Hum. Reprod. 25, 1884–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dulhunty A., Gage P., Curtis S., Chelvanayagam G., Board P. (2001) The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J. Biol. Chem. 276, 3319–3323 [DOI] [PubMed] [Google Scholar]
- 52. Ponsioen B., van Zeijl L., Langeslag M., Berryman M., Littler D., Jalink K., Moolenaar W. H. (2009) Spatiotemporal regulation of chloride intracellular channel protein CLIC4 by RhoA. Mol. Biol. Cell 20, 4664–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taylor M. J., Perrais D., Merrifield C. J. (2011) A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 9, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schappi J. M., Krbanjevic A., Rasenick M. M. (2014) Tubulin, actin, and heterotrimeric G proteins: coordination of signaling and structure. Biochim, Biophys. Acta 1838, 674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perisic L., Lal M., Hulkko J., Hultenby K., Onfelt B., Sun Y., Duner F., Patrakka J., Betsholtz C., Uhlen M., Brismar H., Tryggvason K., Wernerson A., Pikkarainen T. (2012) Plekhh2, a novel podocyte protein downregulated in human focal segmental glomerulosclerosis, is involved in matrix adhesion and actin dynamics. Kidney Int. 82, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 56. Sousa A. D., Berg J. S., Robertson B. W., Meeker R. B., Cheney R. E. (2006) Myo10 in brain: developmental regulation, identification of a headless isoform, and dynamics in neurons. J. Cell Sci. 119, 184–194 [DOI] [PubMed] [Google Scholar]
- 57. Wang J. J., Fu X. Q., Guo Y. G., Yuan L., Gao Q. Q., Yu H. L., Shi H. L., Wang X. Z., Xiong W. C., Zhu X. J. (2009) Involvement of headless myosin X in the motility of immortalized gonadotropin-releasing hormone neuronal cells. Cell Biol. Int. 33, 578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huang X., Cheng H. J., Tessier-Lavigne M., Jin Y. (2002) MAX-1, a novel PH/MyTH4/FERM domain cytoplasmic protein implicated in netrin-mediated axon repulsion. Neuron 34, 563–576 [DOI] [PubMed] [Google Scholar]
- 59. Griffon N., Jeanneteau F., Prieur F., Diaz J., Sokoloff P. (2003) CLIC6, a member of the intracellular chloride channel family, interacts with dopamine D(2)-like receptors. Brain Res. Mol. Brain Res. 117, 47–57 [DOI] [PubMed] [Google Scholar]
- 60. Maeda K., Haraguchi M., Kuramasu A., Sato T., Ariake K., Sakagami H., Kondo H., Yanai K., Fukunaga K., Yanagisawa T., Sukegawa J. (2008) CLIC4 interacts with histamine H3 receptor and enhances the receptor cell surface expression. Biochem. Biophys. Res. Commun. 369, 603–608 [DOI] [PubMed] [Google Scholar]
- 61. Takano K., Liu D., Tarpey P., Gallant E., Lam A., Witham S., Alexov E., Chaubey A., Stevenson R. E., Schwartz C. E., Board P. G., Dulhunty A. F. (2012) An X-linked channelopathy with cardiomegaly because of a CLIC2 mutation enhancing ryanodine receptor channel activity. Human Mol. Genet. 21, 4497–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Berryman M. A., Goldenring J. R. (2003) CLIC4 is enriched at cell-cell junctions and colocalizes with AKAP350 at the centrosome and midbody of cultured mammalian cells. Cell Motil. Cytoskel. 56, 159–172 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.