

Abstract
Branched-chain amino acids (BCAAs) are important nutrient signals that have direct and indirect effects. Frequently, BCAAs have been reported to mediate antiobesity effects, especially in rodent models. However, circulating levels of BCAAs tend to be increased in individuals with obesity and are associated with worse metabolic health and future insulin resistance or type 2 diabetes mellitus (T2DM). A hypothesized mechanism linking increased levels of BCAAs and T2DM involves leucine-mediated activation of the mammalian target of rapamycin complex 1 (mTORC1), which results in uncoupling of insulin signalling at an early stage. A BCAA dysmetabolism model proposes that the accumulation of mitotoxic metabolites (and not BCAAs per se) promotes β-cell mitochondrial dysfunction, stress signalling and apoptosis associated with T2DM. Alternatively, insulin resistance might promote aminoacidaemia by increasing the protein degradation that insulin normally suppresses, and/or by eliciting an impairment of efficient BCAA oxidative metabolism in some tissues. Whether and how impaired BCAA metabolism might occur in obesity is discussed in this Review. Research on the role of individual and model-dependent differences in BCAA metabolism is needed, as several genes (BCKDHA, PPM1K, IVD and KLF15) have been designated as candidate genes for obesity and/or T2DM in humans, and distinct phenotypes of tissue-specific branched chain ketoacid dehydrogenase complex activity have been detected in animal models of obesity and T2DM.
Introduction
Branched-chain amino acids (BCAAs; that is, leucine, isoleucine and valine) are essential amino acids and BCAA supplementation or BCAA-rich diets are often associated with positive effects on the regulation of body weight, muscle protein synthesis and glucose homeostasis. Leucine is a particularly important nutrient signal: levels of this BCAA increase in the circulation after consumption of a protein-containing meal. Despite these effects on metabolic health, studies have highlighted that along with blood sugar, insulin and certain inflammatory markers, increased fasting concentrations of circulating BCAAs are associated with an increased risk of type 2 diabetes mellitus (T2DM) and insulin resistance in humans and in some rodent models. This consistent observation in cross-sectional and prospective human studies, along with limited studies suggesting that BCAA supplementation leads to deterioration in insulin sensitivity, has prompted two questions. Firstly, are BCAAs or BCAA-rich diets harmful, helpful or neutral with respect to insulin and glucose homeostasis? And secondly, what is the aetiology of altered blood levels of BCAAs in the insulin-resistant state? This Review provides evidence that under most conditions, alterations in fasting blood levels of BCAAs in the obese insulin-resistant state result from changes in the rate of appearance and clearance of these metabolites, coupled with decreased activity of catabolic enzymes in some tissues compared with the insulin-sensitive state. Perturbations in BCAA levels probably reflect the insulin resistant and T2DM ‘pathophenotypes’ and BCAAs themselves are probably not necessary or sufficient to trigger disease.
BCAAs and metabolic health
The results from a number of interventional studies have suggested that increasing dietary levels of BCAAs should have a positive effect on the parameters associated with obesity and T2DM, such as body composition, glycaemia levels and satiety. Direct and indirect mechanisms for these positive effects have been proposed. For example, leucine seems to have direct effects on hypothalamic and brainstem processes involved in satiety.1–22
In the gastrointestinal tract and in fat deposits, BCAAs regulate the release of hormones (for example, leptin, GLP-1 and ghrelin) that can potentially affect food intake and glycaemia levels.15,16,20,23–25 BCAAs and insulin are anabolic signals that alter the growth of energy-consuming tissues, mediated in part through their ability to activate the mammalian target of rapamycin complex 1 (mTORC1) and protein kinase Cε (PKCε),26–34 as well as by decreasing protein breakdown through unknown mechanisms.27 Synthesis of muscle protein is thought to underlie the higher level of diet-induced thermogenesis (energy wasting) associated with protein consumption or BCAA infusion than that associated with other nutrients.35–40 Supplementation of BCAAs also seem to result in health benefits in a number of liver diseases.41–45 As a consequence, supplementation with BCAAs or a BCAA-rich diet is believed to improve metabolic health; an increase in the recommended dietary allowance for protein has been proposed, which would effectively increase dietary levels of BCAAs.7,13,20,22,46–51 Nevertheless, the idea that BCAAs or their supplementation might have a positive role in preventing metabolic disease is controversial.
Circulating BCAA levels and poor health
A number of observational studies indicate that elevated circulating levels of BCAAs are associated with poor metabolic health. Given the above cited studies suggesting health benefits of high levels of dietary BCAAs, it might seem paradoxical that levels of BCAAs tend to be increased, not decreased, in insulin-resistant obesity and T2DM (Figure 1).52–71 Such increases are consistently observed in patients with T2DM or obesity and in some rodent models of obesity or T2DM.59,63,72–75 Furthermore, increased levels of BCAAs have been linked to the metabolic syndrome and cardiovascular disease.59,60,76–78 In clinical studies, increased blood levels of BCAAs positively correlate with insulin resistance,63,79 HOMA index79 and levels of HbA1c (Figure 1).66,79 Several longitudinal studies in different cohorts have reported that increased blood levels of BCAAs are predictive of future insulin resistance or T2DM,54,62 which has led to speculation about a potential causative role for BCAAs. Although these associations are consistently observed in human populations, the mechanisms underlying the relationship remain to be fully established. Another issue is that when circulating levels of BCAAs increase, it is possible that they compete with the uptake of amino acid precursors of dopamine and 5-hydroxytryptamine in the brain.80–83 Although speculative, loss of these precursors could contribute to the increased risk of depression in individuals with obesity.84,85
Figure 1.

Plasma BCAA levels and insulin-resistant obesity. a | Association between plasma BCAA levels and insulin-resistant obesity in humans, obese Zucker rats, mice with diet-induced obesity (DIO) and ob/ob mice. Data were compiled from elsewhere and redrawn.63,72,73,75 b | Correlation between plasma levels of leucine and fasting levels of HbA1c in African–American women with obesity and T2DM (blue circles) and those with obesity but no T2DM (green circles). Abbreviations: BCAA, branched-chain amino acid; IR, insulin resistant; T2DM, type 2 diabetes mellitus. Adapted from Fiehn, O. et al. PLoS ONE 5, e15234 (2010),66 which is published under a Creative Commons Licence owned by PLOS ©.
Processes affecting circulating BCAA levels
To understand the seemingly conflicting findings regarding circulating BCAA levels and health it is helpful to appreciate the processes that contribute to BCAA rates of appearance (Ra) and disappearance (Rd) in the blood. Processes contributing to Ra include food intake and tissue protein degradation. However, in the case of the frequently reported association between blood levels of BCAAs and insulin resistance, the phenotype is not due to recent (for example, within a few hours) overeating, as almost all studies were conducted in the overnight-fasted state. That is not to say that overeating is not a culprit in obesity, but rather that other processes affecting the BCAA Ra and/or Rd are responsible for the plasma BCAA concentration in fasting individuals, including those with obesity.
Regulation of protein degradation can occur through changes in autophagy or proteasomal-mediated degradation. Rates of protein degradation in muscle and liver can be inhibited by insulin, insulin-like growth factor 1 (IGF-1) and BCAAs via impairment of autophagy mediated by mTORC1 and AKT (also known as PKB)27,86–92 and the ubiquitin proteasomal pathway.29,87,93–99 Indeed, whereas insulin is able to stimulate protein synthesis in newborn pigs,100 in adult humans there is what has been called a ‘specific effect’ of insulin on protein degradation.101 Consistently, amino acids but not insulin stimulated protein synthesis in leg muscle, whereas insulin but not amino acids attenuated the breakdown of proteins in the leg muscles of humans.99 BCAAs and insulin usually act additively or synergistically to activate mTORC1. This additivity, in addition to the more specific action of insulin on protein degradation, might explain the finding that despite elevated BCAA levels, protein degradation is frequently increased in fasting individuals with obesity and insulin resistance, and in those with poorly controlled T2DM.61,73,102–106 It is tempting to speculate that elevated protein degradation might be attenuated by providing additional BCAAs in the diet (rather than by avoiding them), as even overnight infusion of BCAAs caused sustained decreases in muscle protein degradation.107
The major processes affecting the BCAA Rd include protein synthesis, excretion and BCAA catabolism and/or oxidation. As already mentioned, insulin and amino acids stimulate protein synthesis during growth in newborn pigs.100 In weight-stable adult humans, this effect of insulin is either less important or not apparent (even though protein degradation is affected), whereas amino acids and IGF-1 stimulate protein synthesis in adult humans.101 Counterintuitively, in insulin-resistant obesity and untreated T2DM, several studies have suggested that synthesis of muscle protein is either unchanged or increased.61,102–106 In these situations, muscle mass can decrease because protein degradation is increased more than protein synthesis. BCAA excretion could also be affected by insulin-resistant obesity, owing to the increased levels of circulating amino acids,73 but the degree to which this process is affected in humans remains to be established. BCAA oxidation is discussed further later.
Branched-chain aminoacidaemia and T2DM
Despite evidence that elevated levels of BCAAs predict future insulin resistance or T2DM, it is still unclear whether BCAAs are a causative factor in insulin resistance and T2DM or just a biomarker of impaired insulin action. In terms of the uptake of amino acids by the central nervous system, it is also unclear whether obesity causes depression or whether reverse causality exists;108 causation versus association is an important distinction. If BCAAs improve satiety, cholesterolaemia, glycaemia and lean mass, supplementation with BCAAs might be of therapeutic value. Two potential mechanisms explaining how BCAAs might contribute to insulin resistance in obesity and T2DM have emerged (Figures 2 and 3). The first mechanism proposes that an excess of dietary BCAAs activates mTORC1 signalling, which leads to insulin resistance and T2DM. The second mechanism has its origins in studies of maple syrup urine disease (MSUD) and organic acidurias. This alternative mechanism proposes that in animal models or in individuals with impaired BCAA metabolism (referred to as BCAA dysmetabolism), increased levels of BCAAs are a biomarker of impaired metabolism; however, BCAA dysmetabolism also leads to the accumulation of toxic metabolites that cause mitochondrial dysfunction in pancreatic islet β cells (or elsewhere) and is associated with insulin resistance and T2DM.
Figure 2.

Persistent activation of mTORC1 links increased plasma BCAA levels to insulin resistance. According to this theory,59,109,110 excess nutrients that lead to obesity also result in frequent prandial increases in plasma levels of leucine, which together with insulin activate mTORC1 and S6K1. Persistent activation leads to serine phosphorylation of IRS-1 and IRS-2, which interferes with signalling and might target IRS1 for proteolysis via a proteasomal pathway.109,110 The resulting insulin resistance increases demand on insulin to dispose of excess glucose. Insulin resistance might increase the Ra of BCAAs from protein degradation. Long-term demand for insulin secretion, along with other factors such as lipotoxicity, might negatively affect the function of islets (for example, an initial compensatory increase in β-cell numbers and mass and islet mass, followed by apoptosis), ultimately resulting in a failure to produce sufficient quantities of insulin and leading to the onset of T2DM. Abbreviations: BCAA, branched-chain amino acid; IRS, insulin receptor substrate; mTORC1, mammalian target of rapamycin complex 1; Ra, rate of appearance; Rd, rate of disappearance; S6K1, ribosomal protein S6 kinase β1; T2DM, type 2 diabetes mellitus.
Figure 3.

BCAA dysmetabolism links elevated plasma levels of BCAAs and FFAs to T2DM and obesity-related comorbidities. The schematic shows how obesity might affect a number of factors contributing to elevated circulating BCAA levels via effects on the Ra or Rd of BCAAs. Loss of steps in BCAA metabolism could lead to the accumulation in tissues of BCKAs and BCAA-related acyl-CoAs. Accumulation of these species in inherited disorders can be mitotoxic and might lead to T2DM and other obesity-related comorbidities. A caveat is that while the metabolites of BCAAs are potentially toxic in maple syrup urine disease and organic acidurias, their role in T2DM-associated mitochondrial dysfunction or in activation of stress kinases is unknown. Alternatively, reduced or incomplete valine and isoleucine catabolism could attenuate anaplerosis from these substrates, contributing to anaplerotic stress in one or more tissues affected by T2DM. Abbreviations: BCAA, branched-chain amino acid; BCKA, branched-chain α-keto acid; BCKDC, branched-chain α-keto acid dehydrogenase complex; CoA, coenzyme A; FFA, free fatty acid; KLF15, Krueppel-like factor 15; Ra, rate of appearance; Rd, rate of disappearance; ROS, reactive oxygen species; T2DM, type 2 diabetes mellitus.
Role of mTORC1
Persistent nutrient signalling might cause insulin resistance by BCAA activation of the mTORC1 signalling pathway (Figure 2).59,60,109,110 Persistent activation of the serine kinases S6K1 and mTORC1 promotes insulin resistance through serine phosphorylation of insulin receptor substrate (IRS)-1 and IRS-2, which might occur in response to persistent hyperinsulinaemia or aminoacidaemia. In essence, the theory proposes that over time, the increased demand for insulin from impaired insulin action, along with inflammation and lipotoxicity associated with insulin resistance, elicits hyperinsulinaemia and exhaustion of the β cells. Eventually euglycaemia can no longer be maintained and T2DM becomes evident.
Currently, it is unclear if this putative effect of BCAAs occurs in humans and to what extent other potential mediators besides BCAAs contribute to this scenario. Furthermore, while some experimental evidence supports this model, a number of observations do not support the concept of BCAA activation of mTORC1 as being necessary and sufficient to elicit insulin resistance. Firstly, although increases in BCAAs are associated with mTORC1 signalling in skeletal muscle, this pathway is also activated by exercise, a known factor in the prevention and regression of insulin-resistant and T2DM phenotypes. Secondly, supplementing or increasing circulating levels of BCAAs is associated with metabolic improvements despite increased mTORC1 signalling.21,111 Thirdly, in contrast to studies where large doses of leucine were orally administered,28 it is unclear whether the small changes in BCAA levels observed in patients with obesity and insulin-resistance are sufficient to independently affect serine phosphorylation of IRS-1 and IRS-2 (or to what extent other factors such as insulinaemia or inflammatory mediators might contribute to these phosphorylations). For example, individuals with morbid obesity have raised levels of BCAAs that normalize after gastric bypass surgery;72,112–114 however, mTORC1 activation in muscle did not change after gastric bypass surgery in a longitudinal study.113 Fourthly, isoleucine has been reported to reduce plasma levels of glucose by stimulating glucose uptake in skeletal muscle by an unknown mechanism.115,116 Similarly, an analogue of isoleucine (4-hydroxyisoleucine) increased the glucose Rd in euglycaemic hyperinsulinaemic clamp studies.117 Finally, in adipose tissue, mTORC1 is important for the increase in fat cell mass associated with the positive effects of PPAR-γ agonists on whole body insulin sensitivity.118
Although the focus of the mTORC1 hypothesis is on activation of mTORC1 in skeletal muscle, to the best of our knowledge none of the more recently described candidate genes for obesity, HbA1c and T2DM seem to be involved in activation of the mTORC1 signalling pathway.119–121 Most T2DM candidate genes are thought to exert their actions in pancreatic islets, not in skeletal muscle.119 However, rather than causing T2DM, activation of mTORC1 in β cells has been linked to avoidance of T2DM, which is associated with compensatory increases in islet and β-cell mass.122–128 Adverse effects of the mTORC1 blocker, sirolimus, have consistently included two hallmarks of the metabolic syndrome, hyperglycaemia and dyslipidaemia, along with new onset diabetes after transplantation.122–125,129 Those actions might be secondary to the ability of rapamycin to inhibit mTORC1 and to interfere with mTORC2 assembly over time, which in turn can affect AKT activity.126–128 Both the impaired peripheral resistance that occurs in response to chronic rapamycin treatment and a second effect on islets that impairs their ability to produce sufficient insulin are thought to underlie new onset diabetes after transplantation.129 Thus, a number of studies do not support the notion that sustained activation of mTORC1 promotes islet dysfunction or T2DM.122
BCAA dysmetabolism
In addition to the ‘persistent activation of mTORC1’ mechanism, there is a second hypothetical mechanism explaining how increased levels of BCAAs might be causally linked to insulin resistance and T2DM. Impairments in BCAA metabolism (BCAA dysmetabolism) could result in the accumulation of potentially toxic intermediates that impair cellular function. Individuals or animal models with a phenotype of impaired or incomplete BCAA metabolism might thereby have increased susceptibility to insulin resistance or T2DM. According to this model,75 an accumulation of toxic BCAA metabolites (rather than BCAAs themselves) contributes to β-cell mitochondrial dysfunction and eventually the apoptosis of β cells that accompanies T2DM (Figure 3). Nevertheless, elevated levels of BCAAs would be evident with such dysfunction.
The BCAA metabolic pathway
The first step in the metabolism of BCAAs in most peripheral tissues, except the liver, is catalysed by the mitochondrial isoform of branched-chain-amino-acid transaminase, BCAT(m), encoded by the BCAT2 gene. One piece of evidence supporting a putative mechanism of BCAA dysmetabolism derives from the phenotype of BCAT2−/− mice. Deletion of BCAT2 largely prevents BCAA metabolites from forming in peripheral tissues. Rather than exhibiting insulin resistance as might be expected from the mTORC1 persistent activation mechanism (Figure 2), BCAT2−/− mice exhibit greatly improved glycaemic control, insulin sensitivity, adiposity and lipid profiles, despite overall increased mTORC1 signalling and increased energy expenditure (Box 1).111 One caveat is that at least some of the improvements in glycaemic control in BCAT−/− mice probably result from the loss of gluconeogenic precursors, a reflection of the important role that muscle transaminases have in generating gluconeogenic substrates for the liver (Box 1).
Box 1. Genetic findings in first two steps of BCAA metabolism.
BCAT(m)
BCKDH complex
Human mutations in components of the BCKDH complex lead to MSUD183–186
Individuals with MSUD and MSUD animal models exhibit both elevated levels of BCAAs and BCKAs166,184–187
Addition of BCKAs, which are elevated in MSUD, to cells results in oxidative stress and mitochondrial dysfunction130–133,188
Fibroblasts and neural cells derived from individuals with MSUD undergo apoptosis when BCKAs are added134,135
BCKDHA has been described as a primary susceptibility gene for both obesity and T2DM120
BCKDK
BCKDK transgenic overexpressing cells undergo cell death when leucine is added136
Increasing BCKDK protein levels in animals would be expected to increase plasma concentrations of both BCAAs and BCKAs, but no viable BCKDK transgenic animals have been reported
PPM1K
Ppm1k−/− mice have increased levels of BCAAs and BCKAs137,151
Mutation in human PPM1K leads to an intermittent form of MSUD138
PPM1K has been described as a T2DM susceptibility gene in human islets165
Allelic variation near PPM1K was associated with poorer glycaemic control and body weight response in the POUNDS LOST trial167
Cells from an individual with defective PPM1K and Ppm1k−/− mice display changes frequently linked to obesity, insulin resistance and T2DM, such as increased lipotoxicity and lipid peroxidation, increased oxidative stress (ROS generation and mitochondrial permeability transition pore opening), increased apoptosis and stress kinase activation (JNK and p38)137–140,166
Abbreviations: BCAA, branched-chain amino acid; BCAT(m), branched-chain amino acid transaminase, mitochondrial; BCKA, branched-chain α-keto acid; BCKDH; branched-chain α-keto acid dehydrogenase; JNK, c-Jun N-terminal kinase; MSUD, maple syrup urine disease; p38, mitogen-activated protein kinase p38; ROS, reactive oxygen species; T2DM, type 2 diabetes mellitus.
After BCAT(m), the next step in the BCAA metabolic pathway is rate-controlling and the first irreversible step in BCAA metabolism. This step is catalysed by the multienzyme mitochondrial branched-chain α-ketoacid dehydrogenase complex (BCKDC), which results in the oxidation of BCAAs to their respective ketoacids (Figure 4). BCKDC activity is inhibited by phosphorylation at a single site by the specific kinase, branched-chain α-ketoacid dehydrogenase kinase (BCKDK, Figure 4). Conversely, BCKDC is activated by the mitochondrial isoform of protein phosphatase 1K (PPM1K, also known as PP2CM, Figure 4). A number of metabolic factors altered in obesity, insulin resistance and T2DM affect the activity and expression of these enzymes (Table 1). Mutations in genes encoding subunits of BCKDC or in PPM1K can lead to MSUD—a potentially lethal disease that results in elevated levels of BCAAs and branched-chain α-ketoacids (BCKAs, Box 1), the latter of which are widely believed to be the toxic factor in the disease.
Figure 4.

Mitochondrial genes attributed to BCAA metabolism. The genes involved in mitochondrial BCAA metabolism are shown. Note that BCAT(m) activity is essentially absent from the liver. During reversible BCAT(m) metabolism, an intermediate of isoleucine can tautomerize, leading to alloisoleucine formation.182 Alloisoleucine formation increases when BCKDC activity is impaired and might be useful for identifying individuals with impaired BCKDC activity,75 in addition to its usual use in identifying those with maple syrup urine disease. *Indicates an obesity and/or T2DM susceptibility gene.120,165 ↓ Indicates a literature finding of decreased gene or protein expression observed in human islets or skeletal muscle biopsies from individuals with T2DM, except for HIBADH, which is decreased in skeletal muscle of Goto–Kakizaki rats.154,155,165 Coloured oval shapes represent genes implicated in BCAA metabolism. Abbreviations: BCAA, branched-chain amino acid; BCAT(m), branched-chain-amino-acid aminotransferase, mitochondrial; BCKDC, branched-chain α-ketoacid dehydrogenase complex; KIC, α-ketoisocaproate; KIV, 2-ketoisovalerate; KMV, α-keto-β-methylvalerate. Modified with permission from Herman, M. A. et al. J. Biol. Chem. 285, 11348–11356 (2010)74 © The American Society for Biochemistry and Molecular Biology.
Table 1.
Short and long-term regulation of BCKDC activity
| Regulator | Duration of effect | Effect on BCKDK (activity or expression) | Effect on total BCKDC activity (or subunit expression) | Expected or known effect on BCKDC actual activity or activity state | Organ, tissue, cell affected or in vitro findings |
|---|---|---|---|---|---|
| Clofibrate | Short term and long term | Inhibition of enzyme activity (directly) and ↓protein levels | ↑ Expression of subunits and ↑ total activity | ↑ Percentage of active enzyme | Liver; other PPAR-α agonists have long-term effects but no allosteric effects189 |
| Exercise | Short term | Inhibition of binding to complex | No change | Activation | Skeletal muscle190 and liver191 (↓ binding of BCKDK to BCKDC) |
| Glucocorticoids | Long term | ↓ Protein levels and ↓ gene expression192 | No change | Activation | Kidney cell line (BCKDK studies); other studies on liver hypothesized effects of methylpredisone due to ↑ plasma levels of leucine193 |
| High-protein diet | Long term | ↓ Protein levels | ↑ | ↑ | Liver194–196 |
| Insulin | Long term | ↑ Protein levels | No change | ↓ or no change | No change in healthy animals following insulin administration or in 3T3-L1 cells stimulated with insulin;63 ↓BCKDH activity in liver of methylpredisone-treated rats correlates with plasma levels of leucine;193 clone 9 cells: effects attenuated by mTORC1 and PI3-K inhibition197 |
| Low-protein diet or protein starvation | Long term | ↑ Protein levels | ↓ | ↓ | Liver194,196,198,199 |
| Medium-chain fatty acids (MCFAs) | Short term | Inhibition observed with octanoate on purified kinase | NA | ↑ is expected, but in cells MCFAs also elicit ↓BCKDC activity via ↑NADH:NAD+ ratio and acetyl-CoA143 | Purified kinase;200 uncertainty between the balance of kinase vs FFA inhibition |
| Oleate or palmitoylcarnitine | Short term | NA | NA | ↓ | Liver cells and muscle cells98,175–177 |
| Sepsis, IL-1 and TNF | Long term | NA | NA | ↑ | Skeletal muscle: effect prevented by cortisone treatment201 |
| Starvation* | Short term and long term | Short term: ↑ BCAA Ra and BCKAs inhibit BCKDK Long term: ↑BCKDK activity in liver |
No change | Activation | Liver202,203 |
| STZ-induced diabetes mellitus | Long term | Skeletal muscle: ↓ BCKDK protein levels without affecting gene expression Cardiac muscle: ↑ protein levels ↑ and gene expression Liver: ↓ mass of BCKDK; no change in gene expression |
Skeletal muscle: ↑ activity, ↑protein levels of E1-α, E1-β and E2 subunit, and ↑ gene expression Cardiac muscle: ↓ activity, no effect on protein levels or gene expression of subunits Liver: ↑ total BCKAD activity with diabetes, ↑ mass of each BCKDH subunit, no change in gene expression |
Skeletal muscle: ↑ activity state and ↓ rate of inactivation Cardiac muscle: ↓ activity state and ↑ rate of inactivation Liver: diabetes increases the activity state |
Liver, heart and muscle202,204–207 |
| T3 | Long term | ↑ | Not affected | ↓ | Liver208 |
| α-KIC | Short term | Inhibition | Not affected | ↑ | All tissues tested showed inhibition of the kinase by α-KIC198,209 |
| PPAR-γ agonists | Long term | No effect | ↑ Protein levels and ↑ gene expression | ↑ Predicted | Adipose tissue63,210 |
Starvation for all food and just protein or low-protein diets have opposite effects. Abbreviations: BCKDC, branched-chain α-ketoacid dehydrogenase complex; BCKDK, branched-chain α-ketoacid dehydrogenase kinase; α-KIC, α-ketoisocaproate; mTORC1, mammalian target of rapamycin complex 1; FFA, free fatty acid; NA, not available; PPAR, peroxisome proliferator-activated receptor; PI3-K, phosphoinositide 3-kinase; STZ, streptozotocin; TNF, tumour necrosis factor.
The putative BCAA dysmetabolism mechanism is also supported by studies in which either several BCKAs or the α-ketoacid of leucine, α-ketoisocaproate (α-KIC), are added to cells, and by data from mouse models of altered BCKDC metabolism.130–141 Consistently, the addition of BCKAs to glial cells or to the cerebral cortex increases lipid peroxidation and oxidative stress leading to mitochondrial bioenergetic dysfunction.130,131,141 The ability of BCKAs to cause mitochondrial dysfunction extends beyond the brain. For example, BCKAs inactivate pyruvate dehydrogenase in rat liver and strongly inhibit pyruvate dehydrogenase and α-ketoglutarate dehydrogenase in the heart132,133,141 and cause apoptosis when added to fibroblast and neural cells isolated from patients with MSUD.134,135 Further support for the BCAA dysmetabolism mechanism is derived from studies on human or mouse cells with altered BCKDC activity (Box 1). Cells from patients with classic or intermittent MSUD and mouse cells with disrupted expression of PPm1K and BCKDK exhibit toxic cellular changes, including mitochondrial dysfunction and/or apoptosis (Box 1 and Figure 4). For example, in cells expressing a BCKDK transgene, cell death occurs when leucine is added, presumably due to its rapid conversion to, and the subsequent accumulation of, α-KIC.136 Consistently, mutation or deletion of PPM1K results in a mild increase in levels of BCAAs in humans and mice, which nevertheless increases lipid peroxidation, lipotoxicity, oxidative stress, mitochondrial transition pore opening and apoptosis, along with the activation of the stress kinases JNK and p38.137–140 All of the above-mentioned factors are generally believed to be involved in insulin resistance and in the pathogenesis of T2DM. However, it must be emphasized that blood levels of BCKAs in these conditions are modest compared with those in individuals with MSUD, and elevations in circulating levels of BCKAs in individuals with insulin resistance and in animal models of obesity are not universally observed.63
The products of BCKDC activity are branched-chain acyl-Coenzyme A (CoA) species that are further metabolized by multiple mitochondrial-matrix enzymatic steps, eventually leading to the formation of lipogenic, ketogenic or gluconeogenic substrates (in liver), such as acetoacetyl-CoA, acetyl-CoA and propionyl-CoA (Figure 4). Acyl-CoAs can be converted to acylcarnitines, which in turn can be transported out of the mitochondria and cells. Acylcarnitines can be assayed in plasma or urine as reporters of the status of mitochondrial metabolism involving CoA species. A number of BCAA-derived acylcarnitines are increased in individuals with insulin-resistant obesity or T2DM.59,60,73,113,142–144 Clinically, urine and blood levels of acylcarnitines are used to detect organic acidurias. Similar to MSUD, organic acidurias can result in mitochondrial dysfunction, albeit the mechanism for the latter is not entirely obvious.145 In part, the idea that accumulations of acyl-CoA species are potentially toxic originates from the disorders caused by these inborn errors of metabolism. However, as with BCKAs, it is uncertain whether levels of acyl-CoA species reach sufficient concentrations in insulin-resistant states to cause mitochondrial dysfunction.
Impaired mitochondrial BCAA metabolism
One of the most robust and consistent changes in obesity, regardless of the model, is a marked decrease in adipose tissue expression of genes involved in BCAA metabolism;63,72,74,143,146–150 however, the mechanisms underlying these changes remain to be defined. Changes in adipose tissue concentrations of these proteins in obesity coincide with the above-cited reduced gene expression levels, at least in the first two steps in metabolism that have been examined in Zucker rats and ob/ob mice. In the Zucker rat, the losses in BCAT(m) and BCKDC E1-α are quite large even when changes in adipose tissue mass are considered.72,73 Thus, it has been proposed that decreased BCAA metabolism in fat contributes to increased plasma levels of BCAAs in individuals with insulin-resistant obesity or untreated T2DM,63,72,73,143 and that visceral adipose tissue, in particular, might have an important role in this regard.63 Consistent with the latter view, BCAA catabolic enzyme gene expression in visceral adipose tissue strongly correlates with insulin sensitivity and is reduced in individuals with the metabolic syndrome and obesity compared with control individuals with similar levels of obesity but who do not have the metabolic syndrome.63,150
However, there is a caveat for this proposal given that whole-body BCAA metabolism demonstrates considerable interorgan dependence. A demonstration of interorgan dependence comes from studies in patients with MSUD as well as from mouse models of MSUD. In these studies, transplantation of normal tissue (such as liver or adipose tissue) largely compensates for the loss of BCAA metabolism in the other tissues of the metabolically impaired mice or patients with MSUD and this leads to considerable reductions in plasma levels of BCAAs.74,151,152 This reduction is assisted, in part, by the fact that BCKDC is activated in the transplanted tissue by the elevated circulating α-KIC levels found in patients with MSUD. Thus, in individuals with intact metabolism, a high BCAA intake should be well-tolerated because of the reserve capacity of BCKDC in the body and the fact that BCKDC is activated by excess substrate under normal conditions.153 Loss of BCAA metabolism in one organ, such as fat, might be associated with normal or increased metabolism in other tissues, unless those other tissues exhibit some form of mitochondrial dysfunction, as is common in insulin-resistant obesity or T2DM. As described later, two such phenotypes have been observed in animal models of obesity associated with changes in hepatic BCKDC.75
The issue of whether or not organs other than adipose tissue have altered BCAA metabolism in insulin resistant or T2DM states is starting to be addressed. For example, spectral analysis has revealed decreased expression of two enzymes involved in valine and isoleucine metabolism in muscle of patients with T2DM (Figure 4).154 Similarly, in Goto–Kakizaki rats, expression of 3-hydroxyisobutyrate dehydrogenase, the protein encoded by the Hibadh gene in rats, was reduced by >50% in skeletal muscle (Figure 4).155 This protein catalyses a step in valine metabolism (Figure 4) and impairment of this enzyme in humans is associated with an accumulation of 3-OH-butyryl-CoA and 3-OH-butyrylcarnitine; loss of this enzymatic step can have toxic effects.156 In another study, male individuals with obesity and T2DM, but not similarly affected female individuals, exhibited considerably reduced BCKDC protein concentrations.157
However, several challenges exist in addressing whether whole-body or tissue-specific BCAA metabolism is increased or decreased in states of insulin-resistant obesity and T2DM. Interpretive problems arise when comparing tissues or even whole-body metabolism from individuals with different body compositions (a problem that has also affected the calorimetry field158). For example, the size of the liver is approximately twofold higher in obese compared with lean Zucker rats (livers from obese Zucker rats are also more lipid laden than those from lean Zucker rats) and the contribution of adipose tissue increases even more and the muscle mass declines further in obese Zucker rats than in lean Zucker rats.73 In the case of tissues from individuals with and without obesity, should each tissue be considered as a ‘pool’ of enzyme activity? Thus an important question is which denominator or normalizer is appropriate when measuring tissue levels of BCAA enzymes when the body size and composition are different.
Other issues also present themselves when analysing whole-body BCAA metabolism in individuals with T2DM or insulin resistance, with or without obesity. In addition to the Rd from oxidative metabolism, several processes contributing to the Ra and Rd of BCAAs are likely to be altered. If these changes are dramatic they can confound efforts to develop a simple interpretation of BCAA oxidation data.73 Another issue in whole-body metabolism is related to the choice of using either leucine or α-KIC specific activities for leucine isotope studies. After priming the bicarbonate pool with isotope, α-KIC rather than leucine-specific activity or enrichment is frequently used to monitor muscle or whole-body protein synthesis and turnover,159,160 as α-KIC is believed to better represent the intracellular pool of leucine in skeletal muscle (the frequent focus of this methodology and where there is considerable BCAT(m) activity to interconvert BCAAs and BCKAs).159 However, considerable BCKA oxidation takes place in the liver, which lacks BCAT(m). In Zucker rats, the obese:lean ratios of BCAA and BCKA levels are different in plasma and muscle;73 although the obese:lean BCAA level ratio is similar in liver, the ratio for BCKA levels is far more increased in liver than in plasma and skeletal muscle. Neglecting the specific activity problem, another issue is still what denominator should be used for calculating rates of leucine oxidation and appearance, and the tissue-specific rates of protein synthesis when body compositions are different. Thus, the body composition factors comparing individuals with and without obesity create interpretive dilemmas that need to be considered.
With regard to branched-chain α-keto acid dehydrogenase (BCKDH) activity or phosphorylation (inactivation), there currently seems to be two responses evident in animal models of insulin-resistant obesity (Figure 5). The first phenotype is exemplified by animal models of insulin-resistant obesity, such as Lepob/ob mice, obese Zucker rats, ZDF rats and Otsuka Long–Evans Tokushima Fatty rats.72,73,161,162 Along with the usual reductions in BCAA metabolic pathways in adipose tissue, these animals exhibit impaired active or total BCKDC activity in the liver. Considering that BCKDC activity is regulated by phosphorylation, it is possible to measure its activity in isolated tissues, in comparison to how much the activity might be if the enzyme was fully active—the total activity. Total activity is measured after the enzyme is treated with a phosphatase that removes the inhibitory phosphorylations on the E1-α subunit of BCKDC in homogenates. The impaired hepatic activity of BCKDC in obese Zucker rats is explained in part by increased expression of BCKDK. In obese Zucker rats, a global reduction in BCKDC activity is evident in fat, liver, kidney, skeletal muscle and heart, compared with activity in lean animals.72 This generalized diminution is associated with greatly increased levels of BCKAs and alloisoleucine—the pathognomonic biomarker of MSUD.75
Figure 5.
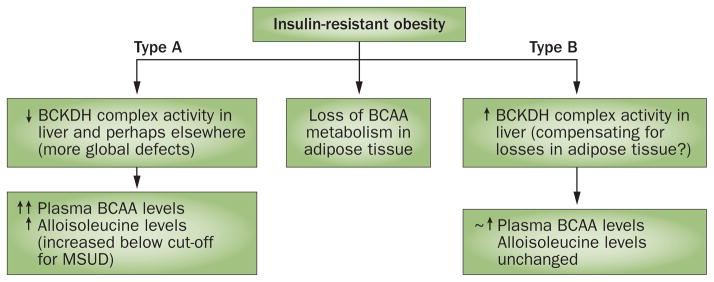
Patterns of altered BCAA metabolism observed in animal models of obesity. Losses of adipose tissue BCAA metabolic gene and protein expression in obesity have consistently been observed. In a rat model of diet-induced obesity,63 reduced levels of BCAA metabolizing enzymes in adipose tissue seem to be compensated for by increased hepatic BCKDH activity163 (termed a type B response). In contradistinction, hepatic BCKDH was also attenuated in other models such as ZDF rats,161,162 Zucker fa/fa rats72,73, ob/ob mice72 and Otsuka Long–Evans Tokushima Fatty rats162 (termed a type A response). Multiple peripheral tissues were examined and found to be affected in Zucker rats.73 These distinct phenotypes are important because uncompensated loss of BCAA metabolism in multiple peripheral tissues could result in a higher range of plasma BCAAs that, when observed in states of obesity, associate to a greater extent with insulin resistance, levels of glycaemia and future T2DM. Alloisoleucine elevations below the level used to screen for MSUD have been proposed as a strategy to distinguish between these phenotypes.75 Levels of BCAAs were considerably increased in models in which metabolism was impaired (↑↑). Abbreviations: BCAA, branched-chain amino acid; BCKDH, branched-chain α-keto acid dehydrogenase; MSUD, maple syrup urine disease. Adapted with permission from John Wiley and Sons © Olson, K. C. et al. Obesity (Silver Spring) 22, 1212–1215 (2014).75
The second putative phenotype is demonstrated by rodents with diet-induced obesity. Whereas this model is characterized by reduced levels of BCAA enzymes in fat,63 other studies show that liver BCKDH activity is actually increased and could compensate for the reduced activity in fat.163 Consistently, the lean versus obese differences in plasma levels of BCAAs are much lower in the model of diet-induced obesity than in Zucker rats.163 Extrapolating, it is tempting to speculate that individuals could also have different phenotypes in this regard. It has been proposed that human studies focusing more carefully on alloisoleucine might be useful in determining where these two phenotypes manifest in patients with insulin-resistant obesity or T2DM.75 This approach would be more practical than measuring BCKDC activity in muscle and liver biopsy samples. However, in a biopsy study, human livers from male individuals with obesity or obesity and T2DM had lower expression levels of BCKDC protein than control lean male individuals; female individuals were not affected.157 Thus, compared to the diet-induced obesity model163 the decrease in BCKDC in male Zucker rats72,73 might more closely model male human obesity and T2DM. The finding that female individuals did not show any changes in BCKDC levels is consistent with the idea that different phenotypes in hepatic BCKDC might occur in various obesity models. Further studies on how BCKDC activity is affected in other tissues of individuals with obesity and T2DM and between different animal models and the effect of sex is needed.
Finally, other evidence that BCAA catabolism is altered in obesity is derived from the observed increases in levels of BCAA-related acylcarnitines mentioned earlier.59,60,73,113,142–144 One interpretation of these increases is that they reflect increased BCAA metabolic flux,59 a reasonable hypothesis given that the substrates for BCAA metabolism are increased in states of obesity. However, a caveat is that, as mentioned earlier, acylcarnitine profiles are used clinically, not to determine accelerated metabolism, but rather to detect defects in metabolism (for example, organic acidurias). Notably, in human skeletal muscle mitochondria isolated from individuals with insulin-resistant obesity, spectral analysis shows a reduction of ~50% in transcript levels of PCCB (encoding propionyl-CoA carboxylase β chain, mitochondrial) and ALDH6A1 (encoding methylmalonate-semialdehyde dehydrogenase [acylating], mitochondrial), consistent with impaired BCAA metabolism (Figure 4).154 Further studies are needed to understand the mechanisms underlying the increased levels of BCAA-related acylcarnitines in states of T2DM and insulin-resistant obesity.
Altered BCAA metabolism in disease states
Attenuation of complete mitochondrial BCAA metabolism, if it should occur in states of insulin resistance or T2DM, could occur by several mechanisms (Figure 3), the simplest of which involves altered gene expression, mutations or epigenetic factors that affect gene expression. Seven independent computational disease-prioritization methods have been applied to 9,556 positional candidate genes for obesity and T2DM.120 This approach identified nine primary candidate genes for T2DM and five for obesity, together with 94 secondary candidates for T2DM and 116 for obesity. BCKDHA, the gene encoding the regulated subunit of BCKDC was only one of two primary susceptibility genes identified that affected the risk of both T2DM and obesity (Figure 4).120 IVD, encoding isovaleryl-CoA dehydrogenase, which catalyses the next step in leucine metabolism, was identified as a secondary T2DM susceptibility gene (Figure 4).
In 2009, PPM1K was identified as the BCKDHA phosphatase.137,164 A later report, attempting to identify T2DM susceptibility genes,165 examined global gene expression profiles of 63 islet donors with or without T2DM and compared this to 48 known genes located near known risk variants of T2DM (Figures 3 and 4). Decreased expression of PPM1K was observed in islets from individuals with T2DM. PPM1K was selected as one of the top 20 candidate genes for further study. Knockdown of PPM1K in rat INS-1 cells impaired glucose-stimulated insulin secretion and activated apoptosis (Figures 3 and 4, Box 1).165 Thus, PPM1K has been designated as a T2DM susceptibility gene in islet β cells. In another study, elevated circulating valine levels and the ratio of BCAA levels to the phenylalanine plus tyrosine concentration were found to be associated with a human single-nucleotide polymorphism called rs1440581.166 RS1440581 is located upstream of PPM1K. Subsequently, an analysis of POUNDS LOST trial participants on an energy-restricted high-fat diet who had the C allele of rs1440581 had poorer weight loss outcomes as well as less improved insulin sensitivity than those missing this allele. (Box 1 and Figure 3).167 Further studies are needed to determine if the rs1440581 polymorphism does indeed affect PPM1K expression.
Obesity and/or insulin resistance are known to disrupt circadian homeostasis.168–172 Krueppel-like factor 15 (encoded by the KLF15 gene) is a master regulator of glucose and amino acid metabolism (including BCAA metabolism)119,173 that is differentially expressed in the muscle and fat of overweight individuals with insulin resistance174 and is thought to regulate circadian nitrogen homeostasis. KLF15 or idiosyncratic responses to it could be another factor in altering BCAA metabolism in insulin-resistant obesity (Figure 3).
Other factors altered by states of obesity and T2DM regulate BCKDC activity in the short and long term (Table 1). Unfortunately, many of these factors have only been studied in a single tissue; an important limitation, as tissue specific regulation has been observed for some factors (Table 1). Thus, it is difficult to predict how some of these regulators affect BCKDC activity in other tissues. It is not immediately obvious from looking at these regulators how the known changes in insulin-resistant states and T2DM might additively work to alter BCAA metabolism or bring about different BCKDC activity phenotypes (Figure 5). However, an interesting point is that long-chain fatty acids and their metabolites, which are elevated in insulin-resistant states and T2DM, inhibit BCKDC activity either by affecting redox states or acetyl-CoA concentrations.98,143,175–181 Increased free fatty acid metabolism has also been linked to the increased generation of reactive oxygen species, which leads to the formation of reactive lipid aldehydes.178–181 Proteomic studies have revealed that enzymes in the BCAA metabolic pathway are carbonylated (for example, by the action of 4-hydroxynonenals) following oxidative stress, which could potentially impair their enzymatic activity (Figure 4).178–181 The increased availability of free fatty acids and their ability to directly or indirectly inhibit BCAA metabolism in mitochondria (for example, through oxidative stress associated carbonylation) could be a factor linking increased free fatty acid levels to the BCAA dysmetabolism model presented here (Figure 3), and discussed elsewhere.143
Conclusions
Although a number of studies have suggested that BCAA supplementation or BCAA-rich diets are beneficial for promoting lean body mass in obesity or catabolic disorders, or for increasing satiety for body weight loss, acceptance of this idea has been tempered by the associations between increased circulating levels of BCAAs and insulin-resistant obesity and T2DM, as well as the observations that these increases might predict future insulin resistance or T2DM. Two mechanisms have been proposed that could explain how elevated levels of BCAAs might be linked to metabolic disease. One involves the persistent activation of the nutrient sensing complex, mTORC1. However, numerous observations indicate that BCAA-associated mTORC1 activation is not necessary or sufficient to trigger insulin resistance. A BCAA dysmetabolism model suggests that those individuals or animal models with the highest plasma levels of BCAAs (that correlate more closely with metabolic dysfunction) might have impaired BCAA metabolism that adds to the contribution of BCAAs resulting from protein degradation in insulin resistant states. In contrast to the mTORC1 mechanism, the BCAA dysmetabolism model assumes that it is the accumulation of BCAA metabolites that result in dysfunction and that the increased BCAA levels are simply reporters of that accumulation. Inborn errors of metabolism and accumulation of BCAA metabolites can cause mitochondrial dysfunction associated with stress kinase activation and β-cell apoptosis—factors that are frequently associated with insulin resistance and T2DM.
Alternatively, BCAA dysmetabolism or incomplete oxidation (of isoleucine and valine, in particular) might promote anaplerotic stress and an anaplerosis–cataplerosis imbalance that contributes to suboptimal mitochondrial function in states of T2DM.66,142 These concepts remain speculative but warrant further study. Even though rodent models of obesity and humans with insulin resistance universally exhibit reduced levels of BCAA metabolic enzymes in fat compared with metabolically healthy controls, (Figure 5) there is emerging evidence that these reductions in adipose tissue BCAA metabolic capacity might either extend to other tissues (phenotype A) or be compensated for by increased BCKDC activity in the liver (phenotype B). If these phenotypes exist in humans (as seems to be the case on the basis of the results of a recent study157)some individuals might have more global reductions in BCAA metabolic capacity that could contribute to increasing circulating levels of BCAAs to the higher ranges that have an increased association with the development of future T2DM and insulin resistance. To determine whether different BCAA metabolic phenotypes affecting multiple tissues exist in humans, a strategy using alloisoleucine has been proposed.75 Further research is needed to elucidate the contributions of various processes, such as tissue uptake, oxidation and lean mass catabolism, in mediating the increased levels of BCAAs found in individuals with insulin resistance and T2DM and to understand the molecular mechanisms underlying the cellular responses to dysfunctional BCAA metabolism.
In summary, although mechanisms have been proposed that might explain how increased BCAA levels could lead to insulin resistance or obesity, it should be appreciated that increased BCAA levels are more likely to be a marker of loss of insulin action and not, themselves, causative.
Key points.
Branched-chain amino acids (BCAAs) have beneficial nutrient signalling effects but paradoxically are associated with obesity, insulin resistance and type 2 diabetes mellitus (T2DM)
BCAAs might be a marker of, rather than, a cause of insulin resistance, as insulin resistance increases the rate of appearance of BCAAs and is linked to reduced expression of mitochondrial BCAA catabolic enzymes
Alternatively, two mechanisms have emerged indicating that a causative link exists between increased plasma concentrations of BCAAs and T2DM or insulin resistance
In the first mechanism, persistent activation of the mammalian target of rapamycin complex 1 signalling pathway uncouples the insulin receptor from the insulin signalling mediator, IRS-1, which leads to insulin resistance
In the second mechanism, abnormal BCAA metabolism in obesity results in accumulation of toxic BCAA metabolites that in turn trigger the mitochondrial dysfunction and stress signalling associated with insulin resistance and T2DM
Factors that alter expression of genes involved in the BCAA metabolic pathway (or post-translational modification of the encoded proteins) are associated with obesity and T2DM; three genes in the pathway are candidate genes for obesity and/or T2DM
Review criteria.
PubMed and Google Scholar were searched for English language articles published between 1968 and 2014 using the following search terms alone and in combination: “ACAD8”, “ACADS”, “ACADSB”, “ACAT1”, “ACAT2”, “ACDASB”, “acylcarnitines”, “adipose tissue”, “ALDH6A1”, “amino acids”, “AUH”, “autophagy”, “BCAT2”, “BCATm”, “BCKDHA”, “BCKDHB”, “BCKDK”, “branched chain amino acids”, “branched chain keto acids”, “branched chain ketoacid dehydrogenase”, “candidate gene”, “cholesterol”, “DBT”, “dehydrogenase”, “diabetes mellitus”, “DLD”, “ECHS1”, “HADHA”, “HbA1c”, “haemoglobin A1C”, “HIBADH”, “HIBCH”, “HMGCL”, “HSD17B10”, “hypothalamic”, “IGF-1”, “inborn”, “inborn errors of metabolism”, “insulin”, “insulin resistance”, “isoleucine”, “lean mass”, “leucine”, “liver”, “MCCC1”, “MCCC2”, “metabolic disease”, “metabolic syndrome”, “metabolism”, “metabolomics”, “mTOR”, “mTORC1”, “mTORC2”, “muscle”, “new onset diabetes”, “nutrient signalling”, “obesity”, “organic acidurias”, “OXCT1”, “OXCT2”, “PCCB”, “PP2Cm”, “IVD”, “PPM1K”, “prediabetes”, “protein degradation”, “protein degradation”, “protein synthesis”, “proteasome”, “rapamycin”, “satiety”, “Sirolimus”, “transaminase”, “transplant”, “turnover”, “type 2 diabetes” and “valine”. The current understanding of the BCAA metabolic pathway was based on annotations from the Kyoto Encyclopaedia of Genes and Genomes (KEGG).
Acknowledgments
C.J.L. acknowledges research support from the NIH (DK091784 and DK084428). S.H.A. acknowledges research support from the USDA–Agricultural Research Service (Intramural Project 5,306-51530-019-00) and the NIH (NIH-NIDDK R01DK078328). The authors wish to thank their many colleagues, collaborators and mentors who have inspired their interest in branched-chain amino acids and metabolic disease.
Footnotes
Competing interests
C.J.L. has received an honorarium for being a panelist for the Protein Summit, Washington, DC, USA, in 2013. S.H.A. declares no competing interests.
Author contributions
C.J.L. and S.H.A. researched data for the article, made substantial contributions to discussions of the content, wrote the article and reviewed and/or edited the manuscript before submission.
Contributor Information
Christopher J. Lynch, Cellular and Molecular Physiology Department, The Pennsylvania State University, 500 University Drive, MC-H166, Hershey, PA 17033, USA
Sean H. Adams, Arkansas Children’s Nutrition Center, and Department of Pediatrics, University of Arkansas for Medical Sciences, 15 Children’s Way, Little Rock, AR 72202, USA
References
- 1.Cota D, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 2.Blouet C, Jo YH, Li X, Schwartz GJ. Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus–brainstem circuit. J Neurosci. 2009;29:8302–8311. doi: 10.1523/JNEUROSCI.1668-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blouet C, Schwartz GJ. Brainstem nutrient sensing in the nucleus of the solitary tract inhibits feeding. Cell Metab. 2012;16:579–587. doi: 10.1016/j.cmet.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz GJ. Central leucine sensing in the control of energy homeostasis. Endocrinol Metab Clin North Am. 2013;42:81–87. doi: 10.1016/j.ecl.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laeger T, et al. Leucine acts in the brain to suppress food intake but does not function as a physiological signal of low dietary protein. Am J Physiol Regul Integr Comp Physiol. 2014;307:R310–R320. doi: 10.1152/ajpregu.00116.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devkota S, Layman DK. Protein metabolic roles in treatment of obesity. Curr Opin Clin Nutr Metab Care. 2010;13:403–407. doi: 10.1097/MCO.0b013e32833a7737. [DOI] [PubMed] [Google Scholar]
- 7.Layman DK, Walker DA. Potential importance of leucine in treatment of obesity and the metabolic syndrome. J Nutr. 2006;136:319S–323S. doi: 10.1093/jn/136.1.319S. [DOI] [PubMed] [Google Scholar]
- 8.Norton LE, Layman DK. Leucine regulates translation initiation of protein synthesis in skeletal muscle after exercise. J Nutr. 2006;136:533S–537S. doi: 10.1093/jn/136.2.533S. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Xu M, Lee J, He C, Xie Z. Leucine supplementation increases SIRT1 expression and prevents mitochondrial dysfunction and metabolic disorders in high-fat diet-induced obese mice. Am J Physiol Endocrinol Metab. 2012;303:E1234–E1244. doi: 10.1152/ajpendo.00198.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Heber D. Sarcopenic obesity in the elderly and strategies for weight management. Nutr Rev. 2012;70:57–64. doi: 10.1111/j.1753-4887.2011.00453.x. [DOI] [PubMed] [Google Scholar]
- 11.Guo K, Yu YH, Hou J, Zhang Y. Chronic leucine supplementation improves glycemic control in etiologically distinct mouse models of obesity and diabetes mellitus. Nutr Metab (Lond) 2010;7:57. doi: 10.1186/1743-7075-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, et al. Increasing dietary leucine intake reduces diet-induced obesity and improves glucose and cholesterol metabolism in mice via multimechanisms. Diabetes. 2007;56:1647–1654. doi: 10.2337/db07-0123. [DOI] [PubMed] [Google Scholar]
- 13.Binder E, et al. Leucine supplementation modulates fuel substrates utilization and glucose metabolism in previously obese mice. Obesity (Silver Spring) 2014;22:713–720. doi: 10.1002/oby.20578. [DOI] [PubMed] [Google Scholar]
- 14.Chen H, Simar D, Ting JH, Erkelens JR, Morris MJ. Leucine improves glucose and lipid status in offspring from obese dams, dependent on diet type, but not caloric intake. J Neuroendocrinol. 2012;24:1356–1364. doi: 10.1111/j.1365-2826.2012.02339.x. [DOI] [PubMed] [Google Scholar]
- 15.Torres-Leal FL, et al. Leucine supplementation improves adiponectin and total cholesterol concentrations despite the lack of changes in adiposity or glucose homeostasis in rats previously exposed to a high-fat diet. Nutr Metab (Lond) 2011;8:62. doi: 10.1186/1743-7075-8-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potier M, Darcel N, Tome D. Protein, amino acids and the control of food intake. Curr Opin Clin Nutr Metab Care. 2009;12:54–58. doi: 10.1097/MCO.0b013e32831b9e01. [DOI] [PubMed] [Google Scholar]
- 17.Lopez N, Sanchez J, Pico C, Palou A, Serra F. Dietary L-leucine supplementation of lactating rats results in a tendency to increase lean/fat ratio associated to lower orexigenic neuropeptide expression in hypothalamus. Peptides. 2010;31:1361–1367. doi: 10.1016/j.peptides.2010.03.028. [DOI] [PubMed] [Google Scholar]
- 18.Nairizi A, She P, Vary TC, Lynch CJ. Leucine supplementation of drinking water does not alter susceptibility to diet-induced obesity in mice. J Nutr. 2009;139:715–719. doi: 10.3945/jn.108.100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevanovic D, et al. Intracerebroventricular administration of metformin inhibits ghrelin-induced hypothalamic AMP-kinase signalling and food intake. Neuroendocrinology. 2012;96:24–31. doi: 10.1159/000333963. [DOI] [PubMed] [Google Scholar]
- 20.Chen Q, Reimer RA. Dairy protein and leucine alter GLP-1 release and mRNA of genes involved in intestinal lipid metabolism in vitro. Nutrition. 2009;25:340–349. doi: 10.1016/j.nut.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macotela Y, et al. Dietary leucine—an environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS ONE. 2011;6:e21187. doi: 10.1371/journal.pone.0021187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qin LQ, et al. Higher branched-chain amino acid intake is associated with a lower prevalence of being overweight or obese in middle-aged East Asian and Western adults. J Nutr. 2011;141:249–254. doi: 10.3945/jn.110.128520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salehi A, et al. The insulinogenic effect of whey protein is partially mediated by a direct effect of amino acids and GIP on β-cells. Nutr Metab (Lond) 2012;9:48. doi: 10.1186/1743-7075-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu G, et al. Gastric mammalian target of rapamycin signaling regulates ghrelin production and food intake. Endocrinology. 2009;150:3637–3644. doi: 10.1210/en.2009-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynch CJ, et al. Leucine in food mediates some of the postprandial rise in plasma leptin concentrations. Am J Physiol Endocrinol Metab. 2006;291:E621–E630. doi: 10.1152/ajpendo.00462.2005. [DOI] [PubMed] [Google Scholar]
- 26.Vary TC, Lynch CJ. Nutrient signaling components controlling protein synthesis in striated muscle. J Nutr. 2007;137:1835–1843. doi: 10.1093/jn/137.8.1835. [DOI] [PubMed] [Google Scholar]
- 27.Dodd KM, Tee AR. Leucine and mTORC1: a complex relationship. Am J Physiol Endocrinol Metab. 2012;302:E1329–E1342. doi: 10.1152/ajpendo.00525.2011. [DOI] [PubMed] [Google Scholar]
- 28.Lynch CJ, et al. Leucine is a direct-acting nutrient signal that regulates protein synthesis in adipose tissue. Am J Physiol Endocrinol Metab. 2002;283:E503–E513. doi: 10.1152/ajpendo.00084.2002. [DOI] [PubMed] [Google Scholar]
- 29.Lang CH, Frost RA, Bronson SK, Lynch CJ, Vary TC. Skeletal muscle protein balance in mTOR heterozygous mice in response to inflammation and leucine. Am J Physiol Endocrinol Metab. 2010;298:E1283–E1294. doi: 10.1152/ajpendo.00676.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vary TC, Deiter G, Lynch CJ. Rapamycin limits formation of active eukaryotic initiation factor 4F complex following meal feeding in rat hearts. J Nutr. 2007;137:1857–1862. doi: 10.1093/jn/137.8.1857. [DOI] [PubMed] [Google Scholar]
- 31.Vary TC, Anthony JC, Jefferson LS, Kimball SR, Lynch CJ. Rapamycin blunts nutrient stimulation of eIF4G, but not PKCε phosphorylation, in skeletal muscle. Am J Physiol Endocrinol Metab. 2007;293:E188–E196. doi: 10.1152/ajpendo.00037.2007. [DOI] [PubMed] [Google Scholar]
- 32.Vary TC, Lynch CJ. Meal feeding enhances formation of eIF4F in skeletal muscle: role of increased eIF4E availability and eIF4G phosphorylation. Am J Physiol Endocrinol Metab. 2006;290:E631–E642. doi: 10.1152/ajpendo.00460.2005. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–369. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- 34.Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126:1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garrow JS. The contribution of protein synthesis to thermogenesis in man. Int J Obes. 1985;9 (Suppl 2):97–101. [PubMed] [Google Scholar]
- 36.Giordano M, Castellino P. Correlation between amino acid induced changes in energy expenditure and protein metabolism in humans. Nutrition. 1997;13:309–312. doi: 10.1016/s0899-9007(97)83052-3. [DOI] [PubMed] [Google Scholar]
- 37.Layman DK, Baum JI. Dietary protein impact on glycemic control during weight loss. J Nutr. 2004;134:968S–973S. doi: 10.1093/jn/134.4.968S. [DOI] [PubMed] [Google Scholar]
- 38.Tsujinaka T, et al. Modulation of thermogenic response to parenteral amino acid infusion in surgical stress. Nutrition. 1996;12:36–39. doi: 10.1016/0899-9007(95)00018-6. [DOI] [PubMed] [Google Scholar]
- 39.Yamaoka I. Modification of core body temperature by amino acid administration. Asia Pac J Clin Nutr. 2008;17 (Suppl 1):309–311. [PubMed] [Google Scholar]
- 40.Pitkänen O, Takala J, Pöyhönen M, Kari A. Branched-chain and mixed amino acid solutions and thermogenesis in postoperative patients. Nutrition. 1994;10:132–137. [PubMed] [Google Scholar]
- 41.Kawaguchi T, Taniguchi E, Sata M. Effects of oral branched-chain amino acids on hepatic encephalopathy and outcome in patients with liver cirrhosis. Nutr Clin Pract. 2013;28:580–588. doi: 10.1177/0884533613496432. [DOI] [PubMed] [Google Scholar]
- 42.Ichikawa K, et al. Branched-chain amino acid-enriched nutrients stimulate antioxidant DNA repair in a rat model of liver injury induced by carbon tetrachloride. Mol Biol Rep. 2012;39:10803–10810. doi: 10.1007/s11033-012-1974-4. [DOI] [PubMed] [Google Scholar]
- 43.Kuwahata M, et al. Supplementation with branched-chain amino acids attenuates hepatic apoptosis in rats with chronic liver disease. Nutr Res. 2012;32:522–529. doi: 10.1016/j.nutres.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 44.Hayaishi S, et al. Oral branched-chain amino acid granules reduce the incidence of hepatocellular carcinoma and improve event-free survival in patients with liver cirrhosis. Dig Dis. 2011;29:326–332. doi: 10.1159/000327571. [DOI] [PubMed] [Google Scholar]
- 45.Holecek M. Three targets of branched-chain amino acid supplementation in the treatment of liver disease. Nutrition. 2010;26:482–490. doi: 10.1016/j.nut.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 46.Layman DK. Dietary guidelines should reflect new understandings about adult protein needs. Nutr Metab (Lond) 2009;6:12. doi: 10.1186/1743-7075-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mojtahedi MC, et al. The effects of a higher protein intake during energy restriction on changes in body composition and physical function in older women. J Gerontol A Biol Sci Med Sci. 2011;66:1218–1225. doi: 10.1093/gerona/glr120. [DOI] [PubMed] [Google Scholar]
- 48.Layman DK, et al. Defining meal requirements for protein to optimize metabolic roles of amino acids. Am J Clin Nutr. doi: 10.3945/ajcn.114.084053. http://dx.doi.org/10.3945/ajcn.114.084053. [DOI] [PMC free article] [PubMed]
- 49.Millward DJ, Layman DK, Tome D, Schaafsma G. Protein quality assessment: impact of expanding understanding of protein and amino acid needs for optimal health. Am J Clin Nutr. 2008;87:1576S–1581S. doi: 10.1093/ajcn/87.5.1576S. [DOI] [PubMed] [Google Scholar]
- 50.Muto Y, et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol Res. 2006;35:204–214. doi: 10.1016/j.hepres.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Norton LE, Wilson GJ, Layman DK, Moulton CJ, Garlick PJ. Leucine content of dietary proteins is a determinant of postprandial skeletal muscle protein synthesis in adult rats. Nutr Metab (Lond) 2012;9:67. doi: 10.1186/1743-7075-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeng YJ, et al. Characteristics and risk factors for hyperglycemia in Chinese female patients with systemic lupus erythematosus. Lupus. 2010;19:1344–1350. doi: 10.1177/0961203310375439. [DOI] [PubMed] [Google Scholar]
- 53.Wurtz P, et al. Metabolic signatures of insulin resistance in 7,098 young adults. Diabetes. 2012;61:1372–1380. doi: 10.2337/db11-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang TJ, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tai ES, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia. 2010;53:757–767. doi: 10.1007/s00125-009-1637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suhre K, et al. Metabolic footprint of diabetes: a multiplatform metabolomics study in an epidemiological setting. PLoS ONE. 2010;5:e13953. doi: 10.1371/journal.pone.0013953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schauder P, Zavelberg D, Langer K, Herbertz L. Sex-specific differences in plasma branched-chain keto acid levels in obesity. Am J Clin Nutr. 1987;46:58–60. doi: 10.1093/ajcn/46.1.58. [DOI] [PubMed] [Google Scholar]
- 58.Pennetti V, Galante A, Zonta-Sgaramella L, Jayakar SD. Relation between obesity, insulinemia, and serum amino acid concentrations in a sample of Italian adults. Clin Chem. 1982;28:2219–2224. [PubMed] [Google Scholar]
- 59.Newgard CB, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–326. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15:606–614. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nair KS, Garrow JS, Ford C, Mahler RF, Halliday D. Effect of poor diabetic control and obesity on whole body protein metabolism in man. Diabetologia. 1983;25:400–403. doi: 10.1007/BF00282518. [DOI] [PubMed] [Google Scholar]
- 62.McCormack SE, et al. Circulating branched-chain amino acid concentrations are associated with obesity and future insulin resistance in children and adolescents. Pediatr Obes. 2013;8:52–61. doi: 10.1111/j.2047-6310.2012.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lackey DE, et al. Regulation of adipose branched-chain amino acid catabolism enzyme expression and cross-adipose amino acid flux in human obesity. Am J Physiol Endocrinol Metab. 2013;304:E1175–E1187. doi: 10.1152/ajpendo.00630.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim MJ, et al. Obesity-related metabolomic analysis of human subjects in black soybean peptide intervention study by ultraperformance liquid chromatography and quadrupole-time-of-flight mass spectrometry. J Obes. 2013;2013:874981. doi: 10.1155/2013/874981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forlani G, et al. Insulin-dependent metabolism of branched-chain amino acids in obesity. Metabolism. 1984;33:147–150. doi: 10.1016/0026-0495(84)90127-6. [DOI] [PubMed] [Google Scholar]
- 66.Fiehn O, et al. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE. 2010;5:e15234. doi: 10.1371/journal.pone.0015234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Felig P, Marliss E, Cahill GF., Jr Plasma amino acid levels and insulin secretion in obesity. N Engl J Med. 1969;281:811–816. doi: 10.1056/NEJM196910092811503. [DOI] [PubMed] [Google Scholar]
- 68.Cheng S, et al. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation. 2012;125:2222–2231. doi: 10.1161/CIRCULATIONAHA.111.067827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caballero B, Finer N, Wurtman RJ. Plasma amino acids and insulin levels in obesity: response to carbohydrate intake and tryptophan supplements. Metabolism. 1988;37:672–676. doi: 10.1016/0026-0495(88)90089-3. [DOI] [PubMed] [Google Scholar]
- 70.Belfiore F, Iannello S, Rabuazzo AM. Insulin resistance in obesity: a critical analysis at enzyme level. A review. Int J Obes. 1979;3:301–323. [PubMed] [Google Scholar]
- 71.Adibi SA. Influence of dietary deprivations on plasma concentration of free amino acids of man. J Appl Physiol. 1968;25:52–57. doi: 10.1152/jappl.1968.25.1.52. [DOI] [PubMed] [Google Scholar]
- 72.She P, et al. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab. 2007;293:E1552–E1563. doi: 10.1152/ajpendo.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.She P, et al. Leucine and protein metabolism in obese Zucker rats. PLoS ONE. 2013;8:e59443. doi: 10.1371/journal.pone.0059443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;285:11348–11356. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Olson KC, Chen G, Xu Y, Hajnal A, Lynch CJ. Alloisoleucine differentiates the branched-chain aminoacidemia of Zucker and dietary obese rats. Obesity (Silver Spring) 2014;22:1212–1215. doi: 10.1002/oby.20691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Batch BC, et al. Branched chain amino acids are novel biomarkers for discrimination of metabolic wellness. Metabolism. 2013;62:961–969. doi: 10.1016/j.metabol.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. 2011;90:220–223. doi: 10.1093/cvr/cvr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Breitman I, et al. The effects of an amino acid supplement on glucose homeostasis, inflammatory markers, and incretins after laparoscopic gastric bypass. J Am Coll Surg. 2011;212:617–625. doi: 10.1016/j.jamcollsurg.2010.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Badoud F, et al. Serum and adipose tissue amino Acid homeostasis in the metabolically healthy obese. J Proteome Res. 2014;13:3455–3466. doi: 10.1021/pr500416v. [DOI] [PubMed] [Google Scholar]
- 80.Fernstrom JD. Branched-chain amino acids and brain function. J Nutr. 2005;135:1539S–1546S. doi: 10.1093/jn/135.6.1539S. [DOI] [PubMed] [Google Scholar]
- 81.Mans AM, DeJoseph MR, Davis DW, Hawkins RA. Regional amino acid transport into brain during diabetes: effect of plasma amino acids. Am J Physiol. 1987;253:E575–E583. doi: 10.1152/ajpendo.1987.253.5.E575. [DOI] [PubMed] [Google Scholar]
- 82.Crandall EA, Fernstrom JD. Effect of experimental diabetes on the levels of aromatic and branched-chain amino acids in rat blood and brain. Diabetes. 1983;32:222–230. doi: 10.2337/diab.32.3.222. [DOI] [PubMed] [Google Scholar]
- 83.Fernstrom MH, Volk EA, Fernstrom JD. In vivo inhibition of tyrosine uptake into rat retina by large neutral but not acidic amino acids. Am J Physiol. 1986;251:E393–E399. doi: 10.1152/ajpendo.1986.251.4.E393. [DOI] [PubMed] [Google Scholar]
- 84.Coppola A, et al. Branched-chain amino acids alter neurobehavioral function in rats. Am J Physiol Endocrinol Metab. 2013;304:E405–E413. doi: 10.1152/ajpendo.00373.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pan A, et al. Bidirectional association between depression and obesity in middle-aged and older women. Int J Obes (Lond) 2012;36:595–602. doi: 10.1038/ijo.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem. 1995;270:2320–2326. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- 87.Maki T, et al. Branched-chain amino acids reduce hindlimb suspension-induced muscle atrophy and protein levels of atrogin-1 and MuRF1 in rats. Nutr Res. 2012;32:676–683. doi: 10.1016/j.nutres.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 88.May ME, Buse MG. Effects of branched-chain amino acids on protein turnover. Diabetes Metab Rev. 1989;5:227–245. doi: 10.1002/dmr.5610050303. [DOI] [PubMed] [Google Scholar]
- 89.Mordier S, Deval C, Bechet D, Tassa A, Ferrara M. Leucine limitation induces autophagy and activation of lysosome-dependent proteolysis in C2C12 myotubes through a mammalian target of rapamycin-independent signaling pathway. J Biol Chem. 2000;275:29900–29906. doi: 10.1074/jbc.M003633200. [DOI] [PubMed] [Google Scholar]
- 90.Mortimore GE, Poso AR. Lysosomal pathways in hepatic protein degradation: regulatory role of amino acids. Fed Proc. 1984;43:1289–1294. [PubMed] [Google Scholar]
- 91.Sugawara T, Ito Y, Nishizawa N, Nagasawa T. Regulation of muscle protein degradation, not synthesis, by dietary leucine in rats fed a protein-deficient diet. Amino Acids. 2009;37:609–616. doi: 10.1007/s00726-008-0180-0. [DOI] [PubMed] [Google Scholar]
- 92.Hong SO, Layman DK. Effects of leucine on in vitro protein synthesis and degradation in rat skeletal muscles. J Nutr. 1984;114:1204–1212. doi: 10.1093/jn/114.7.1204. [DOI] [PubMed] [Google Scholar]
- 93.Adegoke OA, et al. Fed-state clamp stimulates cellular mechanisms of muscle protein anabolism and modulates glucose disposal in normal men. Am J Physiol Endocrinol Metab. 2009;296:E105–E113. doi: 10.1152/ajpendo.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baptista IL, et al. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve. 2010;41:800–808. doi: 10.1002/mus.21578. [DOI] [PubMed] [Google Scholar]
- 95.Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:225–229. doi: 10.1097/mco.0b013e32833862df. [DOI] [PubMed] [Google Scholar]
- 96.Latres E, et al. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–2744. doi: 10.1074/jbc.M407517200. [DOI] [PubMed] [Google Scholar]
- 97.Paula-Gomes S, et al. Insulin suppresses atrophy and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signaling. Horm Metab Res. 2013;45:849–855. doi: 10.1055/s-0033-1347209. [DOI] [PubMed] [Google Scholar]
- 98.Williamson JR, Walajtys-Rode E, Coll KE. Effects of branched chain α-ketoacids on the metabolism of isolated rat liver cells. I Regulation of branched chain α-ketoacid metabolism. J Biol Chem. 1979;254:11511–11520. [PubMed] [Google Scholar]
- 99.Greenhaff PL, et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol Endocrinol Metab. 2008;295:E595–E604. doi: 10.1152/ajpendo.90411.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.O’Connor PM, Bush JA, Suryawan A, Nguyen HV, Davis TA. Insulin and amino acids independently stimulate skeletal muscle protein synthesis in neonatal pigs. Am J Physiol Endocrinol Metab. 2003;284:E110–E119. doi: 10.1152/ajpendo.00326.2002. [DOI] [PubMed] [Google Scholar]
- 101.Fryburg DA, Jahn LA, Hill SA, Oliveras DM, Barrett EJ. Insulin and insulin-like growth factor-I enhance human skeletal muscle protein anabolism during hyperaminoacidemia by different mechanisms. J Clin Invest. 1995;96:1722–1729. doi: 10.1172/JCI118217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Estornell E, Cabo J, Barber T. Protein synthesis is stimulated in nutritionally obese rats. J Nutr. 1995;125:1309–1315. doi: 10.1093/jn/125.5.1309. [DOI] [PubMed] [Google Scholar]
- 103.Guillet C, Masgrau A, Boirie Y. Is protein metabolism changed with obesity? Curr Opin Clin Nutr Metab Care. 2011;14:89–92. doi: 10.1097/MCO.0b013e328341389e. [DOI] [PubMed] [Google Scholar]
- 104.Kumashiro N, et al. Targeting pyruvate carboxylase reduces gluconeogenesis and adiposity and improves insulin resistance. Diabetes. 2013;62:2183–2194. doi: 10.2337/db12-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nilsson MI, et al. Abnormal protein turnover and anabolic resistance to exercise in sarcopenic obesity. FASEB J. 2013;27:3905–3916. doi: 10.1096/fj.12-224006. [DOI] [PubMed] [Google Scholar]
- 106.Welle S, Barnard RR, Statt M, Amatruda JM. Increased protein turnover in obese women. Metabolism. 1992;41:1028–1034. doi: 10.1016/0026-0495(92)90133-U. [DOI] [PubMed] [Google Scholar]
- 107.Louard RJ, Barrett EJ, Gelfand RA. Overnight branched-chain amino acid infusion causes sustained suppression of muscle proteolysis. Metabolism. 1995;44:424–429. doi: 10.1016/0026-0495(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 108.Hung CF, et al. Relationship between obesity and the risk of clinically significant depression: Mendelian randomisation study. Br J Psychiatry. 2014;205:24–28. doi: 10.1192/bjp.bp.113.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Um SH, et al. Absence of S6K1 protects against age and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 110.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 111.She P, et al. Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 2007;6:181–194. doi: 10.1016/j.cmet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kihlberg R, Bark S, Hallberg D. An oral amino acid loading test before and after intestinal bypass operation for morbid obesity. Acta Chir Scand. 1982;148:73–86. [PubMed] [Google Scholar]
- 113.Magkos F, et al. Effect of Roux-en-Y gastric bypass and laparoscopic adjustable gastric banding on branched-chain amino acid metabolism. Diabetes. 2013;62:2757–2761. doi: 10.2337/db13-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Laferrere B, et al. Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci Transl Med. 2011;3:80re2. doi: 10.1126/scitranslmed.3002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doi M, et al. Isoleucine, a blood glucose-lowering amino acid, increases glucose uptake in rat skeletal muscle in the absence of increases in AMP-activated protein kinase activity. J Nutr. 2005;135:2103–2108. doi: 10.1093/jn/135.9.2103. [DOI] [PubMed] [Google Scholar]
- 116.Doi M, Yamaoka I, Nakayama M, Sugahara K, Yoshizawa F. Hypoglycemic effect of isoleucine involves increased muscle glucose uptake and whole body glucose oxidation and decreased hepatic gluconeogenesis. Am J Physiol Endocrinol Metab. 2007;292:E1683–E1693. doi: 10.1152/ajpendo.00609.2006. [DOI] [PubMed] [Google Scholar]
- 117.Broca C, et al. Insulinotropic agent ID-1101 (4-hydroxyisoleucine) activates insulin signaling in rat. Am J Physiol Endocrinol Metab. 2004;287:E463–E471. doi: 10.1152/ajpendo.00163.2003. [DOI] [PubMed] [Google Scholar]
- 118.Blanchard PG, et al. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J Lipid Res. 2012;53:1117–1125. doi: 10.1194/jlr.M021485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Herder C, Roden M. Genetics of type 2 diabetes: pathophysiologic and clinical relevance. Eur J Clin Invest. 2011;41:679–692. doi: 10.1111/j.1365-2362.2010.02454.x. [DOI] [PubMed] [Google Scholar]
- 120.Tiffin N, et al. Computational disease gene identification: a concert of methods prioritizes type 2 diabetes and obesity candidate genes. Nucleic Acids Res. 2006;34:3067–3081. doi: 10.1093/nar/gkl381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vimaleswaran KS, et al. Candidate genes for obesity-susceptibility show enriched association within a large genome-wide association study for BMI. Hum Mol Genet. 2012;21:4537–4542. doi: 10.1093/hmg/dds283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Leibowitz G, Cerasi E, Ketzinel-Gilad M. The role of mTOR in the adaptation and failure of β-cells in type 2 diabetes. Diabetes Obes Metab. 2008;10 (Suppl 4):157–169. doi: 10.1111/j.1463-1326.2008.00952.x. [DOI] [PubMed] [Google Scholar]
- 123.Lopes P, et al. Effects of cyclosporine and sirolimus on insulin-stimulated glucose transport and glucose tolerance in a rat model. Transplant Proc. 2013;45:1142–1148. doi: 10.1016/j.transproceed.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 124.Dandel M, Lehmkuhl HB, Knosalla C, Hetzer R. Impact of different long-term maintenance immunosuppressive therapy strategies on patients’ outcome after heart transplantation. Transpl Immunol. 2010;23:93–103. doi: 10.1016/j.trim.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 125.Gyurus E, Kaposztas Z, Kahan BD. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: a long-term analysis of various treatment regimens. Transplant Proc. 2011;43:1583–1592. doi: 10.1016/j.transproceed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 126.Hughes KJ, Kennedy BK. Rapamycin paradox resolved. Science. 2012;335:1578–1579. doi: 10.1126/science.1221365. [DOI] [PubMed] [Google Scholar]
- 127.Lamming DW, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sarbassov DD, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 129.Pham PTT, Pham PMT, Pham SV, Pham PAT, Pham PCT. New onset diabetes after transplantation (NODAT): an overview. Diabetes Metab Syndr Obes. 2011;4:175–186. doi: 10.2147/DMSO.S19027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bridi R, et al. α-keto acids accumulating in maple syrup urine disease stimulate lipid peroxidation and reduce antioxidant defences in cerebral cortex from young rats. Metab Brain Dis. 2005;20:155–167. doi: 10.1007/s11011-005-4152-8. [DOI] [PubMed] [Google Scholar]
- 131.Funchal C, et al. Morphological alterations and induction of oxidative stress in glial cells caused by the branched-chain α-keto acids accumulating in maple syrup urine disease. Neurochem Int. 2006;49:640–650. doi: 10.1016/j.neuint.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 132.Jackson RH, Singer TP. Inactivation of the 2-ketoglutarate and pyruvate dehydrogenase complexes of beef heart by branched chain keto acids. J Biol Chem. 1983;258:1857–1865. [PubMed] [Google Scholar]
- 133.Walajtys-Rode E, Williamson JR. Effects of branched chain α-ketoacids on the metabolism of isolated rat liver cells. III Interactions with pyruvate dehydrogenase. J Biol Chem. 1980;255:413–418. [PubMed] [Google Scholar]
- 134.Jouvet P, Kozma M, Mehmet H. Primary human fibroblasts from a maple syrup urine disease patient undergo apoptosis following exposure to physiological concentrations of branched chain amino acids. Ann NY Acad Sci. 2000;926:116–121. doi: 10.1111/j.1749-6632.2000.tb05604.x. [DOI] [PubMed] [Google Scholar]
- 135.Jouvet P, et al. Maple syrup urine disease metabolites induce apoptosis in neural cells without cytochrome c release or changes in mitochondrial membrane potential. Biochem Soc Trans. 1998;26:S341. doi: 10.1042/bst026s341. [DOI] [PubMed] [Google Scholar]
- 136.Kasinski A, Doering CB, Danner DJ. Leucine toxicity in a neuronal cell model with inhibited branched chain amino acid catabolism. Brain Res Mol Brain Res. 2004;122:180–187. doi: 10.1016/j.molbrainres.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 137.Lu G, et al. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest. 2009;119:1678–1687. doi: 10.1172/JCI38151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Oyarzabal A, et al. A novel regulatory defect in the branched-chain α-keto acid dehydrogenase complex due to a mutation in the PPM1K gene causes a mild variant phenotype of maple syrup urine disease. Hum Mutat. 2013;34:355–362. doi: 10.1002/humu.22242. [DOI] [PubMed] [Google Scholar]
- 139.Lu G, et al. Functional characterization of a mitochondrial Ser/Thr protein phosphatase in cell death regulation. Methods Enzymol. 2009;457:255–273. doi: 10.1016/S0076-6879(09)05014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lu G, et al. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Amaral AU, et al. α-ketoisocaproic acid and leucine provoke mitochondrial bioenergetic dysfunction in rat brain. Brain Res. 2010;1324:75–84. doi: 10.1016/j.brainres.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 142.Adams SH, et al. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid β-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J Nutr. 2009;139:1073–1081. doi: 10.3945/jn.108.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Adams SH. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv Nutr. 2011;2:445–456. doi: 10.3945/an.111.000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shah SH, et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia. 2012;55:321–330. doi: 10.1007/s00125-011-2356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wajner M, Goodman SI. Disruption of mitochondrial homeostasis in organic acidurias: insights from human and animal studies. J Bioenerg Biomembr. 2011;43:31–38. doi: 10.1007/s10863-011-9324-0. [DOI] [PubMed] [Google Scholar]
- 146.Klimcakova E, et al. Worsening of obesity and metabolic status yields similar molecular adaptations in human subcutaneous and visceral adipose tissue: decreased metabolism and increased immune response. J Clin Endocrinol Metab. 2011;96:E73–E82. doi: 10.1210/jc.2010-1575. [DOI] [PubMed] [Google Scholar]
- 147.Pietilainen KH, et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: pathways behind acquired obesity. PLoS Med. 2008;5:e51. doi: 10.1371/journal.pmed.0050051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Leskinen T, et al. Differences in muscle and adipose tissue gene expression and cardio-metabolic risk factors in the members of physical activity discordant twin pairs. PLoS ONE. 2010;5:e12609. doi: 10.1371/journal.pone.0012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lan H, et al. Gene expression profiles of nondiabetic and diabetic obese mice suggest a role of hepatic lipogenic capacity in diabetes susceptibility. Diabetes. 2003;52:688–700. doi: 10.2337/diabetes.52.3.688. [DOI] [PubMed] [Google Scholar]
- 150.Stancakova A, et al. Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes. 2012;61:1895–1902. doi: 10.2337/db11-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zimmerman HA, Olson KC, Chen G, Lynch CJ. Adipose transplant for inborn errors of branched chain amino acid metabolism in mice. Mol Genet Metab. 2013;109:345–353. doi: 10.1016/j.ymgme.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mazariegos GV, et al. Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative United Network for Organ Sharing experience. J Pediatr. 2012;160:116–121. doi: 10.1016/j.jpeds.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harris RA, Joshi M, Jeoung NH, Obayashi M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr. 2005;135:1527S–1530S. doi: 10.1093/jn/135.6.1527S. [DOI] [PubMed] [Google Scholar]
- 154.Lefort N, et al. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes. 2010;59:2444–2452. doi: 10.2337/db10-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mullen E, Ohlendieck K. Proteomic profiling of non-obese type 2 diabetic skeletal muscle. Int J Mol Med. 2010;25:445–458. doi: 10.3892/ijmm_00000364. [DOI] [PubMed] [Google Scholar]
- 156.Roe CR, et al. Isolated isobutyryl-CoA dehydrogenase deficiency: an unrecognized defect in human valine metabolism. Mol Genet Metab. 1998;65:264–271. doi: 10.1006/mgme.1998.2758. [DOI] [PubMed] [Google Scholar]
- 157.Shin AC, et al. Brain insulin lowers circulating BCAA levels by inducing hepatic branched-chain α keto-acid dehydrogenase. Cell Metab. doi: 10.1016/j.cmet.2014.09.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tschop MH, et al. A guide to analysis of mouse energy metabolism. Nat Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wolfe RR, Chinkes DL. Isotope Tracers in Metabolic Research. Wiley; 2005. pp. 299–323. [Google Scholar]
- 160.Allsop JR, Wolfe RR, Burke JF. Tracer priming the bicarbonate pool. J Appl Physiol. 1978;45:137–139. doi: 10.1152/jappl.1978.45.1.137. [DOI] [PubMed] [Google Scholar]
- 161.Doisaki M, et al. Regulation of hepatic branched-chain α-keto acid dehydrogenase kinase in a rat model for type 2 diabetes mellitus at different stages of the disease. Biochem Biophys Res Commun. 2010;393:303–307. doi: 10.1016/j.bbrc.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 162.Kuzuya T, et al. Regulation of branched-chain amino acid catabolism in rat models for spontaneous type 2 diabetes mellitus. Biochem Biophys Res Commun. 2008;373:94–98. doi: 10.1016/j.bbrc.2008.05.167. [DOI] [PubMed] [Google Scholar]
- 163.Kadota Y, Toyoda T, Kitaura Y, Adams SH, Shimomura Y. Regulation of hepatic branched-chain α-ketoacid dehydrogenase complex in rats fed a high-fat diet. Obes Res Clin Pract. 2013;7:e439–e444. doi: 10.1016/j.orcp.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Joshi M, Jeoung NH, Popov KM, Harris RA. Identification of a novel PP2C-type mitochondrial phosphatase. Biochem Biophys Res Commun. 2007;356:38–44. doi: 10.1016/j.bbrc.2007.02.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Taneera J, et al. A systems genetics approach identifies genes and pathways for type 2 diabetes in human islets. Cell Metab. 2012;16:122–134. doi: 10.1016/j.cmet.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 166.Kettunen J, et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet. 2012;44:269–276. doi: 10.1038/ng.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xu M, et al. Genetic determinant for amino acid metabolites and changes in body weight and insulin resistance in response to weight-loss diets: the preventing overweight using novel dietary strategies (POUNDS LOST) trial. Circulation. 2013;127:1283–1289. doi: 10.1161/CIRCULATIONAHA.112.000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cipolla-Neto J, Amaral FG, Afeche SC, Tan DX, Reiter RJ. Melatonin, energy metabolism, and obesity: a review. J Pineal Res. 2014;56:371–381. doi: 10.1111/jpi.12137. [DOI] [PubMed] [Google Scholar]
- 169.Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Curr Biol. 2013;23:372–381. doi: 10.1016/j.cub.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wu L, Reddy AB. Disrupting rhythms: diet-induced obesity impairs diurnal rhythms in metabolic tissues. Diabetes. 2013;62:1829–1830. doi: 10.2337/db13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Holt RI, Barnett AH, Bailey CJ. Bromocriptine: old drug, new formulation and new indication. Diabetes Obes Metab. 2010;12:1048–1057. doi: 10.1111/j.1463-1326.2010.01304.x. [DOI] [PubMed] [Google Scholar]
- 172.Cano P, et al. Effect of a high-fat diet on 24-hour pattern of circulating adipocytokines in rats. Obesity (Silver Spring) 2009;17:1866–1871. doi: 10.1038/oby.2009.200. [DOI] [PubMed] [Google Scholar]
- 173.Takashima M, et al. Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes. 2010;59:1608–1615. doi: 10.2337/db09-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Elbein SC, et al. Global gene expression profiles of subcutaneous adipose and muscle from glucose-tolerant, insulin-sensitive, and insulin-resistant individuals matched for BMI. Diabetes. 2011;60:1019–1029. doi: 10.2337/db10-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Corkey BE, Martin-Requero A, Walajtys-Rode E, Williams RJ, Williamson JR. Regulation of the branched chain α-ketoacid pathway in liver. J Biol Chem. 1982;257:9668–9676. [PubMed] [Google Scholar]
- 176.Hu H, Jaskiewicz JA, Harris RA. Ethanol and oleate inhibition of α-ketoisovalerate and 3-hydroxyisobutyrate metabolism by isolated hepatocytes. Arch Biochem Biophys. 1992;299:57–62. doi: 10.1016/0003-9861(92)90243-p. [DOI] [PubMed] [Google Scholar]
- 177.Koves TR, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 178.Frohnert BI, Bernlohr DA. Protein carbonylation, mitochondrial dysfunction, and insulin resistance. Adv Nutr. 2013;4:157–163. doi: 10.3945/an.112.003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Frohnert BI, et al. Increased adipose protein carbonylation in human obesity. Obesity (Silver Spring) 2011;19:1735–1741. doi: 10.1038/oby.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Long EK, Olson DM, Bernlohr DA. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radic Biol Med. 2013;63:390–398. doi: 10.1016/j.freeradbiomed.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ruskovska T, Bernlohr DA. Oxidative stress and protein carbonylation in adipose tissue—implications for insulin resistance and diabetes mellitus. J Proteomics. 2013;92:323–334. doi: 10.1016/j.jprot.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mamer OA, Reimer ML. On the mechanisms of the formation of L-alloisoleucine and the 2-hydroxy-3-methylvaleric acid stereoisomers from L-isoleucine in maple syrup urine disease patients and in normal humans. J Biol Chem. 1992;267:22141–22147. [PubMed] [Google Scholar]
- 183.Zhang B, Zhao Y, Harris RA, Crabb DW. Molecular defects in the E1 α subunit of the branched-chain α-ketoacid dehydrogenase complex that cause maple syrup urine disease. Mol Biol Med. 1991;8:39–47. [PubMed] [Google Scholar]
- 184.Strauss KA, et al. Maple syrup urine disease. GeneReviews. [online], http://www.ncbi.nlm.nih.gov/books/NBK1319/(1993)
- 185.Strauss KA, et al. Classical maple syrup urine disease and brain development: principles of management and formula design. Mol Genet Metab. 2010;99:333–345. doi: 10.1016/j.ymgme.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Homanics GE, Skvorak K, Ferguson C, Watkins S, Paul HS. Production and characterization of murine models of classic and intermediate maple syrup urine disease. BMC Med Genet. 2006;7:33. doi: 10.1186/1471-2350-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zinnanti WJ, et al. Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain. 2009;132:903–918. doi: 10.1093/brain/awp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bridi R, et al. Induction of oxidative stress in rat brain by the metabolites accumulating in maple syrup urine disease. Int J Dev Neurosci. 2003;21:327–332. doi: 10.1016/s0736-5748(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 189.Kobayashi R, et al. Clofibric acid stimulates branched-chain amino acid catabolism by three mechanisms. Arch Biochem Biophys. 2002;407:231–240. doi: 10.1016/s0003-9861(02)00472-1. [DOI] [PubMed] [Google Scholar]
- 190.Shimomura Y, et al. Branched-chain 2-oxo acid dehydrogenase complex activation by tetanic contractions in rat skeletal muscle. Biochim Biophys Acta. 1993;1157:290–296. doi: 10.1016/0304-4165(93)90112-l. [DOI] [PubMed] [Google Scholar]
- 191.Shimomura Y, et al. Branched-chain amino acid catabolism in exercise and liver disease. J Nutr. 2006;136:250S–253S. doi: 10.1093/jn/136.1.250S. [DOI] [PubMed] [Google Scholar]
- 192.Wang X, Price SR. Differential regulation of branched-chain α-ketoacid dehydrogenase kinase expression by glucocorticoids and acidification in LLC-PK1-GR101 cells. Am J Physiol Renal Physiol. 2004;286:F504–F508. doi: 10.1152/ajprenal.00296.2003. [DOI] [PubMed] [Google Scholar]
- 193.Block KP, Richmond WB, Mehard WB, Buse MG. Glucocorticoid-mediated activation of muscle branched-chain α-keto acid dehydrogenase in vivo. Am J Physiol. 1987;252:E396–E407. doi: 10.1152/ajpendo.1987.252.3.E396. [DOI] [PubMed] [Google Scholar]
- 194.Popov KM, et al. Dietary control and tissue specific expression of branched-chain α-ketoacid dehydrogenase kinase. Arch Biochem Biophys. 1995;316:148–154. doi: 10.1006/abbi.1995.1022. [DOI] [PubMed] [Google Scholar]
- 195.Kobayashi R, et al. Hepatic branched-chain α-keto acid dehydrogenase complex in female rats: activation by exercise and starvation. J Nutr Sci Vitaminol (Tokyo) 1999;45:303–309. doi: 10.3177/jnsv.45.303. [DOI] [PubMed] [Google Scholar]
- 196.Zhao Y, et al. Effect of dietary protein on the liver content and subunit composition of the branched-chain α-ketoacid dehydrogenase complex. Arch Biochem Biophys. 1994;308:446–453. doi: 10.1006/abbi.1994.1063. [DOI] [PubMed] [Google Scholar]
- 197.Nellis MM, Doering CB, Kasinski A, Danner DJ. Insulin increases branched-chain α-ketoacid dehydrogenase kinase expression in Clone 9 rat cells. Am J Physiol Endocrinol Metab. 2002;283:E853–E860. doi: 10.1152/ajpendo.00133.2002. [DOI] [PubMed] [Google Scholar]
- 198.Harris RA, et al. Regulation of the branched-chain α-ketoacid dehydrogenase and elucidation of a molecular basis for maple syrup urine disease. Adv Enzyme Regul. 1990;30:245–263. doi: 10.1016/0065-2571(90)90021-s. [DOI] [PubMed] [Google Scholar]
- 199.Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4:409–454. doi: 10.1146/annurev.nu.04.070184.002205. [DOI] [PubMed] [Google Scholar]
- 200.Paxton R, Harris RA. Regulation of branched-chain α-ketoacid dehydrogenase kinase. Arch Biochem Biophys. 1984;231:48–57. doi: 10.1016/0003-9861(84)90361-8. [DOI] [PubMed] [Google Scholar]
- 201.Nawabi MD, Block KP, Chakrabarti MC, Buse MG. Administration of endotoxin, tumor necrosis factor, or interleukin 1 to rats activates skeletal muscle branched-chain α-keto acid dehydrogenase. J Clin Invest. 1990;85:256–263. doi: 10.1172/JCI114421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Harris RA, Kobayashi R, Murakami T, Shimomura Y. Regulation of branched-chain α-keto acid dehydrogenase kinase expression in rat liver. J Nutr. 2001;131:841S–845S. doi: 10.1093/jn/131.3.841S. [DOI] [PubMed] [Google Scholar]
- 203.Shimomura Y, Obayashi M, Murakami T, Harris RA. Regulation of branched-chain amino acid catabolism: nutritional and hormonal regulation of activity and expression of the branched-chain α-keto acid dehydrogenase kinase. Curr Opin Clin Nutr Metab Care. 2001;4:419–423. doi: 10.1097/00075197-200109000-00013. [DOI] [PubMed] [Google Scholar]
- 204.Harris RA, et al. In: Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. Patel MS, Packer L, editors. CRC Press; 2008. pp. 101–148. [Google Scholar]
- 205.Gillim SE, Paxton R, Cook GA, Harris RA. Activity state of the branched chain α-ketoacid dehydrogenase complex in heart, liver, and kidney of normal, fasted, diabetic, and protein-starved rats. Biochem Biophys Res Commun. 1983;111:74–81. doi: 10.1016/s0006-291x(83)80119-3. [DOI] [PubMed] [Google Scholar]
- 206.Lombardo YB, Serdikoff C, Thamotharan M, Paul HS, Adibi SA. Inverse alterations of BCKA dehydrogenase activity in cardiac and skeletal muscles of diabetic rats. Am J Physiol. 1999;277:E685–E692. doi: 10.1152/ajpendo.1999.277.4.E685. [DOI] [PubMed] [Google Scholar]
- 207.Lombardo YB, Thamotharan M, Bawani SZ, Paul HS, Adibi SA. Posttranscriptional alterations in protein masses of hepatic branched-chain keto acid dehydrogenase and its associated kinase in diabetes. Proc Assoc Am Physicians. 1998;110:40–49. [PubMed] [Google Scholar]
- 208.Kobayashi R, Shimomura Y, Otsuka M, Popov KM, Harris RA. Experimental hyperthyroidism causes inactivation of the branched-chain α-ketoacid dehydrogenase complex in rat liver. Arch Biochem Biophys. 2000;375:55–61. doi: 10.1006/abbi.1999.1635. [DOI] [PubMed] [Google Scholar]
- 209.Harris RA, Powell SM, Paxton R, Gillim SE, Nagae H. Physiological covalent regulation of rat liver branched-chain α-ketoacid dehydrogenase. Arch Biochem Biophys. 1985;243:542–555. doi: 10.1016/0003-9861(85)90531-4. [DOI] [PubMed] [Google Scholar]
- 210.Hsiao G, et al. Multi-tissue, selective PPARγ modulation of insulin sensitivity and metabolic pathways in obese rats. Am J Physiol Endocrinol Metab. 2011;300:E164–E174. doi: 10.1152/ajpendo.00219.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]