

Abstract
To analyze effects of cellular interaction between human mesenchymal stroma/stem cells (MSC) and different cancer cells, direct co-cultures were performed and revealed significant growth stimulation of the tumor populations and a variety of protein exchanges. More than 90% of MCF-7 and primary human HBCEC699 breast cancer cells as well as NIH:OVCAR-3 ovarian adenocarcinoma cells acquired CD90 proteins during MSC co-culture, respectively. Furthermore, SK-OV-3 ovarian cancer cells progressively elevated CD105 and CD90 proteins in co-culture with MSC. Primary small cell hypercalcemic ovarian carcinoma cells (SCCOHT-1) demonstrated undetectable levels of CD73 and CD105; however, both proteins were significantly increased in the presence of MSC. This co-culture-mediated protein induction was also observed at transcriptional levels and changed functionality of SCCOHT-1 cells by an acquired capability to metabolize 5′cAMP. Moreover, exchange between tumor cells and MSC worked bidirectional, as undetectable expression of epithelial cell adhesion molecule (EpCAM) in MSC significantly increased after co-culture with SK-OV-3 or NIH:OVCAR-3 cells. In addition, a small population of chimeric/hybrid cells appeared in each MSC/tumor cell co-culture by spontaneous cell fusion. Immune fluorescence demonstrated nanotube structures and exosomes between MSC and tumor cells, whereas cytochalasin-D partially abolished the intercellular protein transfer. More detailed functional analysis of FACS-separated MSC and NIH:OVCAR-3 cells after co-culture revealed the acquisition of epithelial cell-specific properties by MSC, including increased gene expression for cytokeratins and epithelial-like differentiation factors. Vice versa, a variety of transcriptional regulatory genes were down-modulated in NIH:OVCAR-3 cells after co-culture with MSC. Together, these mutual cellular interactions contributed to functional alterations in MSC and tumor cells.
Introduction
Human mesenchymal stroma/stem cells (MSC) can be derived as a multipotent stromal population from a large variety of different sources. MSC represent a heterogeneous cell population due to their diverse origin from nearly all vascularized organs and tissues and exhibit migratory capability and regenerative potential [1]. According to their heterogeneity, no specific marker but a broad range of properties are characterized for these stem cells, including the capacity for plastic adherence, simultaneous expression of the CD73, CD90, and CD105 surface molecules with concomitant absence of other cell type-specific markers, including CD14, CD31, CD34 CD45, and HLA-DR, and at least a tri-lineage differentiation potential along the osteogenic, chondrogenic, and adipogenic phenotype [2,3]. Some additional surface markers can be detected in certain subpopulations such as Stro-1 [4], or the chemokine receptors VCAM-1 (CD106) and ICAM-1 (CD54) [5] predominantly found in bone marrow-derived MSC, or the more embryonic-like stem cell markers Oct-4 and Sox2 [6], all of which depend on the local microenvironment and contribute to the multi-facetted functionalities as a part of the heterogeneous MSC population.
MSC can be attracted by inflammatory cytokines/chemokines to migrate toward local tissue injuries in support of tissue regeneration and repair. During this process, MSC get into contact with a variety of different cell types and display mutual cellular interactions, including the release of bioactive molecules [7] and exosomes [8] as well as direct cell-to-cell interactions via integrins and gap junctional intercellular communication (GJIC). At the sites of tissue damage, MSC exhibit immune-modulatory functions predominantly for T cells, NK cells, and macrophages to facilitate repair [9–11]. Moreover, MSC are involved in endothelial cell interactions for the promotion of angiogenesis and neo-vascularization in the damaged area [12,13].
Invasive tumor growth such as breast or ovarian cancer also causes local tissue damage and inflammation and, consequently, attracts immune cells and MSC to contribute to the required repair machinery. Thus, MSC can be detected within the adipose breast tissue and the fibroglandular tissue of the breast, thereby forming close vicinity to normal human mammary epithelial cells (HMEC) and to breast cancer cells within the tumor microenvironment [14–16]. Likewise, MSC are also present in tissues of the ovary and their tumorigenic counterparts. Ovarian cancer similar to breast cancer represents one of the most lethal gynecologic malignancies and can be categorized into different low-grade serous type I tumors in contrast to high-grade type II tumors with aggressive cancer cells predominantly observed in advanced tumor stages [17–19]. Moreover, the small cell carcinoma of the ovary hypercalcemic type (SCCOHT) represents a rare form of an aggressive tumor, which often affects young women during reproductive age. SCCOHT characterizes a separate tumor entity apart from ovarian cancer [20]. However, it remains unclear how MSC interact with these different kinds of breast, ovarian, or other cancer types.
In this study, we established several co-culture models for a variety of MSC populations together with different kinds of tumor cells, including tumor cell lines and primary cells from tumor biopsies of breast and ovarian cancer patients. It was the aim of this study to address potential cell biological effects during direct interaction between the stroma/stem cells and the various tumor cell types. Our co-culture experiments demonstrated elevated growth of the tumor cells in the presence of MSC and mutual exchange of cellular material between MSC and the different tumor cell types.
Materials and Methods
Cell culture
The use of primary cells from human tumor biopsies and the use of primary human mesenchymal stem cells after explant culture have been approved by the Ethics Committee of Hannover Medical School, Project #3916 on June 15th, 2005, and Project #443 on February 26th, 2009, respectively, and informed written consent was obtained from all patients.
Breast cancer cells
Human MCF-7 breast carcinoma cell line was obtained from the American Type Culture Collection and grown in Dulbecco's modified Eagle's medium with medium supplements [10% (v/v) fetal calf serum (FCS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin; all from Sigma Chemie GmbH]. Cultures were maintained at 37°C in a humidified atmosphere with 5% CO2.
Primary human breast cancer-derived epithelial cells (HBCEC) were obtained from explant cultures of human breast cancer biopsies after negative testing for HIV-1, hepatitis B & C, bacteria, yeast, and fungi, respectively, as described [21]. The primary HBCEC 699 were cultured further in serum-free and phenol red-free mammary epithelial cell growth medium (MEGM; Lonza Ltd.).
Ovarian cancer cells
The human NIH:OVCAR-3 ovarian adenocarcinoma cell line (ATCC® #HTB-161™) was commercially obtained in passage 76 (P76) from the Institute for Applied Cell Culture (IAZ). The SK-OV-3 epithelial-like ovarian cancer cells (ATCC #HTB-77™) were commercially obtained in P25 from the ATCC. SCCOHT-1 represent a spontaneously proliferating population derived from a patient with recurrent SCCOHT [22]. These three different cancer cell types were cultivated at about 1,750 cells/cm2 in RPMI 1640 with medium supplements, respectively. NIH:OVCAR-3 and SK-OV-3 cells were subcultured by trypsin/EDTA (Biochrom GmbH) treatment for 5 min at 37°C.
Mesenchymal stroma/stem cells
MSC-like cells were isolated from human umbilical cords as previously reported [23,24]. The cells were obtained from six different patients after delivery of full-term (38–40 weeks) infants either spontaneously or by Cesarean section. MSC were cultured in αMEM supplemented with 10% of allogeneic human AB-serum (HS, commercially obtained from blood bank, University Campus Lübeck, Germany), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine (Sigma) at 37°C in a humidified atmosphere with 5% CO2. For the experiments, MSC primary cultures from the six different donors in different passages (P2 to P6) were used (MSC240113 in P2; MSC280313 in P3, P4 and P5; MSC131113 in P3 and P4; MSC101213 in P5; MSC100314 in P3; and MSC180314 in P3 and P6), respectively.
Human mammary epithelial cells
Primary cultures of normal human mammary epithelial cells (HMEC) were commercially provided by BioWhittaker, Inc. (Lot #1F1012). Juvenile and proliferating HMEC in P13 were cultured at 2,500 cells/cm2 in mammary epithelial cell growth medium (PromoCell) as previously described [25].
Cell line authentication
Cells were tested for mycoplasma by the luminometric MycoAlert Plus mycoplasma detection kit (Lonza, Inc.) according to the manufacturer's recommendations. Moreover, authentication of the cell lines was performed by short tandem repeat (STR) fragment analysis using the GenomeLab human STR primer set (Beckman Coulter, Inc.) demonstrating a similar STR pattern according to the STR database provided by the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ).
Co-culture and proliferation measurement of tumor cell lines and primary cultures with MSC after lentiviral transduction
For discrimination of the different tumor cells in co-culture with MSC and for proliferation measurements, all tumor cell populations were transduced with a third generation lentiviral SIN vector containing the mcherry gene. Likewise, the different MSC populations were similarly transduced with an eGFP gene-containing vector as previously described [26].
A co-culture of 60% GFP-labeled human mesenchymal stem cells (MSCGFP) and 40% mcherry-labeled tumor cells (for MCF-7cherry and MSCGFP the initial ratio was 20% to 80%) were incubated in MSC culture medium till 9 days in cell culture plates (diameter 10 cm; Greiner BioOne GmbH) at an initial density between 500 and 2,000 cells/cm2 as indicated in the experiments.
For proliferation measurement of the co-culture at different time points, the medium was removed and the cells were lysed with 10% SDS. The fluorescence intensity of mcherry (excitation 584 nm/emission 612 nm) and GFP (excitation 485 nm/emission 520 nm) that corresponded to the appropriate cell number of tumor cells and MSC, respectively, was measured in aliquots of the lysate using the Fluoroscan Ascent Fl (Thermo Fisher Scientific). Appropriate mono-cultures of tumorcherry cells and MSCGFP demonstrated no artificial cross-fluorescence.
In an additional independent evaluation of the cell numbers, the different cell cultures were trypsinized at the appropriate time points and the cells were counted after trypan blue staining in a fluorescence microscope (Olympus IX50) using the green and red fluorescence filters, respectively, as well as an FITC/TRIC fluorescence dual band filter.
Analysis of surface markers and cell cycle by flow cytometry
Continuously proliferating mono- and co-culture cells were harvested and analyzed for cell surface marker expression by flow cytometry. After blocking nonspecific binding to Fc-receptors by incubation of 106 cells with 2% bovine serum albumin in phosphate-buffered saline (PBS-BSA) for 30 min at 4°C and washing with PBS-BSA, the cells were incubated with the following appropriately labeled monoclonal anti-human antibodies, respectively: CD73-PE (clone AD2; BD Bioscience); CD90-PE (clone 5E10, IgG1; BioLegend, Inc.); CD105-PE (clone 43A3, IgG1; BioLegend, Inc.); and CD326-PE (=EpCAM-PE, clone G9C4, IgG2b; BioLegend, Inc.). After antibody staining, all samples were washed twice with PBS-BSA and measured by flow cytometry. Appropriately labeled antibodies of the corresponding Ig subclass were used as a control.
For cell cycle analysis, 5×105 cells were fixed in 70% (v/v) ice-cold ethanol at 4°C for 24 h. Thereafter, the fixed cells were stained with CyStain DNA 2 step kit (Partec GmbH) and filtered through a 50 μm filter. Flow cytometry analysis was performed in a Galaxy FACSan (Partec) using FloMax analysis software (Partec).
Analysis of 5′-AMP and adenosine
Steady-state SCCOHT-1 cells and SCCOHT-1 after FACS separation from a 7 days co-culture with MSC were cultivated in PBS with 20 μM 5′-AMP (Sigma) as a substrate for 30 min at 37°C. MSC mono-culture and MSC after FACS separation from a 7 day co-culture with SCCOHT-1 cells were used as a control after incubation with 20 μM 5′-AMP. Supernatants were collected and centrifuged (500 g/5 min) to remove additional cells and debris and cell-free supernatants were analyzed by HPLC-MS/MS using a Shimadzu HPLC-system (Shimadzu) coupled with a QTRAP5500TM triple quadrupole mass spectrometer (ABSCIEX) operating in positive ionization mode to quantify the amount of 5′AMP as the substrate and the level of adenosine as the product.
Immunoblot analysis
Conditioned media (7 days) from mono-cultured MCF-7cherry, SK-OV-3cherry, SCCOHT-1cherry, and NIH:OVCAR-3cherry (initially seeded at 5×104 cells/mL) or MSCGFP (initially seeded at 7.5×104 cells/mL) in comparison to co-cultured cells from MCF-7cherry, SK-OV-3cherry, SCCOHT-1cherry and NIH:OVCAR-3cherry with MSCGFP (cell ratio 40:60; initially seeded at 1.25×105 cells/mL) was used, respectively. In addition, cell lysates of MSCGFP were prepared in reswelling buffer containing 8 M urea (Carl Roth GmbH Co KG), 1% CHAPS (3-[(3-Cholamidopropyl)-dimethylammonio]-1-propanesulfonate; Carl Roth GmbH Co KG), 0.5% (v/v) Pharmalyte 3–10 (GE Healthcare Europe GmbH), 0.002% (w/v) bromophenol blue (SERVA Electrophoresis GmbH) and freshly prepared 0.4% (w/v) DTT (Dithiothreitol; Carl Roth GmbH Co KG). Protein concentration of the MSC lysate was adjusted using the colorimetric BCA-assay (Thermo Scientific). The protein samples (50 μg MSC cell lysate as control) and 40 μL aliquots of all appropriate conditioned media from mono- and co-cultures were subjected to SDS-polyacrylamide gel electrophoresis. In parallel, all conditioned media were concentrated 100-fold by applying Amicon Ultra-4 centrifugal filters (Merck Millipore Ltd.) with a molecular weight cut-off of ∼10 kDa according to the manufacturer's instructions. Gels were transferred to a Amersham™ Protran™ Supported 0.45 μm nitrocellulose membrane (GE Healthcare). The membranes were blocked with PBS containing 5% FCS and 0.05% Tween-20 (PBS/Tween). After washing four times with PBS/Tween, the membranes were incubated with the primary antibodies [monoclonal anti-CD90 (EPR3132, rabbit, dilution 1:250, ab92574; Abcam plc); polyclonal anti-CD105 (N3C3, rabbit, dilution 1:500; Gene Tex, Inc.), and monoclonal anti-β-actin (mouse, dilution 1:1,000, clone AC-15; Sigma-Aldrich)] overnight at 4°C. Thereafter, the membranes were washed four times with PBS/Tween and incubated with the appropriate horseradish peroxidase-conjugated anti-mouse IgG (dilution 1:5,000) or anti-rabbit IgG (dilution 1:5,000) secondary antibody, respectively, (all from GE Healthcare) for 1 h/room temperature. The membranes were washed with PBS/Tween and visualized by autoradiography using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Transcript analysis by reverse transcription PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. One microgram RNA was reverse transcribed into cDNA using 500 μM of dNTP (R0193), 5 μM Oligo(dT)18 primer (S0132), 5 μM Random Hexan primer (S0142), 1 U RiboLock™ RNase Inhibitor (E00381), and 5 U RevertAid™ M-MuLV Reverse Transcriptase (EP0441) in the supplied reaction buffer (all reagents from Thermo Scientific). The cDNA reactions were performed for 10 min/25°C, 1 h/37°C and stopped at 72°C for 10 min. As a template, 2.5 μL of cDNA was used with primers specific for CD73 (sense: 5′-CGC AAC AAT GGC ACA ATT AC-3′; antisense: 5′-CTC GAC ACT TGG TGC AAA GA-3′; amplification product 241 bp [27]), CD90 (sense: 5′-GGA CTG AGA TCC CAG AAC CA-3′; antisense: 5′-ACG AAG GCT CTG GTC CAC TA-3′; amplification product 124 bp [28]), and CD105 (sense: 5′-TGT CTC ACT TCA TGC CTC CAG CT-3′; antisense: 5′-AGG CTG TCC ATG TTG AGG CAG T-3′; amplification product 378 bp [29]). As a control, β-actin polymerase chain reaction (PCR) (sense: 5′-CGG ATG TCC ACG TCA CAC T-3′; antisense: 5′-CCA CTG GCA TCG TGA TGG A-3′; amplification product 427 bp [30]) was performed (all primers customized by Eurofins, MWG GmbH). PCR reactions included 0.2 μM of each primer, 200 μM of dNTP (R0193; Thermo Scientific), and 0.03 U One Taq Hot Start DNA polymerase (New England Biolabs GmbH) in the supplied reaction buffer. PCR cycling conditions were performed for 30 s at 94°C, 1 min at 60°C, and 68°C for 1 min, respectively, including an initial 30 s denaturation step at 94°C and a final 5 min extension step at 68°C (35 cycles). Aliquots of 25 μL of each reverse transcription-PCR product were separated on a 2% agarose gel, including the standard GeneRuler 100 bp DNA Ladder (Thermo Scientific), and visualized by GelRed™ (Biotium, Inc.) staining.
FACS separation, RNA isolation, and microarray analysis of co-cultured MSC and NIH:OVCAR-3 cells
After RNA isolation using the RNeasy mini kit (Qiagen GmbH), 100 ng of total RNA from either steady-state control MSCGFP, control NIH:OVCAR-3cherry, and 7 days co-cultured MSCGFP, or NIH:OVCAR-3cherry cells after FACS separation were used to prepare aminoallyl-UTP-modified (aaUTP) cRNA (Amino Allyl MessageAmp™ II Kit, #AM1753; Life Technologies) as directed by the manufacturer. Labeling of aaUTP-cRNA was performed by CY3 POST-Labelling Reactive Dyes (25-8010-79; GE Healthcare Biosciences). Before the reverse transcription reaction, 1 μL of a 1:10,000 dilution of Agilent's “One-Color spike-in kit stock solution” (#5188-5282; Agilent Technologies) were added to each total RNA sample. cRNA fragmentation, hybridization, and washing steps were carried out as recommended and microarray analysis was performed by use of a refined version of the Whole Human Genome Oligo Microarray 4x44K v2 (AMADID 026652; Agilent Technologies), termed “026652AsQuintuplicatesOn180k” (AMADID 054261) and developed in the Research Core Unit Transcriptomics of Hannover Medical School. Microarray design was defined at Agilent's eArray portal using a 4x180k design format for mRNA expression as a template. All noncontrol probes of AMADID 026652 were printed five times onto one 180k Microarray (on-chip quintuplicates). Control probes required for proper Feature Extraction software algorithms were determined and placed automatically by eArray using recommended default settings. Slides were scanned on the Agilent Microarray Scanner G2565CA (pixel resolution 3 μm, bit depth 20). Data extraction was performed with the “Feature Extraction Software V10.7.3.1” using the extraction protocol file “GE1_107_Sep09.xml.” Processed intensity values of the green channel, “gProcessedSignal” (gPS) were normalized by global linear scaling: All gPS values of one sample were multiplied by an array-specific scaling factor. This factor was calculated by dividing a “reference 75th Percentile value” (set as 1,500 for the whole series) by the 75th Percentile value of the particular microarray (“Array I” in the formula shown next). Accordingly, normalized gPS values for all samples (microarray data sets) were calculated by the following formula:
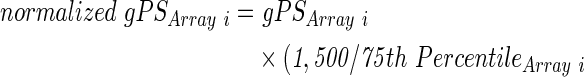 |
Measurements of on-chip replicates (quintuplicates) were averaged using the geometric mean of normalized gPS values. Measurements outside the interval of “1.42× interquartile range” regarding the normalized gPS distribution of the respective on-chip replicate population were excluded from averaging. A lower intensity threshold (surrogate value) was defined as 1% of the reference 75th Percentile value (= 15). All normalized gPS values below this intensity border were substituted by the respective surrogate value of 15. Gene expression levels of more than two-fold difference were compared between control MSC and co-cultured MSC as well as between control NIH:OVCAR-3 and co-cultured NIH:OVCAR-3 cells and stored at the NCBI-GEO database with the accession GSE60035 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60035).
Results
Enhanced cell growth and membrane protein acquisition by cancer cells in the presence of MSC
Co-culture of human MCF-7cherry breast cancer cells with three different individual MSCGFP populations in separate assays was associated with reproducible data demonstrating continuous growth stimulation by MSCGFP within 9 days (Fig. 1A). These growth-stimulatory effects by MSC predominantly required direct intercellular communication, as co-culture experiments performed in transwells carrying a sterile track-etched membrane with 0.4 mm sized-pores revealed no detectable effects in proliferative capacity similar to previous studies (data not shown) [25]. Direct cell counting of MCF-7cherry cells in co-culture with MSCGFP was also performed and compared with an appropriate amount of mono-cultured cells and confirmed a growth induction of the breast cancer cell line in contrast to a reduced MSCGFP cell number during co-culture (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd).
FIG. 1.

MCF-7cherry cells were incubated with three different human primary MSC populations (MSC240113GFP P2, MSC280313GFP P3, and MSC131113GFP P3) in three separate co-cultures at a ratio of 20% MCF-7/80% MSC with 500 cells/cm2 till 9 days. (A) Cell cycle analysis was performed in the co-cultures, and the ratio of the two populations (MCF-7cherry and MSCGFP) was quantified with the corresponding G1 peaks. Data represent the mean±SD from the three separate co-cultures. (B) The percentage of CD90 expression in each population of the co-culture was quantified by flow cytometry. Data represent the mean±SD from the three separate co-cultures. (C) Cell cycle analysis by flow cytometry and CD90 expression demonstrated little, if any, detection in MCF-7cherry and 97.4% in MSCGFP mono-cultures (upper panels), which were compared with an 8 day co-culture (middle panel). After separation of the co-cultured cells into cherry (red fluorescence) and GFP (green fluorescence) populations by sorting via FACS, subsequent flow cytometric analysis for CD90 and cell cycle revealed 91.3% CD90-positive MCF-7 cells. MSC, mesenchymal stroma/stem cells.
Moreover, the MCF-7cherry cells revealed an acquisition of CD90 protein from about 1.4%±0.3% in mono-culture to 92.5%±2.0% (n=3) in MCF-7cherry co-cultured with MSC as evaluated in correspondence to the appropriate G1 cell cycle peaks of MCF-7cherry and MSCGFP (Fig. 1B). The co-cultures were prepared at an initial population ratio of 20% MCF-7cherry and 80% MSCGFP due to the high proliferation rate of the breast cancer cells. Similar data were obtained at an initial co-culture ratio of 40% MCF-7cherry and 60% MSCGFP; however, here MCF-7cherry more rapidly overgrew the MSCGFP representing already 95.7% of the population in co-culture with 57.6% of CD90-positive breast cancer cells after 7 days. These levels increased to 97.3% of MCF-7cherry with 72.6% of CD90-carrying breast cancer cells after 8 days of co-culture (data not shown). The acquisition of CD90 by the breast cancer cells in co-culture was also dependent on the cell density. While MCF-7cherry and MSCGFP initially seeded at 500 cells/cm2 demonstrated more than 90% of CD90 acquisition by the breast cancer cells after 9 days of co-culture (Fig. 1B), a four-fold increased density of 2,000 cells/cm2 was accompanied by 92% of CD90-positive MCF-7 cells already after 3 days of co-culture (data not shown). A direct proof of CD90-carrying breast cancer cells was performed after cell sorting of MCF-7cherry cells and MSCGFP after 8 days of co-culture into cherry- and GFP-positive cells, which revealed more than 90% of CD90 expression in the cherry-labeled MCF-7 cells in contrast to about 1.9% of CD90 in MCF-7 mono-cultured cells (Fig. 1C).
Similar to the MCF-7 breast cancer cell line, CD90 expression was also acquired by primary human breast cancer epithelial cells (HBCEC). HBCEC 699 corresponded to a benign phyllodes breast tumor as previously characterized [31,32] and exhibited little, if any, detectable CD90 expression (Fig. 2A). After mcherry transduction, the resulting HBCEC 699cherry were seeded together with MSCGFP at a ratio of 40% to 60%, respectively. After 10 days of co-culture, a ratio of 39.5% HBCEC 699cherry and 58.7% MSCGFP together with 1.8% of spontaneously fused yellow chimeric/hybrid cells was measured (Fig. 2B), whereby 91.6% of the whole co-culture population expressed CD90 (Fig. 2C), suggesting that the majority of HBCEC 699cherry also acquired this antigen. Cell counting was performed with primary HBCEC 699GFP and MSCcherry and revealed a growth stimulation of the breast cancer cells in co-culture with MSC, whereas the cell number of MSC was decreased in co-culture compared with the appropriate amount of mono-cultured cells (Supplementary Fig. S2).
FIG. 2.

Co-culture of primary MSC with human primary breast cancer epithelial cells at a ratio of 40% HBCEC 699/60% MSC with 500 cells/cm2 till 10 days. (A) Detection and analysis of CD90 expression in mono-cultures of MSC280313 P5 and HBCEC 699. (B) Quantification of the percentage of MSC280313GFP P5 and HBCEC 699cherry and the formation of yellow chimeric/hybrid cells by flow cytometric analysis during a 10 day co-culture. (C) Quantification of the percentage of CD90-positive HBCEC 699 acquired during a 10 day co-culture with MSC. HBCEC, human breast cancer-derived epithelial cells.
Co-culture of MSC with different ovarian cancer cell lines was also associated with growth stimulation of the tumor cells, alterations in the membrane protein composition, and the formation of a small population of spontaneously fused chimeric/hybrid cells. Thus, NIH:OVCAR-3cherry cells increased from an initial population of about 40% to 73.3% in a co-culture with MSCGFP after 7 days and conversely, the initially 60% MSCGFP dropped to 26.7% together with transiently detectable chimeric/hybrid cells as evaluated by fluorescence cell counting using a hemocytometer (Fig. 3A). Similar results were obtained by flow cytometry with 67.0% NIH:OVCAR-3cherry cells, 32.4% MSCGFP, and 0.6% chimeric/hybrid cells after 7 days of co culture (Fig. 3B). CD90 expression was always detectable in about 99% of MSCGFP during co-culture similar to the corresponding MSC mono-cultures (Fig. 3C, D). Conversely, CD90 protein in NIH:OVCAR-3cherry mono-culture was barely detectable (Fig. 3D); however, co-culture with MSCGFP till 7 days continuously increased these levels from about 1.9% to 91.9% (Fig. 3C).
FIG. 3.


Co-culture of primary MSC with human ovarian cancer cells at an initial ratio of 40% NIH:OVCAR-3/60% MSC with 2,000 cells/cm2 till 7 days. (A) Cell counting of MSC131113GFP P4 and NIH:OVCAR-3cherry and yellow chimeric/hybrid cells using a fluorescence microscope (Olympus IX50) with a FITC/TRIC fluorescence dual band filter and calculation of the population percentage during a 7 day co-culture. (B) Quantification of the percentage of MSC131113GFP P4 and NIH:OVCAR-3cherry and the formation of yellow chimeric/hybrid cells by flow cytometric analysis during a 7 day co-culture. (C) Quantification of the percentage of CD90-positive NIH:OVCAR-3 cells acquired during a 7 day co-culture with MSC. (D) Detection and analysis of CD90 expression in mono-cultures of MSC and NIH:OVCAR-3 cells.
Steady-state human SK-OV-3 ovarian adenocarcinoma cells displayed little, if any, CD90 and CD105 expression in contrast to a pronounced presence of more than 95% of these markers in MSC (Fig. 4A). Co-culture of SK-OV-3cherry with MSCGFP till 7 days was associated with an elevated CD105 expression by about 12% (Fig. 4B), and CD90 expression constantly increased over time to about 69.3% in SK-OV-3cherry cells (Fig. 4C). Simultaneously, the ovarian cancer cell population increased from about 40% to 58.7% by cell counting (Fig. 4D) or to 57.0% by flow cytometry analysis (Fig. 4E), whereas MSC declined from initially 60% to 41.3% by cell counting (Fig. 4D) or to 42.7% by flow cytometry analysis (Fig. 4E) within 7 days of co-culture.
FIG. 4.


Co-culture of primary MSC (MSC101213 P5 and MSC 131113 P4) with human ovarian adenocarcinoma cells at an initial ratio of 40% SK-OV-3/60% MSC with 2,000 cells/cm2 till 7 days. (A) Detection and analysis of CD90 and CD105 expression in mono-cultures of SK-OV-3 cells and MSC. (B) Quantification of the percentage of CD105-positive SK-OV-3 cells acquired during a 7 day co-culture with MSC101213. (C) Quantification of the percentage of CD90-positive SK-OV-3 cells acquired during a 7 day co-culture with MSC101213. (D) Cell counting of MSC131113GFP P4 and SK-OV-3cherry and yellow chimeric/hybrid cells using a fluorescence microscope (Olympus IX50) with an FITC/TRIC fluorescence dual band filter and calculation of the population percentage during a 7 day co-culture. (E) Quantification of the percentage of MSC131113GFP P4 and SK-OV-3cherry and the formation of yellow chimeric/hybrid cells by flow cytometric analysis during a 7 day co-culture.
The acquisition of these proteins by the tumor cells primarily required direct cell-to-cell interactions with the MSC, as western blot analysis revealed no detectable CD90 or CD105 proteins in the supernatant of the different mono- and co-cultures in contrast to a clear control staining in protein lysates of MSC (Supplementary Fig. S3).
Primary SCCOHT-1 cells expressed CD105 and functional ecto-5′-nucleotidase (CD73) after co-culture with MSC
The SCCOHT-1 cells, which represent a completely different tumor entity of a small cell hypercalcemic type as compared with other ovarian cancers constitutively, expressed CD90 in more than 99% of the population-like MSC [22]. In contrast, little, if any, CD73 or CD105 was detectable in SCCOHT-1 cells as compared with MSC (Fig. 5A). However, co-culture of SCCOHT-1cherry with MSCGFP was associated with CD73 expression in 10.2% (Fig. 5B) and expression of CD105 in 12.1% of the cancer cell population at day 7, which already appeared after 3 days and remained at about this level (Fig. 5C). The appearance of CD73 (= ecto-5′-nucleotidase) in SCCOHT-1 after co-culture with MSC also demonstrated intact enzymatic activity by the acquired capability of SCCOHT-1 cells to metabolize purine 5′-mononucleotides. Thus, steady-state cultures of SCCOHT-1 demonstrated 5′-AMP substrate concentrations of 2,205±18.7 pmol/100 μL (n=3) during incubation with little detectable production of adenosine similar to a no cell PBS control displaying 2,071.7±185.1 pmol/100 μL (n=3) of the 5′-AMP substrate (Fig. 5D). In contrast, analysis by liquid chromatography coupled with tandem mass spectrometry revealed significantly decreased levels of 1,461.7±86.1 pmol/100 μL (n=3) 5′-AMP paralleled by a more than 13-fold increased adenosine production in co-cultured SCCOHT-1 cells. Indeed, the acquisition of 5′-AMP metabolization was detectable after FACS separation of SCCOHT-1 from a 5 days MSC co-culture with a purity of about 97.5% (Fig. 5E). As a control, constitutively CD73-expressing MSC mono-cultures produced 911.6±100.9 pmol/100 μL (n=3) adenosine with a paralleled decline of the 5′-AMP substrate to 218.0±52.6 pmol/100 μL (n=3). Similar data were obtained from MSC after FACS separation of a 5 days co-culture with SCCOHT-1 cells (Fig. 5D).
FIG. 5.


Co-culture of primary MSC with human small cell ovarian carcinoma cells hypercalcemic type at an initial ratio of 40% SCCOHT-1/60% MSC with 2,000 cells/cm2 till 7 days. (A) Detection and analysis of CD73 and CD105 expression in mono-cultures of SCCOHT-1 cells and MSC. (B) Quantification of the percentage of CD73-positive SCCOHT-1 cells acquired during a 7 day co-culture with MSC101213. (C) Quantification of the percentage of CD105-positive SCCOHT-1 cells acquired during a 7 day co-culture with MSC101213. (D) Quantification of acquired 5′ nucleotidase enzymatic activity by liquid chromatography/tandem mass spectrometry analysis of 5′AMP and adenosine. Data represent the mean±SD of three independent experiments. A statistical analysis between the mono-culture and the corresponding population in co-culture was conducted by unpaired Student's t-test (**P< 0.01). (E) SCCOHT-1 cells previously co-cultured with MSC were separated by fluorescence-activated cell sorting (FACS), and sorted SCCOHT-1 cells were analyzed for purity by a GFP flow cytometry analysis. (F) RT-PCR of CD73 and CD105 transcripts (upper panel) and CD90 transcripts (lower panel) was performed in SCCOHT-1cherry and SK-OV-3cherry mono-cultures as compared with a 7 day co-culture with MSCGFP and subsequent separation by fluorescence-activated cell sorting. (G) RT-PCR of transcripts in MSCGFP and HMEC (P13) mono-cultures were compared with a 7 day co-culture and subsequent separation by fluorescence-activated cell sorting. Unaltered β-actin expression was used as a control. HMEC, human mammary epithelial cells; RT-PCR, reverse transcription PCR.
Transcript analysis of acquired MSC markers during co-culture
RNA isolation and PCR analysis was performed in mono-cultures and after separation of co-cultures by FACS to distinguish between the possibilities of transferring premade proteins as opposed to transferring mRNA and/or transcriptional regulators that induce expression of these proteins in co-cultured cells. Although little, if any, CD73 gene expression was measured in SCCOHT-1 cells and no detectable CD105 mRNAs in SCCOHT-1 and SK-OV-3 cells, both of these transcripts were significantly expressed after co-culture of the tumor cells in the presence of MSC (Fig. 5F, upper panel). Likewise, the absence of CD90 mRNAs in SK-OV-3 cells became detectable after co-culture with MSC (Fig. 5F, lower panel). Altered levels of distinct mRNAs were also observed during co-culture of MSC with HMEC as a nontumorigenic cell population. Although CD73 was constitutively expressed in HMEC (P13), little, if any, CD105 mRNAs were detectable in the HMEC mono-cultures. However, these transcripts were enhanced in HMEC after MSC co-culture (Fig. 5G).
Epithelial cell adhesion molecule (EpCAM/CD326) became detectable in MSC after co-culture with ovarian carcinoma cells
Acquisition of new proteins also worked in the opposite direction that MSC revealed new markers during interactions with cancer cells. There was little, if any, detectable expression of the CD326 epithelial cell adhesion molecule (EpCAM) in MSC populations in contrast to a significant EpCAM expression of more than 99% in the epithelial-like ovarian tumor cells SK-OV-3 and NIH:OVCAR-3 (Fig. 6A). Co-culture of MSC with the ovarian tumor cells, however, was accompanied by continuously increasing EpCAM levels in MSC. About 23% of MSC displayed EpCAM after co-culture with SK-OV-3 cells (Fig. 6B), and about 45% of MSC demonstrated EpCAM expression after intercellular communication with NIH:OVCAR-3 cells till 7 days (Fig. 6C).
FIG. 6.

Co-culture of primary MSC with different human ovarian carcinoma cells (SK-OV-3, NIH:OVCAR-3) at an initial ratio of 40% ovarian carcinoma cells/60% MSC with 2,000 cells/cm2 till 7 days. (A) Detection and analysis of steady-state CD326 (EpCAM) expression in mono-cultures of MSC101213, SK-OV-3, and NIH:OVCAR-3 cells, respectively. (B) Quantification of the percentage of EpCAM-positive MSC acquired during a 7 day co-culture with SK-OV-3 cells. (C) Quantification of the percentage of EpCAM-positive MSC acquired during a 7 day co-culture with NIH:OVCAR-3 cells. EpCAM, epithelial cell adhesion molecule.
Altered protein expression (CD90, CD105) could be abolished by cytochalasin D
Morphological evaluation of the cellular interactions between various MSCGFP populations and different kinds of tumor cells, including MCF-7cherry breast cancer cells (Fig. 7A), revealed cell contacts by the extension of small cytoplasmic protrusions (nanotubes) and exchange of small vesicles (exosomes). Nanotubes (yellow arrows) and GFP-labeled exosomes originating from MSC (white arrows) could be detected between MSC and MCF-7 tumor cells (Fig. 7A). Similar structures were also observed in co-cultures of MSCGFP with NIH:OVCAR-3cherry ovarian adenocarcinoma cells (Supplementary Fig. S4A), SK-OV-3cherry epithelial-like ovarian cancer cells (Supplementary Fig. S4B), and SCCOHT-1cherry small cell hypercalcemic tumor cells of the ovary (Supplementary Fig. S4C), respectively. Furthermore, the release of exosomes and extension of nanotubes was also detectable in MSC mono-cultures (Supplementary Fig. S4D).
FIG. 7.


(A) The morphology during co-culture of MSC180314GFP P3 and MCF-7cherry cells demonstrated various cellular interactions with the extension of nanotubes (yellow arrows), formation of exosomes (white arrows), and overlapping membranes. Scale bars represent 25 μm. (B) Co-culture of MSCGFP with different tumor cell lines demonstrated the formation of yellow chimeric/hybrid cells by simultaneous expression of cherry protein and eGPF (white arrows). Scale bars represent 100 μm. (C) Quantification of acquired CD90 by MCF-7, SK-OV-3, and NIH:OVCAR-3 cells after a 3 and 5 day co-culture with primary MSC180314 P3, respectively, and effect of 50 nM cytochalasin D in these co-cultures. (D) Quantification of acquired CD105 by SK-OV-3 cells within 3 days of co-culture with primary MSC100314 P3 (upper panel) and effect of 50 nM cytochalasin D in these co-cultures (lower panel). (E) Effect of cytochalasin D on the constitutive expression of CD90 and CD105 in 3 and 5 days cultured MSC270114 P2. (F) Flow cytometric analysis of double FACS-separated cells from a 7 day MSC180314GFP P6 and NIH:OVCAR-3cherry co-culture (left panels) and microarray analysis of the separated populations revealed selected prominent changes in gene expression of co-cultured MSCGFP (upper right panel) and co-cultured NIH:OVCAR-3cherry cells (lower right panel). Color images available online at www.liebertpub.com/scd
The formation of distinct chimeric/hybrid cells was observed in each of the co-cultures and demonstrated yellow-colored populations by a simultaneous expression of cherry protein and eGFP (Fig. 7B). This observation indicated spontaneous cell fusion between a mesenchymal stem cell and a corresponding tumor cell as previously described for MDA-MB-231 breast cancer cell hybrids [16,26]. Further characterization of such a chimeric/hybrid cell clone revealed CD90 expression by about 90.4% compared with a parental expression of 97.1% in MSC but undetectable levels of less than 0.1% in parental MDA-MB-231 cells (Table 1). The type-1 membrane glycoprotein CD200 (Ox-2) was detectable in 47.9% of MSC and in 69.3% of MDA-MB-231 cells, which was combined to 74.4% in chimeric/hybrid cells. Moreover, cytokeratins were detectable in 99.7% of chimeric/hybrid cells compared with a parental expression of 99.6% in MDA-MB-231 cells but only about 54.1% in parental MSC (Table 1).
Table 1.
Analysis of Certain Protein Marker Expression by Flow Cytometry
| Protein | MSC (%) | MDA-MB-231 (%) | Chimeric/hybrid cells (%) |
|---|---|---|---|
| CD90 | 97.1 | <0.1 | 90.4 |
| CD200 | 47.9 | 69.3 | 74.4 |
| Pan-cytokeratin | 54.1 | 99.6 | 99.7 |
MSC, mesenchymal stroma/stem cells.
To further explore the cellular interactions and the mutual acquisition of membrane proteins, cytochalasin D (cyt D) was applied to the co-cultures to inhibit a potential exchange of membrane proteins between the co-cultured cell populations via exosomes, formation of nanotubes, or exchange of membrane parts by a process termed trogocytosis [33]. Evaluation of a concentration dependency for cyt D in MSC, MCF-7, SK-OV-3, or NIH:OVCAR-3 cells revealed sublethal concentrations of 50 nM in these cell populations (data not shown). MCF-7 cells demonstrated about 66% and 67.7% CD90 expression after 3 and 5 days of co-culture with MSC, respectively, and incubation of this co-culture in the presence of 50 nM cyt D was associated with a marked reduction of the CD90 levels to 36.2% and 32.4% after 3 and 5 days, respectively (Fig. 7C). About 48.4% and 52.6% of SK-OV-3 cells demonstrated CD90 expression after 3 and 5 days of MSC co-culture; however, cyt D treatment significantly reduced these CD90 levels to about 17.8% and 5.4%, respectively. Likewise, CD90 acquisition during co-culture of MSC with NIH:OVCAR-3 cells was markedly reduced in the presence of cyt D (Fig. 7C). A similar effect of cyt D was observed for the acquisition of CD105. Co-culture of SK-OV-3 cells with MSC till 3 days revealed always more than 99% of CD105 in MSC paralleled by progressively increasing CD105 in SK-OV-3 cells from 1.4% at 0 day to 12.6% after 3 days (Fig. 7D, upper panel), whereas co-culture in the presence of 50 nM cytochalasin D was accompanied by markedly reduced CD105 levels of only 2.6% in SK-OV-3 cells after 3 days while CD105 expression was sustained in more than 99% of MSC (Fig. 7D, lower panel). Together, these effects suggested that cyt D predominantly inhibits the acquisition of new membrane proteins such as CD90 and CD105 by the tumor cells, as no effects were observed on the steady-state expression of these membrane proteins by cyt D in MSC mono-cultures (Fig. 7E).
Microarray analysis of MSC co-culture with NIH:OVCAR-3 cells
More detailed functional alterations were observed after microarray analysis of co-cultured cells (Supplementary Table S1–S4). After a 7 days co-culture of MSCGFP and NIH:OVCAR-3cherry cells, the two cell populations were separated into the appropriate mono-cultures by double FACS to yield about 99% of NIH:OVCAR-3 corresponding cherry-positive and MSC corresponding GFP-positive cells, respectively (Fig. 7F, left panel). Selected microarray data of these co-culture-separated MSC compared with steady-state MSC revealed an increase of epithelial cell-specific transcripts, including the ESPR2 splicing regulator and a variety of cytokeratins (KRT genes). Moreover, transcripts of the epithelial-like differentiation factors dermokine and sciellin that contribute to keratinocyte differentiation were up-regulated in co-cultured MSC. Likewise, regulators of cell–cell interactions, including transcripts of the epithelial cell-specific tight junction factor CRB3, desmosomal periplakin (PPL), and members of the tetraspanin transmembrane components in epithelial cells (UPK1B), were significantly elevated in co-cultured MSC. In addition, EpCAM mRNAs were markedly enhanced by 51.1-fold in co-cultured MSC, suggesting that MSC acquired a variety of epithelial-like cell functionality during co-culture with NIH:OVCAR-3 cells. Furthermore, a variety of chemokine/cytokine transcripts were down-modulated in co-cultured MSC (Fig. 7F, upper right panel).
Selected genes in co-cultured NIH:OVCAR-3 cells compared with steady-state NIH:OVCAR-3 cells demonstrated an up-regulation of growth factors (BMPs), epithelial mitogens (EPGN), and mitotic spindle-associated factors (MZT2A), which promote proliferative capacity. The protein product of the DIRAS3 gene can function as a putative tumor suppressor, whereas the induced kelch domain-containing transcript (KLHDCA8) is associated with elevated tumor aggressiveness. Down-regulated genes in NIH:OVCAR-3 cells included a variety of transcription factor genes such as TAL1, the basic helix-loop-helix family FOS and FOSB, and also HES1 and HES5, which are suggested to promote cancerous development on down-modulation. Likewise, genes of zinc finger transcription factors are down-regulated such as EGR1 and the Kruppel-like factor (KLF10) as a repressor of cell growth (Fig. 7F, lower right panel).
Discussion
Invasive tumor growth causes local tissue injuries that are associated with the attraction of MSC to take part in repair mechanisms. This process is also accompanied by interactions between the adjacent neighboring cells, whereby the close vicinity of mesenchymal stem/stroma cells contributes toward altering the proliferative capacity and a certain functionality of the tumor cells.
Co-culture of different cancer cell populations with six individual MSC primary cultures revealed a growth stimulation of the tumor cells, which was also supported by induction of proliferation-promoting genes in NIH:OVCAR-3 cells and down-modulation of certain transcription factor genes with repressor function of tumorigenic development and cell growth. Furthermore, these findings are substantiated by a previous work demonstrating enhanced tumor cell growth by MSC [34–37], although there are also controversial studies suggesting a reduced tumor cell proliferation in the presence of MSC [38,39]. The divergent effects of MSC on tumor cells may be caused, in part, by a different activation status within the heterogeneous MSC population involving interference with the β-catenin pathway such as DKK-1-mediated depression of Wnt signaling [40]. Moreover, secretory pathways within the tumor cell microenvironment such as MSC-mediated release and activation of distinct matrix metalloproteinases interfere with the migratory and invasive potential of tumor cells and reduce their tumorigenicity [41]. Other work postulated bidirectional effects of naïve or innate MSC on tumors with promotion or inhibition of tumor progression, whereas MSC already primed by inflammatory factors within the tumor microenvironment promote tumor progression [42].
MSC that express CD73, CD90, and CD105 surface markers can transfer, exchange, or induce these proteins during their interactions with tumor cells, thereby altering cellular functionality with the consequence of increased tumor heterogeneity. Little, if any, of the GPI-anchored CD90 antigen was expressed in MCF-7, HBCEC 699, NIH:OVACAR-3, and SK-OV-3 cells; however, co-culture of these human cancer cells with different MSC populations was accompanied by a CD90 induction at both the mRNA and protein levels. Moreover, endoglin (CD105) as a part of the TGFβ receptor complex that is predominantly involved in the regulation of angiogenesis in tumors was acquired by SK-OV-3 and SCCOHT-1 cells on MSC interactions. Furthermore, changes in cellular functionality during co-culture with MSC were also observed for acquisition of CD73 transcripts and expression of the active enzyme by SCCOHT-1 cells. This ecto-5′-nucleotidase is associated to the external face of the plasma membrane via a GPI-anchor and catalyzes the dephosphorylation of purine 5′-mononucleotides, particularly AMP, which increases extracellular levels of nucleosides [43]. Therefore, the acquisition of this property by SCCOHT-1 cells in response to interactions with MSC alters the tumor cell functionality and contributes to changes in nucleoside concentrations within the microenvironment on availability of corresponding substrates. Contrary to these functional changes of the tumor cells, the direct intercellular communication processes also affected MSC. The epithelial cell-specific adhesion molecule (CD326/EpCAM), which functions as a transmembrane glycoprotein mediating Ca2+-independent homotypic cell–cell adhesion in epithelia, was undetectable in MSC. However, co-culture of the ovarian cancer cells with MSC was associated with EpCAM mRNA and protein induction, which suggested certain differentiation processes by adding epithelial cell-like properties to the MSC. Such heterogenic functionality was also discussed in vivo with EpCAM-positive subsets of tumor-initiating cells (cancer stem cells) in patient ovarian cancer ascites also carrying markers of cancer-associated fibroblasts [44]. Other in vivo studies demonstrated cross-talk of MSC with tumor cells and promotion of tumor growth in patients with head and neck cancer, whereby MSC isolated from these patient tumors constitutively produced high levels of IL-6, IL-8, and stromal cell-derived factor (SDF)-1α [45]. In addition, co-transplantation of human adipose tissue-derived MSC with MDA-MB-231 breast cancer cells exhibited partial EMT and was associated with development of tumor xenograft metastases to multiple mouse organs [46].
The alteration in the cellular functionality of tumor cell populations and MSC within the tumor microenvironment contributes to tumor heterogeneity [47] and suggests a process of mutual cellular adaptation whereby MSC acquire tumor cell-specific markers and vice versa. Indeed, enhanced gene expression of epithelial cell-specific markers appeared in MSC after co-culture with ovarian epithelial cancer cells whereas MSC-like properties such as expression of certain chemokine/cytokine genes were down-modulated. These functional alterations can result in MSC differentiation of an altered phenotype, including cancer-associated fibroblasts, perivascular cells, or tumor-associated macrophage-like cells [48]. Moreover, co-culture with injured mesangial cells has demonstrated differentiation of MSC into mesangial cells [49]. In addition, direct co-culture of MSC with human nucleus palposus cells resulted in differentiation of MSC to a nucleus palposus-like phenotype associated with up-regulation of appropriate growth factor and matrix-associated genes [50].
Mutual cellular adaptation was suggested between cells from ductal invasive breast cancer and surrounding MSC [16,51–53], and further studies revealed that disseminated tumor-like cells originating from breast cancer tissue can be long-term hosted in an inactivated (dormant) state in perivascular niches [54], which also provides the stem cell niche for MSC. Therefore, the characterization of a variety of tumor-specific cell types, including cancer-associated fibroblasts or cancer stem cells, may likewise originate from processes of mutual cellular adaptation within the tumor microenvironment. Moreover, altered tumor cell functions during MSC interactions within the tumor microenvironment carry the risk of therapy failure by developing cancer cells displaying certain resistances [55].
According to functional changes by mRNA and/or protein transfer or protein induction via specific communication processes, a previous work confirmed that cellular interactions can promote an MSC-mediated protein expression in MDA-MB-231 breast cancer or natural killer cells, which partially involves GJIC and notch signaling [26,56]. Furthermore, this study demonstrated that the direct interactions include the transfer and exchange of cellular material. This was also substantiated by fluorescence microscopy demonstrating close membrane interactions between MSC and the co-cultured tumor cells together with exosome release and the extension of cytoplasmic protrusions that appear as nanotubes. Exosomes represent the release of extracellular vesicles carrying predominantly proteins, mRNAs, and microRNAs that are exchanged during cellular interactions [57]. Production of exosomes has been described for MSC to transport gene regulatory information to recipient cells that can modulate cell growth and angiogenesis by affecting a variety of cellular pathways [58]. Indeed, the CD73, CD90, and CD105 induction in the different tumor cells was observed at both the protein and mRNA levels, suggesting exchange of proteins and/or transcriptional regulators and/or mRNAs between MSC and co-cultured tumor cells. Moreover, these interactive properties of MSC were also observed in normal cells such as HMEC after MSC co-culture.
Intercellular structures, including membrane channels such as tunneling nanotubes, also enable a direct exchange of biomolecules and small organelles and may also serve as a tool for tumor cell interactions [59]. Thus, a previous work revealed increased regenerative support during nanotubular cross-talk between MSC and damaged cardiomyocytes [60]. Intercellular transport via nanotubes requires actin microfilaments to transmit traction and contraction forces that can be blocked by cytochalasin D for inhibition of actin polymerization. In this context, the transfer between MSC and the different tumor cell populations was significantly reduced in the presence of cytochalasin D. Confirmative studies have demonstrated that formation of nanotubes between MSC and vascular smooth muscle cells enables the exchange of proteins and mitochondria associated with elevated MSC growth that can be abolished by cytochalasin D [61]. These findings suggested that the extent of cellular interactions between MSC and tumor cells is associated with different levels of exchange such as exchange of exosomes or membrane patches or a combination of whole cell membranes by cell fusion. Indeed, all MSC co-cultures revealed a small amount of chimeric/hybrid cells, indicating MSC-tumor cell fusion products. This phenomenon has also been observed in other MSC co-cultures [16,26], and characteristics revealed the acquisition of distinct markers from both parental cell populations. Although most fusion products between MSC and tumor cells are unable to survive due to aberrant signaling by two nuclei, a small amount of chimeric/hybrid cells arrange chromosome/DNA regulation and result in various individual hybrid populations displaying altered expression levels compared with the parental cell types. Fusion of tumor cells with macrophages or bone marrow-derived cells has been discussed in the context of cancer invasion and metastasis [62], whereas the development and progression of chimeric/hybrid populations enhances tumor heterogeneity and complicates therapeutic approaches.
Conclusion
Co-culture of various human cell populations with different individual MSC populations was accompanied by exchange of biological material via different mechanisms, including exosomes and formation of nanotubes. During these cellular interactions, a variety of functional changes were observed, particularly the acquisition of multiple epithelial cell-like properties by MSC after co-culture with ovarian cancer cells and vice versa, an elevated growth of tumor cells. These findings suggested a progressive functional heterogeneity by a process of mutual cellular adaptation.
Supplementary Material
Acknowledgments
The technical assistance of Juliane von der Ohe and the essential support for the GC/tandem MS analysis by the Metabolomics core unit (Dr. Volkhard Kaever) as well as for the cell separation by the FACS core unit (Dr. Matthias Ballmaier) and the microarray analysis by the research core unit Transcriptomics (Dr. Oliver Dittrich-Breiholz) of Hannover Medical School is acknowledged. Yuanyuan Yang is a visiting research fellow from Tongji University, Shanghai, China.
Author Disclosure Statement
The authors declare no financial, personal, or professional conflicts of interest.
References
- 1.Caplan AI. (1991). Mesenchymal stem cells. J Orthop Res 9:641–650 [DOI] [PubMed] [Google Scholar]
- 2.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S. and Marshak DR. (1999). Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147 [DOI] [PubMed] [Google Scholar]
- 3.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop D. and Horwitz E. (2006). Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317 [DOI] [PubMed] [Google Scholar]
- 4.Simmons PJ. and Torok-Storb B. (1991). Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood 78:55–62 [PubMed] [Google Scholar]
- 5.Honczarenko M, Le Y, Swierkowski M, Ghiran I, Glodek AM. and Silberstein LE. (2006). Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 24:1030–1041 [DOI] [PubMed] [Google Scholar]
- 6.Kuroda Y, Kitada M, Wakao S, Nishikawa K, Tanimura Y, Makinoshima H, Goda M, Akashi H, Inutsuka A, et al. (2010). Unique multipotent cells in adult human mesenchymal cell populations. Proc Natl Acad Sci U S A 107:8639–8643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caplan AI. and Correa D. (2011). The MSC: an injury drugstore. Cell Stem Cell 9:11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsuda T, Kosaka N, Takeshita F. and Ochiya T. (2013). The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics 13:1637–1653 [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal S. and Pittenger MF. (2005). Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 105:1815–1822 [DOI] [PubMed] [Google Scholar]
- 10.Hass R, Kasper C, Bohm S. and Jacobs R. (2011). Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal 9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Miguel MP, Fuentes-Julian S, Blazquez-Martinez A, Pascual CY, Aller MA, Arias J. and Arnalich-Montiel F. (2012). Immunosuppressive properties of mesenchymal stem cells: advances and applications. Curr Mol Med 12:574–591 [DOI] [PubMed] [Google Scholar]
- 12.Ruster B, Gottig S, Ludwig RJ, Bistrian R, Muller S, Seifried E, Gille J. and Henschler R. (2006). Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood 108:3938–3944 [DOI] [PubMed] [Google Scholar]
- 13.Nassiri SM. and Rahbarghazi R. (2014). Interactions of mesenchymal stem cells with endothelial cells. Stem Cells Dev 23:319–332 [DOI] [PubMed] [Google Scholar]
- 14.Friedl P. and Alexander S. (2011). Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147:992–1009 [DOI] [PubMed] [Google Scholar]
- 15.Ungefroren H, Sebens S, Seidl D, Lehnert H. and Hass R. (2011). Interaction of tumor cells with the microenvironment. Cell Commun Signal 9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hass R. and Otte A. (2012). Mesenchymal stem cells as all-round supporters in a normal and neoplastic microenvironment. Cell Commun Signal 10:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubeau L. (2008). The cell of origin of ovarian epithelial tumours. Lancet Oncol 9:1191–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurman RJ. and Shih Ie M. (2010). The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 34:433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim A, Ueda Y, Naka T. and Enomoto T. (2012). Therapeutic strategies in epithelial ovarian cancer. J Exp Clin Cancer Res 31:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young RH, Oliva E. and Scully RE. (1995). Small cell carcinoma of the hypercalcemic type in the ovary. Gynecol Oncol 57:7–8 [DOI] [PubMed] [Google Scholar]
- 21.Hass R. and Bertram C. (2009). Characterization of human breast cancer epithelial cells (HBCEC) derived from long term cultured biopsies. J Exp Clin Cancer Res 28:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otte A, Gohring G, Steinemann D, Schlegelberger B, Groos S, Langer F, Kreipe HH, Schambach A, Neumann T, et al. (2012). A tumor-derived population (SCCOHT-1) as cellular model for a small cell ovarian carcinoma of the hypercalcemic type. Int J Oncol 41:765–775 [DOI] [PubMed] [Google Scholar]
- 23.Lavrentieva A, Majore I, Kasper C. and Hass R. (2010). Effects of hypoxic culture conditions on umbilical cord-derived human mesenchymal stem cells. Cell Commun Signal 8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otte A, Bucan V, Reimers K. and Hass R. (2013). Mesenchymal stem cells maintain long-term in vitro stemness during explant culture. Tissue Eng Part C Methods 19:937–948 [DOI] [PubMed] [Google Scholar]
- 25.Bertram C. and Hass R. (2008). MMP-7 is involved in the aging of primary human mammary epithelial cells (HMEC). Exp Gerontol 43:209–217 [DOI] [PubMed] [Google Scholar]
- 26.Mandel K, Yang Y, Schambach A, Glage S, Otte A. and Hass R. (2013). Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev 22:3114–3127 [DOI] [PubMed] [Google Scholar]
- 27.Evans BA, Elford C, Pexa A, Francis K, Hughes AC, Deussen A. and Ham J. (2006). Human osteoblast precursors produce extracellular adenosine, which modulates their secretion of IL-6 and osteoprotegerin. J Bone Miner Res 21:228–236 [DOI] [PubMed] [Google Scholar]
- 28.Montanucci P, Basta G, Pescara T, Pennoni I, Di Giovanni F. and Calafiore R. (2011). New simple and rapid method for purification of mesenchymal stem cells from the human umbilical cord Wharton jelly. Tissue Eng Part A 17:2651–2661 [DOI] [PubMed] [Google Scholar]
- 29.Fonsatti E, Jekunen AP, Kairemo KJ, Coral S, Snellman M, Nicotra MR, Natali PG, Altomonte M. and Maio M. (2000). Endoglin is a suitable target for efficient imaging of solid tumors: in vivo evidence in a canine mammary carcinoma model. Clin Cancer Res 6:2037–2043 [PubMed] [Google Scholar]
- 30.Gomez-Esquer F, Agudo D, Martinez-Arribas F, Nunez-Villar MJ. and Schneider J. (2004). mRNA expression of the angiogenesis markers VEGF and CD105 (endoglin) in human breast cancer. Anticancer Res 24:1581–1585 [PubMed] [Google Scholar]
- 31.Bucan V, Mandel K, Bertram C, Lazaridis A, Reimers K, Park-Simon TW, Vogt PM. and Hass R. (2012). LEF-1 regulates proliferation and MMP-7 transcription in breast cancer cells. Genes Cells 17:559–567 [DOI] [PubMed] [Google Scholar]
- 32.Mandel K, Seidl D, Rades D, Lehnert H, Gieseler F, Hass R. and Ungefroren H. (2013). Characterization of spontaneous and TGF-beta-induced cell motility of primary human normal and neoplastic mammary cells in vitro using novel real-time technology. PLoS One 8:e56591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joly E. and Hudrisier D. (2003). What is trogocytosis and what is its purpose?. Nat Immunol 4:815. [DOI] [PubMed] [Google Scholar]
- 34.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R. and Weinberg RA. (2007). Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449:557–563 [DOI] [PubMed] [Google Scholar]
- 35.Muehlberg FL, Song YH, Krohn A, Pinilla SP, Droll LH, Leng X, Seidensticker M, Ricke J, Altman AM, et al. (2009). Tissue-resident stem cells promote breast cancer growth and metastasis. Carcinogenesis 30:589–597 [DOI] [PubMed] [Google Scholar]
- 36.Roorda BD, Elst A, Boer TG, Kamps WA. and de Bont ES. (2010). Mesenchymal stem cells contribute to tumor cell proliferation by direct cell-cell contact interactions. Cancer Invest 28:526–534 [DOI] [PubMed] [Google Scholar]
- 37.Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S. and Chang C. (2014). Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene 33:2768–2778 [DOI] [PubMed] [Google Scholar]
- 38.Chao KC, Yang HT. and Chen MW. (2012). Human umbilical cord mesenchymal stem cells suppress breast cancer tumourigenesis through direct cell-cell contact and internalization. J Cell Mol Med 16:1803–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kucerova L, Skolekova S, Matuskova M, Bohac M. and Kozovska Z. (2013). Altered features and increased chemosensitivity of human breast cancer cells mediated by adipose tissue-derived mesenchymal stromal cells. BMC Cancer 13:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiao L, Xu ZL, Zhao TJ, Ye LH. and Zhang XD. (2008). Dkk-1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signalling. Cancer Lett 269:67–77 [DOI] [PubMed] [Google Scholar]
- 41.Clarke MR, Imhoff FM. and Baird SK. (2014). Mesenchymal stem cells inhibit breast cancer cell migration and invasion through secretion of tissue inhibitor of metalloproteinase-1 and -2. Mol Carcinog [Epub ahead of print]; DOI: 10.1002/mc.22178 [DOI] [PubMed] [Google Scholar]
- 42.Sun Z, Wang S. and Zhao RC. (2014). The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol 7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Airas L, Niemela J, Salmi M, Puurunen T, Smith DJ. and Jalkanen S. (1997). Differential regulation and function of CD73, a glycosyl-phosphatidylinositol-linked 70-kD adhesion molecule, on lymphocytes and endothelial cells. J Cell Biol 136:421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wintzell M, Hjerpe E, Avall Lundqvist E. and Shoshan M. (2012). Protein markers of cancer-associated fibroblasts and tumor-initiating cells reveal subpopulations in freshly isolated ovarian cancer ascites. BMC Cancer 12:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kansy BA, Dissmann PA, Hemeda H, Bruderek K, Westerkamp AM, Jagalski V, Schuler P, Kansy K, Lang S, Dumitru CA. and Brandau S. (2014). The bidirectional tumor—mesenchymal stromal cell interaction promotes the progression of head and neck cancer. Stem Cell Res Ther 5:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rowan BG, Gimble JM, Sheng M, Anbalagan M, Jones RK, Frazier TP, Asher M, Lacayo EA, Friedlander PL, Kutner R. and Chiu ES. (2014). Human adipose tissue-derived stromal/stem cells promote migration and early metastasis of triple negative breast cancer xenografts. PLoS One 9:e89595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li HJ, Reinhardt F, Herschman HR. and Weinberg RA. (2012). Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov 2:840–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barcellos-de-Souza P, Gori V, Bambi F. and Chiarugi P. (2013). Tumor microenvironment: bone marrow-mesenchymal stem cells as key players. Biochim Biophys Acta 1836:321–335 [DOI] [PubMed] [Google Scholar]
- 49.Wong CY, Tan EL. and Cheong SK. (2014). In vitro differentiation of mesenchymal stem cells into mesangial cells when co-cultured with injured mesangial cells. Cell Biol Int 38:497–501 [DOI] [PubMed] [Google Scholar]
- 50.Strassburg S, Richardson SM, Freemont AJ. and Hoyland JA. (2010). Co-culture induces mesenchymal stem cell differentiation and modulation of the degenerate human nucleus pulposus cell phenotype. Regen Med 5:701–711 [DOI] [PubMed] [Google Scholar]
- 51.Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, Glod JW. and Banerjee D. (2008). Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res 68:4331–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaturvedi S. and Hass R. (2011). Extracellular signals in young and aging breast epithelial cells and possible connections to age-associated breast cancer development. Mech Ageing Dev 132:213–219 [DOI] [PubMed] [Google Scholar]
- 53.Kong D, Li Y, Wang Z. and Sarkar FH. (2011). Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: are they cousins or twins?. Cancers (Basel) 3:716–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, et al. (2013). The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15:807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexander S. and Friedl P. (2012). Cancer invasion and resistance: interconnected processes of disease progression and therapy failure. Trends Mol Med 18:13–26 [DOI] [PubMed] [Google Scholar]
- 56.Chatterjee D, Tufa DM, Baehre H, Hass R, Schmidt RE. and Jacobs R. (2014). Natural killer cells acquire CD73 expression upon exposure to mesenchymal stem cells. Blood 123:594–595 [DOI] [PubMed] [Google Scholar]
- 57.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ. and Lotvall JO. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659 [DOI] [PubMed] [Google Scholar]
- 58.Eirin A, Riester SM, Zhu XY, Tang H, Evans JM, O'Brien D, van Wijnen AJ. and Lerman LO. (2014). MicroRNA and mRNA cargo of extracellular vesicles from porcine adipose tissue-derived mesenchymal stem cells. Gene 551:55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rustom A, Saffrich R, Markovic I, Walther P. and Gerdes HH. (2004). Nanotubular highways for intercellular organelle transport. Science 303:1007–1010 [DOI] [PubMed] [Google Scholar]
- 60.Figeac F, Lesault PF, Le Coz O, Damy T, Souktani R, Trebeau C, Schmitt A, Ribot J, Mounier R, et al. (2014). Nanotubular crosstalk with distressed cardiomyocytes stimulates the paracrine repair function of mesenchymal stem cells. Stem Cells 32:216–230 [DOI] [PubMed] [Google Scholar]
- 61.Vallabhaneni KC, Haller H. and Dumler I. (2012). Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev 21:3104–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pawelek JM. and Chakraborty AK. (2008). Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer 8:377–386 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.