

Prospective comprehensive genomic profiling of 116 predominantly locally advanced or metastatic gastric cancer (GC) cases was performed to identify genomic alterations (GAs) associated with a potential response to targeted therapies or targeted therapy-based clinical trials. Comprehensive genomic profiling of GC identified clinically relevant GAs that suggest benefit from targeted therapy including MET-amplified GC and ERBB2 base substitutions.
Keywords: Gastric cancer, Sequencing, Targeted therapy, Mutation, Profiling, MET
Abstract
Background.
Gastric cancer (GC) is a major global cancer burden and the second most common cause of global cancer-related deaths. The addition of anti-ERBB2 (HER2) targeted therapy to chemotherapy improves survival for ERBB2-amplified advanced GC patients; however, the majority of GC patients do not harbor this alteration and thus cannot benefit from targeted therapy under current practice paradigms.
Materials and Methods.
Prospective comprehensive genomic profiling of 116 predominantly locally advanced or metastatic (90.0%) gastric cancer cases was performed to identify genomic alterations (GAs) associated with a potential response to targeted therapies approved by the U.S. Food and Drug Administration or targeted therapy-based clinical trials.
Results.
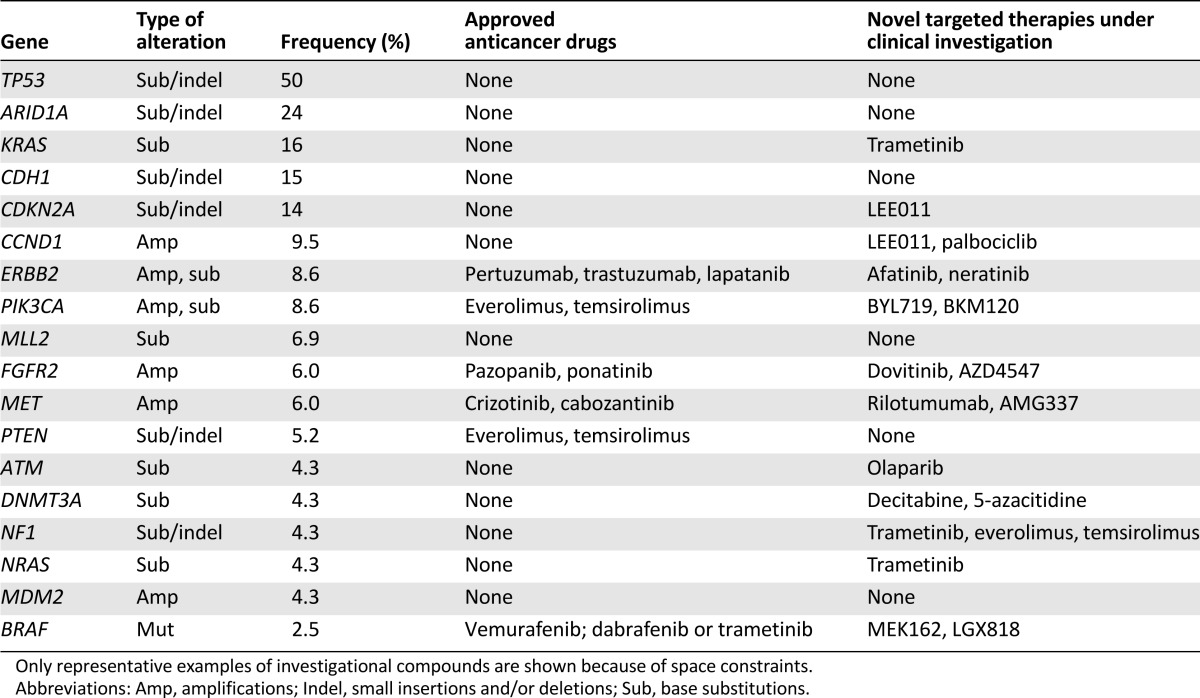
Overall, 78% of GC cases harbored one clinically relevant GA or more, with the most frequent alterations being found in TP53 (50%), ARID1A (24%), KRAS (16%), CDH1 (15%), CDKN2A (14%), CCND1 (9.5%), ERBB2 (8.5%), PIK3CA (8.6%), MLL2 (6.9%), FGFR2 (6.0%), and MET (6.0%). Receptor tyrosine kinase genomic alterations were detected in 20.6% of cases, primarily ERBB2, FGFR2, and MET amplification, with ERBB2 alterations evenly split between amplifications and base substitutions. Rare BRAF mutations (2.6%) were also observed. One MET-amplified GC patient responded for 5 months to crizotinib, a multitargeted ALK/ROS1/MET inhibitor.
Conclusion.
Comprehensive genomic profiling of GC identifies clinically relevant GAs that suggest benefit from targeted therapy including MET-amplified GC and ERBB2 base substitutions.
Abstract
摘要
背景. 胃癌(GC)在全球范围都是严重的癌症负担,同时也是全球癌症相关死亡的第二大原因。化疗联合抗ERBB2(HER2)靶向治疗改善了ERBB2扩增的进展期胃癌患者的生存;但是多数胃癌患者的肿瘤中并没有这一基因变异,因此不能从这一治疗范例的靶向治疗中获益。
材料与方法. 对116例患者进行前瞻性综合性基因组分析,以鉴别与靶向治疗(美国食品和药物管理局批准的靶向治疗,或以靶向治疗为基础的临床试验)部分缓解相关的基因变异(GA),患者主要为局部进展期或转移性胃癌(90.0%)。
结果. 总体而言,78%的胃癌病例含有≥ 1个临床相关性GA。最常见的基因变异见于TP53(50%)、ARID1A(24%)、KRAS(16%)、CDH1(15%)、CDKN2A(14%)、CCND1(9.5%)、ERBB2(8.5%)、PIK3CA(8.6%)、MLL2(6.9%)、FGFR2(6.0%)和MET(6.0%)。20.6%的病例可检测到受体酪氨酸激酶基因变异,主要为ERBB2、FGFR2和MET扩增,而ERBB2变异中扩增和碱基置换各占一半。还观察到少量BRAF突变(2.6%)。一例MET扩增胃癌患者经克唑替尼(ALK/ROS1/MET多靶点抑制剂)治疗5个月有应答。
结论. 对胃癌进行综合性基因组分析可鉴别出临床相关性GA,提示可从针对包括MET扩增胃癌和ERBB2碱基置换在内的靶向治疗中获益。The Oncologist 2015;20:499–507
Implications for Practice:
Despite description of many potentially clinically relevant genomic alterations in retrospective research studies, these alterations are not regularly assessed in a comprehensive manner in clinical practice. This study demonstrates the feasibility of prospective comprehensive genomic profiling (CGP) for advanced gastric carcinoma. It also demonstrates a high frequency of genomic alterations associated with potential benefit from targeted therapies. CGP in this setting may inform therapeutic options beyond standard of care testing by identifying genomic alterations such as point mutations in the kinase domain of ERBB2 and MET amplification. Genotype-directed management is highlighted by the response of a MET-amplified gastric carcinoma patient to crizotinib.
Introduction
Gastric cancer (GC) is the second most frequent cause of cancer-related death worldwide [1]. The majority of patients present with advanced disease, and the overall 5-year survival rate is <28%, compared with <5% for patients presenting with metastatic disease [2–4]. The clinical heterogeneity of GC is highlighted by significant worldwide geographic variations, differences in anatomic origin (proximal vs. distal), risk factors including Helicobacter pylori infection and dietary patterns, and a poorly understood relationship to Asian ethnicity [5–7]. Within the common intestinal histologic subtype, there are differences in ERBB2 amplification frequencies (proximal vs. distal) and association with H. pylori and progression from a metaplastic background (distal vs. proximal, intestinal type) [8–10].
Efforts to identify predictive biomarkers to guide decision making for systemic therapy have yielded inconsistent results. To date, the only validated predictive biomarker for targeted therapy is ERBB2 amplification, which predicts benefit from the anti-ERBB2 (HER2) antibody trastuzumab in advanced disease [8, 11, 12].
Systemic therapy for metastatic, relapsed, or refractory GC is largely based on empiric 5-fluoropyrimidine and platinum combinations, and there are no definitive clinical predictors of response [13]. Although trastuzumab offers improved survival for the 7%–34% of GC patients with ERBB2 amplification, there are no approved molecularly directed therapies for the majority of patients [8, 14]. Although the recent approval of the anti-VEGFR2 antibody ramucirumab increases the GC armamentarium, there are no validated predictive biomarkers to identify patients who may derive benefit from anti-VEGFR targeted therapies [13, 15].
Large-scale retrospective whole-genome sequencing analyses have highlighted recurrent genomic alterations in gastric cancer such as ARID1A, CDH1, RHOA, and FGFR2 [10, 16–18]. Prospective comprehensive genomic profiling based on a clinical next-generation sequencing (NGS) assay in the course of clinical care can identify novel and known clinically relevant genomic alterations (GAs) and increase understanding of the underlying biology and immediately inform patient management options. In this study, we present a large series of primarily relapsed and metastatic gastric carcinoma clinical specimens that underwent prospective comprehensive genomic profiling and highlight therapeutic implications.
Materials and Methods
Comprehensive genomic profiling using a clinical NGS-based assay (FoundationOne) was performed in a Clinical Laboratory Improvements Amendment-certified, College of American Pathologists-accredited laboratory (Foundation Medicine, Cambridge, MA, http://www.foundationmedicine.com) using validated methods [19]. Clinical samples were sent in from both academic and community oncologists for genomic profiling in the context of clinical care, and patient outcomes in selected cases were obtained from the primary treating physician. With the exception of three samples received as extracted DNA, a pathologist reviewed hematoxylin and eosin-stained slides to confirm diagnosis of GC and to ensure adequate formalin-fixed, paraffin-embedded (FFPE) specimen quality: sample volume >1 mm3, nucleated cellularity >80% or >30,000 cells, and >20% tumor nuclei. When required, macrodissection was performed to enrich samples with <20% tumor nuclei.
DNA was extracted from unstained FFPE specimens using the Promega Maxwell 16 Tissue LEV DNA kit (Promega, Madison, WI, http://www.promega.com) and quantified using an Invitrogen PicoGreen fluorescence assay (Thermo Fisher Scientific, Waltham, MA, http://www.thermofisher.com). Library construction was performed with 50–200 ng of DNA sheared by sonication (E210; Covaris, Woburn, MA, http://covarisinc.com) to ∼100–400 base pairs before end repair, dA addition, and ligation of indexed Illumina sequencing adaptors (Illumina, San Diego, CA, http://www.illumina.com). Prior to hybrid selection and sequencing, libraries were amplified with polymerase chain reaction (PCR) for 10 cycles using KAPA HiFi (Kapa Biosystems, Wilmington, MA, http://www.kapabiosystems.com). Solution-based hybrid selection was performed with a custom bait set of 120-bp biotinylated DNA oligonucleotides (Integrated DNA Technology, Coralville, IA, http://www.idtdna.com) covering 3,769 exons of 236 cancer-related genes and 47 introns of 19 genes frequently rearranged in cancer. The Illumina HiSeq 2000 and Illumina HiSeq 2500 platforms were used to perform 49 × 49 paired-end sequencing. Sequence alignment, PCR duplicate-read removal, and local alignment optimization were performed using Burrows-Wheeler aligner bwa-0.5.9 (SourceForge; Slashdot Media, San Francisco, CA, http://slashdotmedia.com), Picard 1.47 (Broad Institute, Cambridge, MA, http://broadinstitute.github.io/picard/), SAM Tools samtools-0.1.12a (SourceForge; Slashdot Media), and GATK 1.0.4705 (Broad Institute).
Variant calling was performed using custom tools. Base substitutions were called using a Bayesian methodology, and short insertions-deletions (indels) were called using local assembly. Somatic variants were annotated using COSMIC, and germline variants were removed using dbSNP. Rearrangements were called using chimeric read pairs clustered by genomic position. Copy number alterations (CNAs) were detected by fitting a statistical copy-number model to normalized coverage and allele frequencies at all exons and ∼3,500 genomewide single nucleotide polymorphisms and accounting for stromal admixture. An extensive validation was performed for base substitutions, short indels, and CNAs. To validate CNA detection, seven tumor cell lines bearing 19 focal gene amplifications (6–15 copies, 15 genes) and 9 homozygous gene deletions (6 genes) with their matched normal cell lines (thereby maintaining consistent genotypes) were pooled to create five ratios ranging from low to high tumor content (20%–75%), creating a total test set of 210 CNAs.
High performance was achieved for both high-level amplifications (copy number ≥8) and homozygous deletions when tumor purity was as low as 30%: sensitivity was 99% (91 of 92) with positive predictive value >99% (127 of 127). Performance was reduced for lower CNAs (6–7 copies) and at lower sample purities (20%–30%), with overall sensitivity >80%. Cancer-related alterations were defined as those that are known sites of somatic mutation, truncations or homozygous deletions of known tumor suppressor genes, and amplifications of oncogenes and fusions of genes known to be rearranged in solid tumors.
Clinically relevant GAs were defined as those that suggested potential response to targeted therapies approved in gastric carcinoma or in other tumor types or that suggested benefit from targeted therapy under development and being administered in the context of a clinical trial. The two-tailed Fisher’s exact test was used to determine statistical significance of all group comparisons. Local site permissions were used to study these samples.
Results
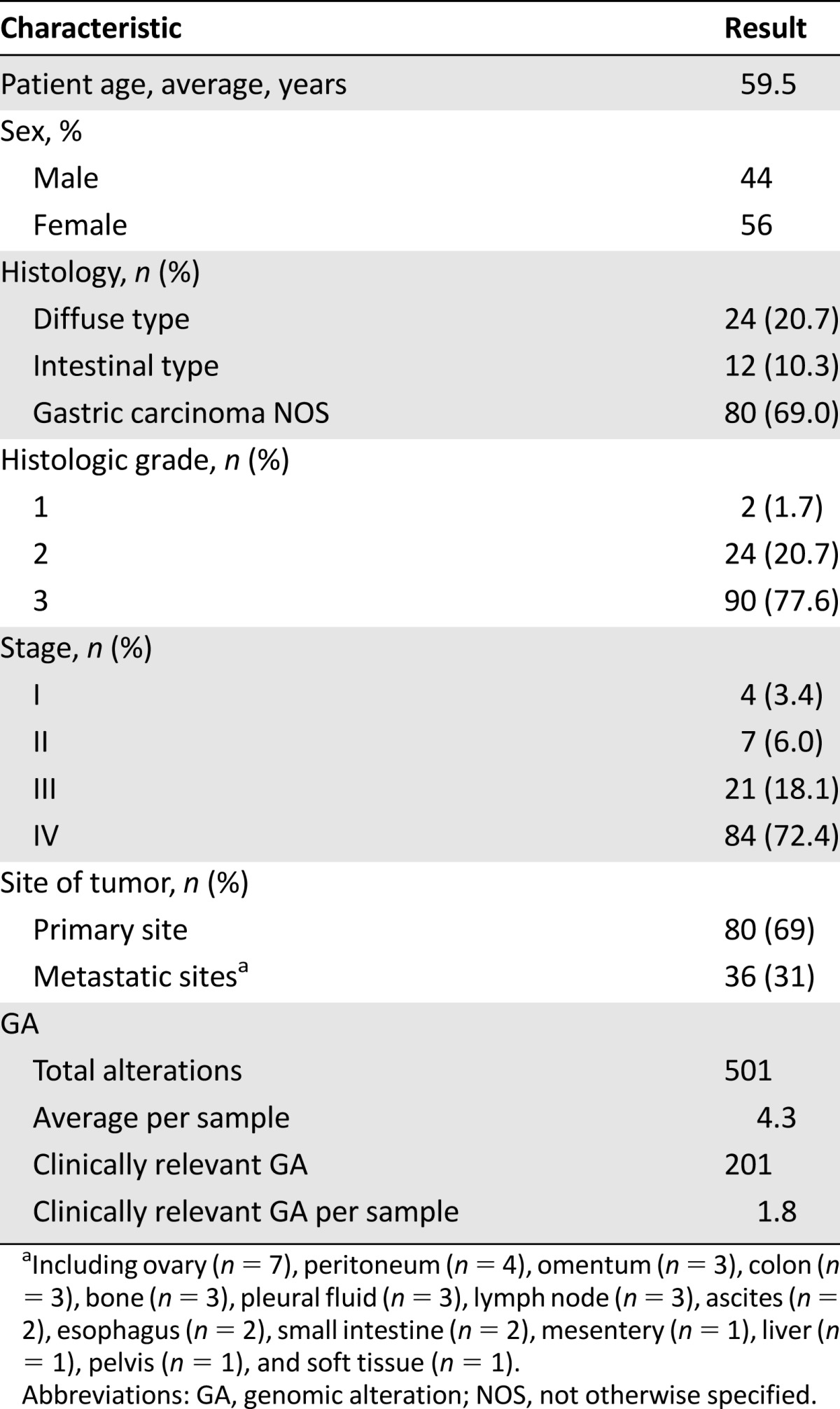
Comprehensive genomic profiling was performed on 116 GC cases. The median patient age at time of testing was 62 years (range: 26–87 years) (Table 1). Sixty-five (56%) specimens were from male patients. The stage distribution is shown in Table 1. Of the samples, 69% (n = 80) were from the primary GC and 31% (n = 36) were from metastatic sites including ovary (n = 7), peritoneum (n = 4), omentum (n = 3), colon (n = 3), bone (n = 3), pleural fluid (n = 3), lymph node (n = 3), ascites (n = 2), esophagus (n = 2), small intestine (n = 2), mesentery (n = 1), liver (n = 1), pelvis (n = 1), and soft tissue (n = 1).
Table 1.
Clinicopathological and genomic characteristics of 116 gastric cancer cases prospectively assayed by a comprehensive genomic profiling assay
Overall, 501 cancer-related genomic alterations were identified in 116 cases, yielding an average of 4.32 alterations per sample (Table 1; Fig. 1). Of 501 GAs identified, 210 (41%) were clinically relevant alterations, yielding an average of 1.8 clinically relevant GAs per case (Table 1). Moreover, 78% of GC cases harbored at least 1 clinically relevant variant associated with targeted therapies approved by the U.S. Food and Drug Administration (FDA) or mechanism-based trials (Table 2). The most common clinically relevant GAs were KRAS, CDKN2A, CCND1, ERBB2, PIK3CA, MLL2, MET, PTEN, ATM, DNMT3A, NF1, NRAS, and MDM2 (Table 2). The most common GAs in the 116 cases were TP53 (58 cases, 50%), ARID1A (28 cases, 24%), and CDH1 (17 cases, 15%).
Figure 1.
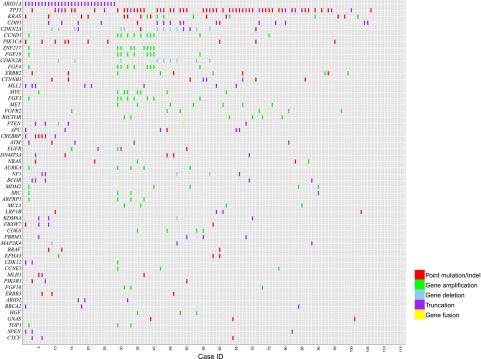
Tile plot of genomic alterations in 116 consecutive gastric cancer cases.
Table 2.
Therapeutic implications of recurrent somatic genomic alterations in 116 clinical gastric cancer cases analyzed by prospective genomic profiling
Twenty-eight cases (24%) harbored loss-of-function ARID1A alterations (Fig. 2). ARID1A-altered samples harbored an increased frequency of CREBBP variants (p < .0005), PIK3CA variants (p = .0017), and MLL2 variants (p = .019) and were less likely to harbor TP53 variants (p < .050). In this series, tumors with ARID1A alterations had less frequent amplifications of cancer-related genes compared with cases with wild-type ARID1A. This finding was statistically significant but was not replicated in an independent set of 192 gastric carcinomas (data not shown). All other ARID1A findings remained significant with a type I error rate of <.05 with a multiple hypothesis correction applied.
Figure 2.
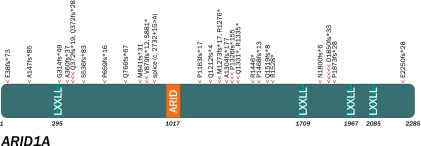
Lollipop plot graphically depicting the location of ARID1A genomic alterations in the 28 ARID1A-altered gastric cancer cases (one arrowhead per genomic alteration [GA] in this series, with some cases harboring several ARID1A GAs).
Somatic CDH1 mutations were found in 6 of 24 (25%) diffuse GC cases compared with 11 of 92 (12%) nondiffuse GC cases (p = .12). Matched normal tissue was not available to investigate germline CDH1 status. Enrichment of APC, CREBBP, and MLL2 alterations was observed in intestinal GC; 3 of 12 cases (25%) contained APC mutations compared with 3 of 104 cases (2.9%) of nonintestinal GC (p = .014). APC variants were not observed in any of the 24 diffuse GC cases. CREBBP was altered in 4 of 12 (33%) intestinal cases compared with 2 of 104 (1.9%) nonintestinal cases (p < .001), and MLL2 was altered in 4 of 12 (33%) intestinal cases compared with 4 of 104 (3.8%) nonintestinal cases (p = .0038).
Alterations of genes involved in mismatch repair were observed in this series at a frequency of 2.6% for MLH1, 0.8% for MSH2, and 0.8% for MSH6. Three of these five cases harbored truncating alterations that are predicted to cause loss of function, and no single case contained more than one alteration in this pathway. No information on microsatellite stability assessed by PCR testing was available.
Alterations in receptor tyrosine kinases (RTKs) were harbored by 24 cases (20.6%). Ten samples (8.6%) harbored ERBB2 alterations, 5 contained somatic base substitutions and 5 harbored amplifications (6–24 copies), with these events being mutually exclusive. ERBB2 base substitutions in this series consisted of R678Q (two cases), S310F (one case), L755S (one case), and V842I (one case). One case harbored both ERBB2 R678Q and MET amplification. CDH1 alteration was not associated with ERBB2 alteration in our GC sample (data not shown).
Seven cases (6%) harbored MET amplifications (7–30 copies) and seven cases (6%) had FGFR2 amplifications (12–32 copies) (Table 2). One patient with MET-amplified gastric carcinoma received crizotinib and achieved disease control (Fig. 3).
Figure 3.

Response to crizotinib in a patient with MET-amplified gastric cancer identified by prospective comprehensive genomic profiling. (A): Precrizotinib FDG-avid hepatic metastasis. (B–D): Ongoing response up to 5 months after crizotinib initiation.
One FGFR2-altered case also harbored a coexisting ARID1A alteration. EGFR alterations were detected in four cases (3.4%) consisting of two amplifications, one point mutation (F795V), and one case with a deletion of exons 2–7 (EGFR viii). Rare RTK alterations identified included FLT3 (amplification; one case), KIT (D716N; one case), and FGFR3 (amplification; one case) (supplemental online Table 1). EGFR amplifications were not exclusive of other RTK alterations because they coexisted with FGFR2 amplification (one case) and both MET and ERBB2 amplifications (one case). No ROS1 alterations, including fusions, were detected. Among clinically relevant alterations in other kinases, BRAF alterations occurred at a frequency of 2.6%, two cases harbored D594X and one case harbored G469V (supplemental online Table 1). Alterations in vascular endothelial growth factor receptors 1–3 (VEGFR1–3) were limited to KDR (VEGFR2) R275* and FLT4 (VEGFR3) S637R in all 116 GC cases, and neither alteration has been linked to benefit from ramucirumab [13, 15].
Discussion
The phase III ToGA trial demonstrated the power of molecular testing to prospectively identify a molecularly defined subgroup of patients who are likely to benefit from anti-ERBB2 (HER2)-directed therapy; the addition of trastuzumab to standard 5-fluorouracil/platinum chemotherapy led to a statistically significant 2.5-month improvement in overall survival for ERBB2-amplified gastric cancer [8]. The methods of molecular testing, however, are also important, as demonstrated by the negative results of the TyTAN trial. In that trial, the addition of lapatinib (an oral HER2 inhibitor) to paclitaxel did not lead to significant improvement in progression-free survival or overall survival (OS) when compared with single-agent paclitaxel as second-line treatment in EBRR2-amplified GC, as determined by fluorescence in situ hybridization (FISH) alone [20]. Subgroup analysis indicated that ERBB2 amplification and immunohistochemistry (IHC) 3+ derived a significant 6.4-month improvement in overall survival. Similarly, early reporting from the LoGIC trial demonstrated OS improvements in Asian patients and those aged <60 years but failed to demonstrate an association between OS and IHC score [21]. Neither of these trials included patients with ERBB2 mutations.
In contrast, the phase III EXPAND and REAL3 trials in an unselected gastric and esophageal cancer population failed to demonstrate improvement in outcomes with the addition of the EGFR-targeted therapies cetuximab or panitumumab to chemotherapy [22, 23]. Retrospective analysis of this trial has not identified any predictive response biomarkers to date [13]. Similar negative results have also been observed in the phase III GRANITE-1 trial of everolimus in unselected advanced GC patients, and few data support empiric use of targeted and biologic agents in advanced GC [24].
Prior reports characterizing the genomic landscape of gastric carcinoma have relied on banked, therapy-naive tissue from primary resections [10, 16–18, 25, 26]. Recent work from the Cancer Genome Atlas has defined four groups of gastric carcinomas, each harboring positivity for Epstein-Barr virus (EBV), microsatellite instability, or multiple copy number amplifications while genomically stable or characterized by chromosomal instability [27]. Characteristic single-gene alterations were often but not perfectly associated with each group; EBV-positive gastric carcinomas often harbored PIK3CA alterations, and the genomically stable group often harbored RHOA alterations [27]. These groups offer insight into common etiologies but do not currently direct therapeutic decision making.
In contrast, the prospective series presented in this study reflects samples characteristic of clinical practice because the series is composed of cases from patients typically with advanced gastric carcinoma. A high percentage of cases (78%) harbor clinically relevant genomic alterations, including 1 in 5 cases (20.6%) with alterations in RTKs, suggesting the utility of comprehensive genomic profiling to match patients with targeted therapies of specific potential benefit in clinical trials (Table 2). For the common genomic alterations KRAS, ARID1A, and TP53, their clinical relevance is best linked to possible benefit from clinical trials with targeted agents. The recent FDA approval of trametinib as a MEK pathway inhibitor for melanoma has resulted in the anecdotal use of trametinib in other tumors types and assessment in clinical trials for other indications [28]. Similarly for TP53 and ARID1A, targeted therapies are in clinical development, such kevetrin (NCT01664000), and inhibitors of chromatin remodeling. A recent development is the paradigm of master trials, such as the Novartis Signature Trial, with multiple agents and genomically defined entry criteria for advanced cancers rather than restriction to a tumor type. Such a trial design can accommodate the addition of future therapies to be developed, and genomic profiling can provide the rationale for entering patients.
We identified five GC cases with ERBB2-activating base substitutions, which cannot be detected by IHC or FISH [29].
The recent description of somatic ERBB2 base substitutions in breast carcinoma and micropapillary urothelial carcinoma suggests that such alterations may also be oncogenic drivers in GC, and at least one such breast carcinoma patient has responded to anti-ERBB2 (HER2)-targeted therapy [29–32]. Both ERBB2 amplifications and base substitutions were observed in our patient series but were mutually exclusive, consistent with observations in breast carcinoma [33]. The low frequency in this series (4.3%) of ERBB2 amplification in contrast with the 20% frequency observed in previous studies is likely is due to a selection bias, that is, cases submitted for genomic profiling were previously tested for ERBB2 amplification and were negative for ERBB2, prompting a search for therapeutic alternatives [34].
Among ERBB2 base substitutions in this series, some have been functionally characterized as activating and sensitive to lapatinib (S310F, V842I) or resistant to lapatinib (L755S) [30]. The frequency of 4.3% was very similar to the frequency reported in other series, confirming that no selection bias was present because standard of care testing does not detect these clinically relevant alterations [34, 35]. Although ERBB2 R678Q was not found to be activating or to confer resistance to anti-ERBB2 (HER2)-targeted therapy, it has been observed multiple times in the context of cancer, which may indicate biologic significance. Activating ERBB2 base substitutions appear to be sensitive to neratinib, suggesting a possible pathway to clinical treatment, and genomically selected basket trials of neratinib (NCT01953926) are ongoing for ERBB2-altered tumors [30].
Previous studies have strongly associated ERBB2-amplified gastric carcinomas with intestinal type histology and proximal gastric location [8, 12]. All five ERBB2-amplified GC cases in this series had histology diagnosed as or at least suggestive of the intestinal subtype but were approximately evenly distributed in site of origin between the proximal and distal stomach (supplemental online Table 1). In contrast, ERBB2 base substitutions were associated with signet ring features in three of four cases with histology available for review. The differing histology of these cases may suggest differing clinicopathologic characteristics of ERBB2-amplified GC compared with ERBB2 base-substituted gastric carcinomas, but this awaits independent confirmation in a larger series.
Alterations in the FGFR family are well recognized as oncogenic drivers [36, 37]. FGFR2 was amplified at 6% in this GC series, similar to a previous study [18, 38]. Limited clinical studies have shown that FGFR2-amplified breast carcinoma patients responded favorably to dovitinib, a multikinase inhibitor that inhibits FGFR family members [39]. For FGFR2-amplified GC, preclinical evidence suggests such tumors are sensitive to FGFR-targeted therapy, and molecularly stratified clinical trials are ongoing (NCT01719549) [40].
Amplification of MET is a known driver of gastroesophageal and lung carcinomas and other tumor types [38, 41, 42]. We identified MET amplification (>6 copies) in 6% of GC cases in this series. Based on the genomic profile, one of the patients with MET amplification (12 copies) was treated with crizotinib and had regression of liver metastasis and disease control for 5 months (Fig. 3). This finding is consistent with previous results for phase I trials for crizotinib in which two advanced gastroesophageal carcinoma patients with MET amplifications (FISH MET/CEN7P ratio of >2.2) had partial response and stable disease with time to progression of 3–4 months [41]. The comprehensive genomic profiling assay in this series used a process-matched normal control to quantitatively estimate the absolute copies of MET while controlling for ploidy. The threshold of six copies for the designation of MET amplification by FoundationOne in cases with a diploid genome can be translated as exceeding a MET/CEP7 ratio of 2.2 (Fig. 4).
Figure 4.
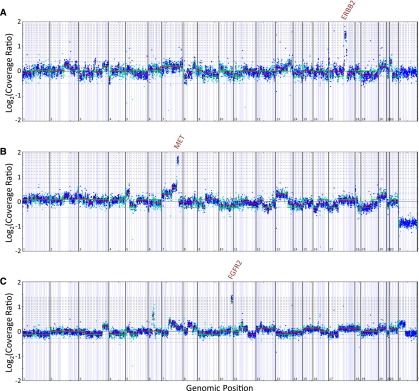
Copy number alteration plots for several cases harboring receptor tyrosine kinase amplifications.
Notably, the comprehensive genomic profiling assay used in this study (FoundationOne) provides quantitative estimates of copy number amplifications (Fig. 4). Copy number estimates made by the genomic profiling assay used do not directly translate to a FISH ratio per se but, as shown by our patient response, provide clinically relevant information that can guide use of targeted therapy (Fig. 3). Comprehensive genomic profiling provides the advantage of simultaneous assessment of many possible clinically relevant copy number amplifications including MET, FGFR2, and ERBB2 while minimizing consumption of the specimen [19]. In contrast, other forms of molecular testing are hypothesis driven, that is, a “hotspot” exon examines only specific exons of genes of interest and is often combined with FISH to assay for amplifications (i.e., ERBB2 and MET). A focused approach offers conceptual simplicity, but for those cases harboring relevant genomic alterations outside the scope of such hotspot testing, genomic profiling could be done to identify potential benefits of targeted therapy instead of expending both time and resources on hotspot testing that might not yield information to guide treatment.
In one of the largest screening studies for KRAS mutations involving GC samples from U.K., Japan, and Singapore, KRAS mutations were found in 29 of 710 GC samples (4.1%). The frequency of KRAS mutations was 5.8% among U.K. patients, 4.0% among Japanese patients, and 1.5% among Singapore Chinese patients. The role of KRAS mutation in GC is unknown, but in this series, KRAS mutations were identified in 16% of GC cases. The most common alterations were G12V (3.4%) and G12D (2.5%), which are both transversions. This most likely reflects a selection bias in this sample population, with patients sent for genomic profiling having poor prognosis and possible KRAS enrichment.
Alterations in the tumor suppressors TP53 and ARID1A are common in gastric cancer. ARID1A encodes the AT-rich interactive domain-containing protein 1A, a member of the SWI/SNF chromatin-remodeling complex. Inactivating alterations in ARID1A are frequent in ovarian clear cell carcinomas, neuroblastomas, and gastric carcinomas and loss of expression in other tumor types, consistent with the hypothesized tumor suppressor role of this protein [43, 44]. Alterations of ARID1A in this series did not cluster around a hotspot (Fig. 2). The statistically significant enrichment of PIK3CA and the paucity of TP53 alterations in the set of ARID1A-altered GCs are consistent with previous findings in gastric carcinoma [43, 45]. In our series, ARID1A-altered cases were also enriched for CREBBP and MLL2 alterations.
Interestingly, our series identified somatic CDH1 mutation rates (25% diffuse, 12% nondiffuse) higher than previously reported (Fig. 5) [46]. The differences in alteration frequency may be related to the interrogation of the entire coding sequence of CDH1 in this assay compared with the limited hotspot assessment of exon 7–10 hotspot interrogation in prior series [46, 47]. CDH1 somatic mutation has been correlated with the shortest survival in GC, and selection bias may underlie increased CDH1 mutation rates in this series because clinicians may be likely to reach for mutational profiling when options are limited [46]. In reporting these results, caveats for directed germline testing are included.
Figure 5.
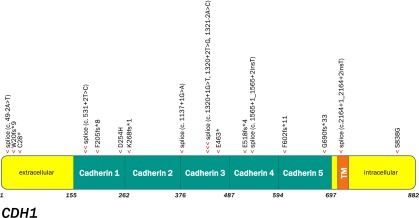
Lollipop plot graphically depicting the location of CDH1 genomic alterations (GAs) in the 17 CDH1-altered gastric cancer cases (one arrowhead per GA in this series).
Abbreviation: TM, transmembrane.
Neither BRAF V600E or V600M alterations previously described in GC were observed in this series, but three nonBRAFV600 mutations were found in this series [13, 48–50] (supplemental online Table 1). The BRAF alterations in these series are variable activators of BRAF.
Interestingly, no alterations in VEGFR1–3 were identified in this series. Ramucirumab has been shown to improve OS in GC in second-line treatment as a single agent or in combination with paclitaxel. Ramucirumab is a monoclonal antibody against VEGFR2, but, like other antiangiogenic therapies, there are no clear predictive biomarkers [51, 52]. Current evidence is insufficient to examine whether VEGFR1–3 alterations serve as biomarkers for ramucirumab.
Identifying clinically relevant alterations in the course of clinical care of gastric carcinoma may drive clinical decisions making, which in turn will generate preliminary data on the efficacy of targeted therapies in gastric carcinoma and care of future patients and will support future systematic investigation through clinical trials. At present, most suggestion of benefit from targeted therapy in gastric carcinoma is guided by analogy to other tumor types. Such reasoning highlights the limitations of this approach, for example, BRAF V600E-mutant colorectal adenocarcinoma has not suggested benefit from vemurafenib monotherapy [53, 54]. Because new approaches such as the combination of vemurafenib and erlotinib confer some benefit to similar patients, as noted, it is hoped that analogous approaches can also benefit gastric carcinoma patients [54].
Conclusion
The high frequency of clinically relevant genomic alterations in this patient population reflective of routine clinical practice is encouraging in a disease that continues to have a poor prognosis with modern chemotherapy. The clinically relevant alterations identified by this assay beyond those detected by standard of case can drive increased clinical trial participation and development (e.g., ERBB2 base substitutions, MET amplifications) and clarify predictive response and resistance biomarkers.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
We acknowledge important contributions from other researchers that could not be cited due to space constraints. No external funding or grant support was involved in this study. This work is dedicated to the patients profiled in this report. G.A.P. is currently affiliated with NantHealth, Culver City, CA.
Author Contributions
Conception/Design: Siraj M. Ali, Eric M. Sanford, Juliann Chmielecki, Vincent A. Miller
Provision of study material or patients: Daniel V. Catenacci, Fadi Braiteh, Sai-Hong Ignatius Ou
Collection and/or assembly of data: Siraj M. Ali, Eric M. Sanford, Kai Wang, Roman Yelensky, Doron Lipson, Daniel V. Catenacci, Fadi Braiteh, Philip J. Stephens, Jeffrey S. Ross
Data analysis and interpretation: Siraj M. Ali, Eric M. Sanford, Kai Wang, Norma A. Palma, Juliann Chmielecki, Roman Yelensky, Gary A. Palmer, Doron Lipson, Daniel V. Catenacci, Jeffrey S. Ross, Sai-Hong Ignatius Ou
Manuscript writing: Siraj M. Ali, Eric M. Sanford, Samuel J. Klempner, Douglas A. Rubinson, Kai Wang, Norma A. Palma, Juliann Chmielecki, Gary A. Palmer, Deborah Morosini, Doron Lipson, Daniel V. Catenacci, Rachel Erlich, Philip J. Stephens, Jeffrey S. Ross, Sai-Hong Ignatius Ou, Vincent A. Miller
Final approval of manuscript: Siraj M. Ali, Juliann Chmielecki, Jeffrey S. Ross, Vincent A. Miller
Disclosures
Siraj M. Ali: Foundation Medicine Inc. (E, OI); Eric M. Sanford: Foundation Medicine Inc. (E, OI); Kai Wang: Foundation Medicine Inc. (E, OI); Norma A. Palma: Foundation Medicine Inc. (E, OI); Juliann Chmielecki: Foundation Medicine Inc. (E, OI); Roman Yelensky: Foundation Medicine Inc. (E, IP); Gary A. Palmer: Foundation Medicine Inc. (E, OI); Deborah Morosini: Foundation Medicine Inc. (E, OI); Doron Lipson: Foundation Medicine Inc. (E, OI, IP); Daniel V. Catenacci: Foundation Medicine Inc. (C/A); Fadi Braiteh: Foundation Medicine, Caris Life Sciences, Genomic Health, Molecular Health (C/A); Amgen, Bayer, Pfizer, BMS, Celgene, Insys, incyte, Caris Life Sciences, Genomic Health (H); Rachel Erlich: Foundation Medicine Inc. (E, OI); Philip J. Stephens: Foundation Medicine Inc. (E, OI); Jeffrey S. Ross: Foundation Medicine Inc. (RF, E, OI, IP); Vincent A. Miller: Foundation Medicine Inc. (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Bertuccio P, Chatenoud L, Levi F, et al. Recent patterns in gastric cancer: A global overview. Int J Cancer. 2009;125:666–673. doi: 10.1002/ijc.24290. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi T, Saikawa Y, Kitagawa Y. Gastric cancer: Current status of diagnosis and treatment. Cancers (Basel) 2013;5:48–63. doi: 10.3390/cancers5010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz RE, Zagala-Nevarez K. Ethnic survival differences after gastrectomy for gastric cancer are better explained by factors specific for disease location and individual patient comorbidity. Eur J Surg Oncol. 2002;28:214–219. doi: 10.1053/ejso.2001.1234. [DOI] [PubMed] [Google Scholar]
- 6.Strong VE, Song KY, Park CH, et al. Comparison of gastric cancer survival following R0 resection in the United States and Korea using an internationally validated nomogram. Ann Surg. 2010;251:640–646. doi: 10.1097/SLA.0b013e3181d3d29b. [DOI] [PubMed] [Google Scholar]
- 7.Strong VE, Song KY, Park CH, et al. Comparison of disease-specific survival in the United States and Korea after resection for early-stage node-negative gastric carcinoma. J Surg Oncol. 2013;107:634–640. doi: 10.1002/jso.23288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 9.Wadhwa R, Song S, Lee JS, et al. Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol. 2013;10:643–655. doi: 10.1038/nrclinonc.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gravalos C, Jimeno A. HER2 in gastric cancer: A new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19:1523–1529. doi: 10.1093/annonc/mdn169. [DOI] [PubMed] [Google Scholar]
- 12.Hu B, El Hajj N, Sittler S, et al. Gastric cancer: Classification, histology and application of molecular pathology. J Gastrointest Oncol. 2012;3:251–261. doi: 10.3978/j.issn.2078-6891.2012.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okines AF, Gonzalez de Castro D, Cunningham D, et al. Biomarker analysis in oesophagogastric cancer: Results from the REAL3 and TransMAGIC trials. Eur J Cancer. 2013;49:2116–2125. doi: 10.1016/j.ejca.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Smyth EC, Cunningham D. Gastric cancer in 2012: Defining treatment standards and novel insights into disease biology. Nat Rev Clin Oncol. 2013;10:73–74. doi: 10.1038/nrclinonc.2012.228. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs CS, Tomasek J, Yong CJ, et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31–39. doi: 10.1016/S0140-6736(13)61719-5. [DOI] [PubMed] [Google Scholar]
- 16.Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 17.Dulak AM, Schumacher SE, van Lieshout J, et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012;72:4383–4393. doi: 10.1158/0008-5472.CAN-11-3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng N, Goh LK, Wang H, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61:673–684. doi: 10.1136/gutjnl-2011-301839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satoh T, Xu RH, Chung HC, et al. Lapatinib plus paclitaxel versus paclitaxel alone in the second-line treatment of HER2-amplified advanced gastric cancer in Asian populations: TyTAN—a randomized, phase III study. J Clin Oncol. 2014;32:2039–2049. doi: 10.1200/JCO.2013.53.6136. [DOI] [PubMed] [Google Scholar]
- 21.Hecht JR, Bang Y-J, Qin S, et al. Lapatinib in combination with capecitabine plus oxaliplatin (CapeOx) in HER2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma (AC): The TRIO-013/LOGiC trial. J Clin Oncol. 2013;31(suppl):LBA4001a. doi: 10.1200/JCO.2015.62.6598. [DOI] [PubMed] [Google Scholar]
- 22.Lordick F, Kang YK, Chung HC, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:490–499. doi: 10.1016/S1470-2045(13)70102-5. [DOI] [PubMed] [Google Scholar]
- 23.Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:481–489. doi: 10.1016/S1470-2045(13)70096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohtsu A, Ajani JA, Bai YX, et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase III GRANITE-1 study. J Clin Oncol. 2013;31:3935–3943. doi: 10.1200/JCO.2012.48.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto H, Watanabe Y, Maehata T, et al. An updated review of gastric cancer in the next-generation sequencing era: Insights from bench to bedside and vice versa. World J Gastroenterol. 2014;20:3927–3937. doi: 10.3748/wjg.v20.i14.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K, Yuen ST, Xu J, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 27.Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- 29.Ross JS. Breast cancer biomarkers and HER2 testing after 10 years of anti-HER2 therapy. Drug News Perspect. 2009;22:93–106. doi: 10.1358/dnp.2009.22.2.1334452. [DOI] [PubMed] [Google Scholar]
- 30.Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross JS, Wang K, Sheehan CE, et al. Relapsed classic E-cadherin (CDH1)-mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19:2668–2676. doi: 10.1158/1078-0432.CCR-13-0295. [DOI] [PubMed] [Google Scholar]
- 32.Ali SM, Alpaugh RK, Downing SR, et al. Response of an ERBB2-mutated inflammatory breast carcinoma to human epidermal growth factor receptor 2-targeted therapy. J Clin Oncol. 2014;32:e88–e91. doi: 10.1200/JCO.2013.49.0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ross JS. Update on HER2 testing for breast and upper gastrointestinal tract cancers. Biomarkers Med. 2011;5:307–318. doi: 10.2217/bmm.11.31. [DOI] [PubMed] [Google Scholar]
- 34.Lee J, Ou SH. Towards the goal of personalized medicine in gastric cancer—time to move beyond HER2 inhibition. Part II: Targeting gene mutations and gene amplifications and the angiogenesis pathway. Discov Med. 2013;16:7–14. [PubMed] [Google Scholar]
- 35.Lee J, Ou SH. Towards the goal of personalized medicine in gastric cancer—time to move beyond HER2 inhibition. Part I: Targeting receptor tyrosine kinase gene amplification. Discov Med. 2013;15:333–341. [PubMed] [Google Scholar]
- 36.Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu YJ, Shen D, Yin X, et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br J Cancer. 2014;110:1169–1178. doi: 10.1038/bjc.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.André F, Bachelot T, Campone M, et al. Targeting FGFR with dovitinib (TKI258): Preclinical and clinical data in breast cancer. Clin Cancer Res. 2013;19:3693–3702. doi: 10.1158/1078-0432.CCR-13-0190. [DOI] [PubMed] [Google Scholar]
- 40.Xie L, Su X, Zhang L, et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin Cancer Res. 2013;19:2572–2583. doi: 10.1158/1078-0432.CCR-12-3898. [DOI] [PubMed] [Google Scholar]
- 41.Lennerz JK, Kwak EL, Ackerman A, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011;29:4803–4810. doi: 10.1200/JCO.2011.35.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013;31:1089–1096. doi: 10.1200/JCO.2012.43.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 44.Jones S, Wang TL, Shih IM, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014 doi: 10.1038/nature13480. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corso G, Carvalho J, Marrelli D, et al. Somatic mutations and deletions of the E-cadherin gene predict poor survival of patients with gastric cancer. J Clin Oncol. 2013;31:868–875. doi: 10.1200/JCO.2012.44.4612. [DOI] [PubMed] [Google Scholar]
- 47.Oliveira C, Sousa S, Pinheiro H, et al. Quantification of epigenetic and genetic 2nd hits in CDH1 during hereditary diffuse gastric cancer syndrome progression. Gastroenterology. 2009;136:2137–2148. doi: 10.1053/j.gastro.2009.02.065. [DOI] [PubMed] [Google Scholar]
- 48.Lee SH, Lee JW, Soung YH, et al. BRAF and KRAS mutations in stomach cancer. Oncogene. 2003;22:6942–6945. doi: 10.1038/sj.onc.1206749. [DOI] [PubMed] [Google Scholar]
- 49.Wu M, Semba S, Oue N, et al. BRAF/K-ras mutation, microsatellite instability, and promoter hypermethylation of hMLH1/MGMT in human gastric carcinomas. Gastric Cancer. 2004;7:246–253. doi: 10.1007/s10120-004-0300-9. [DOI] [PubMed] [Google Scholar]
- 50.van Grieken NC, Aoyama T, Chambers PA, et al. KRAS and BRAF mutations are rare and related to DNA mismatch repair deficiency in gastric cancer from the East and the West: Results from a large international multicentre study. Br J Cancer. 2013;108:1495–1501. doi: 10.1038/bjc.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jubb AM, Oates AJ, Holden S, et al. Predicting benefit from anti-angiogenic agents in malignancy. Nat Rev Cancer. 2006;6:626–635. doi: 10.1038/nrc1946. [DOI] [PubMed] [Google Scholar]
- 52.Van Cutsem E, de Haas S, Kang YK, et al. Bevacizumab in combination with chemotherapy as first-line therapy in advanced gastric cancer: A biomarker evaluation from the AVAGAST randomized phase III trial. J Clin Oncol. 2012;30:2119–2127. doi: 10.1200/JCO.2011.39.9824. [DOI] [PubMed] [Google Scholar]
- 53.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 54.Yaeger R, Cercek A, O’Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.