

Abstract
The presence of SF3B1 gene mutations is a hallmark of refractory anemia with ring sideroblasts (RARS). However, the mechanisms responsible for iron accumulation that characterize the Myelodysplastic Syndrome with ring sideroblasts (MDS-RS) are not completely understood. In order to gain insight in the molecular basis of MDS-RS, an integrative study of the expression and mutational status of genes related to iron and mitochondrial metabolism was carried out. A total of 231 low-risk MDS patients and 81 controls were studied. Gene expression analysis revealed that iron metabolism and mitochondrial function had the highest number of genes deregulated in RARS patients compared to controls and the refractory cytopenias with unilineage dysplasia (RCUD). Thus mitochondrial transporters SLC25 (SLC25A37 and SLC25A38) and ALAD genes were over-expressed in RARS. Moreover, significant differences were observed between patients with SF3B1 mutations and patients without the mutations. The deregulation of genes involved in iron and mitochondrial metabolism provides new insights in our knowledge of MDS-RS. New variants that could be involved in the pathogenesis of these diseases have been identified.
Introduction
Myelodysplastic syndromes (MDS) are clonal hematological disorders characterized by blood cytopenias, ineffective hematopoiesis and hypercellular bone marrow [1]. According to the WHO classification (2008), six subtypes of MDS are distinguished: refractory cytopenia with unilineage dysplasia (RCUD), refractory anemia with ring sideroblasts (RARS), refractory cytopenia with multilineage dysplasia (RCMD), refractory anemia with excess blasts (RAEB-1 and RAEB-2), MDS-unclassified (MDS-U) and MDS associated with isolated del(5q) [2]. Patients with RARS present with isolated anemia, hypochromic erythrocytes, hyperplastic ineffective erythropoiesis and mitochondrial ferritin accumulation in erythroid precursor cells. RARS and RCMD with ring sideroblasts (RCMD-RS) are defined by the presence of more than 15% ringed sideroblasts. The accumulation of ferritin is present in the ring sideroblasts and this is likely to be involved in the increased apoptosis of erythroblasts and, therefore, in ineffective erythropoiesis [3].
Iron is essential for heme synthesis and Fe-S cluster biogenesis in the erythroid cell. Iron is acquired by the erythroid precursors and it is imported into mitochondria by SLC25A37 (Mitoferrin-1) [4]. This protein is a member of the solute carrier family, and is localized in the inner mitochondrial membrane, where it is an essential iron importer. Heme synthesis is initiated in the mitochondrion and the iron that is not incorporated during this process is stored in Fe-S clusters and transported out of the mitochondrion by the ABCB7 membrane protein [5,6,7].
High expression of some heme biosynthesis-related genes, such as ALAS2 and FECH, have been seen in RARS patients [8], whilst low levels of ABCB7 gene expression in patients with RARS compared with other MDS subtypes have also been found [9]. However, no mutations in these genes have been detected in acquired RARS [9,10]. By contrast, several genetic lesions have been identified in inherited sideroblastic anemias, including mutations in the SLC25A38, ALAS2 and ABCB7 genes [11,12,13,14,15].
The presence of recurrent somatic mutations of the splicing factor 3B subunit 1 (SF3B1) gene in a high proportion of patients with RARS (64–83%) or RCMD-RS (57–76%) have been recently demonstrated [16,17,18,19,20,21]. SF3B1 is located on chromosome 2q33.1 and encodes the SF3B1 protein, which plays a role in pre-mRNA splicing and associated transcription [21]. In addition, recent studies have shown a possible role of SF3B1 in the formation of ring sideroblasts in MDS [22,23]. However, some authors suggest that RARS is a disease resulting from a specific alteration in one or more genes involved in mitochondrial function, iron distribution, or both [3]. Thereby, the abnormal mitochondrial iron metabolism that characterizes RARS is not completely understood and the pathogenesis of ring sideroblasts in MDS remains to be clarified.
In order to gain insight in our knowledge of the abnormal iron accumulation, defective mitochondrial function and ineffective heme biosynthesis in low-risk MDS, an integrative study of both expression and mutational status of genes related to iron and mitochondria was carried out. Our study has shown that SLC25A37 and SLC25A38 were over-expressed in RARS patients, and has identified one sequence change in the ALAD gene that could contribute to a better understanding of the pathogenesis of sideroblastic MDS.
Materials and Methods
Patients, samples and cell separation
A total cohort of 231 low-risk MDS patients and 81 controls were included in the studies performed in this work. 69 low-risk MDS patients (30 RARS patients and 39 RCUD patients) and 31 controls without hematological malignancies were included in the gene expression profiling study. Moreover, 6 MDS with ring sideroblasts were analyzed by massive DNA-sequencing techniques, while 175 low-risk MDS patients were analyzed by conventional Sanger sequencing (100 MDS with ring sideroblasts and 75 were other low-risk MDS) (S1 Table). All patients were classified according to the World Health Organization (WHO) 2008 criteria [2] (with the exception of RCMD-RS, which we maintain from the World Health Organization, 2002 [24] as a separate category). In addition, 50 healthy controls were also included in the Sanger sequencing study. Mononuclear cells were isolated from bone marrow (BM) of MDS patients and controls by density gradient (Ficoll). The unfractionated mononuclear cells were used for the expression and sequencing studies. In addition, CD3+ cells from peripheral blood from patients of interest were purified using magnetically activated cell sorting (MACS) CD3 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). The study was approved by the Local Ethical Committee “Comité Ético de Investigación Clínica, Hospital Universitario de Salamanca” and written informed consent was obtained from each patient and their relatives.
RNA and DNA isolation
Total RNA was extracted from cells by homogenization in TRIZOL (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol, treated with RQ1 RNAse-Free DNase (Promega, Madison, USA) to eliminate genomic DNA contamination, and finally purified with RNeasy Minikit (Qiagen, Hilden, Germany). RNA quantity and quality was determined with an Agilent 2100 Bioanalyzer (Santa Clara, CA, USA). Genomic DNA from subject samples was isolated using DNeasy blood and tissue kit following the manufacturer’s protocol (Qiagen).
Gene expression microarray studies
Gene expression profiling (GEP) studies were carried out as part of the Microarray Innovations in LEukemia (MILE) study [25]. GeneChip Human Genome U133 plus 2.0 arrays (Affymetrix, High Wycombe, UK) are gene expression arrays containing 54 613 oligonucleotide probesets that map onto 18 950 human gene loci (obtained using the mapping of microarrays probes to ENSEMBL gene IDs provided by GATExplorer) [26]. RNA was labeled and hybridized according to protocols from Affymetrix. Briefly, 100 ng of total RNA was amplified and labeled using the GeneChip two-cycle cDNA synthesis kit and GeneChip IVT labeling kit (Affymetrix Inc.) and then hybridized onto a Human Genome U133 Plus 2.0 microarray, after quality checking on GeneChip Test3 arrays. Washing and scanning were done with a Fluidics Station 400 and GeneChip Scanner (Affymetrix Inc.) as previously described [27]
Bioinformatic methods for global expression profiling and differential analysis
Robust Microarray Analysis (RMA) algorithm was applied to the raw data of the expression arrays to achieve background correction and intra- and inter-normalization, and to calculate the expression signal [28]. The Significant Analysis of Microarrays (SAM) algorithm was used to identify genes with statistically significant changes differences in expression between different classes [29]. For this purpose, samples were permuted over 100 cycles by using the two-class (unpaired) and multiclass response format, assuming unequal variances for the genes. Significant genes were selected on the basis of a false discovery rate (FDR) of <0.05. To select each gene the p-values of each statistical test were transformed to q-values using the indicated FDR threshold. The algorithms described were applied using the R programming environment and the Bioconductor package.
Validation of the gene expression signature with independent cohort of patients
In order to validate the gene expression signatures obtained with the MDS patient cohorts studied, an independent analysis was performed using gene expression data from bone marrow CD34+ cells of MDS patients and healthy controls obtained from GSE19429 (GEO database: http://www.ncbi.nlm.nih.gov/geo/).
Differential analysis in low-risk MDS with and without SF3B1/SRSF2 mutations
In order to find genes that mark possible differences between low-risk MDS samples with mutated SF3B1/SRSF2 genes (n = 22) and low-risk MDS samples without mutations (n = 13), a recursive algorithm written in R was designed and applied. This alogarithm produces multiple subsets of samples from each class and run differential expression analysis for each one of these subsets using LIMMA [30]. In this way, random selected groups of 7 versus 7 samples were analyzed running a total of 10000 differential expression tests. The recursive algorithm identified and counted the genes that gave a significant differential expression in each one of the contrasts (using as cutoff: q-value < 0.01, that corresponds to the p-value adjusted by FDR). In this way, a gene signature of 200-top genes was selected. Once these genes were selected we used an outcome/response algorithm called Global Test [31] to check further if the genes selected were able to predict the two classes investigated.
The same strategy was used to compare MDS-RS with mutated SF3B1 (n = 13) and non-mutated SF3B1 (n = 6). However in this case, we random selected groups of 4 versus 4 samples running a total of 7425 differential expression analyses and a p-value adjusted by FDR < 0.10 was used. A gene signature of 75-top genes was selected, being genes that appeared as significant in at least ten different contrasts.
Functional enrichment analysis
To analyze the functional enrichment of the selected gene lists the bioinformatic resource DAVID (http://david.abcc.ncifcrf.gov/) [32] and the web-delivered bioinformatics tool set IPA (Ingenuity Pathway Analysis 9.0; http://www.ingenuity.com) were used. Both tools enable the functional modules and the most relevant biological processes present in the gene lists to be identified by statistical enrichment analysis based on contingency tests.
Targeted Sequence Capture and DNA Sequencing assay
Array-based sequence capture (Roche NimbleGen) followed by next-generation sequencing (Roche GS FLX Titanium sequencing platform) was used to analyze 93 genes related to hematological malignancies. Details of the design of the array, 454 sequencing, coverage statistics and data analysis are provided in the Supplementary Methods (S1 Methods).
Real-Time PCR
The expression levels of selected genes were analyzed by Real-Time PCR. First-strand cDNA was generated from 1 μg of total RNA using poly-dT as primer and the M-MLV reverse transcriptase (Promega). Real-time PCR was performed in triplicate and was analyzed as previously described [33]. The primers were designed for specific sequences (S2 Table) and checked by the BLAST algorithm [34].
Sanger sequencing
To elucidate and validate the presence of possible genes variations, Sanger re-sequencing was carried out. Oligonucleotide primers were designed against all exons of SLC25A37 and a genomic fragment of exons 6 and 7 in the ALAD gene. A pair of primers to amplify a 1 696-bp genomic fragment and a second reverse internal primer for the sequencing in ALAD was used. All primers were designed using Primer3 (http:/frodo.wi.mit.edu/primer3/) (S3 Table). In addition, the previously published primers against the exons more frequently mutated of SLC25A38 in congenital sideroblastic anemia [14] and against the exon 14 and 15 of SF3B1 were used [17]. Genomic DNA was amplified with the Fast Start High Fidelity PCR System (Roche, Basel, Switzerland) following the manufacturer’s instructions and including some variations of the annealing temperature and magnesium concentration (S3 Table). DNA sequences were evaluated using Scanner v1.0 (Applied Biosystems, Carlsbad, CA, USA) and Accelrys DS Gene v1.5 software. Data were analysed using annotations of genome version GRCh37 (hg19).
Bioinformatics analysis of missense variants
The effects of amino acid changes on protein function were predicted with SIFT using the protein sequences of human ALAD as the input. Homologous protein sequences of the human ALAD gene were retrieved from the NCBI genome database with BLASTP.
Results
Gene expression analysis reveals an iron-related profile in RARS patients
The gene expression profile (GEP) from the BM of 30 RARS patients was compared with that of 31 healthy individuals. A total of 1 145 genes showed significant differences (q-value<0.05) in mRNA expression levels between the two groups: 700 and 445 genes were over and under-expressed, respectively, in the RARS samples (S1 Fig and S4 Table). The most over-expressed gene in RARS was GDF15 (p-value < 0.0001; R fold = 20.27). In addition, genes associated with iron and mitochondrial metabolism represented the largest function group of genes involved: 38% (266 molecules) of the over-expressed genes were identified in this study. Of these genes, 106 were related to mitochondria function, 13 were related to iron binding, including CYBRD1, STEAP3 and ACO1, while 11 were involved in the heme biosynthetic process, six of which (ALAD, HMBS, UROS, UROD, CPOX, PPOX) had a direct role in heme formation. In addition, ABCB6, whose function is to move coproporphyrinogen III from the cytoplasm into the mitochondria during heme biosynthesis, was over-expressed in RARS patients. Nine genes were related to cellular iron ion homeostasis, of which TF, TFR2, TFRC, FXN, SLC25A37 and SCL25A38 were up-regulated in RARS patients (Table 1).
Table 1. Most representative deregulated cellular functions in RARS patients respect to the control group.
| Fuction | Number of genes | p Value | Over-expressed genes |
|---|---|---|---|
| MITOCHONDRION | 106 | 5,70658E-46 | |
| Membrane and Mitochondrion | 32 | 0,00000000 | VDAC3, NDUFV3, SLC25A15, SLC25A36, SLC25A38, ATP7B, BCS1L, C18orf55, HAX1, TIMM50, NDUFB6, APOO, HTRA2, CYB5A, MTX2, NDUFS2, TMEM14C, ABCB6, SLC25A37, C3orf1, CCDC90A, ABCB10, STOML2, STXBP1, HK1, NDUFS4, MRPS17, PPOX, TIMM44, MAOA, SLC25A20, CLIC4 |
| Mitochondrial inner membrane | 23 | 0,00000000 | VDAC3, CHCHD3, NDUFV3, SLC25A15, SLC25A36, SLC25A38, DCI, NME4, BCS1L, NDUFB6, COX15, APOOL, NDUFS2, SLC25A37, SHMT2, HSPD1, FDXR, ABCB10, GCDH, STOML2, GCAT, NDUFS4, SLC25A20 |
| Cytoplasm and Mitochondrion | 21 | 0,00000002 | LONP1, GART, PPP2CA, ATP7B, CAPRIN2, APOO, CYB5A, TP53, DARS2, TXN2, HSPD1, HEBP1, STOML2, SOD1, STXBP1, ISOC2, THG1L, AIFM1, OAT, CLIC4, HMBS |
| Mitochondrial matrix | 16 | 0,00000003 | DNAJA3, DCI, ETFA, COQ3, ACAT1, MIPEP, PCCB, DARS2, HSPD1, FDXR, FXN, GCDH, SOD1, HSPE1, TIMM44, OAT |
| Nucleus and Mitochondrion | 14 | 0,00000128 | GART, PPP2CA, C14orf156, HAX1, TIMM50, REXO2, HTRA2, TP53, APEX2, BRD8, TSFM, SOD1, ISOC2, AIFM1 |
| Nucleolus and Mitochondrion | 4 | 0,00077681 | GART, TP53, TXN2, BRD8 |
| PROTEIN BINDING | 25 | 0,00000000 | VDAC3, CHCHD3, LONP1, DLAT, GART, MRPS9, PPP2CA, ATP7B, PMAIP1, TIMM50, ACAT1, TUFM, SHMT2, FDXR, FXN, GCDH, STXBP1, ISOC2, TIMM44, ATP5B, ARG2, MAOA, AIFM1, OAT, CLIC4 |
| NUCLEOTIDE BINDING | 17 | 0,00000000 | VDAC3, LONP1, GART, ATP7B, NME4, C14orf156, BCS1L, TRAP1, DARS2, TUFM, ABCB6, HSPD1, ABCB10, MRPL39, HK1, TIMM44, ATP5B |
| ATP BINDING | 15 | 0,00000006 | LONP1, GART, ATP7B, NME4, BCS1L, TRAP1, TP53, DARS2, ABCB6, HSPD1, ABCB10, HSPE1, HK1, TIMM44, ATP5B |
| TRANSPORT | 15 | 0,00000027 | NDUFV3, SLC25A15, SLC25A36, SLC25A38, ETFA, NDUFB6, CYB5A, NDUFS2, TXN2, ABCB6, FDXR, ABCB10, NDUFS4, MRPS17, SLC25A20 |
| IRON ION BINDING | 13 | 0,00003287 | PPAT, RRM2, PPP2CA, LIAS, DOHH, RFESD, CYBRD1, MIPEP, ACO1, SLC11A2, NDUFS2, SLC25A37, STEAP3 |
| ELECTRON CARRIER ACTIVITY | 13 | 0,00028086 | ETFA, RFESD, NDUFS2, TXN2, TSTA3, FDXR, STEAP3, GCDH, TXNL3, PPOX, MAOA, AIFM1, SUOX |
| HEME BIOSYNTHETIC PROCESS | 11 | 0,00000001 | ALAD, UROS, FXN, PPOX, CPOX, UROD, HMBS, COX15, EPRS, BLVRB, ABCB6 |
| ELECTRON TRANSPORT CHAIN | 11 | 0,00000514 | NDUFV3, FADS2, ETFA, CYBRD1, NDUFB6, CYB5A, TXNL1, NDUFS2, TXN2, FDXR, NDUFS4 |
| CELLULAR IRON ION HOMEOSTASIS | 9 | 0,00000053 | MYC, TF, ABCB6, FXN, TFR2, SOD1, TFRC, SLC25A37, SLC25A38 |
| HYDROLASE ACTIVITY | 8 | 0,00009485 | MTHFD2, PPP2CA, ATP7B, REXO2, HINT2, THEM2, ATP5B, ARG2 |
| METABOLIC PROCESS | 8 | 0,00017248 | DLAT, C5orf33, DCI, LIAS, ATP7B, COQ3, ACAT1, ISOC2 |
| OXIDOREDUCTASE ACTIVITY | 8 | 0,00062130 | RFESD, TSTA3, FDXR, STEAP3, PPOX, MAOA, AIFM1, SUOX |
| ACYLTRANSFERASE ACTIVITY | 6 | 0,04443590 | DLAT, ESCO2, KS, NAT13, GCAT, TGM2 |
| ION TRANSPORT | 5 | 0,00039908 | ATP7B, TF, SLC25A37, ATP5B, CLIC4 |
To determine whether the genes involved in mitochondrial metabolism were exclusive to the RARS expression profile, this group of patients was compared with other low-risk MDS cases (RCUD group). The comparative analysis of the gene expression profile of both groups identified a set of 192 differentially expressed genes: 128 genes were up-regulated in RARS patients while 64 were down-regulated (S2 Fig and S5 Table). Interestingly, 33% (42 genes) of the over-expressed genes were related to iron and mitochondrial metabolism as inferred from the functional enrichment analyses. 33 genes were related to mitochondria, five were associated with iron binding and eight were involved in heme formation. Thus, ALAD, HMBS, UROS, UROD, CPOX and PPOX were over-expressed in RARS patients with respect to the other low-risk MDS (Table 2).
Table 2. Most representative deregulated cellular functions in RARS patients respect to the RCUD group.
| Fuction | Number of genes | p Value | Over-expressed genes | |
|---|---|---|---|---|
| MITOCHONDRION | 33 | 0,000000 | ||
| Integral to membrane | 15 | 0,047316 | GBGT1, ST6GALNAC4, SLC25A38, AADACL1, CYBRD1, TSPAN17, RHBDD1, APOO, TMEM14C, KCNH2, MS4A7, PPAPDC1A, STEAP3, ABCB10, AC079061.8 | |
| Cytoplasm and mitochondrion | 7 | 0,000042 | ATP7B, CAPRIN2, APOO, HSPD1, HEBP1, ISOC2, HMBS | |
| Membrane | 6 | 0,002383 | SLC25A38, ATP7B, APOO, TMEM14C, ABCB10, PPOX | |
| Mitochondrial inner membrane | 4 | 0,009138 | SLC25A38, NME4, HSPD1, ABCB10 | |
| HEME BIOSYNTHETIC PROCESS | 8 | 0,000000 | ALAD, UROS, PPOX, CPOX, UROD, HMBS, EPRS, BLVRB | |
| NUCLEOTIDE BINDING | 6 | 0,000019 | ATP7B, NME4, C14orf156, TRAP1, HSPD1, ABCB10 | |
| ATP BINDING | 5 | 0,000078 | ATP7B, NME4, TRAP1, HSPD1, ABCB10 | |
| PROTEIN BINDING | 5 | 0,002258 | MRPS9, ATP7B, ACAT1, ISOC2, ARG2 | |
| IRON ION BINDING | 5 | 0,000516 | PPAT, PIR, RFESD, CYBRD1, STEAP3 | |
Validation of the RARS expression signature with independent datasets
In order to validate the expression signatures found in our unfractionated cohort of MDS samples, a comparison between the present studies and an independent cohort of MDS patients where CD34+ cells were isolated (published by Pellagatti et al. 2010) [35] was carried out. Odds ratio was used to measure how strongly is the overlapping of the altered genes in both analysis. Analysing the Pellagatti et al. dataset (GSE19429), a total of 5 840 genes showed significant differences (FDR ≤ 0.05) in mRNA expression levels between CD34+ cell from RARS patients (using only MDS-RS samples with normal karyotype) and their control group: 3 697 and 2 143 genes were over and under-expressed, respectively, in the RARS samples. This signature had a significant overlap of 457 genes with our signature for the same RARS vs controls comparison (only considering the up-regulated genes); which corresponds to an odds ratio for this comparison of 7.14 (Lower CI 95% = 8.38).
Further analysis of the Pellagatti data (34) for the CD34+ differential expression between RARS and RCUD groups identified a set of 404 differentially expressed genes (FDR ≤ 0.05): 284 genes were up-regulated in RARS patients while 120 were down-regulated. A very significant overlap was also observed between this up-regulated gene set and the set of 128 genes that we discovered up-regulated in our RARS patients versus the RCUD group (Odds ratio = 31.83; Lower CI 95% = 21.51).
A targeted genome capture and next-generation sequencing strategy identifies gene variants in MDS with ring sideroblasts
In order to identify any gene variants in MDS-RS, a capture and sequence approach was done on 93 genes from a group of 6 MDS with ring sideroblasts using a custom NimbleGen array was carried out: 39 of the gene targets were related to iron and mitochondrial metabolism (S6 Table). The enrichment assay followed by NGS detected a total of 8 230 variants in all patients analyzed (median 1 367 variants per sample, range 1 078–1 672). All putative variants were first compared with published single nucleotide polymorphism (SNP) data from dbSNP130. The 2 217 known SNPs (56%) were discarded along with the new variants found at non-coding regions (85%) and, finally, those that did not give rise to an amino-acid change in their protein sequence (58%)
As a result, a missense variation was detected in the ALAD gene in one case with ring sideroblasts (Chr9: 116,152,735); therefore the incidence of this variation, and known SNPs, were examined, by Sanger sequencing, in a larger cohort of 100 MDS patients with ring sideroblasts. In addition, conventional mutational analysis as alternative approach was also performed for the SLC25A37 and SLC25A38 genes.
RARS patients present a frequent haplotype in exon 6 and a new variant in exon 7 of the ALAD gene
Two known polymorphisms have been reported in exon 6 of the ALAD gene: rs8177807 (T10728C) and rs2228083 (C10679T). The SNPs were located at positions Chr9: 116,152,891 and Chr9: 116,152,940, respectively, 49 base pairs from each other. The study revealed two possible haplotypes for these variants: “common haplotype” (T10728 and C10679) and “variant haplotype” (C10728 and T10679). Interestingly, the “variant haplotype” was present in 12 of 100 MDS with ring sideroblasts (12%), while it was only found in 5 of 100 controls and RCUD patients (5%) (p = 0.07).
In addition, exon 7 of the ALAD gene was analyzed in a larger cohort of MDS with ring sideroblasts (n = 100) and the change previously identified by the capture and sequence approach was confirmed by conventional sequencing (Fig 1A). The positively charged arginine residue (R174) was replaced by an uncharged cysteine residue (Fig 1B). The three-dimensional structure showed that R174 residue is completely buried into the monomeric structure (Fig 2) and the protein was predicted to be potentially damaging.
Fig 1. Variation in the ALAD gene in a RARS case.
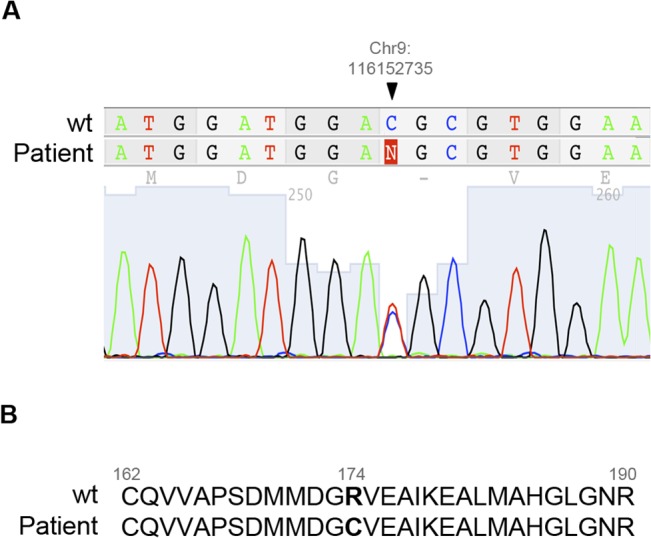
(A) New missense variation in the ALAD gene in the BM of one RARS patient found by massive sequencing. The variation is heterozygous and is located at Chr9: 116,152,735 position in exon 7. (B) Protein sequences from wild-type and RARS patient. Amino-acid change in the protein sequence of ALAD in a RARS patient. Arginine (R174) is replaced by cysteine y the mutant protein. (RARS: refractory anemia with ring sideroblasts)
Fig 2. Three-dimensional structure of the ALAD protein.
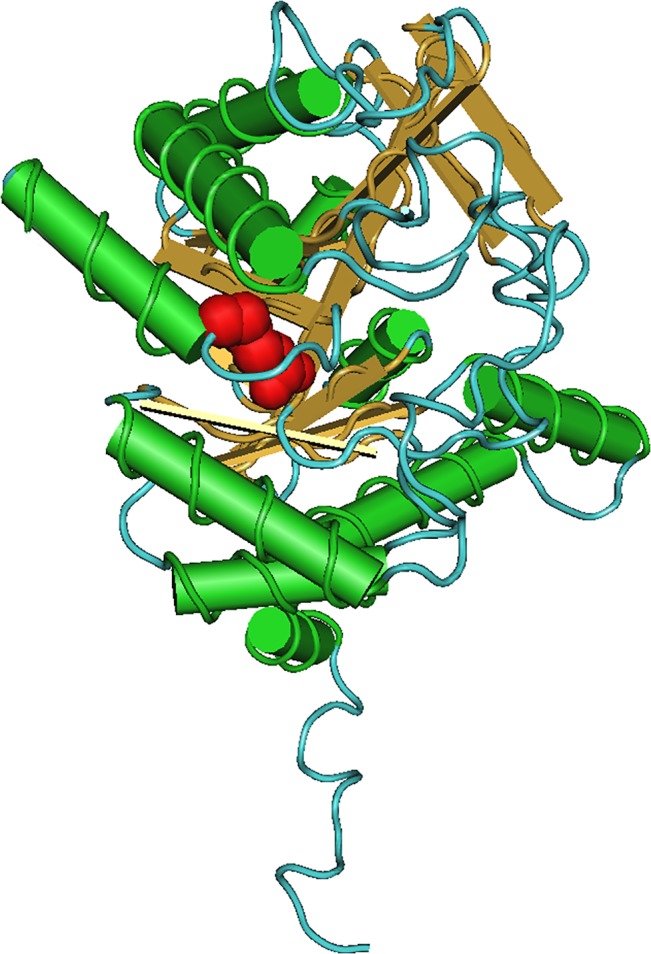
Monomeric structure of the ALAD protein. The red color indicates the position of the arginine 174. The amino acid is completely buried into the monomeric structure of the ALAD protein.
Furthermore, the variant was found in the CD3+ cells of PB from the same patient suggesting the presence of a germ line mutation. The variant was not found in any of the control samples or in those analysed from RCUD patients.
SLC25A37 (Mitoferrin-1) is un-mutated in acquired RARS
SLC25A37, that encodes a mitochondrial iron transporter that specifically mediates iron uptake in developing erythroid cells, was over-expressed in RARS patients (R Fold = 1.70). Therefore, a complete sequence analysis of the SLC25A37 gene was performed in the group of 50 patients MDS with ring sideroblasts: 62% of all the cases analyzed had at least one known polymorphism in their sequence. Specifically, the polymorphisms were located in exons 2 and 4 of SLC25A37 with 6% of the patients having the rs1047384 polymorphism (exon 4). The majority of polymorphisms (56%) were in exon 2; furthermore, 38%, 14% and 4% of cases exhibited one, two or three polymorphisms in this exon, respectively (S3 Fig). All polymorphisms were homozygous. No new variations were observed in the SLC25A37 gene in the patients with ring sideroblasts.
SLC25A38 showed a new mutation in one patient with RCUD
SCL25A38, recently described as a mutated gene in congenital sideroblastic anemia, was found to be over-expressed in RARS patients in our study (R Fold = 1.78). We further analyzed this gene by sequencing in the group of low-risk MDS patients (n = 175). A new missense mutation was observed in the BM from one of the RCUD patients. The mutation, in exon 4, was located at position Chr3: 39,432,957 and resulted in the amino-acid change of valine for alanine (V97A) (Fig 3A–3B).
Fig 3. SLC25A38 mutation in a RCUD patient.

(A) New missense mutation in exon 4 of a RCUD patient detected in BM by Sanger sequencing. The mutation is heterozygous and is located at the Chr3: 39,432,957 position. (B) Amino-acid change in the protein sequence of SLC25A38 in the RCUD patient. Valine 97 is replaced by alanine in the mutant protein. (RCUD: refractory cytopenia with unilineage dysplasia).
In addition, three known polymorphisms (rs1995236, rs870843 and rs9877539) were observed in the SCL25A38 sequence in the group of low-risk MDS. They were located close to the coding sequence for 4, 6 and 7 exons. Interestingly, both rs1995236 and rs9877539 polymorphisms were homozygous for the less common variants in the population (100% and 97.6% of all the cases with these variations, respectively) (S7 Table).
Recurrent SF3B1 and SRSF2 Mutations in low-risk MDS patients
SF3B1, recently described as a mutated gene in a high proportion of MDS patients with ring sideroblasts, was analyzed in the same cohort of patients with MDS-RS (n = 100). SF3B1 was mutated in 82 of 100 cases with ring sideroblasts (82%). Exon 15 was more frequently mutated (47%) than exon 14 (32%). The mutations in exon 15 were located in the same codon (700) while those in exon 14 were located in four codons: 622 (7%), 625 (1%), 662 (12%) and 666 (12%). All patients had a single mutation in the gene, except for three cases (3%), which had two changes. One of them showed the mutations in the same exon (codons 662 and 666) while the mutations in the other two patients were located in different exons (codons 666 and 700). However, the clinical characteristics of these patients did not differ from those with a single mutation.
No relationships between the presence of polymorphisms in SCL25A37 or SLC25A38 and SF3B1 mutations were found. The case with ALAD variation showed a mutation in the SF3B1 gene (exon 14, codon 662).
In addition, a mutational study of SF3B1 and SRSF2 in all RCUD patients with available expression profile information was carried out. 30% of the cases showed mutations in SF3B1, 10% carried variations in SRSF2 and 60% showed non-mutations in any of the genes analyzed. The mutations detected in RCUD were located in hot-spots previously described by other groups. Specifically, they were observed in codons 622, 625, 662, 666 and 700 in the SF3B1 gene and codon 95 in SRSF2.
Gene expression profile showed significant differences between mutated patients and non-mutated cases
A supervised analysis of GEP between mutated low-risk MDS cases (RARS and RCUD) and non-mutated low-risk MDS patients was carried out. Herein, GDF15 was the most up-regulated gene in low-risk MDS patients with mutation in spliceosome-related genes. ALAD, SLC25A37, SLC11A2, PCK2, MRPS9, AIFM2 and HK1, all of them involved in iron and mitochondrial metabolism, were also over-expressed and were included in the Top200 more differentially expressed genes (S8 Table). In addition, a functional study with this gene Top200 gene set showed that cell cycle (Benjamini-Hochberg value < 0.0003) and mitosis (Benjamini-Hochberg value < 0.005) were the most frequently deregulated molecular functions, involving 24 and 21 differentially expressed genes, respectively.
A supervised analysis between the gene expression levels of SF3B1 mutations patients and non-mutated MDS-RS cases was carried out. An over-expressed gene-signature of 71 genes was identified between both sub-groups (S9 Table). Interestingly, GDF15 was overexpressed in patients showing SF3B1 mutations. In addition, other genes such as PPP2R5B, PPP1R16A and DDIT4L, related to SF3B1 and GDF15, were up-regulated in the mutated group. A functional analysis with this gene set showed two deregulated pathways: porphyrin biosynthesis and heme biosynthesis (p< 0.001). ALAS2, PPOX and UROD, all of them involved in heme formation, were also over-expressed in the mutated patients.
Discussion
The integrative analysis of the massive gene analysis has provided new insights in the knowledge of the pathogenesis of MDS [27]. In the present study, an over-expression of iron and mitochondrial metabolism related genes was observed in patients with RARS. Therefore a custom sequence capture array was designed in order to identify genes that could play a role in the pathogenesis of MDS with ring sideroblasts.
The use of unfractionated compared to fractionated cells in this type of expression or sequencing study has become a matter of debate in recent years. The approach taken in the present study is supported by previous studies that have been able to identify biological and prognostic characteristics in MDS and AML by studying the gene expression profile and the use of unfractionated mononuclear cells [27,36,37]. Furthermore, the results of the present study are supported by previous reports in CD34+ cells [8] due to a significant overlap between both analysis. The use of CD34+ cells is of great scientific value while the analysis of unfractionated samples could allow the identification of the interaction between the different cell types. In this respect, the present study has suggested that the erythroid lineage displays a robust gene expression signature that allows the identification from the global set of mononuclear cells.
Interestingly, the most significant functional category from the gene expression signature was iron and mitochondrial metabolism. This category had the highest proportion of genes over-expressed in RARS patients when compared to the controls or the RCUD group, representing 38% and 33% of all over-expressed genes, respectively. Of note, six key enzymes in the heme biosynthesis pathway showed increased expression, some members of this pathway are also over-expressed in CD34+ cells of RARS patients [8]. In addition to the enzymes that catalyze heme formation, our study highlighted the over-expression of the ABCB6 gene in RARS patients. This molecule is ideally located in the outer membrane, where it can move coproporphyrinogen III from the cytoplasm into the mitochondrion using ATP hydrolysis as the source of energy [7,38]. The deregulation of ABCB6 expression may contribute to the impaired heme biosynthesis found in MDS with ring sideroblasts.
The ALAD gene encodes a cytosolic enzyme that catalyses the condensation of two molecules of d-aminolevulinic acid (ALA) to form porphobilinogen (PBG) in the second step of the heme biosynthetic pathway [39]. Our analysis of the ALAD gene identified two polymorphisms in exon 6 located 49 bases from each other and, interestingly, the presence of one of them was always determined by the presence of the other one. The occurrence of both polymorphisms (“variant haplotype”) was more frequent in MDS with ring sideroblasts (12%) than in members of the other groups analyzed (5%). Therefore, our study showed a trend to an association of the variant haplotype with the ring sideroblasts. The presence of haplotypes has been linked to the deregulation of the genes in different hematological malignances, such as chronic lymphocytic leukemia and acute lymphoblastic leukemia [40,41]. Thus, these findings could be related to the deregulation of the ALAD gene and consequently to the abnormal iron and mitochondrial metabolism in MDS with ring sideroblasts.
The capture and sequencing study identified a non-described sequence change in the ALAD gene in one RARS patient showing a SF3B1 mutation; the ALAD gene was also up-regulated in the gene expression studies. The variants of this gene were also found in the CD3+ population in the PB. In addition, the variant led to amino-acid change in the ALAD protein. Therefore this variant could have a possible role in the predisposition to disease (as a first event) as well as contributing to the pathogenesis of RARS where SF3B1 mutations are the trigger cause.
In addition, we have identified a new missense mutation in exon 4 of the SCL25A38 gene in one RCUD patient. Thus, the mutation led to the 97V>A amino-acid change in the protein sequence. SLC25A38 gene has been previously linked to congenital sideroblastic anemia [14]. These findings would indicate that mutations in the SLC25A38 gene might be associated to the low-risk MDS.
SLC25A37 is a member of the mitochondrial solute carrier family. Some authors have shown that SLC25A37 contributes to mitochondrial iron acquisition in mammalian cells, since decreases in SLC25A37 severely reduce mitochondrial iron-consuming processes, such as Heme and Fe-S cluster synthesis. In fact, mitochondrial iron transport is reduced by more than 90% in cells silenced for SLC25A37, suggesting that SLC25A37 is a major contributor to mitochondrial iron acquisition [42,4]. Our results showed up-regulation of SLC25A37 in RARS patients with respect to the control group. These findings led us to hypothesize that this over-expression could be responsible of iron accumulation. For these reasons, we sought out to determine whether MDS with ring sideroblasts cases were characterized by SLC25A37 somatic mutations. No mutations were found for this gene in cases with ring sideroblasts. Therefore, other mechanisms give rise to the over-expression of the SLC25A37 gene in RARS patients. In addition, it should be noted that no relationships between the presence of polymorphisms in SCL25A37 or SLC25A38 and SF3B1 mutations were found.
The GEP from the BM of SF3B1 mutated patients was compared to that from the BM of non-mutated individuals. 71 genes showed over-expression in mRNA levels in mutated cases. Interestingly, two pathways observed in the functional analysis were related to mitochondrial metabolism. GDF15, a cytokine from the TGFβ family, was one of the most highly differentially expressed gene. GDF15 is expressed at high levels in patients with ineffective erythropoiesis. In contrast to the low levels of GDF15 expressed during normal erythropoiesis, ineffective erythropoiesis causes high-level expression of GDF15 [43]. In addition, it has been suggested that over-expression of GDF15 in patients with RARS might be involved in the systemic iron overload by suppressing hepcidin secretion [43,44], being sensitive to iron depletion and this response is specifically antagonized by the reprovision of iron [45]. Therefore, the GDF15 over-expression could be related to the presence of mutations in SF3B1 gene, and therefore, to a higher percentage of ring sideroblasts previously described [17]. These findings also suggest that the up-regulation of ALAS2, PPOX and UROD, all of them involved in heme formation, could be related to the presence of SF3B1 mutations. Furthermore, the comparison between the GEP of mutated low-risk MDS patients and non-mutated cases showed the cell cycle and mitosis as the most frequently deregulated pathways. This would support the hypothesis that a mutation in spliceosome-related genes could be the trigger cause, and support the presence of a second event for the deregulation of iron and mitochondrial metabolism.
In summary, our integrated expression and sequencing approaches has identified both the deregulation of genes involved in iron and mitochondrial metabolism and a new variant in the ALAD gene. Both potential mechanisms provide new insights into the pathogenesis of MDS with ring sideroblasts and, specifically, in patients with SF3B1 mutations.
Supporting Information
700 genes were over-expressed and 445 genes were under-expressed in the RARS cases. Each point represents the log2 of R.fold value from each gene.
(PDF)
128 genes were up-regulated and 64 were down-regulated in RARS cases. Each point represents the log2 of R.fold value from each gene.
(PDF)
56% of the analyzed cases had some polymorphism in exon 2. The patients showed one, two or three polymorphisms in this exon in 38%, 14% and 4% of the cases respectively. The most common polymorphism was rs2942194 as isolated variation. The analysis showed two possible combinations for the patients with two polymorphisms: rs2942194 and rs10992 or rs10992 and rs3736032. The first combination was more frequent than the second combination. The combination between rs2942194 and rs3736032 was not found in any patient.
(TIF)
(DOC)
(XLS)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
The authors would like to thank Irene Rodríguez, Sara González, Teresa Prieto, Mª Ángeles Ramos, Almudena Martín, Ana Díaz, Ana Simón, María del Pozo, Vanesa Gutiérrez, Isabel M Isidro and Sandra Pujante from the Centro de Investigación del Cáncer, Salamanca, Spain, for their technical assistance. We further thank Dr. Carlos R. Vázquez de Aldana for support in data analysis.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was partially supported by grants from: the Spanish Fondo de Investigaciones Sanitarias (PI14/1971, PI12/00281); Proyectos de Investigación del SACYL (BIO/SA52/14, BIO/SA10/14, BIO/SA31/13, GRS 994/A/14); Consejería Educación, Junta Castilla y León (HUS272U13); COST Action "EuGESMA" (BM0801); Red Temática de Investigación Cooperativa en Cáncer (RTICC), Instituto de Salud Carlos III (ISCIII), Spanish Ministry of Economy and Competitiveness & European Regional Development Fund (ERDF) "Una manera de hacer Europa" (Innocampus) (RD12/0036/0069, RD12/0036/0029, RD12/0036/0044); European Union's Seventh Framework Programme (FP7/2007-2013 nº 306242- NGS-PTL). MDR is fully supported by a "Grant from Fundación Española de Hematología y Hemoterapia". MA was fully supported by a "Grant from the Spanish Consejo Superior de Investigaciones Científicas, Junta para la Ampliación de Estudios (JAE Predoctoral) [09-02402]". MH is fully suported by an "Ayuda predoctoral de la Junta de Castilla y León" from "European Regional Development Fund". DMZ was funded by the Spanish Ministry of Science and Innovation (PI082025) and Junta de Castilla y León (CSI224A11-2, SAN/103/2011). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Cazzola M, Malcovati L (2005) Myelodysplastic syndromes—coping with ineffective hematopoiesis. N Engl J Med 352: 536–538. [DOI] [PubMed] [Google Scholar]
- 2. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114: 937–951. 10.1182/blood-2009-03-209262 [DOI] [PubMed] [Google Scholar]
- 3. Nikpour M, Pellagatti A, Liu A, Karimi M, Malcovati L, Gogvadze V, et al. (2010) Gene expression profiling of erythroblasts from refractory anaemia with ring sideroblasts (RARS) and effects of G-CSF. Br J Haematol 149: 844–854. 10.1111/j.1365-2141.2010.08174.x [DOI] [PubMed] [Google Scholar]
- 4. Shaw GC, Cope JJ, Li L, Corson K, Hersey C, Ackermann GE, et al. (2006) Mitoferrin is essential for erythroid iron assimilation. Nature 440: 96–100. [DOI] [PubMed] [Google Scholar]
- 5. Cavadini P, Biasiotto G, Poli M, Levi S, Verardi R, Zanella I, et al. (2007) RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 109: 3552–3559. [DOI] [PubMed] [Google Scholar]
- 6. Cuijpers ML, Raymakers RA, Mackenzie MA, de Witte TJ, Swinkels DW (2010) Recent advances in the understanding of iron overload in sideroblastic myelodysplastic syndrome. Br J Haematol 149: 322–333. 10.1111/j.1365-2141.2009.08051.x [DOI] [PubMed] [Google Scholar]
- 7. Krishnamurthy P, Xie T, Schuetz JD (2007) The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol Ther 114: 345–358. [DOI] [PubMed] [Google Scholar]
- 8. Pellagatti A, Cazzola M, Giagounidis AA, Malcovati L, Porta MG, Killick S, et al. (2006) Gene expression profiles of CD34+ cells in myelodysplastic syndromes: involvement of interferon-stimulated genes and correlation to FAB subtype and karyotype. Blood 108: 337–345. [DOI] [PubMed] [Google Scholar]
- 9. Boultwood J, Pellagatti A, Nikpour M, Pushkaran B, Fidler C, Cattan H, et al. (2008) The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS One 3: e1970– 10.1371/journal.pone.0001970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steensma DP, Hecksel KA, Porcher JC, Lasho TL (2007) Candidate gene mutation analysis in idiopathic acquired sideroblastic anemia (refractory anemia with ringed sideroblasts). Leuk Res 31: 623–628. [DOI] [PubMed] [Google Scholar]
- 11. Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM (1999) Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet 8: 743–749. [DOI] [PubMed] [Google Scholar]
- 12. Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, et al. (2000) Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 96: 3256–3264. [PubMed] [Google Scholar]
- 13. Cotter PD, Rucknagel DL, Bishop DF (1994) X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood 84: 3915–3924. [PubMed] [Google Scholar]
- 14. Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, et al. (2009) Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat Genet 41: 651–653. 10.1038/ng.359 [DOI] [PubMed] [Google Scholar]
- 15. Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD (2007) Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood 109: 3567–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Damm F, Thol F, Kosmider O, Kade S, Loffeld P, Dreyfus F, et al. (2012) SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implication. Leukemia 26: 1137–1140. 10.1038/leu.2011.321 [DOI] [PubMed] [Google Scholar]
- 17. Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, la Porta MG, Pascutto C, et al. (2011) Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118: 6239–6246. 10.1182/blood-2011-09-377275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384–1395. 10.1056/NEJMoa1103283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, et al. (2012) SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 119: 569–572. 10.1182/blood-2011-09-377994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. (2012) SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26: 542–545. 10.1038/leu.2011.232 [DOI] [PubMed] [Google Scholar]
- 21. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- 22.Nikpour M, Scharenberg C, Liu A, Conte S, Karimi M, Mortera-Blanco T, et al. (2012) The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia [DOI] [PMC free article] [PubMed]
- 23. Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, et al. (2012) SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 120: 3173–3186. 10.1182/blood-2012-05-430876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vardiman JW, Harris NL, Brunning RD (2002) The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100: 2292–2302. [DOI] [PubMed] [Google Scholar]
- 25. Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. (2010) Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol 28: 2529–2537. 10.1200/JCO.2009.23.4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Risueno A, Fontanillo C, Dinger ME, De Las Rivas J (2010) GATExplorer: genomic and transcriptomic explorer; mapping expression probes to gene loci, transcripts, exons and ncRNAs. BMC Bioinformatics 11: 221– 10.1186/1471-2105-11-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. del Rey M, O'Hagan K, Dellett M, Aibar S, Colyer HA, Alonso ME, et al. (2013) Genome-wide profiling of methylation identifies novel targets with aberrant hypermethylation and reduced expression in low-risk myelodysplastic syndromes. Leukemia 27: 610–618. 10.1038/leu.2012.253 [DOI] [PubMed] [Google Scholar]
- 28. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wettenhall JM, Smyth GK (2004) limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics 20: 3705–3706. [DOI] [PubMed] [Google Scholar]
- 31. Goeman JJ, van de Geer SA, de KF, van Houwelingen HC (2004) A global test for groups of genes: testing association with a clinical outcome. Bioinformatics 20: 93–99. [DOI] [PubMed] [Google Scholar]
- 32. Huang dW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. (2007) DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35: W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. del Rey M, Pericacho M, Velasco S, Lumbreras E, Lopez-Novoa JM, Hernandez-Rivas JM, et al. (2013) Alteration in endoglin-related angiogenesis in refractory cytopenia with multilineage dysplasia. PLoS One 8: e53624– 10.1371/journal.pone.0053624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, et al. (2010) Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 24: 756–764. 10.1038/leu.2010.31 [DOI] [PubMed] [Google Scholar]
- 36. Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, et al. (2009) Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood 114: 1063–1072. 10.1182/blood-2008-10-187203 [DOI] [PubMed] [Google Scholar]
- 37. Theilgaard-Monch K, Boultwood J, Ferrari S, Giannopoulos K, Hernandez-Rivas JM, Kohlmann A, et al. (2011) Gene expression profiling in MDS and AML: potential and future avenues. Leukemia 25: 909–920. 10.1038/leu.2011.48 [DOI] [PubMed] [Google Scholar]
- 38. Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, et al. (2006) Identification of a mammalian mitochondrial porphyrin transporter. Nature 443: 586–589. [DOI] [PubMed] [Google Scholar]
- 39. Sassa S (1982) Delta-aminolevulinic acid dehydratase assay. Enzyme 28: 133–145. [DOI] [PubMed] [Google Scholar]
- 40. Martin-Guerrero I, Enjuanes A, Richter J, Ammerpohl O, Colomer D, Ardanaz M, et al. (2011) A putative "hepitype" in the ATM gene associated with chronic lymphocytic leukemia risk. Genes Chromosomes Cancer 50: 887–895. 10.1002/gcc.20912 [DOI] [PubMed] [Google Scholar]
- 41. Rousseau J, Gagne V, Labuda M, Beaubois C, Sinnett D, Laverdiere C, et al. (2011) ATF5 polymorphisms influence ATF function and response to treatment in children with childhood acute lymphoblastic leukemia. Blood 118: 5883–5890. 10.1182/blood-2011-05-355560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J (2009) Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol 29: 1007–1016. 10.1128/MCB.01685-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ramirez JM, Schaad O, Durual S, Cossali D, Docquier M, Beris P, et al. (2009) Growth differentiation factor 15 production is necessary for normal erythroid differentiation and is increased in refractory anaemia with ring-sideroblasts. Br J Haematol 144: 251–262. 10.1111/j.1365-2141.2008.07441.x [DOI] [PubMed] [Google Scholar]
- 44. Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al. (2007) High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 13: 1096–1101. [DOI] [PubMed] [Google Scholar]
- 45. Lakhal S, Talbot NP, Crosby A, Stoepker C, Townsend AR, Robbins PA, et al. (2009) Regulation of growth differentiation factor 15 expression by intracellular iron. Blood 113: 1555–1563. 10.1182/blood-2008-07-170431 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
700 genes were over-expressed and 445 genes were under-expressed in the RARS cases. Each point represents the log2 of R.fold value from each gene.
(PDF)
128 genes were up-regulated and 64 were down-regulated in RARS cases. Each point represents the log2 of R.fold value from each gene.
(PDF)
56% of the analyzed cases had some polymorphism in exon 2. The patients showed one, two or three polymorphisms in this exon in 38%, 14% and 4% of the cases respectively. The most common polymorphism was rs2942194 as isolated variation. The analysis showed two possible combinations for the patients with two polymorphisms: rs2942194 and rs10992 or rs10992 and rs3736032. The first combination was more frequent than the second combination. The combination between rs2942194 and rs3736032 was not found in any patient.
(TIF)
(DOC)
(XLS)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.