

Short summary
We report the first patients with a homozygous loss of function mutation in the RAC2 gene, presenting with clinical features of common variable immunodeficiency. In addition, the patients suffered from glomerulonephritis, coagulopathy, multiple hormone deficiencies potentially on the autoimmune basis and abnormalities of neutrophil granules.
Keywords: Ras-related C3 botulinum toxin substrate 2 (RAC2), common variable immunodeficiency (CVID), kappa-deleting recombination excision circles (KRECs), T-cell receptor excision circles (TRECs), neutrophil granules
To the Editor,
Common Variable Immunodeficiency (CVID) is a heterogeneous group of disorders with variable immunological and clinical phenotypic features, including hypogammaglobulinemia, recurrent infections and autoimmunity1. Over the past decades, a number of genetic defects have been identified which are associated with CVID. However, these alterations only account for the etiologies of a minority of CVID cases (<15%)1. In this paper, we present a novel genetic defect in the RAC2 gene in two siblings previously diagnosed with CVID.
The proband, a 21-year-old woman of Iranian descent born to first-degree consanguineous parents, presented at 6 months of age with recurrent pneumonia, followed by edema, proteinuria and membranous glomerulonephritis six months later, at which time serum IgA was undetectable and IgG was elevated (Table E1 in this article's Online Repository at www.jacionline.org). Based on the clinical presentation and an increased anti-streptolysin O (ASO) titer, a diagnosis of post-streptococcal glomerulonephritis (PSGN) was made along with a tentative diagnosis of selective IgA deficiency (SIgAD). At 1 year of age, the patient had a normal percentage of CD3+ T-cells, a slightly reduced ratio of CD4+/CD8+ T-cells, but a decreased percentage of CD19+ B-cells (Table 1). Coagulation factor XI deficiency (activity <1%) was diagnosed at 2 years of age. Dermatological features developed three years later, including urticaria (induced by exposure to sunlight), recurrent erythematous plaques and food allergy, requiring corticosteroid therapy. Her neutrophil markers and the nitrobluetetrazolium (NBT) test were normal, but IgG level decreased to 430 mg/dl by 7 years of age, thus, a diagnosis of CVID was made and intravenous immunoglobulin (IVIG) treatment was initiated (Tables 1 and E1). Other morbidities in the following years included arthralgia, bronchiectasis, hypothyroidism with anti-TPO autoantibodies and hyperparathyroidism (Table 1). Her PSGN progressed to end-stage renal disease, requiring renal transplantation. She died as a result of graft rejection and possible cerebral hemorrhage at the age of 21 years.
Table 1.
Laboratory and immunological data of two patients with RAC2 deficiency
| Laboratory test | Proband | Sibling | Reference values* |
|---|---|---|---|
| Complete blood count | At age of 1 yr | At age of 7 yrs | |
| WBC/mm3 | 9200 | 7800 | 4000-11000 |
| Neutrophils (%) | 62 | 66 | 20-70 (for age of 1 yr) |
| 35-80 (for age of 7 yrs) | |||
| Lymphocytes (%) | 27↓ | 25↓ | 45-75 (for age of 1 yr) |
| 30-55 (for age of 7 yrs) | |||
| CD3+ (%) | 60 | 62 | 30-78** |
| CD4+ (%) | 30 | 25 | 22-58** |
| CD8+ (%) | 33 | 35 | 10-37** |
| CD4+/CD8+ ratio | 0.9↓ | 0.7↓ | 1-4** |
| CD19+ (%) | 2.5↓ | 8.5 | 3-14** |
| Serum complement factors | At age of 1 yr | At age of 7 yrs | |
| C3 (mg/dl) | 79↓ | 110 | 83-177 |
| C4 (mg/dl) | 27 | 20 | 15-45 |
| Neutrophil analyses | At age of 7 yrs | At age of 28 yrs | |
| CD18+ (%) | 70 | n.a. | 70-100 |
| CD11a+ (%) | 92↑ | n.a. | 60-90 |
| CD11b+ (%) | 98↑ | n.a. | 60-90 |
| CD11c+ (%) | 99↑ | n.a. | 45-80 |
| NBT test (%) | 98 | n.a. | 95-100 |
| Chemotaxis without CF (μm) | n.a. | 10↓ | 22-54 |
| Chemotaxis with CF (μm) | n.a. | 45↓ | 77-125 |
| Serum immunoglobulins | At age of 7 yrs | At age of 10 yrs | |
| IgM (mg/dl) | 30↓ | 28↓ | 40-230 |
| IgG (mg/dl) | 430↓ | 640↓ | 700-1600 |
| IgA (mg/dl) | 5↓ | 19↓ | 41-297 (for age of 7 yrs) |
| 51-297 (for age of 10 yrs) | |||
| Vaccine antibodies | At age of 27 yrs | ||
| Antibody level pre-Pneumovax 23 (U/ml) | n.a. | 1↓ | 1.2-2 |
| Antibody 3 weeks post-Pneumovax 23 (U/ml) | n.a. | 8↓ | 10-14 |
| Antibody 1 year post-Pneumovax 23 (U/ml) | n.a. | 8 | 6-9 |
| Anti-tetanus (IU/ml) | n.a. | 1.56 | > 0.1 |
| Anti-diphtheria (IU/ml) | n.a. | 0.4 | > 0.1 |
| Autoantibodies and thyroid markers | At age of 7-16yrs | At age of 10-16 yrs | |
| ANA | Negative | Negative | Negative |
| ANCA | Negative | Negative | Negative |
| Anti-dsDNA | Negative | Negative | Negative |
| Anti-TPO (IU/ml) | 68↑ | 81↑ | 0-35 |
| TSH (mU/l) | 11.4↑ | 11.0↑ | 0.4-5.5 |
| T3 (μg/dl) | 30 | 33 | 25-36 |
| T4 (nmol/l) | 8 | 4↓ | 5-14 |
| PTH (pg/ml) | 194↑ | n.a. | 15-65 |
| GH (ng/ml) | n.a. | 1.3↓ | 10-40 |
| Anti-IgA (U/ml) | n.a. | 2.0 | 1.5-2.9 |
| Other tests | At age of 18 yrs | At age of 25 yrs | |
| KRECs | 2↓ | 8↓ | (85, 22)*** |
| TRECs | 24↓ | 10↓ | (67, 23)*** |
Abbreviations used: yr, year; WBC, white blood cells; n.a., not analyzed; NBT, nitroblue tetrazolium; CF, chemotactic factor; ANA, anti-nuclear antibody; ANCA, anti-neutrophil cytoplasmic antibody; Anti-dsDNA, anti-double stranded DNA antibody; Anti-TPO, anti-thyroperoxidase antibody; TSH, thyroid stimulating hormone; PTH, parathyroid hormone; GH, growth hormone; KRECs, kappa-deleting recombination excision circles; TRECs, T-cell receptor excision circles.
Reference: age-appropriate reference range or value from healthy Iranian individuals
The local hospital reference range is not stratified for age between 1-7 years
Due to the lack of appropriate reference values for the adult population, the TRECs and KRECs analyses were compared to two age-matched healthy controls.
Her 28-year-old brother, presented at 2 years of age with recurrent sinopulmonary infections and failure to thrive. Urticaria and sinusitis occurred at age 7 years when SIgAD was diagnosed with normal T- and B-cell percentages in peripheral blood (Tables 1 and E1). At 8 years of age, he developed pneumonia and subsequent PSGN (with increased ASO titer). Coagulation factor XI deficiency was diagnosed at the age of 10 years. His IgM and IgG serum levels were also reduced at this age (Tables E1 and E2 in this article's Online Repository at www.jacionline.org), when a diagnosis of CVID was established and IVIG replacement was commenced. Between 10 to 16 years of age he developed submandibular reactive lymphadenopathy, bronchiectasis, hypothyroidism with anti-TPO antibodies and growth hormone deficiency (Table 1). A recent extended lymphocyte immunophenotyping showed severe B-lymphopenia and abnormalities in T-cell subpopulations, with reversed ratio of CD4+/CD8+ T-cells, decreased percentages of naïve CD4+ and CD8+ T-cells as well as reduced percentages of regulatory T-cells and recent thymic emigrant cells (Table E3 in this article's Online Repository at www.jacionline.org).
A novel homozygous nonsense mutation in codon 56 (W56X) was identified in the RAC2 gene in the proband by whole exome sequencing (WES) analysis. Mutations in F11, encoding the coagulation factor XI, was excluded by both Sanger sequencing and WES data (detailed information on methods is provided in the Methods section in this article's Online Repository at www.jacionline.org). All known causative gene defects responsible for CVID were furthermore excluded in this patient based on the WES data. Sanger sequencing confirmed the homozygous RAC2 mutation in the proband, her brother, and in a heterozygous form in their mother (Fig. 1a, b). Western blot analysis was subsequently carried out on fibroblast cells transfected with either wild type RAC2 plasmid or a RAC2 mutant (RAC2W56X) containing plasmid. The expression of RAC2 was completely absent in cells transfected with the mutant identified in the patients (Fig. 1c).
Figure 1.
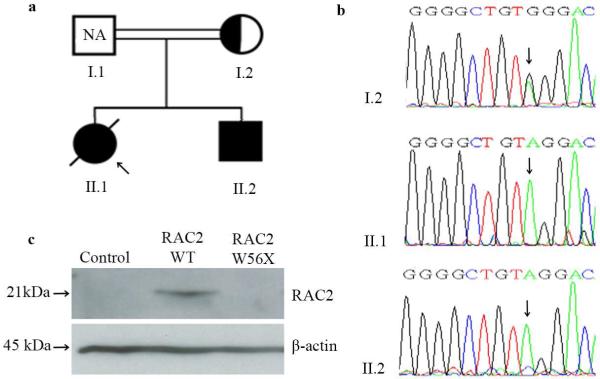
a) Family pedigree (solid fill, homozygous; half fill, heterozygous carrier). The proband is indicated by an arrow. The father's sample was not available (NA). b) Sequence analysis of the RAC2 gene. c) Detection of RAC2 expression by Western blot analysis. Control, fibroblast cells without transfection.
The affected gene encodes RAC2 (Ras-related C3 botulinum toxin substrate 2), a hematopoietic-specific member of the Rho family of guanosine triphosphatases (Rho GTPase) that are crucial regulators of cell signaling and actin cytoskeleton. As deficiency of RAC2 has mainly been associated with neutrophil dysfunction2, the neutrophils from the proband were thus analyzed by transmission electron microscope (TEM). The number of primary (azurophilic) and secondary (specific) granules per μm2 of cytoplasm were significantly reduced in the proband (3.8±0.7 vs. 5.9±1.3 in the control; Student's t-test, p=2.3×10−5). Furthermore, the shape of the secondary granules in the proband was often more elongated or collapsed (Fig. 2c) as compared to the control (Fig. 2d). Cytoplasmic inclusions were also more frequent in the proband (Fig. 2a). The majority of inclusions were very dense and composed of a multi-membrane layer surrounded by a double membrane (Fig. 2e) and were interpreted as autophagosomes. Morphological differences between the primary granules of the proband and the control were not observed. Finally a chemotactic defect was observed in the neutrophils derived from the affected brother (Table 1).
Figure 2.

TEM images of neutrophils. a) proband: cytoplasmic inclusions (arrow). b) control, normal ultrastructure. c) proband: fewer cytoplasmic granules, with most secondary granules displaying an elongated or collapsed shape (arrow). d) control: normal, rounded secondary granules (arrow). e) proband: cytoplasmic inclusion, showing dense multi membrane layered structure (arrow) surrounded by a double membrane (arrowhead). f) control: normal ultrastructure.
To date, only de novo, dominant negative (DN) mutations affecting RAC2 (D57N) have been reported in 2 male infants3-5. The first case presented with a complex neutrophil dysfunction disease, characterized by multiple and progressive soft-tissue infections during the first few weeks of life, neutrophilia and a neutrophil chemotaxis defect3, 4. The second case was an apparently healthy 2-week-old infant, who exhibited reduced numbers of T-cell receptor excision circles (TRECs) in the Wisconsin statewide newborn screening for T-lymphopenia5. Further testing revealed a leukocytosis, neutrophilia, CD4+ T-lymphopenia and reduced serum levels of IgA and IgM5. He later developed fever, omphalitis and a paratracheal abscess and neutrophil chemotaxis was severely reduced5. Both infants underwent successful hematopoietic cell transplantations (HCT)4, 5.
We describe here, for the first time, a homozygous loss of function RAC2 mutation in two patients with early onset and progressive hypogammaglobulinemia. As shown in Table E4, in this article's Online Repository at www.jacionline.org, the clinical and laboratory findings of our patients differ from the previously reported cases in many respects. Notably, our patients did not present severe clinical abnormalities in the neonatal period associated with the neutrophil dysfunction. This may be explained by the fact that the previously reported cases involved expression of a DN protein, which affected the GTPase activity not only of RAC2, but also of RAC1, the other major RAC GTPase expressed in human neutrophils6, 7. Nevertheless, neutrophils from our patients showed reduced chemotaxis activity and as revealed by TEM analysis, reduced numbers of neutrophil granules as well as morphological changes of the secondary granules, suggesting that the neutrophil functions were still affected by the RAC2 loss of function mutation. The antibody deficiency observed in our patients supports an important role of RAC2 in T- and B-cell development, as also suggested by murine studies8, as well as some of the immunological features observed in the second infant carrying the D57N mutation5. No Guthrie cards were available from our patients that would allow a retrospective analysis of TRECs. However we did observe a reduced number of TRECs and kappa-deleting recombination excision circles (KRECs) in the patients’ peripheral blood as compared to age-matched controls, which in line with the immunophentyping data, may suggest a decreased number of recent thymic emigrants and a relative B-lymphopenia in both patients (Table 1 and E2). It is also of note that the antibody deficiency in our patients and the B-lymphopenia in the brother was progressive, a feature that was not possible to evaluate in the previous cases, as they received HCT already at the age of 10 and 3 months, respectively. The urticaria in our patients may be further related to the mast cell defects as observed in Rac2−/− bone marrow-derived mast cells9. However, pinpointing a specific mechanism will require additional investigations. Other phenotypes in our patients, such as multiple hormone deficiencies, coagulation factor XI deficiency and PSGN may not attributable directly to the RAC2 mutation, since expression of RAC2 is highly regulated and considered to be hematopoietic-specific. However, these phenotypes might be secondary to the overall increased susceptibility to infections (especially for PSGN) and/or autoimmunity in our patients (evident by an increased serum level of BAFF in the brother; data not shown). Thus, the polyendocrinopathies, coagulopathy and kidney disease observed might be part of a syndromic entity related to the RAC2 deficiency as is the case in selected forms of primary immunodeficiency with autoimmune features.
Our patients illustrate that different types of mutations in a given gene may be associated with vastly different clinical phenotypes10. Further investigation of the mutation identified in this report may help to elucidate the function of this gene in relation to this CVID-like, but more complex immunodeficiency.
Supplementary Material
Acknowledgments
Declaration of all sources of funding:
This work was supported by the Swedish Research Council, the Swedish Cancer Society, the Jeffrey Modell Foundation, the European Research Council (242551-ImmunoSwitch), the German Federal Ministry of Education and Research (BMBF 1315883) and the National Institutes of Health grant (NIH 5R01DK062757-12). All authors have no conflicts of financial interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 2.Troeger A, Williams DA. Hematopoietic-specific Rho GTPases Rac2 and RhoH and human blood disorders. Exp Cell Res. 2013;319:2375–83. doi: 10.1016/j.yexcr.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci U S A. 2000;97:4654–9. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurkchubasche AG, Panepinto JA, Tracy TF, Jr., Thurman GW, Ambruso DR. Clinical features of a human Rac2 mutation: a complex neutrophil dysfunction disease. J Pediatr. 2001;139:141–7. doi: 10.1067/mpd.2001.114718. [DOI] [PubMed] [Google Scholar]
- 5.Accetta D, Syverson G, Bonacci B, Reddy S, Bengtson C, Surfus J, et al. Human phagocyte defect caused by a Rac2 mutation detected by means of neonatal screening for T-cell lymphopenia. J Allergy Clin Immunol. 2011;127:535–8. e1–2. doi: 10.1016/j.jaci.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96:1646–54. [PubMed] [Google Scholar]
- 7.Gu Y, Jia B, Yang FC, D'Souza M, Harris CE, Derrow CW, et al. Biochemical and biological characterization of a human Rac2 GTPase mutant associated with phagocytic immunodeficiency. J Biol Chem. 2001;276:15929–38. doi: 10.1074/jbc.M010445200. [DOI] [PubMed] [Google Scholar]
- 8.Walmsley MJ, Ooi SK, Reynolds LF, Smith SH, Ruf S, Mathiot A, et al. Critical roles for Rac1 and Rac2 GTPases in B cell development and signaling. Science. 2003;302:459–62. doi: 10.1126/science.1089709. [DOI] [PubMed] [Google Scholar]
- 9.Gu Y, Byrne MC, Paranavitana NC, Aronow B, Siefring JE, D'Souza M, et al. Rac2, a hematopoiesis-specific Rho GTPase, specifically regulates mast cell protease gene expression in bone marrow-derived mast cells. Mol Cell Biol. 2002;22:7645–57. doi: 10.1128/MCB.22.21.7645-7657.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buckley RH. Variable phenotypic expression of mutations in genes of the immune system. J Clin Invest. 2005;115:2974–6. doi: 10.1172/JCI26956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.