

SUMMARY
Transcription through immunoglobulin switch (S) regions is essential for class switch recombination (CSR) but no molecular function of the transcripts has been described. Likewise, recruitment of activation-induced cytidine deaminase (AID) to S regions is critical for CSR; however, the underlying mechanism has not been fully elucidated. Here, we demonstrate that intronic switch RNA acts in trans to target AID to S region DNA. AID binds directly to switch RNA through G-quadruplexes formed by the RNA molecules. Disruption of this interaction by mutation of a key residue in the putative RNA-binding domain of AID impairs recruitment of AID to S region DNA, thereby abolishing CSR. Additionally, inhibition of RNA lariat processing leads to loss of AID localization to S regions and compromises CSR; both defects can be rescued by exogenous expression of switch transcripts in a sequence-specific manner. These studies uncover an RNA-mediated mechanism of targeting AID to DNA.
INTRODUCTION
Following antigen receptor assembly, mature B cells home to peripheral lymphoid organs where they encounter antigens and undergo immunoglobulin (Ig) heavy chain (Igh) class switch recombination (CSR). CSR is a deletional-recombination event that exchanges the default Cμ constant region gene (CH) for one of several downstream CH segments (Cγ, Cε or Cα). The reaction proceeds through the introduction of DNA double-strand breaks (DSBs) into transcribed, repetitive DNA elements, called switch (S) regions that precede each CH gene segment. End-joining of DSBs between a donor (Sμ) and a downstream acceptor S region deletes the intervening DNA and juxtaposes a new CH gene to the variable region gene segment. The B cell thereby “switches” from expressing IgM to one producing IgG, IgE or IgA, with each secondary isotype having a distinct effector function during an immune response (Matthews et al., 2014).
The single-strand DNA-specific cytidine deaminase AID is essential for CSR (Muramatsu et al., 2000; Revy et al., 2000). AID deaminates cytosines within transcribed S regions (Chaudhuri et al., 2003; Maul et al., 2011) and the deaminated DNA engages the ubiquitous base-excision and mismatch repair machineries to generate DSBs that are required for CSR (Petersen-Mahrt et al., 2002). A failure to efficiently recruit AID to S regions impairs CSR (Nowak et al., 2011; Pavri et al., 2010; Xu et al., 2010). Conversely, mistargeting of AID activity to non-Ig genes has been implicated in chromosomal translocations and pathogenesis of B cell lymphomas (Nussenzweig and Nussenzweig, 2010; Pasqualucci et al., 2008). While AID is phosphorylated at multiple residues, including at Serine-38, phosphorylation is not required for DNA binding (Matthews et al., 2014).
Thus, the molecular mechanisms by which AID is specifically targeted to S regions continue to be an active area of investigation. Transcription through S regions is essential for CSR and is closely linked to the mechanism by which AID specifically binds and gains access to S regions during CSR (Matthews et al., 2014). Each of the CH genes is organized as individual transcription units comprising of a cytokine inducible promoter, an intervening I-exon, S region and CH exons. Splicing of the primary transcript joins the I- and CH exons to generate a non-coding mature transcript and releases the intronic switch sequence. Transcription through S regions, 1–12 kb long repetitive DNA elements with a guanine-rich non-template strand, predisposes formation of RNA:DNA hybrid structures such as R-loops that expose single-stranded DNA substrates for AID (Matthews et al., 2014). Germline transcription is also required for the binding of AID at S regions through the ability of AID to interact with components of RNA polymerase II (Nambu et al., 2003; Pavri et al., 2010). Both R-loop formation and RNA polymerase II-mediated recruitment of AID relies on the process of transcription, but the role of germline switch transcripts themselves in the recombination reaction has yet to be identified.
Several intriguing reports have suggested that germline switch transcripts might have mechanistic roles in CSR. Deletion of the Iγ1 exon splice donor site, which inhibits splicing of the primary switch transcripts, specifically abrogated CSR to IgG1, even though transcription through Sγ1 was unaffected (Lorenz et al., 1995). Additionally, increasing levels of Sα transcripts by expression from a plasmid enhanced CSR to IgA in a cell line (Muller et al., 1998). Furthermore, while neither the specificity of the interaction nor the physiological significance of the binding was ascertained, AID was shown to bind various RNA in vitro, including tRNA and RNA from insect cells (Bransteitter et al., 2003; Dickerson et al., 2003) suggesting that RNA:AID interactions might be relevant for CSR. Finally, AID was also shown to preferentially mutate small RNA genes when expressed in yeast in a manner that suggests RNA might have a role in its recruitment (Taylor et al., 2014). Interestingly, RNA-guided processes have been shown to regulate DNA rearrangements in ciliates (Mochizuki et al., 2002; Nowacki et al., 2008) and to localize proteins to specific parts of the genome to modify chromatin (Tsai et al., 2010; Zhao et al., 2008). Switch RNAs are complementary to the S region DNA templates and would be ideal to provide specificity as a targeting factor to guide AID to its target DNA (S regions) during CSR. Thus, these observations led us to examine the possibility that switch transcripts can serve as molecular guides that target AID to S region DNA during CSR.
RESULTS
Switch RNA binds AID
For switch transcripts to serve as targeting factors for AID, we reasoned that switch RNAs and AID may interact, and therefore examined this notion in a series of RNA pull-down assays. Purified biotinylated in vitro transcribed (IVT) RNAs were allowed to fold into secondary/tertiary structures and examined for their ability to interact with AID present in extracts of CH12 cells stimulated for CSR. The mouse CH12 B lymphoma cell line switches at a high frequency from IgM to IgA with anti-CD40, IL-4 and TGF-β (henceforth referred to as CIT) stimulation, and has been used as a model system to study CSR (Nowak et al., 2011; Pavri et al., 2010). As sense and anti-sense transcription have been reported through the S regions (Perlot et al., 2008), both sense (forward, F) and anti-sense (reverse, R) switch transcripts were analyzed in this assay. Sense switch μ (SμF) and switch α (SαF) RNAs specifically bound AID, while their anti-sense counterparts (SμR, SαR) did not (Figure 1A). Neither brain cytoplasmic RNA (Bc1), which has been described to form complexes with proteins (Zalfa et al., 2003), nor a fragment of RNA derived from one of the GAPDH introns, associated with AID (Figure 1A). In contrast, interaction of these transcripts with Apobec3, a member of the cytidine deaminase family, was not detected (Figure 1A). An RNA dot blot assay showed that all RNAs were recovered by streptavidin beads at comparable levels (Figure 1A). To assess the interaction between switch RNA and AID in vivo, we generated CH12 cells that stably express Sα transcripts in either orientation (SαF or SαR) with an S1 tag, an RNA aptamer with affinity for streptavidin (Srisawat and Engelke, 2001). S1-tagged RNA from CIT-stimulated cells was recovered on streptavidin beads and probed for AID (Figure 1B). AID only co-purified with S1SαF RNA, even though more S1SαR RNA was recovered (Figure 1B). Apobec3 did not co-purify with either of the Sα transcripts (Figure 1B). Thus, switch RNA can bind to AID both in vitro and in vivo.
Figure 1. Switch RNA can bind AID.
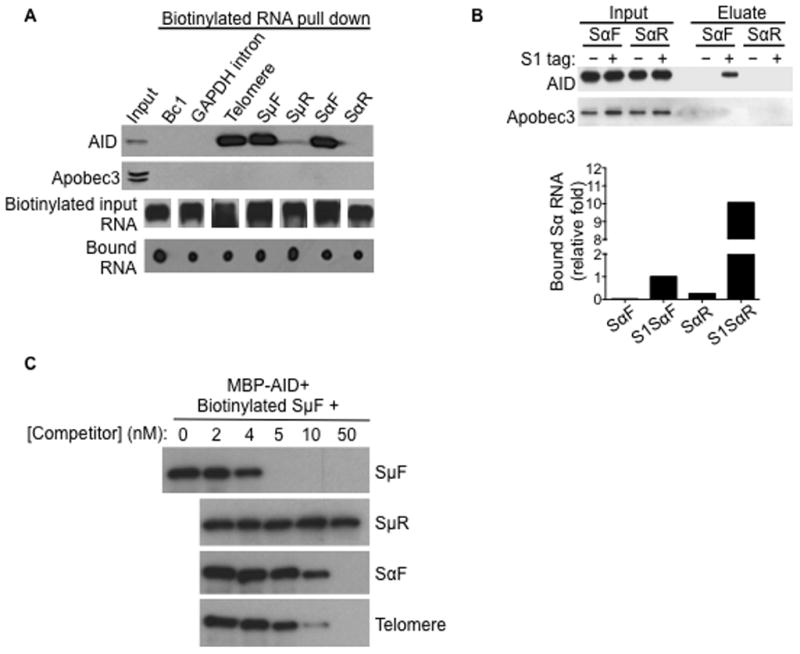
(A) In vitro transcribed (IVT) telomeric and switch RNAs bind to AID. IVT biotinylated RNAs were folded and incubated with whole cell extracts from stimulated CH12 cells, followed by pull-down with streptavidin beads. Proteins recovered were analyzed by immunoblot with AID or Apobec3 antibodies; while bound RNAs were analyzed by dot blot using streptavidin-HRP. Input RNAs were verified to be biotinylated by streptavidin-HRP-northern blot. SμF, SαF; forward/sense switch μ and α RNA. SμR, SαR; reverse/anti-sense switch μ and α RNA. Result shown is representative of three independent pull-down experiments.
(B) Switch RNA interacts with AID in vivo. CH12 cells stably expressing S1-aptamer tagged Sα transcripts, in either the forward/sense (S1SαF) or the reverse/anti-sense (S1SαR) orientation, were stimulated. Untagged SαF and SαR expressing cells were used as controls. The S1-aptamer tag has affinity for streptavidin and ribonucleoprotein complexes were isolated on streptavidin beads. RNA in the eluates was reverse transcribed and analyzed by qPCR (RT-qPCR) for amounts of Sα transcripts relative to S1SαF, while proteins in the eluates were analyzed by immunoblot. Result shown is representative of three independent pull-down experiments.
(C) Competition RNA binding assay. RNA pull-down was performed with 1 nM biotinylated SμF RNA and 100 ng MBP-AID(WT) protein, in the presence of increasing concentrations of non-biotinylated competitor RNAs. Bound MBP-AID(WT) recovered by pull-down with streptavidin beads were analyzed by immunoblot with an AID antibody. Data shown is representative of three experiments.
To determine the binding affinity between AID and switch RNAs, recombinant maltose-binding protein (MBP)-tagged mouse AID was purified and its binding to SμF RNA was examined. Consistent with the interactions observed with cell extracts, in vitro binding assays showed that MBP-AID associated with SμF (Figure 1C), indicating that the AID:RNA interaction is direct. As switch RNA molecules appeared to form large extensive structures that did not migrate into even low percentage gels, Kd values for the interaction between AID and RNAs could not be determined using conventional gel-shift assays. We therefore examined the relative binding affinities in competition assays. Inhibition of binding can be achieved in the nanomolar range (SμF, ~4–5nM; SαF, ~5–50nM) (Figure 1C), suggesting that these interactions are highly specific and would likely have a physiological role.
G-quadruplex structures in switch RNA
Interestingly, AID also co-purified with telomeric RNA (Figure 1A), which like switch transcripts, is G-rich and consists of repetitive sequences (Azzalin and Lingner, 2007). Competitive binding experiments indicate that the affinity of AID for telomeric RNA is comparable to the binding affinities for sense switch RNAs (relative affinities, SμF> telomere > SαF) (Figure 1C). This suggests that AID can associate with sense switch RNAs and similarly G-rich, repetitive RNAs through secondary and/or tertiary structures that are common to these transcripts. Nucleic acid sequences that are rich in guanine tracts can form G-quadruplex structures, wherein guanine residues are arranged in a planar conformation through Hoogsteen base pairing interactions to form a four-stranded structure that is highly stabilized by a central K+ ion, but to a much lesser extent by Li+ (Lane et al., 2008; Sen and Gilbert, 1988; Williamson et al., 1989). Indeed, similar to telomeric RNA, computational analysis indicates that sense switch RNA sequences have strong G-quadruplex forming potential, while their anti-sense counterparts, Bc1 and GAPDH intron do not (Figure 2A).
Figure 2. G-quadruplex structures in switch RNA.
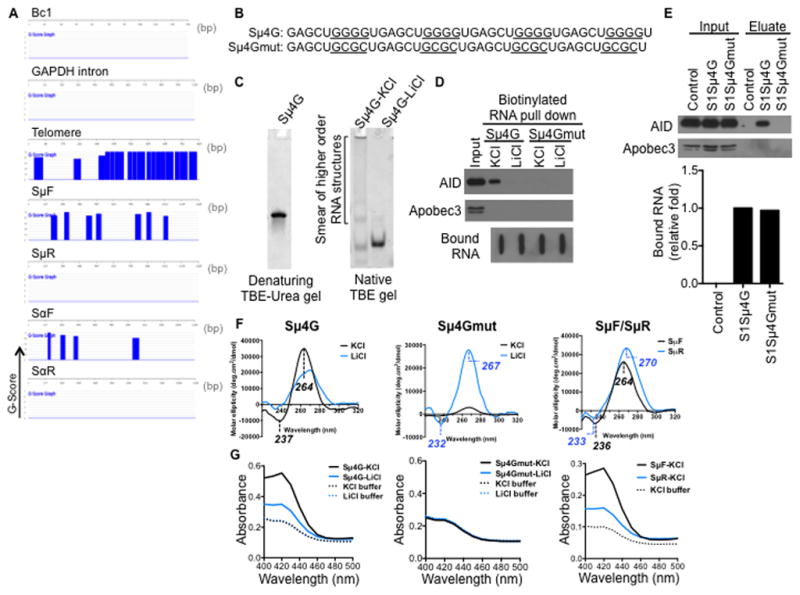
(A) Sense switch RNAs and telomere RNA are predicted to form G-quadruplex structures. QGRS Mapper software was used to assess the G-quadruplex forming potential of the RNA sequences. The probability of G-quadruplex structure formation is reported as a G-score and represented over the corresponding regions in blue.
(B) Sequences of synthetic RNA oligonucleotides. Sμ4G, four Sμ repeats in tandem; mutant Sμ4G (Sμ4Gmut), with G-to-C mutations that abolish guanine tracts. Guanine tracts in Sμ4G and corresponding regions in Sμ4Gmut are underlined.
(C) Sμ4G was resolved on a denaturing gel, or folded in either KCl- or LiCl-containing buffer and resolved on a native gel, and stained with SYBR GOLD following electrophoresis.
(D) Sμ4G associates with AID while Sμ4Gmut does not. RNA pull-down was performed with biotinylated Sμ4G and Sμ4Gmut (folded in either KCl- or LiCl-containing buffer) and stimulated CH12 extracts. Recovered proteins were analyzed by immunoblot and bound RNAs by streptavidin-HRP blot. Result shown is representative of three independent pull-down experiments.
(E) Sμ4G interacts with AID in vivo. CH12 cells stably expressing S1-aptamer tagged Sμ4G or Sμ4Gmut transcripts were stimulated and ribonucleoprotein complexes were isolated on streptavidin beads. CH12 cells expressing anti-sense S1Sμ4G, which is unable to bind streptavidin, were used as control. RNA in the eluates was analyzed by RT-qPCR for amounts of exogenous transcripts relative to S1Sμ4G, while proteins in the eulates were analyzed by immunoblot. Result shown is representative of two independent pull-down experiments.
(F) Circular dichroism (CD) analysis of G-quadruplex structures. CD spectra of Sμ4G, Sμ4Gmut, SμF and SμR RNAs (folded in either KCl- or LiCl-containing buffer). Wavelengths of observed peaks are indicated. Peaks characteristic of parallel G-quadruplexes are (positive- ~260 nm, negative- ~240 nm).
(G) Colorimetric assay of G-quadruplexes. RNAs were folded and incubated with hemin. G-quadruplexes bind hemin and the resultant complex exhibits peroxidase-like activity, which can be detected as an increase in absorbance around 420 nm when substrate is added (Haeusler et al., 2014; Li et al., 2013).
See also Figure S1.
To assess if switch RNAs can form these higher order G-quadruplex structures, we used a synthetic RNA oligonucleotide representing four Sμ repeats in tandem (Sμ4G) (Figure 2B). Analysis of Sμ4G mobility by gel electrophoresis showed that it migrated as a single species under denaturing conditions but as a higher molecular weight smear under native conditions (Figure 2C), indicating the formation of higher order RNA structures. These higher order structures were lost when Sμ4G was folded in the presence of Li+ (Figure 2C). Similar to what was observed for the longer ~1kb SμF IVT RNA (Figure 1A), Sμ4G interacted with AID from extracts of CIT-stimulated CH12 cells in the RNA binding assays (Figure 2D). In comparison, no binding of Sμ4G to Apobec3 was observed in this assay (Figure 2D). Strikingly, when Sμ4G was folded in the presence of Li+, the interaction with AID was lost. In addition, a mutant form of Sμ4G containing G-to-C mutations that abolished the tandem guanine tracts (Sμ4Gmut) (Figure 2B) also did not bind AID (Figure 2D). Furthermore, in CH12 cells that stably expressed either S1-tagged Sμ4G or Sμ4Gmut transcripts, endogenous AID interacted with Sμ4G but not with Sμ4Gmut RNA (Figure 2E), Taken together, these results suggest that switch RNAs can form G-quadruplexes and may bind AID through these structures.
To further demonstrate the ability of switch RNA to form G-quadruplex structures, we performed circular dichroism spectroscopy. Sμ4G, but not Sμ4Gmut, displayed the characteristic spectrum of parallel G-quadruplexes (Kumari et al., 2007; Xu et al., 2008), with a positive peak at ~260 nm and a negative peak at ~240 nm, both of which are reduced in the presence of Li+ (Figure 2F). These results are consistent with that observed for the well-characterized G-quadruplex-forming C9orf72 hexanucleotide repeat expansion (HRE) RNA (Haeusler et al., 2014) (Figure S1A). This signature spectrum was also evident in SμF RNA indicating that it also forms parallel G-quadruplexes, while SμR showed shifts in peaks to wavelengths of 270 nm and 233 nm that are not typical of these structures (Figure 2F). Formation of G-quadruplex structures was additionally verified in a ligand-binding colorimetric assay that is based on the ability of G-quadruplexes to bind hemin (Li et al., 2013). G-quadruplex:hemin complexes exhibit peroxidase-like activity that is detected as a green coloration when exposed to substrate (Figure S1B). This can be measured as an absorbance signal upon a spectral scan from 400 to 500 nm (Li et al., 2013) with a maxima at 420 nm as seen for the known G-quadruplex-forming C9orf72 HRE RNA (Haeusler et al., 2014) (Figure S1C). In this assay, Sμ4G RNA, when folded in the presence of K+ also showed the characteristic absorbance maxima at 420 nm, which was clearly reduced when Sμ4G RNA was folded in the presence of Li+ (Figure 2G). Sμ4Gmut RNA did not exhibit any detectable absorbance signal above the buffer control (Figure 2G). Similar to Sμ4G, SμF also exhibited the characteristic absorbance maxima at 420 nm that was greatly reduced for SμR (Figure 2G). Finally, the biotinylated Sμ4G used in the in vitro interaction experiments also exhibited G-quadruplex forming ability (Figure S1D, S1E). Taken together, these data strongly suggest that switch RNA can form G-quadruplexes and these structures might mediate the interaction with AID.
AID(G133V) does not bind switch RNA and cannot mediate CSR
Next, we investigated the G-quadruplex binding domain in AID. No RNA-binding domain in AID has been described; however, sequence alignment revealed that amino acids 130–138 of mouse AID shares some sequence homology with the RNA-binding domain of the well-characterized G-quadruplex RNA binding protein RHAU (Creacy et al., 2008; Vaughn et al., 2005) (Figure S2A). Mutations of the two glycine residues to proline in RHAU greatly reduced its binding to G-quadruplex RNA (Lattmann et al., 2010). Likewise, mutation of the corresponding glycine-133 and glycine-137 in mouse AID to prolines completely abolished the ability of AID to restore CSR when expressed in AID-deficient mouse splenic B cells (Figure S2B–D). Furthermore, a G133V mutation in AID has been identified in two hyper-IgM patients with severe CSR defects (Mahdaviani et al., 2012). As proline mutations can be disruptive to overall protein structure, and that the glycine-133 residue is conserved in AID across all species, mouse AID with the less bulky G133V mutation was characterized further.
Recombinant MBP-tagged wild-type (WT) and G133V mouse AID were purified (Figure 3A) and assessed for their ability to interact with switch RNAs. MBP-AID(G133V) was substantially impaired in its ability to bind switch RNAs compared to MBP-AID(WT) (Figure 3B), providing further support that the AID:RNA interaction is specific and direct. To determine if the failure to bind switch RNA is due to a general misfolding of MBP-AID(G133V), we carried out deamination assays on single-stranded DNA (ssDNA) substrates. While mouse AID is known to display weak deaminase activity in vitro (Chaudhuri et al., 2003), the activity of MBP-AID(G133V) was comparable to MBP-AID(WT) (Figure 3C). Additionally, the ssDNA binding ability of MBP-AID(G133V) was similar to the wild-type protein (Figure 3D). Thus, the defect in RNA binding is unlikely to be due to general loss of the structural integrity of the MBP-AID(G133V) protein.
Figure 3. Glycine-133 of AID is critical for RNA binding and CSR.
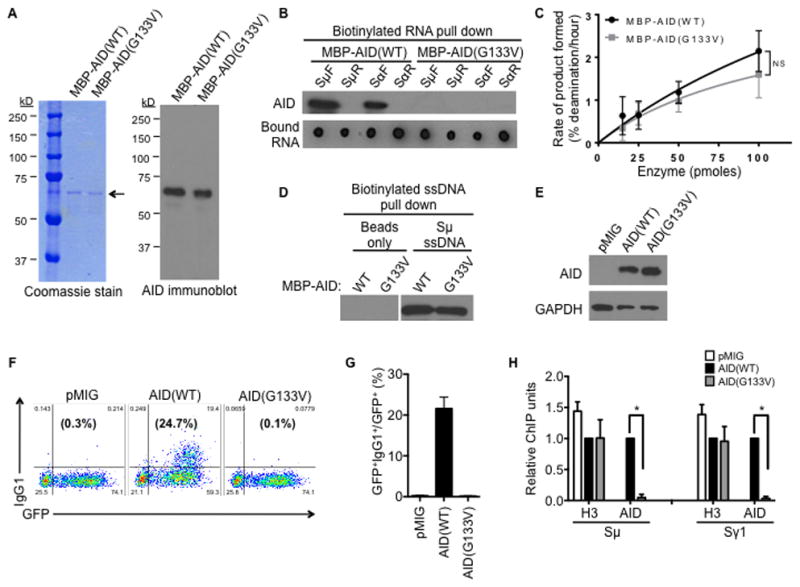
(A) Purified recombinant MBP-tagged, wild-type (WT) or mutant AID(G133V) proteins were analyzed by Coomassie stain and immunoblot with AID antibody. Arrow indicates the size corresponding to full-length MBP-AID proteins on the Coomassie stained gel.
(B) Purified proteins were used in the RNA pull-down assay with IVT biotinylated switch RNAs, followed by analysis of recovered proteins by immunoblot with an AID antibody, and bound RNAs by streptavidin-HRP dot blot. Result shown is representative of three independent pull-down experiments.
(C) Enzymatic activity of purified proteins was determined by a deamination assay. The rate of deamination was determined as a function of protein concentration as described in Experimental Procedure. The average ± SD of three independent protein preparations is shown; NS, p=not significant, p≥0.05 at 25, 50 and 100 pmoles enzyme concentrations.
(D) The G133V mutation does not affect binding of AID to single-stranded DNA (ssDNA). Purified proteins were incubated with biotinylated ssDNA, followed by pull-down with streptavidin beads and analysis by immunoblot with an AID antibody. Data shown is representative of three experiments.
(E–H) Splenic B cells were isolated from AID−/− mice and transduced with vector control (pMIG), or vectors to express AID(WT) or AID(G133V).
(E) Expression of AID proteins was verified by immunoblot with AID or GAPDH (control) antibodies.
(F) CSR to IgG1 was determined by flow cytometry. A representative experiment is shown. The numbers in the corners indicate the percentage of cells in each quadrant, while the numbers in parentheses indicate the percentage of IgG1+ cells within the GFP+ gate.
(G) The average percentage of IgG1+ cells within the GFP+ gate from three independent experiments ± SD is shown.
(H) Localization of AID proteins to S regions was determined by ChIP, using AID or H3 antibodies. Sμ and Sγ1 DNA in ChIP samples was measured by qPCR, normalized to input DNA and the AID(WT) control. The mean of three independent experiments ± SD is shown.
See also Figure S2.
To determine the functional relevance of AID(G133V) in CSR, the mutant protein was expressed in AID-deficient splenic B cells. AID(G133V) was expressed at similar levels as AID(WT) (Figure 3E) and was present in the nucleus at comparable levels (Figure S2E), once again suggesting that the mutation did not have a gross effect on protein structure. Strikingly, AID(G133V) was completely inactive in inducing CSR in AID-deficient B cells. While AID(WT) resulted in CSR in over 20% of the transduced cells, CSR in AID(G133V)-expressing cells was similar to that of the vector control (Figure 3F, 3G). Expression of AID(G133V) did not adversely affect the level of germline switch transcripts compared to AID(WT) (Figure S2F). To assess if impaired CSR is due to reduced ability of AID(G133V) to bind DNA, chromatin immunoprecipitation (ChIP) was carried out with an AID antibody that can immunoprecipitate both wild-type and mutant proteins equally (Figure S2G). The ChIP experiments showed that AID(G133V) was significantly impaired in its ability to bind S region DNA (Figure 3H). While AID(WT) efficiently associated with Sμ and Sγ1 (the two S regions that recombine upon anti-CD40 plus IL-4 stimulation), binding of AID(G133V) to these S regions was profoundly reduced (Figure 3H). Neither the wild-type nor mutant AID protein associated with the control non-target locus Cγ1 region (Figure S2H). The binding of histone H3 remained unaltered (Figure 3H and Figure S2H), demonstrating the specificity of the ChIP assays. This indicates that the failure of AID(G133V) to mediate CSR is due to a loss of binding to S region DNA. It is interesting to note that despite the abundance of telomeric RNA in splenic B cells (Figure S2J) and the ability of AID to interact with telomeric RNA in vitro (Figure 1A), AID did not bind telomere DNA (Figure S2I), probably because the telomeres are protected by a large number of proteins and tightly packed into heterochromatin, which might render them inaccessible to AID. Overall, the observation that a point mutation in AID that impairs its ability to bind G-quadruplex RNA also markedly reduces its ability to bind DNA, strongly supports the notion that switch RNAs can guide AID to DNA.
Debranching of RNA lariats is required for CSR
The direct association of AID with switch (or “guide”) RNAs, and the defect in CSR when the interaction is impaired, led us to hypothesize that perturbations to the processing of germline switch transcripts could interfere with the generation of the guide RNAs that recruit AID to S region DNA. As S region sequences lie within the intronic region of the germline switch transcripts, we tested this hypothesis by depleting the lariat debranching enzyme, DBR1, to perturb the processing of the switch RNAs without affecting transcription and splicing, upstream events that are known to be important for CSR (Matthews et al., 2014). DBR1 is responsible for debranching intronic lariats by catalyzing the hydrolysis of the 2′,5′-phosphodiester bond at the branchpoint (Arenas and Hurwitz, 1987; Ruskin and Green, 1985). We generated DBR1 “gene-trapped” mice in which a truncated, non-functional, fusion protein, which is missing ~80% of the DBR1 protein, is produced (Figure S3A). No live homozygous (DBR1−/−) mutant mice were obtained from breeding DBR1+/− mice, indicating that DBR1 is required for embryonic development. However, DBR1+/− splenic B cells activated for CSR exhibited a significant decrease in expression of full length DBR1 mRNA (Figure 4A) with concomitant expression of the gene-trapped fusion mRNA (Figure S3B). DBR1+/− B cells expressed similar levels of AID protein (Figure S3C) and germline transcripts (Figure S3D), and proliferated at comparable rates (Figure S3E, S3F) to DBR1+/+ littermate controls. DBR1+/− B cells stimulated ex vivo showed a small but significant reduction in CSR to IgG1 (Figure 4B and Figure S3G). The CSR defect was rescued by expression of Xpress-tagged DBR1 (XpDBR1) (Figure 4C, 4D). Exogenous expression of XpDBR1 did not affect levels of AID (Figure 4C) or germline switch transcripts (Figure 4E).
Figure 4. DBR1+/− splenic B cells exhibit reduced CSR upon ex vivo stimulation.

Splenic B cells were isolated from heterozygous (+/−) DBR1 gene-trapped mice and wild-type (+/+) littermate controls, and stimulated in culture with anti-CD40 and IL-4 for 72 h.
(A) Expression of full length DBR1 is reduced in DBR1+/− B cells. The level of DBR1 mRNA was measured by RT-qPCR using primers downstream of the gene trap insertion, normalized to β-actin mRNA and DBR1+/+ control. The average of four pairs of DBR1+/− mice and their littermate DBR1+/+ controls ± SD are shown; *p<0.05.
(B) CSR to IgG1 was determined at 72h post-stimulation by flow cytometry. Data shows the mean of 10 pairs of DBR1+/− mice and their DBR1+/+ littermates ± SD. *p<0.05.
(C–E) Splenic B cells from DBR1+/+ and DBR1+/− mice were transduced with retroviral vector control (pMIR) or vector expressing Xpress-tagged DBR1 (XpDBR1).
(C) Expression of XpDBR1 and AID were verified by immunoblot by anti-Xpress, anti-AID and anti-GAPDH (control) antibodies.
(D) CSR to IgG1 was determined by flow cytometry. The average percentage of IgG1+ cells within the mCherry+ gate from three independent experiments ± SD is shown. *p<0.05.
(E) Expression of XpDBR1 does not adversely affect levels of μ- and γ1-germline switch transcripts (GLT) compared to pMIR. Levels of μ- and γ1-GLT were determined by qRT-PCR, normalized to β-actin mRNA and pMIR control. Data represents the average of three independent experiments ± SD; NS, p=not significant, p≥0.05.
See also Figure S3.
Impaired CSR in DBR1+/− B cells was accompanied by a slightly reduced, though not statistically significant, frequency of mutations in Sμ (Figure S3H). Somatic hypermutation (SHM) in Peyer’s patches B cells remained unaffected (Figure S3I). While these data suggest that DBR1 influences CSR, the relative difficulty of performing experimental manipulations in short-lived ex vivo cultured mouse splenic B cells, as well as the modest CSR and Sμ mutation frequency defects in DBR+/− mice, led us to use the CH12 cell line as a model system to further elucidate the roles of switch RNAs in CSR.
Knockdown of DBR1 was achieved in CH12 cells using shRNA (Figure 5A) and was accompanied by an increase in the RNA lariat levels of Sμ (Figure 5B and Figure S4B) and β-actin intron 3 (Figure S4A, S4C) as compared to the scrambled shRNA control, confirming a reduction in DBR1 enzymatic activity. When DBR1 knockdown cells were stimulated with CIT, we observed a significant reduction in CSR to IgA compared to control cells at all time points assayed by flow cytometry (Figure 5C) and by semi-quantitative measurement of the Iα-Cμ circle transcripts produced from the excised extrachromosomal DNA (Figure S4D). Levels of mature germline transcripts were not altered in DBR1 knockdown cells (Figure S4E), suggesting that transcription and splicing were not affected. DBR1 knockdown did not affect expression of AID mRNA (Figure S4F) or protein (Figure 5D). DBR1 knockdown cells proliferated at rates comparable to the scrambled control (Figure S4G, S4H). Thus, DBR1 knockdown significantly impaired CSR without affecting transcription, splicing of primary germline switch transcripts, AID expression or proliferation.
Figure 5. Debranching of intronic RNA lariats is required for targeting of AID to S regions and efficient CSR.
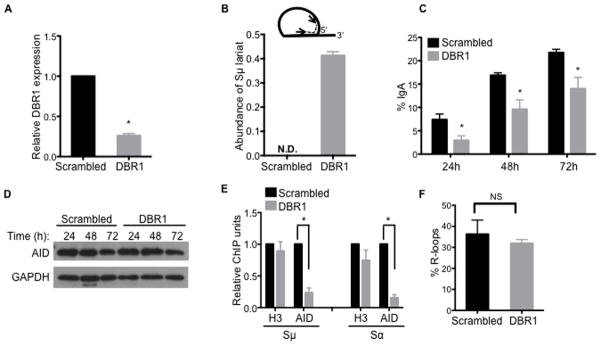
(A–F) Knockdown of DBR1 impairs CSR and AID localization to S regions. CH12 cells were transduced with shRNA against DBR1 or scrambled control shRNA.
(A) Knockdown of DBR1 transcripts was determined by RT-qPCR.
(B) Accumulation of Sμ lariats following DBR1 knockdown was determined by RT-qPCR using primers (arrows) positioned across the branchpoint as shown (inset). N.D., not detected. Data in (A) and (B) were normalized to β-actin mRNA and the scrambled control; the average of at least three independent knockdown experiments ± SD are shown; *p<0.05.
(C) CSR to IgA was assayed by flow cytometry at indicated times following stimulation. Data shows the mean of three independent knockdown experiments ± SD. *p<0.05.
(D) AID protein expression was analyzed at indicated times following stimulation by immunoblot with AID or GAPDH (control) antibodies.
(E) Localization of AID to S regions was determined by ChIP on cells 48 h post-stimulation using AID or H3 antibodies. Sμ and Sα DNA in ChIP samples were measured by qPCR and normalized to input DNA and the scrambled control. Data represents mean of three independent knockdown experiments ± SD. *p<0.05.
(F) R-loop formation is unaffected by DBR1 knockdown. Genomic DNA was prepared from cells 24 h post-stimulation and treated with the bisulfite modification assay. Sα was examined and the number of molecules containing R-loops is represented as a percentage of the total number of DNA amplicons sequenced. The mean of three independent knockdown experiments ± SD is shown; NS, p=not significant, p≥0.05.
To determine if the CSR defect in DBR1 knockdown CH12 cells is due to impaired targeting of AID to S region DNA, ChIP assays were performed. DBR1 knockdown led to a significant reduction in AID localization (~5-fold) to both Sμ and Sα, while the binding of histone H3 was unaffected (Figure 5E). AID did not associate with the control non-target locus Cγ1 (Figure S5A), demonstrating the specificity of the assay. The defect in the ability of AID to bind S regions in DBR1 knockdown cells was not due to altered stability or abundance of R-loops at S region DNA (Kao et al., 2013) (Figure 5F and Figure S5B). The localization of RNA polymerase II and Spt5 to S regions were unaffected by DBR1 knockdown (Figure S5C), indicating that the loss of AID binding at S regions was not due to perturbations to these known factors of AID targeting (Pavri et al., 2010). These data suggest that post-transcriptional factors (guide RNAs) facilitate AID targeting to S regions in addition to previously characterized co-transcriptional factors. It is noteworthy that DBR1 knockdown occasionally resulted in a more severe defect in CSR (Figure S5D), which correlated with a larger reduction of AID localization (>100-fold) to S regions (Figure S5E, S5F). Despite the drastic loss of AID targeting in this instance, AID binding to S regions was not completely abrogated in DBR1 knockdown cells, as evident from the quantitative PCR (qPCR) results that showed enrichment of Sμ and Sα fragments in anti-AID ChIPs as compared to the non-specific IgG control (Figure S5G). Although it is unclear why the magnitude of the defect varies, DBR1 knockdown consistently led to a reproducible reduction in AID localization to S region DNA.
Expression of switch RNA in trans rescues CSR defect in DBR1 depleted cells
Given that DBR1 is responsible for debranching all intronic lariats in the cell, it remains to be determined if the loss of AID targeting can be attributed to impaired processing of switch transcripts alone. To determine if switch transcripts can guide AID to S region DNA during CSR, we expressed Sα transcripts in either the sense (exoSαF) or anti-sense (exoSαR) orientation in CH12 cells and examined whether these exogenous switch transcripts could rescue AID localization to S regions and CSR in DBR1 knockdown cells (Figure 6A). Exogenous Sα transcripts were readily detected (Figure S6A) and the cells expressed similar levels of AID upon CIT-stimulation (Figure S6B). ChIP analyses showed that expression of exoSαF, but not exoSαR, restored binding of AID to endogenous Sα DNA (Figure 6B), despite exoSαR being expressed to higher levels than exoSαF (Figure S6C). The qPCR quantification in the ChIP analyses was performed using primers that detected the endogenous Sα locus, but not the exogenously transduced sequence, indicating that the observed results are a rescue of AID targeting to the endogenous Sα locus. Interestingly, neither exoSαF nor exoSαR could rescue AID localization to the non-complementary endogenous Sμ DNA in DBR1 knockdown cells (Figure 6C), suggesting that switch RNAs can target AID to S region DNA in a sequence-specific manner. As expected from the selective rescue of AID localization to only the Sα DNA, exogenous expression of Sα transcripts could not restore CSR to IgA (Figure 6D). Additionally, higher expression of exoSαR compared to exoSαF (Figure S6C) was not detrimental to CSR, as cells expressing exoSαR switched at a level comparable to cells expressing exoSαF and those infected with the empty vector control (Figure 6D).
Figure 6. Localization of AID to S regions can be restored by exogenous expression of switch transcripts.
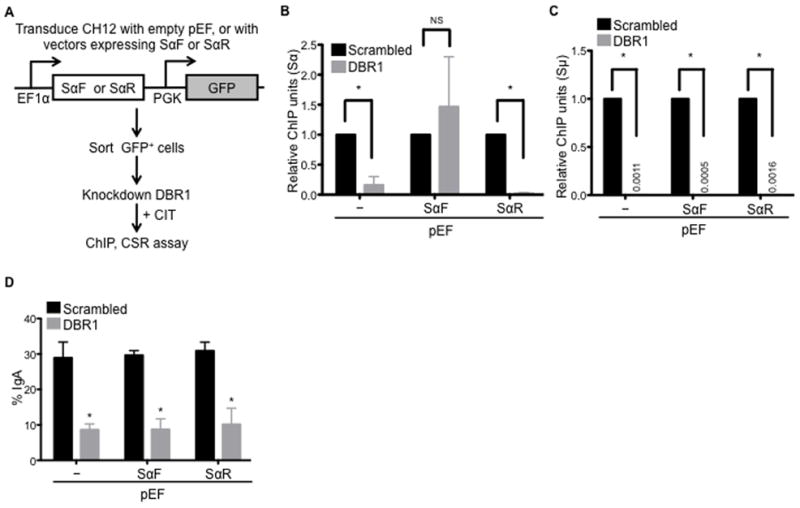
(A) Experimental design. CH12 cells were transduced with empty vector (pEF) or vectors to express forward/sense or reverse/anti-sense switch α RNA (pEF-SαF or pEF-SαR, respectively). Transduced cells were sorted, infected with scrambled control shRNA or shRNA to knockdown DBR1, and stimulated with CIT.
(B–C) Expression of exogenous SαF rescues AID localization to Sα at the endogenous IgH locus, but not to the non-complementary Sμ. ChIP was performed on cells 48 h post stimulation by immunoprecipitation with anti-AID antibodies. Sα (B) and Sμ (C) DNA in ChIP samples was quantified by qPCR, and normalized to input and scrambled control.
(D) Expression of exogenous SαF does not rescue CSR in DBR1 knockdown cells. CSR to IgA was assayed 72 h after stimulation by flow cytometry. Data in (B)–(D) represent the mean of three independent experiments ± SD. *p<0.05; NS, p=not significant, p≥0.05.
See also Figure S6.
The inability of exogenous Sα alone to rescue IgA levels suggests that restoration of AID targeting to both participating switch loci, Sμ and Sα, is required before productive CSR to IgA can occur. To test this hypothesis, we transduced CH12 cells to co-express sense (For: exoSμF + exoSαF) or anti-sense (Rev: exoSμR + exoSαR) transcripts of Sμ and Sα (Figure 7A). Cells expressing exogenous switch transcripts were then knocked-down for DBR1 expression (Figure S6D). DBR1 depletion increased lariat accumulation (Figure S6E) but did not affect steady state levels of exogenous switch transcripts (Figure S6F) or AID expression (Figure S6G). Strikingly, co-expression of exoSμF and exoSαF transcripts significantly rescued switching to IgA in DBR1 knockdown cells (Figure 7B). In contrast, dual expression of exoSμR and exoSαR transcripts had no effect in restoring IgA levels in DBR1 knockdown cells (Figure 7B). Thus, expression of sense Sμ and Sα transcripts together can functionally reconstitute CSR in cells where debranching of intronic RNA lariats was inhibited. It is interesting to note that co-expression of SμF and SαF transcripts was unable to increase switching to IgA in the absence of DBR1 knockdown (Figure 7B). This suggests that endogenous guide RNAs are not limiting during CSR, or perhaps the rate of the switching reaction is at a maxima and the supply of exogenous guide RNAs could not increase CSR to IgA any further in these cells. Taken together, these data provide strong experimental support for a model (Figure 7C) wherein switch RNA, through its ability to fold into G-quadruplex structures, can bind AID and target AID to S region DNA during CSR.
Figure 7. Co-expression of sense Sμ and Sα transcripts rescues CSR to IgA in DBR1 knockdown cells.

(A) Experimental design. CH12 cells were transduced with empty vectors (EF) or vectors expressing forward/sense (For) or reverse/anti-sense (Rev) switch transcripts. Doubly transduced cells were sorted, infected with scrambled control shRNA or shRNA to knockdown DBR1, and stimulated with CIT.
(B) CSR to IgA was assayed by flow cytometry 72 h after stimulation. The average of three independent experiments ± SD is shown. *p<0.05; NS, p=not significant, p≥0.05.
(C) Model for RNA-mediated targeting of AID during CSR. When B cells are stimulated to undergo class switching, transcription occurs at each of the recombining S regions to produce primary switch transcripts. Primary transcripts are spliced, with the intronic switch region sequences (Sx) spliced out as a lariat intermediate. Debranching enzyme (DBR1) catalyzes the release of the lariat and debranches the switch transcript into its linear form. The linear switch transcript, free of exonic sequences, can function as guide RNAs by forming G-quadruplex or G-quadruplex-like structures, which allows association with AID. AID, bound to guide RNAs, can be targeted specifically to the complementary S region DNA based on sequence information provided by the guide RNAs.
See also Figure S6.
DISCUSSION
Collectively, our results provide strong evidence for the existence of an RNA cofactor in the targeting of AID to S regions during CSR. In this model, guide RNAs derived from the intronic region of germline switch transcripts can form G-quadruplexes or G-quadruplex-like RNA structures that allow association with AID, thereby guiding AID preferentially to the complementary S region DNA in a sequence-specific manner (Figure 7C). Identification of two patients with hyper-IgM syndrome, who carry the G133V mutation (Mahdaviani et al., 2012) in the putative G-quadruplex RNA binding domain of AID that disrupts binding to switch RNAs, further highlights the relevance of the switch RNA:AID interaction in CSR.
The importance of sequence information encoded by guide RNAs for their function indicates that base-pairing mediated recognition is likely involved. Yet, according to the prevailing R-loop-based model for CSR (Chaudhuri et al., 2007), the template DNA strand of S regions is stably hybridized to the nascent primary transcript. Thus, the interaction of the RNA:AID complex with the template DNA strand likely requires displacement of the nascent transcripts by the guide RNA molecules. Transcription through the switch regions may be temporally regulated, with a wave of transcription to generate the guide RNAs, followed by a period of transcriptional quiescence at the locus to free the template strand to interact with guide RNAs bound to AID molecules. Alternatively, anti-sense transcription (Perlot et al., 2008) through the IgH locus could expose the sense strand to base-pair with guide RNAs. Finally, it is also possible that following R-loop collapse (maybe after RNaseH action or RNA exosome activity (Basu et al., 2011)), the complementary DNA strands misalign due to the repetitive nature of S regions and results in exposed stretches of ssDNA (Yu and Lieber, 2003), providing an ideal seed-sequence for guide RNA binding. RNA:RNA base-pairing could also allow guide RNAs to find the S region DNA. Interestingly, the anti-sense switch transcripts appear to be inert in that while they do not promote CSR, they do not act as decoys and decrease CSR (Figure 7B). This suggests that when expressed in trans, anti-sense switch transcripts do not interact with or affect the activity of the guide RNAs and sense germline transcripts. Nevertheless, the possibility remains that anti-sense transcripts may have a role in cis, and that nascent anti-sense transcripts at the switch locus could serve as docking sites for guide RNAs to find their target DNA region. Alternatively, guide RNAs may interact with the nascent sense switch transcripts, perhaps by the same interactions that allow switch RNAs to form G-quadruplex structures (Figure 2F, 2G), and thus be localized to the vicinity of their target DNA.
RNA-mediated recruitment of AID to DNA may also have implications for the aberrant targeting of AID to regions outside the immunoglobulin locus. As AID has the ability to associate with RNA by binding G-quadruplex structures (Figure 1, 2), AID could potentially associate with other cellular RNA that fold into similar structures, thus mis-targeting AID to these other genomic loci. For instance, c-Myc has been reported to be a hotspot for AID activity outside the immunoglobulin locus and contributes to the c-Myc/IgH translocations seen in B cell lymphomas (Nussenzweig and Nussenzweig, 2010). G-quadruplex structures in DNA have been implicated in CSR (Dempsey et al., 1999) and have also been found in the first exon and intron of c-Myc, which correspond to common breakpoints in c-Myc/IgH translocations that involve AID (Duquette et al., 2004; Duquette et al., 2005). Interestingly, examination of a subset of genes that are bound by AID in ChIP-seq analysis (Yamane et al., 2011) showed that the primary transcripts derived from these genes have greater potential to form G-quadruplexes than RNA transcribed from genes that are not targeted by AID (Figure S7). While AID activity at these non-Ig loci has been attributed to secondary structures in the DNA, it is tempting to speculate that the G-quadruplex RNA molecules derived from this region mediates mistargeting of AID. Additionally, recent studies further revealed that anti-sense and convergent transcription at super-enhancers, especially at these non-Ig hotspots, can facilitate mistargeting by providing single-stranded DNA substrates for AID (Meng et al., 2014; Pefanis et al., 2014; Qian et al., 2014). The high binding affinity of AID for switch RNA that is in the nanomolar range could potentially facilitate AID to distinguish the switch RNAs from the other RNA species in the physiological setting of the cell. A detailed landscape of transcriptome-wide association of AID will be important to better establish a global map of AID:RNA interactions. However, these studies await generation of CLIP-seq or RIP-seq grade antibodies and better sequencing approaches to map repetitive sequences before such experiments can be meaningfully undertaken.
Recruitment of AID to S regions by interaction with RNA polymerase II in a co-transcriptional step and by binding to switch RNA molecules in a post-transcriptional step provides two distinct mechanisms by which AID could be efficiently and specifically targeted to the IgH locus during CSR. This two-step recruitment not only ensures the localization of a high density of AID molecules at S regions required for CSR but also provides a means by which this general mutator is sequestered from other regions of the genome. In this regard, S regions in Xenopus are not G-rich and RNA transcribed from the Xenopus IgH locus is not predicted to form G-quadruplex structures. It is likely that CSR in Xenopus occurs through a SHM-like mechanism that does not require defined RNA structures (Zarrin et al., 2004).
In summary, we have uncovered a novel mechanism for the targeting of AID specifically to S regions at the IgH locus during CSR that is based on sequence information encoded in RNA. This study specifies a role for germline transcripts independent of transcription and provides an explanation for the long-standing link between the requirement of splicing and CSR (Lorenz et al., 1995). RNA-guided processes are emerging as an efficient mechanism to target proteins to defined genomic regions (Tsai et al., 2010; Zhao et al., 2008) and our findings reveal CSR to be yet another example of this versatile system.
EXPERIMENTAL PROCEDURES
RNA folding and in vitro RNA pull-down assay
RNA folding and pull-down assay were performed as described in (Booy et al., 2012). Briefly, RNAs were folded by heating at 95 °C, then allowed to cool passively to room temperature. Purified AID proteins or whole cell extracts from stimulated CH12 cells were incubated with folded biotinylated RNAs, followed by pull-down with streptavidin beads. Bound proteins and RNAs were analyzed by immunoblot and dot blot respectively. For details, see Extended Experimental Procedures.
Purification of S1-tagged ribonucleoprotein complexes
Whole cell extracts were prepared from stimulated CH12 cells that stably express S1-tagged switch RNAs. S1-tagged RNAs and associated proteins were recovered by pull-down with streptavidin beads, and analyzed by RT-qPCR and immunoblot respectively. See Extended Experimental Procedures for more details.
Deamination assay
Deamination assay was performed as described (Nabel et al., 2012), using a 5′-radiolabeled 30 bp oligonucleotide with a single cytosine as substrate. Following incubation with MBP-AID proteins, UDG was added and DNA at abasic sites was cleaved by heating in 0.1N NaOH. Samples were resolved and analyzed by autoradiography; percentage product formed over time was calculated and normalized to the 0 h control. The rate of the reaction was calculated from the slope of the curve, and plotted against concentration of enzyme. See Extended Experimental Procedures for more details.
Computational analysis of RNA sequences
The G-quadruplex prediction software QGPRS Mapper (http://bioinformatics.ramapo.edu/QGRS/analyze.php) (Kikin et al., 2006) was used to assess the G-quadruplex forming potential of RNA sequences. Parameter used are as follows: max length=45, min G-group size=3, loop size=0–36. The highest G-score for each primary transcript was noted as the G-score for that sequence.
Circular dichroism (CD) spectroscopy
RNA oligonucleotides (C9orf72 HRE, Sμ4G and Sμ4Gmut) were folded at 10 μM, while SμF and SμR RNA were folded at 0.5 μM. CD spectra were obtained using an Aviv 202 CD spectrometer 62DS at 25 °C, with wavelength scan range of 220–320 nm and path length of 1mm. Spectra were subtracted for buffer controls and smoothing was performed using Prism software by averaging 4 neighboring points.
Supplementary Material
Acknowledgments
The authors wish to thank T. Honjo for AID−/− mice and CH12 cells, S. Kabir (Rockefeller University) for telomere-specific probes and S. Vasudevan (Harvard University) and T. Agarwal for their helpful technical advice. We thank members of the Chaudhuri laboratory for their comments and discussion. This work was supported by grants from the National Institutes of Health (1RO1AI072194) and the Starr Cancer Foundation (I4-A447 and I7-A767) to JC.
Footnotes
AUTHOR CONTRIBUTIONS
S.Z., B.Q.V., and J.C. developed ideas, designed experiments and wrote the manuscript. S.Z. performed most of the experiments described in the manuscript, B.Q.V. carried out some of the ChIP assays, retroviral reconstitution, SHM and protein fractionation studies, B.V. purified MBP-AID proteins, performed the XpDBR1 experiments, deamination and colorimetric assays. J-Y. L. and F-T. H. carried out the R-loop assays. B.Q.V and B.V. contributed equally to the work.
The authors declare that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arenas J, Hurwitz J. Purification of a RNA debranching activity from HeLa cells. The Journal of biological chemistry. 1987;262:4274–4279. [PubMed] [Google Scholar]
- Azzalin CM, Lingner J. Molecular biology: damage control. Nature. 2007;448:1001–1002. doi: 10.1038/4481001a. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booy EP, Meier M, Okun N, Novakowski SK, Xiong S, Stetefeld J, McKenna SA. The RNA helicase RHAU (DHX36) unwinds a G4-quadruplex in human telomerase RNA and promotes the formation of the P1 helix template boundary. Nucleic Acids Res. 2012;40:4110–4124. doi: 10.1093/nar/gkr1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Creacy SD, Routh ED, Iwamoto F, Nagamine Y, Akman SA, Vaughn JP. G4 resolvase 1 binds both DNA and RNA tetramolecular quadruplex with high affinity and is the major source of tetramolecular quadruplex G4-DNA and G4-RNA resolving activity in HeLa cell lysates. J Biol Chem. 2008;283:34626–34634. doi: 10.1074/jbc.M806277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey LA, Sun H, Hanakahi LA, Maizels N. G4 DNA binding by LR1 and its subunits, nucleolin and hnRNP D, A role for G-G pairing in immunoglobulin switch recombination. J Biol Chem. 1999;274:1066–1071. doi: 10.1074/jbc.274.2.1066. [DOI] [PubMed] [Google Scholar]
- Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID Mediates Hypermutation by Deaminating Single Stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette ML, Pham P, Goodman MF, Maizels N. AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation. Oncogene. 2005;24:5791–5798. doi: 10.1038/sj.onc.1208746. [DOI] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao YP, Hsieh WC, Hung ST, Huang CW, Lieber MR, Huang FT. Detection and characterization of R-loops at the murine immunoglobulin Salpha region. Mol Immunol. 2013;54:208–216. doi: 10.1016/j.molimm.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Kikin O, D’Antonio L, Bagga PS. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006;34:W676–682. doi: 10.1093/nar/gkl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S, Bugaut A, Huppert JL, Balasubramanian S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nat Chem Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, Chaires JB, Gray RD, Trent JO. Stability and kinetics of G-quadruplex structures. Nucleic Acids Res. 2008;36:5482–5515. doi: 10.1093/nar/gkn517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattmann S, Giri B, Vaughn JP, Akman SA, Nagamine Y. Role of the amino terminal RHAU-specific motif in the recognition and resolution of guanine quadruplex-RNA by the DEAH-box RNA helicase RHAU. Nucleic Acids Res. 2010;38:6219–6233. doi: 10.1093/nar/gkq372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Liu J, Fang Y, Qin Y, Xu S, Liu Y, Wang E. G-quadruplex-based ultrasensitive and selective detection of histidine and cysteine. Biosens Bioelectron. 2013;41:563–568. doi: 10.1016/j.bios.2012.09.024. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267:1825–1828. doi: 10.1126/science.7892607. [DOI] [PubMed] [Google Scholar]
- Mahdaviani SA, Hirbod-Mobarakeh A, Wang N, Aghamohammadi A, Hammarstrom L, Masjedi MR, Pan-Hammarstrom Q, Rezaei N. Novel mutation of the activation-induced cytidine deaminase gene in a Tajik family: special review on hyper-immunoglobulin M syndrome. Expert Rev Clin Immunol. 2012;8:539–546. doi: 10.1586/eci.12.46. [DOI] [PubMed] [Google Scholar]
- Matthews AJ, Zheng S, Dimenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, Gramlich HS, Schatz DG, Woodgate R, Wilson DM, 3rd, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol. 2011;12:70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng FL, Du Z, Federation A, Hu J, Wang Q, Kieffer-Kwon KR, Meyers RM, Amor C, Wasserman CR, Neuberg D, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159:1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in tetrahymena. Cell. 2002;110:689–699. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- Muller JR, Giese T, Henry DL, Mushinski JF, Marcu KB. Generation of switch hybrid DNA between Ig heavy chain-mu and downstream switch regions in B lymphocytes. J Immunol. 1998;161:1354–1362. [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8:751–758. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, Yokota Y, Shimizu A. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- Nowacki M, Vijayan V, Zhou Y, Schotanus K, Doak TG, Landweber LF. RNA-mediated epigenetic programming of a genome-rearrangement pathway. Nature. 2008;451:153–158. doi: 10.1038/nature06452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC, 3rd, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefanis E, Wang J, Rothschild G, Lim J, Chao J, Rabadan R, Economides AN, Basu U. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature. 2014 doi: 10.1038/nature13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlot T, Li G, Alt FW. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci U S A. 2008;105:3843–3848. doi: 10.1073/pnas.0712291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G, Tang Z, Mathe E, Benner C, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159:1524–1537. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Ruskin B, Green MR. An RNA processing activity that debranches RNA lariats. Science. 1985;229:135–140. doi: 10.1126/science.2990042. [DOI] [PubMed] [Google Scholar]
- Sen D, Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988;334:364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- Srisawat C, Engelke DR. Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA. 2001;7:632–641. doi: 10.1017/s135583820100245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BJ, Wu YL, Rada C. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. eLife. 2014;3:e03553. doi: 10.7554/eLife.03553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn JP, Creacy SD, Routh ED, Joyner-Butt C, Jenkins GS, Pauli S, Nagamine Y, Akman SA. The DEXH protein product of the DHX36 gene is the major source of tetramolecular quadruplex G4-DNA resolving activity in HeLa cell lysates. J Biol Chem. 2005;280:38117–38120. doi: 10.1074/jbc.C500348200. [DOI] [PubMed] [Google Scholar]
- Williamson JR, Raghuraman MK, Cech TR. Monovalent cation-induced structure of telomeric DNA: the G-quartet model. Cell. 1989;59:871–880. doi: 10.1016/0092-8674(89)90610-7. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kaminaga K, Komiyama M. Human telomeric RNA in G-quadruplex structure. Nucleic Acids Symp Ser (Oxf) 2008:175–176. doi: 10.1093/nass/nrn089. [DOI] [PubMed] [Google Scholar]
- Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, et al. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nature structural & molecular biology. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair (Amst) 2003;2:1163–1174. doi: 10.1016/j.dnarep.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C. The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell. 2003;112:317–327. doi: 10.1016/s0092-8674(03)00079-5. [DOI] [PubMed] [Google Scholar]
- Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, Tian M. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol. 2004;5:1275–1281. doi: 10.1038/ni1137. [DOI] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.