

Abstract
Epigenetic mechanisms may regulate the expression of pro-angiogenic genes, thus affecting reparative angiogenesis in ischemic limbs. The enhancer of zest homolog-2 (EZH2) induces thtrimethylation of lysine 27 on histone H3 (H3K27me3), which represses gene transcription. We explored (i) if EZH2 expression is regulated by hypoxia and ischemia; (ii) the impact of EZH2 on the expression of two pro-angiogenic genes: eNOS and BDNF; (iii) the functional effect of EZH2 inhibition on cultured endothelial cells (ECs); (iv) the therapeutic potential of EZH2 inhibition in a mouse model of limb ischemia (LI). EZH2 expression was increased in cultured ECs exposed to hypoxia (control: normoxia) and in ECs extracted from mouse ischemic limb muscles (control: absence of ischemia). EZH2 increased the H3K27me3 abundance onto regulatory regions of eNOS and BDNF promoters. In vitro RNA silencing or pharmacological inhibition by 3-deazaneplanocin (DZNep) of EZH2 increased eNOS and BDNF mRNA and protein levels and enhanced functional capacities (migration, angiogenesis) of ECs under either normoxia or hypoxia. In mice with experimentally induced LI, DZNep increased angiogenesis in ischaemic muscles, the circulating levels of pro-angiogenic hematopoietic cells and blood flow recovery. Targeting EZH2 for inhibition may open new therapeutic avenues for patients with limb ischemia.
Introduction
Chromatin regulation contributes to the modulation of endothelial gene expression during vascular development and under different physiological and pathological conditions (reviewed in 1). Ischemic disease is a condition characterized by impaired blood perfusion. Therapeutic induction of the growth of new blood vessels is regarded as a possibility for improving the perfusion of ischemic tissue. Therefore, understanding the molecular mechanism behind ischemia-initiated blood flow recovery is important. Surgical mouse models based on the obstruction of blood flow in the femoral, coronary or cerebral arteries, respectively leading to limb ischemia (LI), myocardial infarct or ischemic stroke, have significantly contributed to better understanding of the cellular and molecular mechanisms behind postischemic revascularization.2 In particular, the postischemic vascular regeneration requires establishment and regulations of angiogenic pathways, which act in concert to form a functional vascular network in the ischemic areas.3 Enhanced expression of angiogenic genes during hypoxia/ischemia is a primary requisite for vascularization and tissue regeneration (reviewed in 3). Evidence for a role of chromatin modifications in the regulation of the angiogenesis process are emerging4,5,6 and the epigenetic machinery behind endothelial gene expression and cell homeostasis during hypoxia/ischemia merits better understanding.7 N-terminal histone (H) tails are subject to posttranslational modification, including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation.8 Hypoxia-induced chromatin changes on gene expression could impact on clinical outcome in ischemic patients.3,9
In this study, we have focused on EZH2 methyltransferase (enhancer of zeste homolog-2), the catalytic component of the Polycomb Repressor Complex 2 (PRC2).10 EZH2 is the only enzyme capable to induce histone H3 bi (me2)- and tri (me3)- methylation of Lys 27 (H3K27me2 and H3K27me3) in mammalian cells.11 Increased presence of H3K27me3 mark leads to transcriptional repression, whereas tri-methylation of H3 on lysine 4 (H3K4me3) positively associates with active transcription.12 Gene promoter regions commonly enriched for both H3K27me3 and H3K4me3 are known as bivalent chromatin domains,12 which concur with the PRC2 occupancy.10,13 The interplay between H3K27/H3K4 trimethyl marks, and PRC2 recruitment, is of potential mechanistic significance for re-activation of pro-angiogenic genes.14 Amongst several genes targeted by EZH2, in our study, we have focused at endothelial nitric oxide synthase (eNOS/NOS3)14 and brain-derived neurotrophic factor (BDNF).15,16 eNOS is highly expressed in endothelial cells (EC), which have restricted potency and it defines their exclusive endothelial identity through its promoter being stably integrated into the genome with a distinct chromatin signature.17 As a master regulator of endothelial function, eNOS is needed for endothelial homeostasis,18 and its activity is required for angiogenesis and blood flow recovery from limb ischemia.19 Moreover, eNOS in concert with Akt is part of the cell survival and pro-angiogenic pathways activated by several pro-angiogenic growth factors, including the BDNF.20,21 BDNF is expressed persistently at high levels in adult ECs22 and is known to promote vascular development and endothelial cell sprouting and to induce angiogenesis in vitro and in vivo in mice with LI.23,24,25 Our laboratory has a specific interest in the cardiovascular actions of neurotrophins and this additionally contributed to the selection of BDNF for this study. EZH2 requires noncatalytic protein subunits for its methylation activity: Suz12 (Suppressor of zeste-12 homolog), EED (embryonic ectoderm development), and histone-binding proteins RbAp48/46.10 Additionally, EZH2 reportedly regulates gene expression in ECs26 and has been proposed to regulate the transcriptional program leading to endothelial lineage commitment of stem cells,14 cardiovascular developmental commitment27 and cardiac homeostasis.28 Moreover, EZH2 is regulated by hypoxia in tumor microenvironment,29 where EZH2 appears to induce angiogenesis by a non-cell-autonomous mechanism.30 However, the role of EZH2 in postischemic angiogenesis has not yet been investigated. Despite the individual evidence for eNOS and BDNF expression being under control of EZH214,16 and hypoxia,31,32 a link between EZH2 and hypoxia in regulating the expression of these two genes has not been previously established. Based on the information from the epigenetic silencing mechanisms, we reasoned that removal of H3K27me3 using either EZH2 silencing or EZH2 pharmacological inhibition by 3-deazaneplanocin (DZNep), an S-adenosylhomocysteine hydrolase inhibitor,33 could remodel chromatin surrounding these two genes which are important for endothelial function and repair, thus providing a setting in which the endothelial-genome is permissive to gene transcription and driving angiogenesis.
Results
EZH2 inhibition increases the expression of eNOS and BDNF in HUVECs exposed to hypoxia
The EZH2 inhibitor DZNep has decreased levels of EZH2, H3K27me3, H3K27me2, and SUZ12 in HUVECs (Figure 1a) (Supplementary Figure S1i–iii). Similar results were obtained using small interfering RNA (siRNA)-mediated transient knockdown of EZH2 (siEZH2) (Figure 1a) (Supplementary Figure S1ii–iv). Next, we studied the effect of hypoxia (<2% pO2 for 48 hours) on H3K27me3, H3K27me2 levels and EZH2 expression in HUVECs. PCR primers for these analyzes are presented in Supplementary Table S1. Hypoxia increased the global levels of H3K27me3 in HUVECs but not H3K27me2 (Figure 1a) (Supplementary Figure S1v). As shown in Figure 1b, hypoxia also increased EZH2 mRNA levels in HUVECs (P < 0.01 versus normoxia) and as expected, siEZH2 reduced EZH2 under both normoxia and hypoxia (P < 0.01 for both comparisons). Moreover, hypoxia reduced eNOS (Figure 1c) and increased BDNF (Figure 1d) mRNA levels. PCR primers for these analyzes are presented in Supplementary Table S1. Next, we investigated if EZH2 is directly involved in the expressional regulation of eNOS and BDNF. siEZH2 maintained eNOS mRNA levels under hypoxia (Figure 1c), whereas DZNep was not tested in this experiment. Hypoxia reduced eNOS protein (Figure 1e–f) (Supplementary Figure S2i,ii) levels in HUVECs, with this effects being prevented by either siEZH2 (Figure 1e) (Supplementary Figure S2i) or DZNep (Figure 1f) (Supplementary Figure S2ii). Hypoxia increased BDNF mRNA (Figure 1d) and protein (Figure 1g–h) levels. Moreover, EZH2 inhibition by either siEZH2 (Figure 1g) or DZNep (Figure 1h) increased the release of BDNF by HUVECs under hypoxia.
Figure 1.
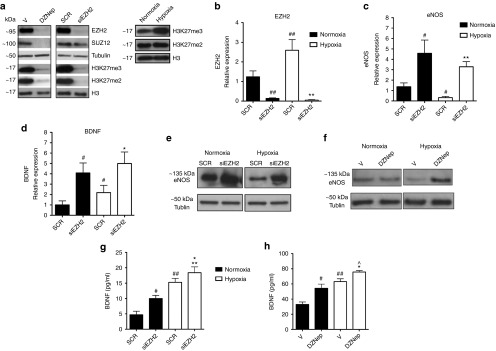
Hypoxia regulates PCR2, H3K27me3, eNOS, and BDNF levels and EZH2 inhibits eNOS and BDNF expression in HUVECs. (a) Western blot analyses of the PRC2 components EZH2 and SUZ12 and of the H3K27me3 and H3K27me2 epigenetic mark in HUVECs previously transfected with siRNA for EZH2, treated with the EZH2 inhibitor DZNep, or cultured in hypoxia (<2% O2) for 48 hours. Tubulin and histone H3 were used as housekeeping protein controls. Densitometry quantification of all Western blot analyses is presented in Supplementary Figure S1i–v. (b–d) mRNA expression levels of (b) EZH2 (c) eNOS, and (d) BDNF were measured in HUVECs using 18S as an internal control. Cells were transfected with EZH2 siRNA or scramble (SCR) control and then cultured in normoxia (21% O2) or hypoxia (<2% O2) for 48 hours. (e–f) eNOS protein levels were detected using Western blot analysis to examine the effect of (e) EZH2 siRNA versus SCR or (f) DZNep versus Vehicle (V) on HUVECs under hypoxia against tubulin control. Densitometry quantification of Western blot analyses is presented in Supplementary Figure S2. (g–h) BDNF protein levels in HUVEC conditioned medium were detected using an ELISA to examine the effect of (g) EZH2 siRNA or (h) DZNep. All data were expressed as mean ± SEM. ##P < 0.01 and #P < 0.05 versus normoxia; **P < 0.01 versus hypoxia; +P < 0.0 versus siEZH2 in normoxia; ^P < 0.05 versus DZNep in normoxia; Experiments were performed in duplicate and repeated at least three times.
PRC2 occupies the promoters of eNOS and BDNF
Taking advantage of the UCSC ENCODE Genome Browser (Human Feb. 2009, GRCh37/hg19),34 we analyzed chromatin marks at promoter region of two key endothelial-enriched genes using data generated by the ENCODE Consortium.35 Both eNOS and BDNF reportedly undergo transcriptional regulation by H3K27me3.16,36 Thus, we performed chromatin immunoprecipitation using antibodies individually targeting PRC2 components (EZH2, SUZ12, and EED) and RNA polymerase II (RNA Pol II, which catalyses DNA transcription into mRNA). The characterization of eNOS and BDNF promoter region spanning around 0.5 kb sequence upstream of transcription start site (TSS) was performed using PCR primers for each of different promoter regions (Supplementary Table S2). Notably, RNA Pol II and PRC2 occupy gene promoters in a mutually exclusive manner.37 Indeed, our data show that displacement of PRC2 components from the promoter region of eNOS (Figure 2c) or BDNF (Figure 2d) was occurring together with the recruitment of RNA Pol II within TSS of each of the examined gene. At the same promoter regions, the histone mark of active gene transcription, H3K4me3 has previously been reported to be enriched and dominant at TSS.14 We subsequently observed enrichment of repressive mark H3K27me3, further upstream from the TSS, and it coincided with the PRC2 components at the examined genes (Figure 2e,f).
Figure 2.
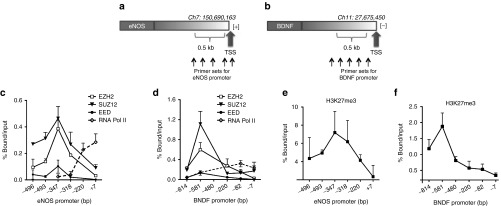
PCR2 binds to the eNOS and BDNF gene promoters, which are also exposed to histone 3 modifications. Binding of the PCR2 to the eNOS and BDNF gene promoters. Quantitative ChIP analysis of PRC2 complex proteins (EZH2, SUZ12, and EED) and RNA Pol II was performed in HUVECs to check their occupancy within 0.5 kbp locus on (a) eNOS (−496 to +7 bp from TSS, transcribed in [+] direction) and (b) BDNF (−814 to −7 bp from TSS, transcribed in [−] direction) within the promoter region. The binding of each PRC2 component and RNA polymerase are shown at the (c) eNOS and (d) BDNF gene promoters. IgG was used as a negative control. Data were calculated as % bound/input. All data were expressed as mean ± SD and are representative of at least n = 3 independently performed experiments.
Of note, using UCSC Genome Browser, we identified YY1 putative DNA cis-element within the same region of eNOS (−121/−117 bp) and BDNF (−738/−733 bp) promoters, respectively. Presence of YY1 DNA cis-elements in the promoter has previously been suggested to participate in the formation of nucleoprotein complexes,38 thus further allowing recruitment of PRC2 at these sites.10 Interestingly, we observed an enrichment of YY1 protein (Supplementary Figure S3) in the same region of eNOS and BDNF promoters, respective with enrichment of EZH2 and H3K27me3.
Effect of hypoxia and DZNep on PRC2 and RNA polymerase II occupancy on eNOS and BDNF promoters in HUVECs
The PRC2 binding regions on eNOS (−493/−318 bp) and BDNF (−753/−480 bp) promoters were further investigated for the effect of hypoxia and DZNep on the binding of EZH2. Quantitative ChIP analysis showed an increased occupancy of the eNOS proximal promoter by EZH2 in HUVECs exposed to hypoxia and treated with the vehicle or DZNep (Figure 3a). Yet, hypoxia reduced the EZH2 occupancy of BDNF promoter (Figure 3c), corresponding to an increased BDNF gene expression. The enrichment profiles of EZH2 and H3K27me3 at the eNOS promoter were similar (Figure 3a,b). Moreover, at the BDNF promoter, hypoxia induced a loss of both EZH2 and H3K27me3 in vehicle-treated HUVECs (Figure 3c,d). Next, we investigated the occupancy of the eNOS and BDNF promoters by EZH2 and H3K27me3 in the presence of DZNep in normoxic and hypoxic HUVECs. As shown in Figure 3a–d, DZNep prompted loss of EZH2 and reduced H3K27me3 binding at both eNOS (Figure 3a,b) and BDNF (Figure 3c,d) promoters. Collectively, our results suggest the hypothesis that enrichment of EZH2 under hypoxia on the eNOS and BDNF promoters may modulate the transcriptional responses of endothelial genes to hypoxic environment.
Figure 3.

Effect of DZNep and hypoxia on EZH2 enrichment at eNOS and BDNF gene promoters. (a–d) Quantitative ChIP-PCR analyses of occupancy by (a,c) EZH2 and the (b,d) associated H3K27me3 modification of genomic regions (a,b) −493/−318 bp on eNOS and (c,d) −753/−480 bp on BDNF gene promoter in HUVECs. The promoter occupancy was determined in the presence of DZNep or vehicle control (V) under normoxia and hypoxia and expressed as % chromatin bound/input (for EZH2) or % total bound/H3 (for H3K27me3). 1% of total chromatin sample was used as input. IgG was used as a negative control. All data were expressed as mean ± SD and are representative of at least n = 3 independently performed experiments. #P < 0.05 versus vehicle under normoxia; *P < 0.05; **P < 0.01 versus vehicle under hypoxia.
EZH2 regulates functional responses of vascular endothelial cells to hypoxia
To reveal the functional role of EZH2 in endothelial cells, we investigated the effect of siEZH2 on HUVECs either cultured under normoxic condition or exposed to prolonged (48 hours) hypoxia. As shown in Figure 4a–c, hypoxia hampered the angiogenic capacity of HUVECs when tested by Matrigel assay. In fact, prolonged hypoxia has reduced both the endothelial cell tube-like length (Figure 4a) and the number of branching points (Figure 4b). Use of siEZH2 to knockdown EZH2 gene has improved HUVEC angiogenic potential under both normoxia and hypoxia. Moreover, hypoxia decreased the migratory capacity of HUVECs prompted in a scratch assay (Figure 4d,e), whereas siEZH2 enhanced HUVEC migration under both normoxia and hypoxia. These results are in line with the increased expression of eNOS and BDNF in EZH2-silenced HUVEC (Figure 1c,d). The functional results of EZH2 knockdown were corroborated by the use of pharmacological inhibitor of EZH2, DZNep. In fact, as shown in Supplementary Figure S4i–v, DZNep enhanced the HUVEC migratory and angiogenic potential under hypoxia.
Figure 4.
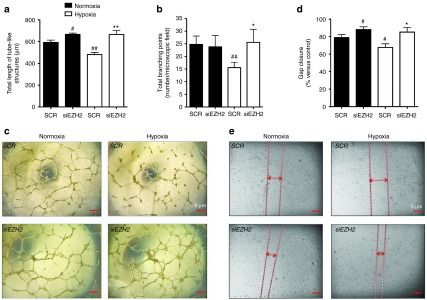
EZH2 inhibition increases the in vitro networking and migratory capacities of endothelial cells. (a–e) siRNA-mediated EZH2 knockdown (control: scramble –SCR) was conducted in HUVECs to investigate the functional role of EZH2 using (a–c) Matrigel and (d,e) scratch assays. Functional property of HUVECs treated with DZNep or vehicle (V) control is presented in the Supplementary Figure S3i–iii. Cells were studied under (c) normoxia or hypoxia Matrigel and (e) scratch assay images and were taken at 5× magnification. Scale bar: 5 µm. For the Matrigel assay, (a) the average total length of tubes and (b) the total number of tubes branching points have been calculated. For the scratch assay, (d) the % gap closure was calculated. All measurements are mean ± SEM. #P < 0.05 versus SCR under normoxia; **P < 0.01 and *P < 0.05, versus SCR under hypoxia. Experiments were performed in triplicate and repeated at least three times.
To then validate the role of EZH2 in the observed pro-angiogenic phenotype of HUVECs in Figure 4a,e and Supplementary Figure S4i–v, we performed a rescue experiment using adenovirus delivery of the wild type gene. Toward this end, we initially knocked down EZH2 in HUVEC, which again resulted in increased cell migration and network formation on Matrigel. We then performed a rescue experiment using adenovirus delivery of the wild type EZH2 cDNA (Supplementary Figure S5i–ii). We find that overexpression of the wild-type EZH2 adenovirus alone (SCR+AdwtEZH2) has reduced both the endothelial cell tube-like length (Supplementary Figure S6i) and the number of branching points (Supplementary Figure S6ii) when tested by Matrigel assay (Supplementary Figure S6iii), and further reduced cell migration as compared with the Null control (SCR+AdNull), Supplementary Figure S6iv–v. Thus, reintroduction of a wild-type EZH2 adenovirus into pro-angiogenic siEZH2-transfected HUVECs (siEZH2+AdwtEZH2) has impaired their functional capacity by reducing cell ability to form network on Matrigel and cell migration (Supplementary Figure S6iii,v), as compared with the siEZH2 Null control (siEZH2+AdNull).
EZH2 inhibition by DZNep regulates eNOS and BDNF mRNA levels in endothelial cells of limb muscle
On the basis of our in vitro findings, we next asked if EZH2 is regulated by ischemia in vivo and if it modulates eNOS and BDNF expressional levels in the endothelial cells of limb muscles. To address these points, we used a mouse model of unilateral limb ischemia (LI). Mice were treated every 2 days with DZNep or vehicle control starting the day before induction of LI by left femoral artery occlusion and they were sacrificed at 2 and 7 days postsurgery. DZNep was well tolerated in these mice and we could not observe any weight loss or other gross side effects. DZNep reduced the levels of EZH2 protein (Figure 5a) and of H3K27me3 and H3K27me2 (Figure 5b) in the ischemic limb muscles. In the vehicle group, ischemia increased EZH2 mRNA levels in the limb muscles (Figure 5c) and in muscular CD146pos endothelial cells (Figure 5d), prepared using a previously published protocol of ours.39 However, DZNep abolished these responses to ischemia (Figure 5c,d) and additionally reduced EZH2 mRNA levels. Moreover, as shown in Figure 5e, in the vehicle group, muscular eNOS mRNA expression was reduced at 7 days after LI induction. Notably, DZNep reversed this response to ischemia. In ECs extracted from either nonischemic muscles (from sham-operated mice) or muscles at 2 days after induction of ischemia in vivo DZNep resulted in increased eNOS mRNA levels (Figure 5f). Additionally, DZNep increased BDNF mRNA expression in the ischemic muscles (Figure 5g) and in the muscular CD146pos ECs (Figure 5h).
Figure 5.

Ischemia regulates the expression of EZH2, eNOS and BDNF in limb muscle and muscular ECs and DZNep increases eNOS and BDNF levels. Limb ischemia (LI) or sham operation was induced in mice. The ischemic limb adductor muscles were harvested from mice treated with DZNep or vehicle (V) and sacrificed at 3 days after surgery. Nonischemic muscle controls were taken from sham-operated mice. For protein analysis, total muscular nuclear fractions of protein were prepared and separated by Western blot to detect EZH2 protein levels against (a) Lamin A/C control or H3K27me3 and H3K27me2 levels against total histone H3. Densitometry quantification of Western blot analyses is presented in Supplementary Figure S1. (b) mRNA levels of (c,d) EZH2, (e,f) eNOS, and (g,h) BDNF were measured by qPCR using 18S as an internal control in the (c,e,g) total muscle or (d,f,h) muscular endothelial cells (CD146pos). Data are presented as mean ± SEM. #P < 0.05 versus vehicle in nonischemia; **P < 0.01 and *P < 0.05 versus vehicle in ischemia; +P < 0.05 versus DZNep in nonischemia; n = 5–8 mice/group.
EZH2 inhibition by DZNep improves postischemic blood flow recovery and angiogenesis
Next, we asked if EZH2 inhibition by DZNep could improve blood flow recovery and angiogenesis after LI. Mice were injected with DZNep or vehicle (as described above) and the limb blood flow was monitored weekly before sacrificing mice at 21 days post-LI. DZNep was well tolerated for all this time and we could not observe further side effects. DZNep consistently improved the blood flow recovery (**P < 0.01 at day 7 and *P < 0.05 at days 14 and 21 for comparisons versus vehicle) (Figure 6a). In addition, DZNep increased the density of both capillaries (Figure 6b–d) and small (<50 µmol/l diameter) arterioles (Figure 6c,d) in the ischemic adductor muscle at 3 weeks after LI (*P < 0.05 versus vehicle for both comparisons).
Figure 6.
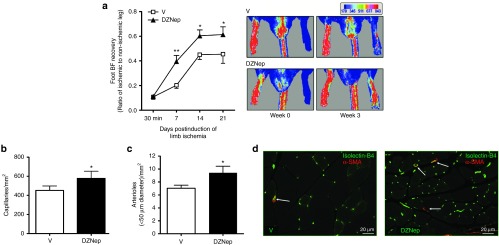
EZH2 inhibition improves postischemic blood flow recovery and angiogenesis in mice. (a–d) Unilateral limb ischemia was induced in mice (n = 12–16 mice/group), which were treated with either DZNep or vehicle (V) and studied for sequential blood flow recovery by (a) color laser Doppler before being sacrificed at 21 days postischemia for analyses of (b,d) capillary and (c,d) arteriole density in their ischemic muscles. Histology was performed in muscles from n = 7 mice/group (randomly selected from the mice undergoing the Doppler analyses). (a) Graph shows the time course of blood flow recovery to the ischemic foot. Pictures are representative of foot blood flow at 3 weeks postischemia, in a color scale with dark-blue being the lowest and dark-red the highest perfusion levels. (b) Capillary and (c) small arteriole densities are expressed per mm2 of muscle transverse section. (d) The representative immunofluorescent pictures were taken (at 20× magnification; size bar: 20 µm) after staining of ischemic adductor muscle sections with green fluorescent isolectin-B4 (revealing endothelial cells) and a red-fluorescence conjugated antibody targeting alpha smooth muscle actin (to identify smooth muscles cells which are present in arterioles, but not in capillaries). All data are mean ± SEM compared between DZNep treated and V control group. *P < 0.05 and **P < 0.01 versus V.
DZNep increases the abundance of circulating pro-angiogenic cells in mice with limb ischemia
Bone marrow-derived pro-angiogenic cells contribute to postischemic angiogenesis. eNOS allows for the egression of pro-angiogenic cells from the bone marrow in response to an ischemic event.40 Moreover, BDNF reportedly promotes the liberation of hematopoietic Sca-1posCD11bpos cells in mice with LI.23 DZNep increased the circulating levels of pro-angiogenic Sca-1posCD11bpos cells (Figure 7a) and LinnegSca-1posc-kitpos (LSK) cells (Figure 7b) at 2 days after LI-induction (**P < 0.01 for both comparisons versus vehicle control). Supplementary Figure S7 shows the gating strategies for these flow cytometric analyses. Concomitantly, DZNep reduced the abundance of both Sca-1posCD11bpos cells (Figure 7c) and LSK cells (Figure 7d) in the bone marrow of ischemic mice. Taken together, our in vivo data are compatible with the hypothesis that EZH2 inhibition may act as an aiding tool to promote postischemic vascular regeneration by directly targeting the resident endothelial cells within the ischemic muscles and additionally stimulating the bone marrow to release pro-angiogenic cells into the systemic circulation.
Figure 7.

EZH2 inhibition increased the abundance of circulating levels of hematopoietic pro-angiogenic cells in mice. (a–d) The abundance of hematopoietic (a,c) Sca-1posCD11bpos cells and (b,d) LinnegSca-1posc-kitpos cells in the (a,b) peripheral blood and (c,d) bone marrow was measured by flow cytometric analyses at 2 days after limb ischemia or sham-operation in mice treated with DZNep or vehicle control (V). Flow cytometry gating strategies are presented in Supplementary Figure S7. The number of cells in the blood and bone marrow was established using fluorescent counting beads. Data are presented as mean ± SEM, and compared with the relevant control. #P < 0.05 versus V in nonischemia, **P < 0.01 versus V in ischemia (LI); ++P < 0.01 and +P < 0.05 versus DZNep in nonischemia; n = 6–8 mice/group.
Discussion
Despite many exciting basic science discoveries, the field of therapeutic angiogenesis has not yet been able to translate the preclinical potential into clinical success (reviewed in 9,41). Loss of endothelial cell survival and function after injury impairs and delays the growth of blood vessels.41 In ischemic tissues, reduced blood perfusion and the low oxygen levels are unable to satisfy the metabolic needs.3 Fittingly, the importance of epigenetic mechanisms and hypoxia in endothelial gene regulation is gaining more appreciation6,7 as our understanding of such mechanisms is still in its infancy. The driving force in our research was the hope to develop a novel approach to stimulate postischemic angiogenesis productive for tissue reperfusion based on unlocking the expression of pro-angiogenic genes, through modulation of the epigenetic landscape. In this study, we provide the first evidence that the histone H3 methyltransferase EZH2, the catalytic subunit of the PRC2 complex, represses angiogenesis under hypoxia and ischemia. We found increased EZH2 in the endothelial cells exposed to hypoxia. This is in line with the fact that EZH2 contains hypoxia response element (HRE).29 Moreover, we showed that in endothelial cells, hypoxia increases EZH2 to the regulatory regions of eNOS and BDNF gene promoters and augment the abundance of H3K27me3 at these locations, which perhaps results in chromatin condensation and gene silencing.10 Expression of eNOS was shown to decrease in hypoxia,31 which is in line with our results. The hypoxia-induced increase in H3K27me3 on gene promoters (characteristic of a closed chromatin structure) can either counterbalance high levels of transcriptional activity (the case with eNOS in ECs) or poise active transcription at genomic regions (the case with BDNF under hypoxia).37 Accordingly, EZH2 depletion removed the repressive H3K27me3 epigenetic mark from both eNOS and BDNF gene promoters, hence increasing their expression in cultured endothelial cells exposed to hypoxia. This correlated with improved functional capacities of endothelial cells. Moreover, we have shown that the EZH2 inhibitor DZNep stimulates postischemic blood flow recovery and reparative neovascularisation in the mouse LI model in vivo. It is conceivable that vascular endothelial cells can be epigenetically controlled to accelerate their pro-angiogenic capacity by modifying chromatin on the “key-regulatory” genes. Furthermore, our results emphasize two mechanistic features of PRC2, namely, its ability (i) to regulate eNOS and BDNF expression in human ECs and (ii) to add the H3K27me3 mark onto the promoter of these two genes under hypoxia. Our work fits with the hypothesis that under hypoxia increased or unchecked PRC2 activity affects the global and site-specific (on eNOS and BDNF promoter regions) level of H3K27me3 in endothelial cells and represses the transcription of eNOS and BDNF. In line with this hypothesis, ECs extracted from the ischemic limb muscles of mice treated with DZNep presented higher eNOS and BDNF mRNA levels. Thus, the resetting of epigenetic marks by inhibiting EZH2 in the endothelial cells exposed to hypoxia could be consistent with gene transcriptional reactivation of additional angiogenic genes.
Using published UCSC Human Genome Browser resources,34 we found that eNOS and BDNF gene promoter regions within 1 kb of TSS contain H3K27me3 mark. The eNOS promoter lacks a typical TATA box and it is not rich in CpG islands,17 yet the activation of the BDNF promoter is region-specific and it depends on the type of stimulus.42 There are YY1 DNA binding cis-element (5′-CCATT-3′, 5′-GCCAT-3′ or 5′-CGCCATNTT-3′) present within both of these gene promoters, which could participate in the binding of nucleoprotein complexes like PRC2 and consequently trimethylate chromatin on H3K27.38 We observed an increased recruitment of PRC2 complex at YY1-regions in both eNOS and BDNF promoters. Whether YY1, through interaction with different transcription factors or with noncoding RNAs at its recognition sequences,38 could be involved in writing H3K27 methylation11 under hypoxia it still remains to be investigated.
We have determined that in normoxia all three components of PRC2 bind chromatin of eNOS and BDNF promoters. While SUZ12 typically occupies genes that control transcription in development and differentiation,43 it is possible that it may assist eNOS and BDNF transcription along with EZH2, but its direct role was not studied in here, and preliminary work of ours shows loss of EC-function upon depletion of SUZ12 (data not shown). The RNA Pol II occupies genes controlling broader cell proliferation, and its activity on both eNOS and BDNF promoters was prominent with the disappearance of PRC2 complex as already shown for pluripotency genes.37 Although we show that both genes are regulated by PRC2 enzymatic activity, additional layers of epigenetic regulation could be involved in determining deposition of H3K27me3 to directly influence eNOS and BDNF expression in ECs and under hypoxia. Moreover, angiogenic responses under ischemia may require a reset of other epigenetic marks. For instance, the deposition of H3K27me2 mark, could ensure structural function and contribute to the activity of PRC2 to write H3K27me3.11 Deposition of regulatory permissive modifications like H3K36me3 along with the removal of EZH2/methylation of H3K27 at the poised genes, may be linked with higher mobility of histones and active gene transcription.11 It is yet unknown if demethylation of H3K27 correlates with the increase in H3K36me3 or H3K27 acetylation at the selected genes during ischemia. It is also possible that the homolog of EZH2, EZH1, can contribute to writing trimethylation of H3K27.44 In fact, this may explain the difference observed in the levels of H3K27me3 at eNOS and BDNF promoters upon treatment of HUVEC with DZNep between normoxia and hypoxia. Additionally, the demethylase Jmjd3 is suggested to be time-dependently increased by hypoxia and postinduction of limb ischemia in mice, whereby it contributes to the eNOS regulation.14 This is in line with the increased levels of H3K27me3 on eNOS promoter under hypoxia that could allow binding of Jmjd3. Since H3K27me3 is the only known substrate for Jmjd3, the latter enzyme could erase this repressive mark under hypoxia45 or ischemia, thus paving the way for the RNAPol II and active chromatin marks to further direct endothelial gene expression. The accumulation of H3K27 methylation by PRC2 at the studied genes could also be a way of endothelial cell-type-specific maintenance of gene transcriptional repression. Thus, EZH2 inhibition may be one of the strategies for the reversal of chromatin changes in order to unlock endothelial cell angiogenic potential and to promote vascular regeneration in ischemic tissue.
Bone marrow mobilization of angiogenic progenitor cells is eNOS-dependent.40 In addition, eNOS levels regulate the postischemic regenerative capacity of BM-derived progenitor cells.40 Moreover, Kermani et al. showed that treatment with BDNF promotes the mobilization of pro-angiogenic hematopoietic Sca-1posCD11bpos cells to the peripheral blood and it induces postischemic recovery in mice with LI.23 Accordingly, we have shown that EZH2 inhibition by DZNep increases the circulatory levels of Sca-1posCD11bpos and LSK cells after LI induction in mice. It is possible that EZH2 inhibition promotes angiogenesis by directly acting on differentiated endothelial cells, which are resident within the ischemic muscle and through promoting the liberation of BM cells, which have been shown to support angiogenesis, mainly through paracrine activities. In conclusion, EZH2 could represent a novel therapeutic target for preservation and restoration of function following limb ischemia, and the use of EZH2 inhibitors may be a tractable option for healing ischemic tissues.
Our in vivo data were produced in the young mice undergoing acute LI-induction, as such we could think of a clinical scenario with young patients who might have injured their limbs in accidents, where these findings would be relevant. Further studies are necessary to investigate the clinical potential of EZH2 inhibition in the critical limb ischemia.
Finally, we cannot exclude the possibility that DZNep could induce angiogenesis in the nonischemic tissues. However, DZNep was shown to reduce cancer angiogenesis in mouse xenograft models, thus discouraging this hypothesis.30,46
Materials and Methods
Cell culture and treatments. Human umbilical cord vein endothelial cells (HUVECs, Lonza, Slough, UK) were cultured at 37 °C with 5% CO2 in EBM-2 endothelial cell basal medium (Lonza) with addition of SingleQuot Kit (EGM-2 medium, Lonza) as instructed. For hypoxia experiments, cells were exposed to hypoxia (<2% pO2) for 48 hours (normoxia control: 21% pO2). The treatment with EZH2 inhibitor 3-deazaneplanocin, (DZNep, an S-adenosylhomocysteine hydrolase inhibitor, 2.5 µmol/l, CAS 102052-95-9, Cayman; Cambridge Biosciences, Cambridge, UK) or DMSO control (0.2%, Sigma, Gillingham, UK) was performed in EGM-2 medium between 6 and 72 hours.
siRNA transfection and adenoviral transduction. For siRNA transfection, the cells were seeded on six-well plate at a density of 2 × 105 cells per well in 2 ml complete medium and grown for 1–3 days until 80% confluent. Lipofectamine 2000 (Life Technologies, Paisley, UK) was used to transfect HUVECs and conduct siRNA-mediated transient knockdown of EZH2 (120 nmol/l flexitube siRNA, Qiagen, Manchester, UK). A scramble sequence (Qiagen) was used as control. For rescuing EZH2 expression, HUVECs were seeded on six-well plate at a density of 2 × 105 cells and grown until 80% confluent. Transduction was performed using adenovirus at concentration of 250 MOI, as previously described.47
Quantitative analysis of mRNA. Total RNA was isolated and reversely transcribed using the Quantitect reverse-transcription kit (Qiagen) from cells or tissue. cDNA (equivalent to 300 ng total RNA) was incubated in triplicate to assess gene mRNA expression of EZH2, eNOS and BDNF and 2 × ABI Power SYBR green mastermix (Life Technologies) PCR cycling and detection of fluorescent signal was carried out using a Roche Light Cycler 480 (Roche, West Sussex, UK). Results were corrected for the expression of 18S and expressed as the fold change. All primer sequences were obtained from PrimerBank, provided in Supplementary Table S1, and ordered from Quick Start (Sigma design).
Western blotting. Cells were treated with a lysis buffer (50 mmol/l Tris, pH 7.5, 150 mol/l NaCl, and 1 mol/l EDTA, 1% Triton X-100) containing 1 mmol/l sodium orthovanadate, 1 mmol/l sodium pyrophosphate, 1 mmol/l sodium fluoride (all from Sigma) and mini-EDTA complete tablet (Roche). Lysates were prepared from frozen tissues by extraction into ice-cold lysis buffer (described above). Nuclear extracts were prepared using ice-cold S300 extraction buffer (9 mol/l urea, 25 mmol/l Tris-HCl pH6.8, 1 mmol/l EDTA, and 10% glycerol). The details of primary antibodies used are provided in Supplementary Table S3 and they were used at following dilutions: EZH2 (clone RbAb AC22 1:1,000), pan H3 1:2,000 (Active motif, La Hulpe, Belgium), SUZ12 1:2,000, EED 1:1,000 (Abcam, Cambridge, UK), H3K27me3, H3K27me2, H3K4me3, H3Ac (1:2,000 Merck Millipore, Hertfordshire, UK), eNOS 1:2,000, β-actin 1:1,000 (Santa Cruz Biotechnology, Middlesex, UK), and BDNF (Promega, Southampton, UK; catalog number G164B). α/β-tubulin (2148S, 1:1,000) served as the loading control. Proteins were visualized with a goat anti-rabbit or donkey anti-goat secondary antibody (GE Healthcare, Little Chalfont, UK) and HRP substrate (Immobilon Western Chemiluminescent, Merck Millipore). We detected the chemiluminescent signal with X-ray film (GE Healthcare). The same membranes were usually stripped (with 20% Azide in 5% milk) and reused for detection of loading controls; band intensities were quantified using the densitometry and ImageJ analysis.
BDNF ELISA. An ELISA for (BDNF (Emax ImmunoAssay System, Promega) was carried out on HUVEC conditioned medium. Cells were either incubated with 2.5 µmol/l DZNep (control: vehicle, 0.2% DMSO) or have been previously transfected with siRNA-EZH2 (control: scramble). Cells were exposed to hypoxia (<2% pO2) for 48 hours (normoxia control: 21% pO2). The conditioned medium was collected from a confluent six-well plate (1 ml/well, 6 × 106 cells/well) and frozen at −80 °C until use, following manufacturer's instructions.
In vitro angiogenesis and migration assay. HUVECs were transfected with siRNA for EZH2 or scramble control; or treated with DMSO versus DZNep (2.5 µmol/l), and seeded in 48-well plates coated with a growth factors-enriched Matrigel (BD Biosciences, Oxford, UK). Endothelial network formation was quantified in randomly captured microscopic fields (magnification 5×) by calculating the length of cellular network. Cell migration was assayed by scratching the cell monolayer of confluent HUVECs. Cells were incubated with EGM-2 containing 2 mmol/l of hydroxyurea (Sigma) to arrest cell proliferation. Pictures were taken immediately after scratching 8 hours thereafter. The migration distance (in μm) as the reduction of the width of the open area was calculated.
Chromatin immunoprecipitation (ChIP). HUVECs (Lonza) were plated in T150 cm plates at a density of 2 × 106 cells per dish and treated with DMSO or DZNep (2.5 µmol/l) for 48 hours. Fully confluent cells were cross-linked for 10 minutes with formaldehyde (1%) and quenched with glycine (0.125 mol/l) for further 5 minutes. Chromatin was sonicated (3 × 9 cycles 30”ON/30”OFF) to an average of 300–500 bp DNA length using Bioruptor sonicator (Diagenode, Seraing, Belgium). ChIP assays were carried out as described before.43 Immunoprecipitation was performed in 1 ml volume with addition of 3–5 µg of the following antibodies; EZH2 (clone mAb AC22), pan H3 (Active motif), SUZ12, EED (Abcam), H3K27me3 (#07-449), H3K27me2 (#07-452), H3K4me3 (#07-473), anti-H3KAc (#06-599) (Merck Millipore) or YY1 (sc-281), RNA Polymerase II (sc-889X) and IgG control (Santa Cruz Biotechnology). ChIP enriched DNA and input DNA was subjected to qRT-PCR analysis with ABI Power SYBR Green qPCR Master Mix (Life Technology) using specific primers sequence for eNOS and BDNF as reported (Supplementary Table S2). Enrichment by ChIP assay on the specific genomic regions was assessed relative to the input DNA and IgG control, or total histone H3. Within each ChIP experiment, a negative control ChIP was performed using 5 micrograms of polyclonal rabbit anti-mouse immunoglobulin (IgG). To verify the ChIP conditions, a qPCR was performed on enriched DNA using promoter primers against GAPDH for absence of histone modifications, or MYT-1 and Neurogenin D2 promoter region as known targets of polycomb group proteins. Associated DNA was then purified by extraction using QIA Quick qPCR kit (Qiagen). Quantitative PCR was used to determine recovery of specific DNA fragments. The promoter sequences for BDNF (Accession number 8188) and eNOS (Accession number 26552) were retrieved from the “Transcriptional Regulatory Element Database” website (http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home) and the transcription start sites were retrieved from the “DBTSS: Database of Transcriptional Start Sites” website (http://dbtss.hgc.jp/index.html?nmid=DBTSS:NM_000603) and the “Human (Homo sapiens) Genome Browser Gateway” website (http://genome-euro.ucsc.edu/cgi-bin/hgGateway).
In vivo experiments. The experiments involving mice were covered by project and personal licenses issued by the United Kingdome Home Office and they were performed in accordance with the Guide for the Care and Use of Laboratory Animals (the Institute of Laboratory Animal Resources, 1996). CD1 male mice age 14 weeks were injected with DZNep (1.5 mg/kg/day, i.p. every 2 days) or vehicle (1% DMSO in 0.9% saline) starting 1 day before undergoing surgery to induce unilateral limb ischemia under general anesthetic, Avertin, after pre-operative analgesia (Vetergesic, 0.05 ml/30 g animals). Limb ischemia was obtained by occlusion of the left femoral artery, as we reported previously.48 Sham-operated mice underwent all surgical procedures apart from femoral artery occlusion. For mobilization and molecular biology analyses, mice were sacrificed at 2 or 7 days after surgery, respectively. For the efficacy testing of DZNep, mice were studied for 3 weeks after surgery. The superficial blood flow to both feet was measured using high resolution laser color Doppler imaging system (Moor LDI2, Moor Instruments, Devon, UK) at 30 minutes and days 2, 7, 14, and 21 after limb ischemia. Blood flow recovery was calculated (n = 12–16 mice/group) as a ratio of ischemic over contralateral foot blood flow. After the last Doppler analysis (at day 21 after surgery), mice were perfusion-fixed under terminal anesthesia and limb muscles were harvested for histological and immunohistochemical analyses.
Immunohistochemistry. Functional impact of DZNep treatment in CD1-ischemic mice was assessed by measuring capillary and arteriole densities in the adductor muscle. Isolectin B4-FITC-conjugated bovine anti-sheep IgG (Life Technologies) was used to detect endothelial cells, followed by staining with a red-conjugated rabbit polyclonal anti- α-smooth muscle actin (Sigma) used to stain smooth muscle cells (which are present in arterioles, but not in capillaries). Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). The slides were mounted using a mounting medium (Vector Labs, Peterborough, UK). The relative amount of positive cells was counted in 10 randomly selected high-power fields (magnification 40×) using Zeiss inverted fluorescent microscope. Analyses were performed using muscles from eight mice per group and these mice were randomly selected from the ones used for laser color Doppler analyses.
Flow cytometry analyses. At three days from surgery to induce limb ischemia or sham-operation, blood and bone marrow cells were extracted from mice that have been treated with either DZNep or vehicle (as described above). Cells were stained with fluorescently labeled antibodies against cell lineages cocktails (Cat#55971), CD11b (clone M1/70; cat#55567), Sca1 (clone D7; cat#553108), and c-kit (clone 2B8; cat#553356) (all from BD Biosciences). Absolute count of cells per each sample was obtained using AccuCheck Flow Cytometry Counting Beads (Life Technologies). As shown in Supplementary Figure S7 (for peripheral blood), the major population of lympho-mononuclear cells was resolved on a dot plot of linear forward light scatter (FSC) versus linear side light scatter (SSC) and it was gated into a region P1 to exclude contaminating events such as red blood cells, platelets, and cell debris. The cell populations were analyzed on a double-laser BD FACSCANTO system (BD Biosciences). For detecting CD11posSca-1pos cells, the Sca-1 events were established on a dot plot of Sca-1 staining versus FSC and they were gated into a region designated P3, where they were analyzed for CD11b staining, with CD11bpos cells being gated into a region designated P4. For detecting linnegSca-1posc-kitpos cells, lineage negative events were established on a dot plot of lineage negative staining versus FSC, and were gated into a region designated P3, where they were analyzed for Sca-1 and c-kit staining, with CD11bposc-kitpos cells being gated into a region designated P4. An equivalent approach was used for bone marrow analyses.
CD146pos cell isolation from mouse limb muscles. Adductor muscles were collected at 2 days after surgery from mice previously treated with DZNep or vehicle as reported above. Muscles were rinsed in PBS and digested with collagenase II (600 U/ml final, Worthington) and DNase I (1 mg/ml, Sigma) using gentle MACS dissociator (Miltenyi Biotech, Surrey, UK) M tubes (130-093-236) and following the manufacturer's protocol. Next, endothelial cells were sorted using an immunomagnetic CD146 antibody (clone ME-9F1, Miltenyi Biotech), as previously reported.39
Computer analysis. Genome-wide searching approaches have been used to predict the importance of the PRC2 complex in the regulation of transcription of endothelial targets through H3K27 methylation. To identify the family or individual transcription factors that potentially bind to the identified sequences, genomic regions from UCSC ENCODE genome browser34,49 were compared against a comprehensive library of known motifs consisting of motif matrices from the JASPAR database (http://jaspar.genereg.net/).50 The motifs derived from published transcription factor ChIP-seq data were compared against the motifs that are not represented in the known UCSC genomic sequence of genes of interest. Further analysis of characterized Polycomb/Trithorax Response Elements (PRE/TREs or PREs for short) was performed using jPREdictor tool http://bibiserv.techfak.uni-bielefeld.de/jpredictor.
Statistical analysis. Data are expressed as means ± SEM. Data were analyzed using GraphPad Prism (version 5; GraphPad Software, San Diego, CA). Two-group comparisons were performed with Student t test with P ≤ 0.05 taken as statistically significant. Multi-group analysis was performed using two-way analysis of variance to compare the combined and individual effect of ischemia and DZNep treatment, or combined individual effect of hypoxia and siEZH2. Statistical significance was determined at a value of P ≤ 0.05 with Bonferroni posttest.
SUPPLEMENTARY MATERIAL Figure S1. Quantification of Western blot analyses presented in the Figure 1a. Figure S2. Quantification of Western blot analyses presented in the Figure 1e. Figure S3. Binding of YY-1 to the eNOS and BDNF gene promoters. Figure S4. DZNep increases the in vitro networking and migratory capacities of endothelial cells. Figure S5. Overexpression of wild type EZH2 into HUVECs and quantification of Western blot analyses. Figure S6. Rescuing the EZH2 protein levels in HUVECs worsens the endothelial cell function. Figure S7. Flow cytometry strategies behind the data presented in the Figure 7a,b. Table S1. Sequence of qPCR primers used in this study. Table S2. Sequence of primer used for chromatin immunoprecipitation (ChIP) qPCR analysis in this study. Table S3. The list of antibodies used in the study.
Acknowledgments
The authors are thankful to Dr Betty Descamps for assistance with revision. This work was supported by the European Federation for the Study of Diabetes (EFSD) (project grant to C.E. and T.M. and Albert Renold travel fellowship to T.M.), the National Institute for Health Research (NIHR) through the Bristol Cardiovascular Biomedical Research Unit (BRU), and the British Heart Foundation (BHF) Regenerative Medicine centre for vascular biology. T.M. was recipient of a Society for Endocrinology Early Career Grant; A.C. is a BHF Intermediate Fellow and The University of Edinburgh Chancellor's Fellow. C.E. is a senior BHF research fellow and G.D.A. is a BHF Professor. The other authors declare no conflict of interest. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Supplementary Material
References
- Han P, Hang CT, Yang J, Chang CP. Chromatin remodeling in cardiovascular development and physiology. Circ Res. 2011;108:378–396. doi: 10.1161/CIRCRESAHA.110.224287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeddu P, Emanueli C, Spillmann F, Meloni M, Bouby N, Richer C, et al. Murine models of myocardial and limb ischemia: diagnostic end-points and relevance to clinical problems. Vascul Pharmacol. 2006;45:281–301. doi: 10.1016/j.vph.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Smadja DM, Lévy BI. Postischemic revascularization: from cellular and molecular mechanisms to clinical applications. Physiol Rev. 2013;93:1743–1802. doi: 10.1152/physrev.00006.2013. [DOI] [PubMed] [Google Scholar]
- Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22:3549–3560. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosson CE, Mani SK, Husain S, Alsarraf O, Menick DR. Inhibition of histone deacetylase protects the retina from ischemic injury. Invest Ophthalmol Vis Sci. 2010;51:3639–3645. doi: 10.1167/iovs.09-4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AB, Denko N, Barton MC. Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat Res. 2008;640:174–179. doi: 10.1016/j.mrfmmm.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illi B, Colussi C, Rosati J, Spallotta F, Nanni S, Farsetti A, et al. NO points to epigenetics in vascular development. Cardiovasc Res. 2011;90:447–456. doi: 10.1093/cvr/cvr056. [DOI] [PubMed] [Google Scholar]
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochot-Przeczek A, Dulak J, Jozkowicz A. Therapeutic angiogenesis for revascularization in peripheral artery disease. Gene. 2013;525:220–228. doi: 10.1016/j.gene.2013.03.097. [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stützer A, et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014;53:49–62. doi: 10.1016/j.molcel.2013.10.030. [DOI] [PubMed] [Google Scholar]
- Apostolou E, Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature. 2013;502:462–471. doi: 10.1038/nature12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Willson TA, Wakefield MJ, Trounson E, Hilton DJ, Blewitt ME, et al. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39:7415–7427. doi: 10.1093/nar/gkr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, Vlachojannis GJ, Koyanagi M, Boeckel JN, Urbich C, Farcas R, et al. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ Res. 2011;109:1219–1229. doi: 10.1161/CIRCRESAHA.111.247304. [DOI] [PubMed] [Google Scholar]
- Lopez JP, Mamdani F, Labonte B, Beaulieu MM, Yang JP, Berlim MT, et al. Epigenetic regulation of BDNF expression according to antidepressant response. Mol Psychiatry. 2013;18:398–399. doi: 10.1038/mp.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–459. doi: 10.1038/nbt.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res. 2008;102:873–887. doi: 10.1161/CIRCRESAHA.107.171025. [DOI] [PubMed] [Google Scholar]
- Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol, Cell Physiol. 2006;291:C803–C816. doi: 10.1152/ajpcell.00457.2005. [DOI] [PubMed] [Google Scholar]
- Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, et al. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci USA. 2005;102:10999–11004. doi: 10.1073/pnas.0501444102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuchel LW, Thompson MA, Cassivi SD, Pabelick CM, Prakash YS. Neurotrophins induce nitric oxide generation in human pulmonary artery endothelial cells. Cardiovasc Res. 2011;91:668–676. doi: 10.1093/cvr/cvr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–794. doi: 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Kermani P, Rafii D, Jin DK, Whitlock P, Schaffer W, Chiang A, et al. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653–663. doi: 10.1172/JCI200522655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, et al. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127:4531–4540. doi: 10.1242/dev.127.21.4531. [DOI] [PubMed] [Google Scholar]
- Caporali A, Emanueli C. Cardiovascular actions of neurotrophins. Physiol Rev. 2009;89:279–308. doi: 10.1152/physrev.00007.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreger H, Ludwig A, Weller A, Stangl V, Baumann G, Meiners S, et al. Epigenetic regulation of cell adhesion and communication by enhancer of zeste homolog 2 in human endothelial cells. Hypertension. 2012;60:1176–1183. doi: 10.1161/HYPERTENSIONAHA.112.191098. [DOI] [PubMed] [Google Scholar]
- He A, Ma Q, Cao J, von Gise A, Zhou P, Xie H, et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ Res. 2012;110:406–415. doi: 10.1161/CIRCRESAHA.111.252205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Olguín P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44:343–347. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo KH, Hennighausen L. EZH2 methyltransferase and H3K27 methylation in breast cancer. Int J Biol Sci. 2012;8:59–65. doi: 10.7150/ijbs.8.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Zhang W, Bakken T, Schutten M, Toth Z, Jung JU, et al. Cancer angiogenesis induced by Kaposi sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 2012;72:3582–3592. doi: 10.1158/0008-5472.CAN-11-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JE, Yan MS, Matouk CC, St Bernard R, Ho JJ, Ho JJ, Jr, et al. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem. 2010;285:810–826. doi: 10.1074/jbc.M109.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Zhang P, Zhang J, Chen X, Lu H, Tian Y, et al. Adenovirus-mediated brain-derived neurotrophic factor expression regulated by hypoxia response element protects brain from injury of transient middle cerebral artery occlusion in mice. Neurosci Lett. 2009;465:220–225. doi: 10.1016/j.neulet.2009.08.049. [DOI] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42 Database issue:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantzoulis-Fegaras F, Antoniou H, Lai SL, Kulkarni G, D'Abreo C, Wong GK, et al. Characterization of the human endothelial nitric-oxide synthase promoter. J Biol Chem. 1999;274:3076–3093. doi: 10.1074/jbc.274.5.3076. [DOI] [PubMed] [Google Scholar]
- Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011;25:742–754. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Olson SY, Garbán H. Transcription regulator Yin-yang 1: from silence to cancer. Crit Rev Oncog. 2011;16:227–238. doi: 10.1615/critrevoncog.v16.i3-4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali A, Meloni M, Völlenkle C, Bonci D, Sala-Newby GB, Addis R, et al. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. 2011;123:282–291. doi: 10.1161/CIRCULATIONAHA.110.952325. [DOI] [PubMed] [Google Scholar]
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- Dragneva G, Korpisalo P, Ylä-Herttuala S. Promoting blood vessel growth in ischemic diseases: challenges in translating preclinical potential into clinical success. Dis Model Mech. 2013;6:312–322. doi: 10.1242/dmm.010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics. 2007;90:397–406. doi: 10.1016/j.ygeno.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J. 2004;23:4061–4071. doi: 10.1038/sj.emboj.7600402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011;25:485–498. doi: 10.1101/gad.2019811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani K, Zhao C, Dobreva G, Manavski Y, Kluge B, Braun T, et al. Jmjd3 controls mesodermal and cardiovascular differentiation of embryonic stem cells. Circ Res. 2013;113:856–862. doi: 10.1161/CIRCRESAHA.113.302035. [DOI] [PubMed] [Google Scholar]
- Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011;43:285–298. doi: 10.1016/j.molcel.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzenda A, Lomberk G, Svingen P, Mathison A, Calvo E, Iovanna J, et al. Functional characterization of EZH2ß reveals the increased complexity of EZH2 isoforms involved in the regulation of mammalian gene expression. Epigenetics Chromatin. 2013;6:3. doi: 10.1186/1756-8935-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanueli C, Salis MB, Stacca T, Gaspa L, Chao J, Chao L, et al. Rescue of impaired angiogenesis in spontaneously hypertensive rats by intramuscular human tissue kallikrein gene transfer. Hypertension. 2001;38:136–141. doi: 10.1161/01.hyp.38.1.136. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42 Database issue:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.