

Abstract
The advent of induced pluripotent stem cells—generated via the ectopic overexpression of reprogramming factors such as OCT4, SOX2, KLF4, and C-MYC (OSKM) in a differentiated cell type—has enabled groundbreaking research efforts in regenerative medicine, disease modeling, and drug discovery. Although initial studies have focused on the roles of nuclear factors, increasing evidence highlights the importance of signal transduction during reprogramming. By utilizing a quantitative, medium-throughput screen to initially identify signaling pathways that could potentially replace individual transcription factors during reprogramming, we initially found that several pathways—such as Notch, Smoothened, and cyclic AMP (cAMP) signaling—were capable of generating alkaline phosphatase positive colonies in the absence of OCT4, the most stringently required Yamanaka factor. After further investigation, we discovered that cAMP signal activation could functionally replace OCT4 to induce pluripotency, and results indicate that the downstream exchange protein directly activated by cAMP (EPAC) signaling pathway rather than protein kinase A (PKA) signaling is necessary and sufficient for this function. cAMP signaling may reduce barriers to reprogramming by contributing to downstream epithelial gene expression, decreasing mesenchymal gene expression, and increasing proliferation. Ultimately, these results elucidate mechanisms that could lead to new reprogramming methodologies and advance our understanding of stem cell biology.
Introduction
In 2006, it was discovered that overexpression of four transcription factors—OCT4, SOX2, KLF4, and C-MYC (OSKM)—was sufficient to revert mouse embryonic fibroblasts to a pluripotent, embryonic stem cell (ESC)-like state capable of self-renewal, and these induced cells have the ability to differentiate into any adult cell type.1 The induction of pluripotent stem cells from adult cells has powerful clinical implications for drug discovery and personalized medicine, and understanding the mechanisms of cellular reprogramming and finding novel reprogramming targets also hold basic significance for stem cell and developmental biology.
Signal transduction pathways, protein and second messenger networks that convey extracellular signals to the nucleus, are well known to regulate ESC function. For example, LIF and STAT3 signaling play key roles in murine ES cell self-renewal.2 In addition, activation of the WNT-β-catenin signaling pathway, or inhibition of its antagonist GSK3β, has been shown to promote ESC self-renewal by potentially upregulating STAT3 transcription.3,4,5 In addition, β-catenin interacts with the transcription factor TCF3 to activate pluripotent gene transcription.6
To date, several signaling pathways have also been implicated in reprogramming. Similar to ESC biology, the WNT-β-catenin pathway has utility in iPS cell reprogramming. For example, WNT3A can improve reprogramming in the absence of C-MYC,7 and the GSK3β inhibitor CHIR99021 was shown to stabilize partially reprogrammed cells.8 As another example, TGFβ signal inhibition can promote reprogramming.9 Because signaling pathways offer numerous targets for pharmacological and genetic intervention, identifying new roles for signaling pathways in reprogramming can lead to the development of new reagents and approaches for both reprogramming and stem cell biology.
Here, we have systematically screened major cellular signaling pathways for their ability to replace individual reprogramming factors, particularly OCT4, SOX2, and KLF4. During OCT4 screening, we found several signaling pathways induced alkaline phosphatase positive colony formation, including the Notch, Smoothened, and cyclic AMP (cAMP) signaling pathways. In addition, this approach revealed that the activation of cAMP signaling via the adenylyl cyclase signaling pathway was sufficient to generate OCT4 positive colonies in the absence of the Oct4 transgene. Furthermore, we found that activating cAMP signaling with forskolin, consistent with an earlier report,10 along with the addition of GSK3β and MEK inhibitors (CHIR99021 and PD 0325901, or 2i) could replace OCT4 at relatively high efficiency, as determined by the percentage of OCT4-postitive colonies formed from cells infected with reprogramming factors. In addition, investigation of downstream cAMP signaling effectors indicated that EPAC was sufficient for OCT4 replacement and that its downstream effector RAP1 is necessary during both four-factor reprogramming and OCT4 replacement. Moreover, a small molecule cAMP analog that specifically activates the EPAC pathway could replace OCT4 in reprogramming. Finally, cAMP signaling may reduce the barriers for reprogramming by regulating downstream mesenchymal-to-epithelial transition genes, particularly by upregulation of Epcam and downregulation of mesenchymal markers, and by promoting cellular proliferation.
Results
Modulation of signaling pathways can generate alkaline phosphatase colonies in the presence of only three reprogramming factors
To study the role of key signaling pathways during reprogramming, we had previously constructed 38 lentiviral vectors encoding factors that upregulate or downregulate major signal transduction pathways (see Supplementary Table S1).11 As C-MYC was determined to be dispensable for reprogramming,12,13 we asked whether any signaling pathways were able to replace the other three reprogramming factors: OCT4, SOX2, or KLF4. To reduce variability inherent in infection with multiple viruses, we adapted the STEMCCA loxP cassette14 to generate three vectors, each encoding three Yamanaka factors: STEMCCA-SKM loxP (SKM), STEMCCA-OKM loxP (OKM), and STEMCCA-OSM loxP (OSM), which removed Oct4, Sox2, and Klf4 from the viral transgene, respectively (see Supplementary Figure S1).
As an initial screen for each signaling factor's ability to replace a reprogramming factor, murine embryonic fibroblasts (MEFs) were plated and infected with STEMCCA loxP vectors multiplicity of infection (MOI) of 0.3 viruses/cell resulting in ~26% of cells infected) and lentiviruses encoding constitutively-active (CA), dominant-negative, or wild-type signal transduction proteins (MOI of 1.0 resulting in ~63% of cells infected). Cells were passaged into mouse ESC conditions 2 days postinfection. Once colony morphology was apparent, cultures were fixed and stained for alkaline phosphatase expression, an early reprogramming marker, 10 days postinfection for OSM (Klf4-negative), 16 days postinfection for OKM (Sox2-negative), and 13 days postinfection for SKM (Oct4-negative). In addition, by using a robust, early marker of the reprogramming process, the screen also elucidated potential signaling pathways that could lead to a partially reprogrammed state. In the event that factors yielding only partially reprogrammed cells were initially found, we could subsequently screen for additional factors to enable full pluripotency. High-content imaging and analysis were used to quantify the number of alkaline phosphatase positive colonies (see Supplementary Table S2).
As estimated by the viral titer and a Poisson distribution, ~410 cells per well were coinfected with both the stem cell cassette and a signaling factor. Since reprogramming is a rare process,15 it is likely that an insufficient number of cells would be infected for a statistical determination of reprogramming efficiencies. However, as OCT4, SOX2, and KLF4 are necessary for the reprogramming of mouse embryonic fibroblasts,1,9,16,17 even rare alkaline phosphatase positive colony formation can be a promising initial indicator of the potential to replace a reprogramming factor. Consistent with expectations, colony formation with a maximum of 1–2 colonies per condition was observed, with the exception of CA HRAS in KLF4 replacement, and led to preliminary experiments of several signaling pathways. Example colonies from the conditions are shown in Supplementary Figures S2–S4.
Adenylyl cyclase activators produce OCT4 positive colonies in the absence of the Oct4 transgene
During subsequent in-depth analysis of the initially hit signaling pathways (using higher MOIs and greater number of cells), the activation of the adenylyl cyclase/cAMP signaling pathway by CA GNAS resulted in robust and reproducible OCT4 positive colony formation. To accurately quantify this OCT4 replacement, we calculated reprogramming efficiencies (percentage of infected cells that produced colonies expressing OCT4). As GNAS activates adenylyl cyclase and cAMP signaling, we also studied the small molecule agonist forskolin in parallel reprogramming experiments. Nine days postinfection, colonies expressing OCT4 were present in both the CA GNAS and the forskolin conditions (Figure 1a), and the number of OCT4 positive colonies was quantified via high-content imaging and analysis (Figure 1b and also see Supplementary Figure S5). In addition, differences in cell division rates in the interval between infection and passage into mouse ESC conditions were taken into consideration and used for normalization (see Supplementary Figure S6). For example, cells infected with SKM and GNAS or SKM and the lentiviral control had different proliferation rates (P < 0.05) (see Supplementary Figure S6). Efficiencies for various conditions, including controls, are included in Supplementary Table S3.
Figure 1.

The cAMP activators, constitutively-active (CA) GNAS and forskolin, induced OCT4 positive colonies in the absence of the Oct4 transgene. (a) Example colony images from the Sox2, Klf4, c-Myc, and CA GNAS condition and the Sox2, Klf4, c-Myc, and forskolin (FSKM) condition are shown. Scale bars are 300 µm. (b) Example images of the entire 24 wells are shown. The minimum colony size measured in the colony mask was 15,000 µm2. Scale bars are 3 mm. (c) The number of OCT4 positive colonies was measured from high-content imaging analysis for CA GNAS, the infection control pHIV CTRL, and forskolin with Sox2, Klf4, and c-Myc. Conditions with and without 2i small molecule inhibitors in serum-free media are shown. (d) The number of OCT4 positive colonies with forskolin, dimethyl sulfoxide (DMSO), and Oct4 conditions is shown. The Oct4 conditions were normalized to the SKM conditions due to variability of infection. Statistical significance was measured using a Student's t-Test (two-tailed, homoscedastic) with *P < 0.05 and **P < 0.005. Negative controls are P < 0.005. The minimum colony size was 15,000 µm2. Error bars represent SD (n = 3). All conditions were cultured in 0.22% DMSO and fixed with paraformaldehyde 9 days postinfection.
With SKM expression, the activation of cAMP signaling via CA GNAS or forskolin significantly increased the number of OCT4 positive colonies (P < 0.05) compared with the negative control (Figure 1c). In addition, forskolin resulted in a greater increase (2.2-fold) in colony number compared to CA GNAS, potentially due to more potent cAMP activation. Since both CA GNAS and forskolin still yielded significantly fewer colonies than with ectopic OCT4 expression (P < 0.005, 12-fold and 6-fold reduction, respectively) (Figure 1d), we next investigated additional treatments to further increase efficiency. The 2i inhibitors (CHIR99021 and PD 0325901) have been shown to aid reprogramming,8 and their addition with cAMP activators during reprogramming yielded a further ~4-fold increase in the number of OCT4 positive colonies in both forskolin and CA GNAS conditions (Figure 1c).
Subsequently, we wanted to study the individual roles of 2i components CHIR99021 and PD 0325901, which activate the WNT-β-catenin signaling pathway and inhibit the MAPK signaling pathway, respectively. In addition, to minimize differentiation signals, serum-free media was used with the 2i inhibitors,8 and we also quantified the effect of the serum-free medium during OCT4 replacement. When SKM and forskolin (FSKM) reprogramming was conducted in serum-free medium, the 3.9-fold increase was replicated, indicating that the medium was sufficient for a similar increase in colony number as the 2i inhibitors (Figure 1d), which supports previous evidence that serum-free conditions promote reprogramming.18 However, when added alone, PD 0325901 could also significantly increase the number of OCT4 positive colonies compared to the 2i condition (see Supplementary Figure S7). Forskolin activates PKA, which has been shown in some cases to activate the MAPK pathway during cell differentiation.19,20,21 PD 0325901 inhibition of MAPK signaling could conceivably prevent PKA-mediated differentiation during reprogramming and thereby result in an increase in reprogramming efficiency. Furthermore, as CHIR99021 did not significantly affect reprogramming (see Supplementary Figure S7), the cAMP-mediated replacement of OCT4 is apparently independent of the WNT-β-catenin signaling pathway.
As additional controls, we also varied the colony cut-off size in the imaging analysis as well as analyzed NANOG expression, a late stage pluripotency marker, to complement OCT4 staining (see Supplementary Figures S8 and S9). Although NANOG staining was not as robust and not ideal for automated imaging analysis, the same trends for forskolin replacement of OCT4 were observed (see Supplementary Figure S9a).
cAMP activation with 2i inhibition results in pluripotency without the Oct4 transgene
Next, to determine if the resultant cells (with 2i) were pluripotent, colonies were isolated and passaged to form cell lines. These iPS lines, including the SKM FK 2i-06 line, were stained for pluripotency markers SSEA1, OCT4, and NANOG (Figure 2, see Supplementary Figures S10 and S11). SSEA1 expression was present although not uniform across all colonies (see Supplementary Figure S10). However, cAMP signaling-induced lines had consistent endogenous OCT4 and NANOG expression (Figure 2, see Supplementary Figures S10 and S11). For ease of culture, the iPS cell lines could also be transitioned to feeder-free conditions, and pluripotency marker expression persisted (Figure 2). In addition, the cell lines were stable and maintained colony morphology in culture for at least 19 passages (see Supplementary Figure S12).
Figure 2.

SKM FK 2i-06 cell line expresses pluripotency markers SSEA1, OCT4, and NANOG in feeder-free conditions. SKM FK 2i-06 was transitioned to serum without small molecules at two passages postisolation. SKM FK 2i-06 is shown eight passages postisolation and two passages in feeder free conditions. Scale bars are 50 µm.
To test the ability of these cell lines to undergo differentiation, embryoid bodies (EBs) were formed in hanging drops in the absence of LIF, a growth factor used in mouse embryonic stem cell maintenance. These EBs were then cultured in suspension with retinoic acid (to enhance neuronal differentiation) and allowed to attach to culture surfaces to enable cell migration and differentiation (Figure 3a). The resulting cultures stained positive for the mesodermal marker smooth-muscle actin, the ectodermal marker β-III tubulin, and the endoderm marker HNF3β (Figure 3b, see Supplementary Figure S13).
Figure 3.

Forskolin 2i cell lines form embryoid bodies and produce three germ layers in vitro. (a) The line SKM FK 2i-06 formed embryoid bodies after initial culture for 2 days in hanging drops. Pictures are shown 2, 3, 5, and 6 days in differentiation conditions. Scale bars are 40 µm. (b) SKM FK 2i-06 differentiates into three germ layers. Images are of different regions expressing smooth-muscle actin, βIII-tubulin, or HNF3β, respectively, in embryoid body outgrowth. Retinoic acid (5 µmol/l) was added to the differentiation media during embryoid body suspension culture. Scale bars are 25 µm.
cAMP acts through the EPAC-RAP1 pathway to induce reprogramming
We determined that the activation of adenylyl cyclase/cAMP signaling was sufficient to replace OCT4 during reprogramming; however, it is mechanistically unclear which downstream effectors mediate this effect. Adenylyl cyclases catalyze the conversion of ATP to 3′, 5′—cAMP, which subsequently signals through two primary effectors, PKA and EPAC.22,23 We thus modulated these two downstream pathways to assess their importance in OCT4 replacement and reprogramming (Figure 4).
Figure 4.
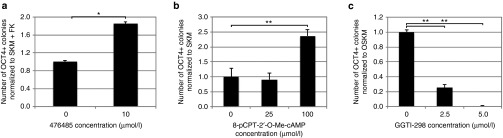
EPAC-RAP1 is a key pathway for replacement of Oct4 and reprogramming. (a) The PKA inhibitor, 476485, increased the number of OCT4 positive colonies in forskolin-induced reprogramming. (b) Adding the EPAC analog, 8-pCPT-2′-O-Me-cAMP at higher concentrations increased the number of OCT4 positive colonies compared to a SKM control. (c) The RAP1 inhibitor GGTI-298 decreased the number of colonies in OSKM reprogramming. Statistical significance was measured using a Student's t-test (two-tailed, homoscedastic) with *P < 0.05 and **P < 0.005. The minimum colony size was 15,000 µm2. Error bars represent SD (n = 3). All conditions were cultured in 0.22% DMSO and fixed with paraformaldehyde nine days postinfection.
First, to understand whether the PKA pathway was necessary to replace OCT4, the PKA inhibitor 476485 was added during FSKM reprogramming. This inhibition yielded a significant increase (P < 0.05) in the number of OCT4 positive colonies (Figure 4a), indicating that cAMP signaling does not act via PKA to replace OCT4. Interestingly, PKA has been shown to accelerate mouse ESC differentiation via methylation of pluripotency genes,24 consistent with our observation that PKA inhibition actually enhances reprogramming. To determine whether a second cAMP effector, EPAC, was sufficient to replace OCT4, the EPAC-specific cAMP analog 8-pCPT-2′-O-Me-cAMP25 was added to SKM during reprogramming (CSKM condition), and we found this compound to significantly increase (P < 0.005) the number of OCT4 positive colonies compared to the negative control (Figure 4b), indicating that EPAC pathway activation may be sufficient to replace OCT4. Because of a high proliferation rate of the starting MEF cells in this particular experiment (Figure 4b), the negative control did have some OCT4-positive colonies; however, EPAC signaling substantially increased the number of colonies. All other experiments resulted in negligible colonies with the SKM negative control.
To determine whether the EPAC pathway is necessary during reprogramming, we inhibited an EPAC effector, RAP1.22 Titration of the RAP1 inhibitor GGTI-298 on MEFs indicated that concentrations ≤5 µmol/l did not compromise MEF viability (see Supplementary Figure S14a). Addition of 2.5 µmol/l GGTI-298 to OSKM reprogramming induced a significant decrease in colony number (3.9-fold, P < 0.005), and 5 µmol/l of the compound drastically decreased OSKM reprogramming (158-fold) (Figure 4c). Similarly, increasing the concentration of GGTI-298 in FSKM reprogramming reduced reprogramming levels back to the negative control condition, SKM (see Supplementary Figure S15), again indicating that the EPAC signaling pathway is necessary for reprogramming and OCT4 replacement.
Finally, we investigated the activation of RAP1 during reprogramming (Figure 5). RAP1 was increasingly activated between 3 and 12 days postinfection for OSKM, FSKM, and CSKM-mediated reprogramming, with activated levels peaking and declining after day 9 through day 12 (Figure 5a). The highest level of activated RAP1 was seen in the CSKM condition 12 days postinfection. Although activated RAP1 was also seen in the SKM condition, which did not support substantive reprogramming (Figure 1c,d), it was at a significantly lower level and appeared later compared to the other conditions. In parallel, NANOG expression was measured over the same time period and appeared to gradually increase for OSKM, FSKM, and CSKM conditions but remained low for the SKM condition (Figure 5b).
Figure 5.

RAP1 and NANOG are activated during reprogramming. Mean fluorescence intensity of antibody-stained (a) activated RAP1 and (b) NANOG is shown 3, 6, 9, 12, and 15 days postinfection in SKM, FSKM, CSKM, and OSKM reprogramming conditions and normalized to the SKM day 3 condition. Normalized mean fluorescence intensities ± propagated SDs (n = 3) from positively stained colonies were calculated in ImageJ. Statistical significance with respect to the SKM condition for each day was measured using a Student's t-test (two-tailed, homoscedastic) with *P< 0.05 and **P < 0.005.
EPAC-specific cAMP analog results in pluripotency without the Oct4 transgene
To assess the pluripotency of cells reprogrammed with SKM and the EPAC-specific cAMP analog 8-pCPT-2′-O-Me-cAMP (CSKM condition), colonies were isolated and passaged to form cell lines. The pluripotency markers OCT4, NANOG, and SSEA1 were expressed in the resulting cell lines (Figure 6a). In addition, marker expression was maintained after an additional 10 passages in medium free of the EPAC-specific cAMP (see Supplementary Figure S16). Furthermore, to demonstrate pluripotency and the potential to generate the three different germ layers, EBs were formed from CSKM cell lines and differentiated in vitro. The resulting cultures stained positive for α-smooth muscle actin (mesoderm), HNF3β (endoderm), and βIII-tubulin (ectoderm) (Figure 6b).
Figure 6.

Cell lines created with the EPAC agonist are pluripotent and form three germ layers in vitro. (a) CSKM-06 cells express pluripotency markers OCT4, NANOG, and SSEA1. Cells were imaged two passages after isolation from feeder cultures. (b) CSKM-06 cells differentiate into the three germ layers. Images are from different regions showing α-smooth muscle actin (mesoderm), HNF3β (endoderm), and βIII-tubulin (ectoderm). Embryoid bodies were generated from passage 2 cells, and images were taken 10–20 days after differentiation. Scale bars are 100 μm.
To further assess pluripotency, a teratoma assay was conducted in female NOD/SCID mice using passage 4 iPS cells generated under the CSKM and FKSM conditions. Pluripotency marker expression was confirmed before implantation (see Supplementary Figure S17). Tumors appeared in mice 3–4 weeks after cell injection, and H&E staining of tumor sections showed the presence of endodermal, ectodermal and mesodermal structures for both conditions (Figure 7). Overall, these assays demonstrate that EPAC-specific cAMP pathway activation alone can successfully replace OCT4 and generate pluripotent stem cells.
Figure 7.

CSKM and FSKM conditions create iPS cells capable of forming teratomas in NOD/SCID mice. SKM FK 2i-06 and CSKM-06 cells at passage 4 were subcutaneously injected into the hind flank of mice. H&E stained sections show the presence of endoderm, mesoderm, and ectoderm for both SKM FK 2i-06 (a-c) and CSKM-06 (d-f). Images are representative of three animals per group. Scale bars are 250 μm.
cAMP signaling increases cellular division rates and results in transcriptional changes for genes involved in the mesenchymal to epithelial transition
After determining the role of cAMP effectors in OCT4 replacement, we next investigated which reprogramming process this pathway may modulate. During reprogramming, we noted that forskolin addition induced early morphological changes in MEFs compared to the positive and negative controls (OSKM and SKM infected cells, respectively) (Figure 8a). For instance, colonies were visible 5 days postinfection with forskolin, and although SKM and OSKM conditions also yielded colonies, they were comparatively less distinguishable from surrounding cells. Because of this early presence of colonies in the reprogramming, we investigated whether cell proliferation was augmented, especially as cell division is a barrier to reprogramming.26,27,28 DNA synthesis was assayed by measuring 5-ethynyl-2′-deoxyurinide (EdU) incorporation during DNA synthesis 24, 48, and 72 hours after forskolin addition. To analyze solely the proliferation of SKM lentivirus infected fibroblasts, an antibody against SOX2 was also used to probe for viral transgene expression. The addition of the cAMP agonist forskolin to SKM significantly increased the percentage of dividing cells 24 hours after addition (24% versus 17% of the cells dividing with or without forskolin, respectively, P < 0.005) (Figure 8b). The cell division remained higher 48 and 72 hours after forskolin addition (Figure 8b), which offers a potential mechanism by which cAMP decreases a barrier to reprogramming.26
Figure 8.

cAMP activation induces cell division and gene expression changes that may lower the barriers to reprogramming. (a) Brightfield images show the change in morphology of mouse embryonic fibroblasts 3, 5, and 7 days postinfection with SKM, FSKM, and OSKM. Conditions contain 0.1% DMSO. Scale bars are 40 µm. (b) Forskolin causes an increase in cell division rate hours after addition. Mouse embryonic fibroblasts were infected with SKM. Two days postinfection, cells were split in mouse ESC conditions with forskolin or DMSO. EdU was incubated for 1 hour, and the cells were fixed. To account for noninfected cells, the cells were costained against SOX2, which is present in the viral transgene. Error bars represent SD (n = 3). (c–i) mRNA levels of pluripotency genes (c–e), epithelial genes (f–g), and mesenchymal genes (h–i) were analyzed at early time points in reprogramming. Each gene expression was normalized to the internal control gene, Hprt. Each condition was subsequently normalized to the SKM day 3 control condition. Error bars represent SD (n = 2, technical).
In addition, due to the early changes in cell morphology, we investigated whether cAMP signaling altered expression of mesenchymal and epithelial marker genes, particularly as the mesenchymal to epithelial transition is also an early barrier to reprogramming.29,30 Quantitative RT-PCR was used to analyze key marker expression 3, 5, or 7 days postinfection. Forskolin increased the expression of epithelial genes Cdh1 and Epcam (Figures 8f,g) and decreased the expression of mesenchymal genes Cdh2 and Slug (Figures 8h,i) when compared with the SKM negative control. However, expression for Epcam, Cdh2, and Slug differed from corresponding levels in OSKM cultures, indicating that the role of cAMP signaling differs from OCT4 expression by decreasing mesenchymal gene expression and results in an increase in Epcam expression during the early stages of reprogramming.
As cAMP signaling was sufficient to functionally replace OCT4 in reprogramming, we also studied the pathway's effects on pluripotency gene expression. Forskolin resulted in an increase in Oct4 expression but failed to upregulate Nanog as high as OSKM 7 days postinfection (2-fold, 14-fold, and 348-fold increase for Nanog with SKM, FKSM, and OSKM, respectively) (Figures 8c,e). As OCT4 and SOX2 are exogenously expressed in OSKM reprogramming and bind to the promoter region of Nanog,31,32,33,34 it is unsurprising that Nanog expression is higher in conditions with OCT4 (and SOX2). Interestingly, forskolin decreased the expression of Klf4 during the timeframe studied (Figure 8d), even though earlier studies indicated Klf4 expression as a target of forksolin activity.35,36 Thus, activated cAMP does not upregulate Klf4 but is capable of upregulating Oct4 during reprogramming during the time frame studied. Thus, cAMP signaling can replace OCT4 through the EPAC pathway and ultimately leads to changes in Oct4, mesenchymal, and epithelial gene expression in addition to cellular proliferation, which may reduce the barriers to reprogramming.
Discussion
Reprogramming somatic cells into induced pluripotent stem cells has immense potential for the regenerative medicine, and by investigating underlying mechanisms, we can discover novel reprogramming methodologies and further understand both reprogramming and stem cell biology. Here, we conducted a screen of major signaling pathways for reprogramming factor replacement. By screening for replacements of OCT4, SOX2, and KLF4, we discovered that adenylyl cyclase activation and cAMP elevation could functionally replace OCT4 during reprogramming.
In the initial approach, we screened 38 genes that upregulate or downregulate major signal transduction pathways. As signaling pathways can be manipulated with small molecules or growth factors, they offer advantages in reprogramming research compared to transcription factors, which are challenging to pharmacologically manipulate. For example, transient small molecule addition during reprogramming can result in pluripotent cells without genetic modification as shown earlier,10 which may be safer than overexpression of transcription factors that are known oncogenes, such as Klf4 and c-Myc and potentially OCT4 (ref. 37). Small molecule replacements have been discovered for SOX2 (refs. 9,16,38,39) and KLF4 (refs. 17,40,41,42) in reprogramming, in some cases through the use of small molecules libraries. In addition, recent work by Hou et al. screened up to 10,000 small molecules to discover a replacement by OCT4 and identified forskolin.10 Although small molecule screens in stem cell and reprogramming biology effectively build on pharmacology and toxicology screening infrastructure, the approach does have limitations, including an abundance of inhibitors rather than activators, unknown cellular targets in some libraries, lack of target specificity in others, cellular toxicity, and challenges in adapting complex cell cultures to high throughput format. Here, the signal perturbation library we generated contained 250-fold fewer members, yet implicated cAMP signaling plus additional pathways that can be explored in future studies (Supplementary Table S3).
The ability of cAMP signaling to aid in the generation of induced pluripotent stem cells also raises questions on the mechanistic role of this pathway in reprogramming. To date, few studies have investigated cAMP signaling in ESCs and reprogramming. As examples, a study has reported that the cAMP-analog, 8-Br-cAMP, can increase human fibroblast reprogramming twofold,43 and in addition, forskolin in combination with the 2i inhibitors (CHIR99021 and PD 0325901 (ref. 8)) and LIF is sufficient to transform human ESCs to a naive, mouse embryonic-like state.35 By analyzing the downstream effectors of cAMP, we discovered the importance of the EPAC signaling pathway. This pathway has been relatively unstudied in the ESC biology, with a report of a role for RAP1 in human ESC self-renewal44 and unstudied in reprogramming until now. In this study, we linked increasing RAP1 activation to reprogramming with or without using small molecules. In addition, a cAMP analog, 8-pCPT-2′-O-Me-cAMP, that specifically activates the EPAC pathway successfully replaced OCT4 to generate pluripotent stem cells capable of forming the three germ layers in EB differentiation and produce teratomas in immunodeficient mice. Cells were passaged up to 12 times, 10 of which were free of 8-pCPT-2′-O-Me-cAMP, and maintained pluripotency marker expression. Thus, cAMP signal effectors including EPAC and RAP1 are interesting targets for future reprogramming and stem cell studies.
By studying downstream transcriptional targets during the early stages of reprogramming, we found that activation of cAMP signaling increased the expression of the epithelial markers Epcam and Cdh1 compared with that of SKM conditions, and Epcam was significantly increased with cAMP signaling compared to OSKM. Epcam has been shown to be important to mouse ESC renewal45 and serves as a pluripotency marker for human ESCs,46 and it thus may aid reprogramming in the absence of OCT4. In addition, cells infected with the Cdh1 transgene (i.e., E-cadherin) in addition to SOX2, KLF4, and C-MYC have been previously shown to replace OCT4 in reprogramming.47 As RAP1 has been shown to interact with CDH1 in human ESCs to maintain self-renewal,44 cAMP signaling could be activating RAP1 and CDH1 to replace OCT4.
In summary, this work furthers our understanding of the cAMP signaling pathway in reprogramming and in OCT4 replacement and could indicate potential signaling targets to study within stem cell biology. Moreover, the screening methodology presented here may be useful to study the importance of signaling pathways within mammalian cell biology, including pluripotency reprogramming, direct lineage reprogramming, and/or differentiation.
Materials and Methods
Cell culture and plasmid constructs. HEK293T cells were maintained in Iscove's modified Dulbecco's medium (IMDM) with 10% FBS and 1% penicillin/streptomycin. Strain 129 MEF cells were a kind gift of Lin He (University of California, Berkeley, CA) and isolated as described earlier.11 Murine embryonic fibroblasts were maintained in Dulbecco's modified Eagle medium (DMEM) (high glucose), 10% FBS, 1% GlutaMAX, and 1% penicillin/streptomycin (Invitrogen, Grand Island, NY). Cells undergoing reprogramming were maintained in serum-containing, mouse ESC (mESC-FBS) conditions: DMEM (high glucose), 15% FBS (Hyclone, SH3007003E), 0.5% penicillin/streptomycin, 1% GlutaMAX, 1% sodium pyruvate, 1% MEM NEAA, 0.1 mmol/l 2-mercaptoethanol (Invitrogen), and 1,000 U/ml LIF (Millipore, Billerica, MA ESG1106). Feeder-free cell lines were maintained with GMEM (Sigma-Aldrich, St. Louis, MO) instead of DMEM. 15% KnockOut Serum Replacement (Invitrogen) was used in place of FBS in serum-free conditions (mESC-KSR). Differentiating EBs were maintained in mESC-FBS media without LIF. Cells were maintained at 37 °C with 5% CO2.
STEMCCA-SKM loxP (SKM), STEMCCA-OKM loxP (OKM), and STEMCCA-OSM loxP (OSM) were constructed from the STEMCCA loxP vector.14 In brief, STEMCCA-SKM loxP was constructed by creating a NotI-Klf4-IRES-NdeI PCR fragment (NotI-Klf4 Forward: 5′—GTAATATGCGGCCGCCATGGCTGTCAGCGACGCTCTG—3′, Nde-IRES Reverse: 5′—CCCCCCCCCCATATGTGTGGCCATATTATCATCGTGTTTTTCAAAGGAAAACC—3′, restriction sites in bold). After enzymatic digestion, the PCR fragment was inserted into a NotI-NdeI digested STEMCCA loxP subsequently removing the Oct4 gene. STEMCCA-OKM loxP was constructed by creating an NdeI-cMyc-ClaI PCR fragment (NdeI-cMyc Forward: 5′—GGATAGCATATGATGCCCCTCAACGTGAACTTC—3′, ClaI-cMyc Reverse: 5′—CGATCTATCGATTTATGCACCAGAGTTTCGAAGCTGTTC—3′). After enzymatic digestion, the PCR fragment was inserted into an NdeI-ClaI digested STEMCCA loxP subsequently removing the Sox2 gene. STEMCCA-OSM loxP was constructed through overlap-extension PCR. Two PCR fragments, NotI-Oct4-STOP-IRES and Oct4-IRES-NdeI, were created (NotI-Oct4-STOP-IRES Forward: 5′—GAATAAGCGGCCGCCATGGCTGGACACCTGGCTTCAGACTTCGCCTTCTCACC—3′, NotI-Oct4-STOP-IRES Reverse: 5′—GCCAGTAACGTT AGGGGGGGGGGAGTCAGTTTGAATGCATGGGAGAGCCCAGA—3′, Oct4-IRES-NdeI Forward: 5′—GGGCTCTCCCATGCATTCAAACTGACTCCCCCCCCCCTAACGTTACTGGCCGA—3′, Oct4-IRES-NdeI Reverse: 5′—GGGGGGCATATGTGTGGCCATATTATCATCG TGTTTTTCAAAGGAAAACCACGTCCCCGTGGTTCGGGGG—3′, sequences located in primer in italics). The two fragments were used in a subsequent PCR with the NotI-Oct4-STOP-IRES Forward and the Oct4-IRES-NdeI Reverse primers creating a NotI-Oct4-IRES-NdeI PCR fragment. After enzymatic digestion, the PCR fragment was inserted into a NotI-NdeI digested STEMCCA loxP subsequently removing the Klf4 gene.
pCDNA3.1+ Gαs Q227L was obtained from Missouri S&T cDNA Resource Center (www.cdna.org). Using standard cloning techniques, all genes were inserted into the murine leukemia virus retroviral vector CLGPIT48 and subsequently transferred to the lentiviral vector pHIV IG loxP. pHIV IG loxP was created by insertion of three oligo constructs into the pHIV EGFP backbone.49 The oligos encoded the lox66 site, a multiple cloning site, or the lox71 site. Cloning information for the 38 signaling genes (see Supplementary Table S2) has been described earlier.11 DNA was prepared with QIAGEN Plasmid Midi or Maxi Kit (Qiagen, Venlo, The Netherlands) per manufacturer's instructions. Plasmid sequences were verified by restriction enzyme digest and sequencing.
Viral production and titer. CA GNAS lentivirus was produced by HEK293T cells using calcium phosphate transfection and concentrated as described earlier without a sucrose layer.48 For the reprogramming cassettes, the lentiviral transfection and centrifugation were performed as described earlier.14,50 The STEMCCA loxP viruses are second-generation lentiviruses, and CLPIT Tat-mCherry was thus added during transfection to promote viral genomic mRNA expression. 0.45 μm bottle top filters (Thermo Scientific, Waltham, MA, 09-740-28D) were used to filter the virus, and no sucrose layer was used in ultracentrifugation. The concentrated virus was aliquoted and stored at −80 °C. Aliquots were only thawed once.
To quantify the amount of lentivirus, 1,600 MEFs were plated in 0.1% gelatin-coated (Millipore, ES-006-B) black-walled 96-well plates (E+K Scientific, Santa Clara, CA). After cell attachment, varying volumes of virus were added to the media. Seventy two hours postinfection, viral transgene expression was analyzed. After fixation with 4% paraformaldehyde, antibodies were used to amplify the protein expression: for STEMCCA loxP viruses containing Oct4, an anti-OCT4 antibody (Santa Cruz Biotechnology, Dallas, TX, sc-5279, 1:100 dilution) and a secondary antibody (Invitrogen, A-21235, 1:250) were used; for STEMCCA-SKM loxP, an anti-SOX2 antibody (Santa Cruz Biotechnology, sc-17320, 1:200) and a secondary antibody (Jackson ImmunoResearch, West Grove, PA, 705-165-147, 1:250) were used; for CA GNAS, an anti-GFP primary antibody (Abcam, Cambridge, MA, ab13970, 1:250) and a secondary antibody (Jackson Immunoresearch, 703-545-155, 1:250) were used. DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen, 1:2,000) was used as a nuclear stain. OCT4, SOX2, or GFP expression was imaged with the ImageXpress Micro (Molecular Devices, Sunnyvale, CA) and analyzed with MetaXpress software. Infectious titers were calculated as described earlier.50
iPS cell reprogramming—factor replacement (SKM, OSM, OKM). 7,500 passage 3 MEF cells were plated into 48-well plates. The following day one signal transduction virus (24–650 µl) was added at an MOI of 1.0 (63% cells infected) and STEMCCA-loxP virus was added at an MOI of 0.3 (26% cells infected) to each condition. The following day, the media were replaced with fresh MEF media. Each 48-well plate was split into triplicate onto MEF feeder layers 48 hours postinfection (GlobalStem, Gaithersburg, MD GSC-6101M) in mouse ESC media. Media were changed every day. The six-channel multichannel adjustable pipette (Rainin, Oakland, CA, LA6-1200XLS) was used to passage cells and change media.
Nine days postinfection, cells were fixed with 4% paraformaldehyde for 15 minutes. Alkaline phosphatase expression was assayed using the ELF Phosphatase Detection Kit (ATCC, Manassas, VA, SCRR-3010) per manufacturer's instructions. Note that ATCC kit has been discontinued, and we have found the ELF 97 Endogenous Phosphatase Detection Kit (Invitrogen, E-6601) to be an appropriate substitute. HCS NuclearMask Red stain (Invitrogen, H10326, 1:1,000 dilution) was used to visualize nuclei.
The 24-well plates were imaged with a ImageXpress Micro (Molecular Devices) high-throughput imager. A custom filter cube (Semrock BrightLine filters, Rochester, NY, Excitation: FF01-377/50-25, Dichroic: FF409-Di03-25x36, Emission: FF01-536/40-25) was necessary to detect alkaline phosphatase expression. For image analysis, a custom journal was created in the MetaXpress software to merge the images and identify colonies/regions of alkaline phosphatase expression.
iPS cell reprogramming and efficiency calculations. 4,000 3 mouse embryonic fibroblasts were plated per sample and combined into four wells for infection: SKM, OSKM, SKM, and CA GNAS, or SKM and pHIV CTRL, with cell densities between 9,473 and 12,631 cells/cm2. Approximately 12 hours after passage, the wells were infected with SKM or OSKM at an MOI of 0.47 infectious particles/cell. In addition, virus expressing CA GNAS or the infection control, pHIV CTRL, was added to their respective wells at an MOI of 1.2. The media was replaced with MEF media 24 hours postinfection. Cells were passed to 24-well plates with mitomycin-c treated MEF feeder layers 48 hours postinfection (GlobalStem GSC-6101M) with mESC-FBS media with or without forskolin (10 µmol/l, Enzo Life Sciences, Farmingdale, NY, BML-CN100-0010). Media and small molecules were changed daily. At 5 days postinfection, the small molecules CHIR99021 (3 µmol/l, Cayman Chemical, Ann Arbor, MI, 13122) and PD 0325901 (1 µmol/l, Cayman Chemical, 13034) were added to the appropriate wells. All conditions containing CHIR99021 and/or PD 0325901 were cultured in serum-free (mESC-KSR) media on small molecule addition. The small molecules and media were changed daily. After passage, all wells were maintained in 0.22% DMSO. The wells were fixed 9 days postinfection.
To account for differences in cell division between the four viral infections, three wells per viral infection were also plated on a 24-well plate with feeder layers in mESC-FBS medium with 0.22% DMSO during the passage. After 8 hours, the cells were fixed with 4% paraformaldehyde. The wells were stained for SOX2 or SOX2 and GFP expression with an anti-Sox2 antibody and an anti-GFP antibody (antibodies described earlier). The wells were imaged with the ImageXpress Micro (Molecular Devices) and analyzed with MetaXpress software. By normalizing between the numbers of cells infected per condition, the number of resulting OCT4 colonies is comparable. Reprogramming efficiency was calculated as the number of OCT4 positive colonies normalized to the number of infected cells at passage on day 2.
The fixed reprogrammed cells were stained for OCT4 and NANOG expression: an anti-OCT4 antibody (Santa Cruz Biotechnology, sc-5279, 1 : 100) and an anti-NANOG antibody (Abcam, ab70482, 1:250) were incubated for 1 hour at room temperature. The secondary antibodies (Jackson ImmunoResearch, 715-585-150 and 711-605-152, 1:500) were incubated for 2 hours at room temperature. For image analysis, a custom journal was created in the MetaXpress software to merge the images and identify colonies/regions of OCT4 or NANOG expression. The colony numbers were normalized to the SKM condition. Significance was measured using a Student's t-Test (two-tailed, homoscedastic).
Cell line isolation and pluripotency immunostaining. Cells were reprogrammed as previously described. Twelve or more days postinfection, cells were manually selected under a microscope (EVOS xl core, AMG, Grand Island, NY), trypsinized, and added to separate wells in a 48-well plate with mitomycin-c treated MEF feeder layers. The SKM FK 2i cell lines were maintained in mESC-KSR media with forskolin, CHIR99021, and PD 0325901 for 1–3 passages and then transferred to mESC-FBS media without small molecules. The cells were passed every 2–3 days in mESC-FBS media on feeder layers.
To transfer the cells to feeder-free conditions, the cells were plated into gelatin-coated plates with GMEM-based mESC media that had been conditioned with mitomycin-c treated MEF feeder layers, harvested, and supplemented with LIF (GMEM-CM). The cells were incubated with 50% GMEM-CM and 50% GMEM mESC media after 48 hours. The following day the cells were passaged. The cells were maintained in 100% GMEM mESC media 48 hours after passage.
To probe for expression of pluripotency proteins, confluent cells were fixed and stained with the following antibodies: SSEA1, OCT4, and NANOG (described earlier). For SSEA1, the primary antibody (Millipore, MAB4301, 1:200) and secondary antibody (Invitrogen, A-21045, 1:1,000) were used. As SSEA1 is a cell-surface marker, no Triton-X100 was used to permeabilize the cells. The staining was imaged on the ImageXpress Micro (Molecular Devices).
EB differentiation. iPS cell lines were trypsinized and incubated on 0.1% gelatin-coated tissue culture plates in EB medium for 1 hour at 37 °C with 5% CO2 to separate the MEF feeder layers from the iPS cells. The unattached cells were collected and diluted to 2.25 × 104 cells/ml. Twenty microliter drops were pipetted onto a 15-cm lid. PBS buffer of 10 ml was added to the plate and the lid was gently placed on top of the plate. The hanging drops were incubated at 37 °C with 5% CO2 for 2 days. The drops were collected and the embryoid body suspension was added to a bacterial-grade sterile 10-cm dish. The embryoid bodies were incubated for ≥3 days before pipetting individual embryoid bodies into the wells of a 24-well plate coated with gelatin. The following day, the media was partially changed. The media was changed every other day or every day on confluency. The embryoid bodies were fixed when the EBs had attached, and cells had migrated and appeared differentiated (12–20 days after hanging drop).
To improve neural differentiation in cultures, retinoic acid (5 µmol/l, Enzo Life Sciences, BML-GR100-0500) was added during the transfer of the hanging drops to the suspension culture. Every other day, 50% of the media was changed with retinoic acid. The retinoic acid treatment was stopped when the EBs were transferred for attachment.
To visualize the expression of differentiation markers, the wells were fixed and stained with one the following antibodies: for mesoderm, anti-α-smooth muscle actin (Sigma-Aldrich, A2547, 1:500) and the secondary (Invitrogen, A-11003, 1:1,000); for endoderm, anti-HNF3β (also known as FOXA2) (Millipore, 07-633, 1:500); and for ectoderm, anti-βIII-tubulin (Covance, Princeton, NJ, MRB-435P, 1:500). The secondary antibody (Invitrogen, A11-012, 1:1,000) was used for HNF3β and βIII-tubulin staining. The staining was imaged on the ImageXpress Micro (Molecular Devices).
Small molecule reprogramming assay. Cells were reprogrammed, stained, and analyzed as described earlier. Small molecules were added 2 days postinfection: 476485 (Millipore, 476485), GGTI-298 (Santa Cruz Biotechnology, sc-221673), and 8-pCPT-2′-O-Me-cAMP (Sigma-Aldrich, C8988). The small molecules and media were changed daily. After passage, all wells were maintained in 0.22% DMSO. The wells were fixed 9 days postinfection.
Proliferation assay. Cells were reprogrammed as described earlier. The cells were incubated with 10 µmol/l EdU (Invitrogen, E10187) for 1 hour 3, 4, or 5 days postinfection. The cells were washed, fixed, and subsequently stained for SOX2 expression (antibody described earlier) with a secondary antibody (Jackson ImmunoResearch, 705-545-147, 1:500). The cells were then stained with the Click-iT EdU Alexa Fluor 594 Imaging Kit (Invitrogen, C10339). Nuclei were visualized with DAPI (1:2000). The SOX2 and EdU staining was imaged with the ImageXpress Micro (Molecular Devices) and analyzed with MetaXpress software.
Quantitative RT-PCR. Cells were reprogrammed as described earlier. In brief, 6,000 passage 3 MEFs per sample were combined for SKM or OSKM infection. The following day, the cells were infected at an MOI of 0.3 infectious particles/cell. The passage and media changes remained the same as the previous protocol. All conditions were maintained in 0.1% DMSO after passage. RNA was isolated using the RNeasy Micro Kit (Qiagen, 74004) per manufacturer's instructions, 3, 5, or 7 days postinfection. RNA was quantified, normalized, and reverse-transcribed using the ThermoScript RT-PCR for First-Strand cDNA Synthesis (Invitrogen, 11146) per manufacturer's instructions. The samples were analyzed for gene expression on the BioRad iQ5 with standard curves, water samples, and melt curve controls. qPCR primers are listed in Supplementary Table S4. Data were analyzed by the ΔΔCt method compared to the Hprt control gene and the SKM day 3 control, respectively. Statistics were calculated as described in the ABI manual.
CSKM reprogramming and pluripotency assays. Cells were reprogrammed using SKM as described earlier. After passage onto MEF feeder layers, cells were maintained in mESC-FBS with 100 μmol/l 8-pCPT-2′-O-Me-cAMP with media changes every other day (CSKM condition). After 10 days of culture, colonies were isolated. Cells were maintained in mESC-FBS with 8-pCPT-2′-O-Me-cAMP for two passages and then transferred to mESC-FBS media without small molecules. The cells were then passed every 2–3 days in mESC-FBS media on feeder layers. To probe for expression of pluripotency markers, confluent cells were fixed and stained for SSEA1, OCT4, and NANOG. Images were taken on a Nikon TE2000-E microscope.
In addition, embryoid bodies were cultured and differentiated without retinoic acid. After 10–20 days of differentiation in 2D culture, cells were fixed and stained for the germ layer markers, α-smooth muscle actin (mesoderm), HNF3β (endoderm) and βIII-tubulin (ectoderm), using antibodies as described earlier. Images were taken on a Nikon TE2000-E microscope.
Furthermore, a teratoma formation experiment was conducted in 6-week-old female NOD/SCID mice (Jackson Labs, Bar Harbor, ME, 005557) using FSKM and CSKM iPS cell lines. At passage 4, cells were harvested and resuspended in Matrigel (Corning, Corning, NY), and 0.5 million cells in 100 μl were injected subcutaneously in the hind flanks of mice. Tumor growth was monitored regularly, and 3–4 weeks after cell implantation, animals were euthanized and the tumors were excised and fixed in 4% paraformaldehyde. Tumors were sequentially dehydrated in increasing concentrations of ethanol, de-fatted in xylene, embedded in paraffin, and sectioned into 10-μm slices on a Microm HM 355 microtome. Sections were stained with Hematoxylin and Eosin on a Varistain Gemini ES automated slide stainer (Thermo Scientific) and imaged with an Olympus IXTVAD camera attached to an Olympus IX50 microscope.
RAP1 activation during reprogramming. Cells were reprogrammed as described earlier. Four different reprogramming cocktails were used: OSKM, SKM, SKM with forskolin (FSKM), and SKM with 8-pCPT-2′-O-Me-cAMP (CSKM). After passage, small molecules and media supplemented with 0.1% DMSO were replenished every other day. On days 3, 6, 9, 12, and 15 postinfection, cells were fixed using 4% paraformaldehyde and stained using primary antibodies against activated RAP1 (Neweast Biosciences, King of Prussia, PA, 26912) or NANOG (Abcam, ab70482), with secondary antibodies against mouse (Invitrogen, A-11003) and against rabbit IgG (Invitrogen, A-11012). Cells were imaged on a Nikon TE2000-E microscope and were analyzed with ImageJ software.
SUPPLEMENTARY MATERIAL Figure S1. STEMCCA loxP viral vectors. Figure S2. Signaling pathways can induce alkaline phosphatase colonies in the absence of KLF4. Figure S3. Signaling pathways can induce alkaline phosphatase colonies in the absence of SOX2. Figure S4. Signaling pathways can induce alkaline phosphatase colonies in the absence of OCT4. Figure S5. Negative controls have little OCT4 expression. Figure S6. The number of infected cells plated in each condition can be used in determining the efficiency of reprogramming. Figure S7. Serum-free conditions and PD 0325901 improve the reprogramming efficiency for Oct4 replacement. Figure S8. Measuring larger OCT4 colonies results in similar trends in reprogramming efficiency. Figure S9. NANOG staining reveals similar trends for forskolin conditions, but fewer colonies compared to OCT4 staining. Figure S10. SKM FK 2i-01 cell line expresses pluripotency markers, SSEA1, OCT4, and NANOG. Figure S11. The cell line, SKM FK 2i-02, was stained for pluripotency markers, SSEA1, OCT4, and NANOG. Figure S12. SKM FK 2i-06 maintains embryonic stem cell colony morphology 19 passages after isolation. Figure S13. SKM FK 2i-02 produces three germ layers in vitro. Figure S14. GGTI-298 is toxic to mouse embryonic fibroblasts at higher concentrations. Figure S15. The RAP1 inhibitor, GGTI-298, decreases the number of OCT4 positive colonies in forskolin reprogramming at increased concentrations. Figure S16. The cell line, CSKM-06, was stained for pluripotency markers, SSEA1, OCT4, and NANOG. Figure S17. The cell lines (a) CSKM-06 and (b) SKM FK 2i-06 were stained for pluripotency markers SSEA1, OCT4, and NANOG. Table S1. Signal transduction genes. Table S2. Alkaline phosphatase positive colonies are present in the absence of the KLF4, SOX2, or OCT4 transgene. Table S3. Calculated reprogramming efficiencies based on OCT4 expression. Table S4. Primers used for qPCR. Supplementary Methods.
Acknowledgments
STEMCCA loxP was a kind gift of Gustavo Mostoslavsky (Boston University). Lin He and Yong Jin Choi (UC Berkeley) kindly provided MEFs. The authors would like to thank Lin He, Timothy Downing, Yong Jin Choi, and Teppei Yamaguchi for their helpful discussions about reprogramming. The authors would also like to thank Paula Gedraitis (Molecular Devices) and Mary West (Stem Cell Center, University of California, Berkeley). This work was funded by National Science Foundation Graduate Fellowship (ALF) and NIH R01 ES020903. The authors also thank the CIRM/QB3 Berkeley Shared Human Embryonic Stem Cell Facility. The authors declare that they have no conflicts of interest. A.L.F., M.M.A., and D.V.S. designed the experiments; A.L.F. and M.M.A. performed the experiments; A.L.F., M.M.A., and S.R.M. analyzed the data; A.L.F. and M.M.A. prepared the figures; A.L.F. wrote the manuscript with revision with M.M.A. and editorial feedback and revision with D.V.S.
Supplementary Material
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Li TG, Qi X, Zhao DF, Zhao GQ. WNT/beta-catenin pathway up-regulates Stat3 and converges on LIF to prevent differentiation of mouse embryonic stem cells. Dev Biol. 2006;290:81–91. doi: 10.1016/j.ydbio.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Nishinakamura R, Iwamatsu Y, Shimosato D, Niwa H. Synergistic action of Wnt and LIF in maintaining pluripotency of mouse ES cells. Biochem Biophys Res Commun. 2006;343:159–166. doi: 10.1016/j.bbrc.2006.02.127. [DOI] [PubMed] [Google Scholar]
- Cole MF, Johnstone SE, Newman JJ, Kagey MH, Young RA. Tcf3 is an integral component of the core regulatory circuitry of embryonic stem cells. Genes Dev. 2008;22:746–755. doi: 10.1101/gad.1642408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Foreman R, Chevalier B, Bilodeau S, Kahn M, Young RA, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3:132–135. doi: 10.1016/j.stem.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Barrandon O, Nichols J, Kawaguchi J, Theunissen TW, Smith A. Promotion of reprogramming to ground state pluripotency by signal inhibition. PLoS Biol. 2008;6:e253. doi: 10.1371/journal.pbio.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida JK, Blanchard J, Lam K, Son EY, Chung JE, Egli D, et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell. 2009;5:491–503. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- Fritz AL, Mao SR, West MG, Schaffer DV. A medium-throughput analysis of signaling pathways involved in early stages of stem cell reprogramming. Biotechnol Bioeng. 2015;112:209–219. doi: 10.1002/bit.25336. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Sommer CA, Sommer AG, Longmire TA, Christodoulou C, Thomas DD, Gostissa M, et al. Excision of reprogramming transgenes improves the differentiation potential of iPS cells generated with a single excisable vector. Stem Cells. 2010;28:64–74. doi: 10.1002/stem.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Eggan K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest. 2010;120:51–59. doi: 10.1172/JCI40553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Hochedlinger K. Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr Biol. 2009;19:1718–1723. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, Foreman RK, Staerk J, Garcia M, Mathur D, Markoulaki S, et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc Natl Acad Sci USA. 2009;106:8912–8917. doi: 10.1073/pnas.0903860106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blelloch R, Venere M, Yen J, Ramalho-Santos M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell. 2007;1:245–247. doi: 10.1016/j.stem.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, York RD, Misra-Press A, Carr DW, Stork PJ. The cyclic adenosine monophosphate-dependent protein kinase (PKA) is required for the sustained activation of mitogen-activated kinases and gene expression by nerve growth factor. J Biol Chem. 1998;273:8240–8247. doi: 10.1074/jbc.273.14.8240. [DOI] [PubMed] [Google Scholar]
- Yao H, Labudda K, Rim C, Capodieci P, Loda M, Stork PJ. Cyclic adenosine monophosphate can convert epidermal growth factor into a differentiating factor in neuronal cells. J Biol Chem. 1995;270:20748–20753. doi: 10.1074/jbc.270.35.20748. [DOI] [PubMed] [Google Scholar]
- Kim SS, Choi JM, Kim JW, Ham DS, Ghil SH, Kim MK, et al. cAMP induces neuronal differentiation of mesenchymal stem cells via activation of extracellular signal-regulated kinase/MAPK. Neuroreport. 2005;16:1357–1361. doi: 10.1097/01.wnr.0000175243.12966.f5. [DOI] [PubMed] [Google Scholar]
- Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 2008;40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Ostrom RS. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol. 2003;23:305–314. doi: 10.1023/A:1023684503883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamizu K, Fujihara M, Tachibana M, Katayama S, Takahashi A, Hara E, et al. Protein kinase A determines timing of early differentiation through epigenetic regulation with G9a. Cell Stem Cell. 2012;10:759–770. doi: 10.1016/j.stem.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, et al. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghule PN, Medina R, Lengner CJ, Mandeville M, Qiao M, Dominski Z, et al. Reprogramming the pluripotent cell cycle: restoration of an abbreviated G1 phase in human induced pluripotent stem (iPS) cells. J Cell Physiol. 2011;226:1149–1156. doi: 10.1002/jcp.22440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Panopoulos AD, Herrerías A, Bissig KD, Lutz M, Berggren WT, et al. A high proliferation rate is required for cell reprogramming and maintenance of human embryonic stem cell identity. Curr Biol. 2011;21:45–52. doi: 10.1016/j.cub.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda T, Tada M, Kubota H, Kimura H, Hatano SY, Suemori H, et al. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol. 2005;25:2475–2485. doi: 10.1128/MCB.25.6.2475-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, et al. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci USA. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godmann M, Kosan C, Behr R. Krüppel-like factor 4 is widely expressed in the mouse male and female reproductive tract and responds as an immediate early gene to activation of the protein kinase A in TM4 Sertoli cells. Reproduction. 2010;139:771–782. doi: 10.1530/REP-09-0531. [DOI] [PubMed] [Google Scholar]
- Kumar SM, Liu S, Lu H, Zhang H, Zhang PJ, Gimotty PA, et al. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. 2012;31:4898–4911. doi: 10.1038/onc.2011.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhou H, Abujarour R, Zhu S, Young Joo J, Lin T, et al. Generation of human induced pluripotent stem cells in the absence of exogenous Sox2. Stem Cells. 2009;27:2992–3000. doi: 10.1002/stem.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Desponts C, Do JT, Hahm HS, Schöler HR, Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Chen J, Liu J, Yang J, Chen Y, Chen J, Ni S, et al. BMPs functionally replace Klf4 and support efficient reprogramming of mouse fibroblasts by Oct4 alone. Cell Res. 2010;21:205–212. doi: 10.1038/cr.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang Q, Yin X, Yang W, Du Y, Hou P, et al. Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res. 2011;21:196–204. doi: 10.1038/cr.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Adjaye J. A cyclic AMP analog, 8-Br-cAMP, enhances the induction of pluripotency in human fibroblast cells. Stem Cell Rev. 2011;7:331–341. doi: 10.1007/s12015-010-9209-3. [DOI] [PubMed] [Google Scholar]
- Li L, Wang S, Jezierski A, Moalim-Nour L, Mohib K, Parks RJ, et al. A unique interplay between Rap1 and E-cadherin in the endocytic pathway regulates self-renewal of human embryonic stem cells. Stem Cells. 2010;28:247–257. doi: 10.1002/stem.289. [DOI] [PubMed] [Google Scholar]
- González B, Denzel S, Mack B, Conrad M, Gires O. EpCAM is involved in maintenance of the murine embryonic stem cell phenotype. Stem Cells. 2009;27:1782–1791. doi: 10.1002/stem.97. [DOI] [PubMed] [Google Scholar]
- Ng VY, Ang SN, Chan JX, Choo AB. Characterization of epithelial cell adhesion molecule as a surface marker on undifferentiated human embryonic stem cells. Stem Cells. 2010;28:29–35. doi: 10.1002/stem.221. [DOI] [PubMed] [Google Scholar]
- Redmer T, Diecke S, Grigoryan T, Quiroga-Negreira A, Birchmeier W, Besser D. E-cadherin is crucial for embryonic stem cell pluripotency and can replace OCT4 during somatic cell reprogramming. EMBO Rep. 2011;12:720–726. doi: 10.1038/embor.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Schaffer DV. Selection of novel vesicular stomatitis virus glycoprotein variants from a peptide insertion library for enhanced purification of retroviral and lentiviral vectors. J Virol. 2006;80:3285–3292. doi: 10.1128/JVI.80.7.3285-3292.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008;2:90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier J, Schaffer DV. Viral packaging and transduction of adult hippocampal neural progenitors. Methods Mol Biol. 2010;621:103–116. doi: 10.1007/978-1-60761-063-2_7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.