

Abstract
Human genome analyses have revealed that increasing gene copy number alteration is a driving force of incurable prostate cancer (PC). Since most of the affected genes are hidden within large amplifications or deletions, there is a need for fast and faithful validation of drivers. However, classic genetic PC engineering in mouse makes this a daunting task because generation, breeding based combination of alterations and non-invasive monitoring of disease are too time consuming and costly. To address the unmet need, we recently developed RapidCaP mice, which endogenously recreate human PTEN-mutant metastatic PC based on Cre/Luciferase expressing viral infection, that is guided to PtenloxP/Trp53loxP prostate.
Here we use a sensitized, non-metastatic Pten/ Trp53-mutant RapidCaP system for functional validation of human metastasis drivers in a much accelerated time frame of only 3-4 months. We used in vivo RNAi to target three candidate tumor suppressor genes FOXP1, RYBP and SHQ1, which reside in a frequent deletion on chromosome 3p and show that Shq1 cooperates with Pten and p53 to suppress metastasis. Our results thus demonstrate that the RapidCaP system forms a much needed platform for in vivo screening and validation of genes that drive endogenous lethal PC.
1. Introduction
1.1 Metastatic prostate cancer
Cancer of the Prostate (CaP) is the second leading cause of cancer deaths of men in the United States [1]. The vast majority of CaP deaths are related to metastatic disease and more than 70% of deaths are due to complications resulting from late-stage tumors that have spread to distant sites. CaP typically first spreads to the tissues immediately adjacent to the prostate, including the seminal vesicles and nearby lymph nodes. In a majority of advanced prostate disease, metastasis is found particularly in bone, which is a well known and leading cause of mortality and morbidity. Bone metastasis is most commonly seen in the lower spine, the pelvis, and the upper legs, though CaP can spread to bones anywhere in the body. The second most common metastatic site is liver, followed by the lungs and adrenal glands. In order to form a metastatic lesion, initiating cells have to retain proliferation properties subsequent to their migration to a secondary site. A basic presumed sequence of metastasis is initiated by local invasion to seminal vesicles and the lymph nodes, intravasation, survival in circulation, extravasation and colonization of distant organs [2]. For a particular malignancy to progress to metastatic disease a complex series of biological properties must be acquired. In this vein, it has been observed that only a small portion of cells from a primary tumor are able to initiate secondary growth [2]. There is also evidence that tumor cell dissemination occurs early in disease progression for a variety of cancers, including CaP, but the vast majority of these cells can not establish metastases. It had indeed been reported that the genetic aberrations in metastatic cells from bone marrow samples did not resemble those in the primary tumor, and this was explained by tumor cells disseminating very early and evolving independently in metastasis and in the primary tumors [3, 4]. In contrast however, recent whole genome analysis of CaP evolution suggested a clonal origin of lethal metastasis [5] that may in some cases even be not derived from the major genetic population of cells that characterize the tumor [6]. In a landmark study however, single nucleus sequencing analysis of breast cancer evolution has suggested a tractable evolutionary relationship between primary and metastatic cells [7].
1.2 The need for fast and faithful in vivo validation of human candidate cancer genes
Genetically Engineered Mouse (GEM) models have been pivotal in answering fundamental questions of cancer development [8]. Classic GEM models remain a gold standard for validation and understanding of individual or synergistic contributions of gene alterations that were first identified in patients [9]. Because this germline based technology requires long animal generation and analysis times, a typical experiment takes 4-5 years, it is now falling far behind the ever increasing speed of human genome sequencing. Furthermore, the insights from high-throughput (HT) genomics studies pose new challenges for candidate cancer gene validation. In prostate, for example, landmark studies have revealed that cancer and metastases are characterized by the accumulation of broad structural chromosomal rearrangements and only rarely by gene mutations [ 10, 11-14]. While cancer associated missense mutations can immediately point to genes of interest whose biology can be studied on a case by case basis [11], gene deletions mostly encompass large regions of heterozygous loss, so that only few already known driver deletions, such as TP53 or RB1 are identifiable within broad deletions. As a consequence, the number of candidate cancer driver events that need to be tested is completely overwhelming the validation capacity of traditional germline GEM models [15].
We have developed RapidCaP, a new mouse model to study autochthonous (endogenous, “mouse-made”) metastatic prostate cancer [16]. Our approach makes use of existing GEMs with conditionally expressed tumor suppressor alleles. After viral delivery of Cre- and luciferase transgenes through intra-prostatic injection, disease progression can be monitored non-invasively with bioluminescence imaging. We have used the RapidCaP system to analyze and treat metastatic prostate cancer, which it rapidly generates at very high penetrance [16]. For instance, we found that primary and metastatic disease show variable response to castration, suggesting that additional spontaneous genetic changes beyond loss of Pten and Trp53 are important for the emergence of castration resistance. Beyond these important applications for cancer therapy, further advantages make the RapidCaP system very well suited to tackle the problem of cancer gene validation: (1) its high flexibility allows for straightforward testing of candidate genes by viral transgene transfer, (2) its speed allows for a typical experiment to be concluded within less than a hundred days.
We have previously shown how spontaneously amplified genes, such as Myc, can be validated as drivers of metastasis in RapidCaP using cDNA transgenic virus [16]. Here we present technology for identification and validation of tumor suppressors that lie in regions of human genomic deletions. We focus on candidate genes that cooperate with loss of PTEN, as the flexibility of RapidCaP allows for testing candidate genes in a specific genetic context. We achieved this combination by using RNA-interference techniques against genes in a candidate tumor suppressor locus and established the Pten-mutant genetic context using mice harboring conditional Pten alleles.
2. Approach
2.1. Analysis of the chr. 3p14 deletion in human prostate cancer
Comprehensive analysis of the human prostate cancer genome [13] revealed a frequent deletion on chromosome 3 (Figure 1A). This deletion is significantly associated with loss of PTEN and TP53 (p= 1E-3, and p=3.5E-6, respectively), a genetic setting that forms a hallmark of human prostate metastasis [17] and successfully recapitulates highly penetrant metastasis in the RapidCaP system [16]. This locus spans the 3p14.1-3p13 region and contains some 10 annotated protein coding genes and integration of RNA expression and deletion patterns highlighted 3 genes as most likely targets ([13], see also Figure 1A). These encode for the Forkhead family transcription factor FOXP1, RYBP, a polycomb family protein and the ribonucleoprotein (RNP) binding SHQ1 protein. Studying this data set, neither the individual deletion frequencies (FOXP1: 14.7%, RYBP: 15.6%, SHQ1: 14.7%) nor the expression differences between the three genes could point to one being a favorite target. Also, analysis of the smallest deleted regions did not point to a favorite (Figure 1B). Mutation analysis of these genes across all cancer genome studies (as curated at cbioportal.org [18, 19]) suggested no strong differences in mutation (frequencies of up to 5%, 2% and 3% respectively, for FOXP1, RYBP and SHQ1). Thus, it has remained unclear if an individual gene would be critical for tumor suppression in the PTEN/ TP53-loss context, or if suppression of all of the genes in the locus was required.
Figure 1. Analysis of a focal deletion in human prostate cancer.
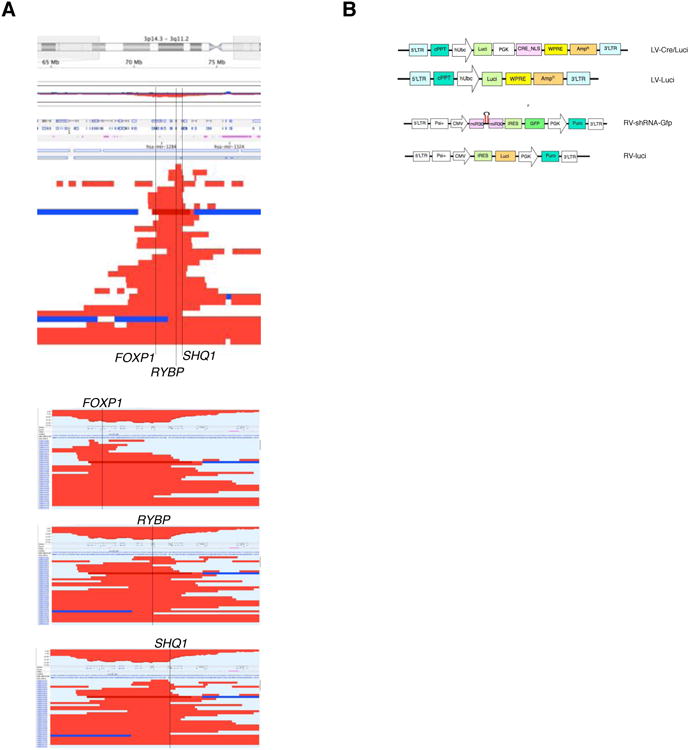
(A) Top panel: overview of focal deletions targeting the chromosome 3p14-3p13 region. Each line represents a patient sample published in Ref. [13]. Location of the three genes FOXP1, RYBP, SHQ1 is indicated. Bottom panels: sorting of deletions based on the three genes does not reveal a single candidate tumor suppressor based on focality.
(B) Maps of plasmids used for viral transduction of prostate in RapidCaP.
We therefore tested if suppression of any of the three targets alone or combined could accelerate tumor initiation and progression to metastasis in the RapidCaP system.
2.2. Generation of a sensitized RapidCaP screening platform
In the RapidCaP system we trigger disease by lentiviral Cre- and luciferase delivery (LV Cre-Luci, see Ref. [16] and Figure 1B) through intra-prostatic injection and monitor progression of disease, which expresses the luciferase transgene. This approach allows for non-invasive visualization using bioluminescence imaging and produces focal primary disease that retains intact histopathology of surrounding stroma and normal tissue [16]. In contrast, the tissue specific Probasin promoter, which deletes the Pten/ Trp53 genes in the entire prostatic epithelium, results in lethal primary disease burden within months [20-22]. The focal loss of Pten/ Trp53 in RapidCaP mice then progresses to metastatic disease within only 100 days, which is consistent with insights from human metastatic prostate cancer [17]. Thus, the system can provide a platform for validation of candidate cancer genes that are discovered through prostate cancer genomics [11-13]. To make this system amenable for screening of candidate genes, it is necessary to establish a wide screening window, in which negative control hairpins show no effects and can clearly be separated from the effects of positive control hairpins. It has previously been shown that progression of prostate cancer after loss of Pten is dramatically accelerated by complete loss of p53 [20], which forms the basis of the regular RapidCaP system. To limit the fast progression speed of this model and thus make it dependent on additional hits that are delivered by RNAi, we have developed a sensitized version of Pten-deficient the RapidCaP system that retains one copy of the Trp53 tumor suppressor. Injection of LV Cre-luci into PtenloxP/loxP; Trp53 +/ loxP mice results in a residual allele of Trp53 which suffices to prevent emergence of stable primary disease, and also blocks development of secondary metastases after more than 300 days (Figure 2A, LV Cre/Luci and LV Cre/ Luci RV…). Importantly, co-injection of LV Cre/Luci with a p53-targeting short hairpin RNAi virus (RV-shp53-Gfp) revealed efficient take of disease and expansion to distant sites well within 100 days in this sensitized model (Figure 2A, shp53), thus showing disease kinetics that are very similar to the non-sensitized, published Pten/ Trp53 double-deficient model [16]. These data therefore suggested that the sensitized RapidCaP mice may provide a sufficiently large screening window to attempt in vivo prostate tumor suppressor discovery and validation.
Figure 2. Targeting of candidate tumor suppressors in RapidCaP.
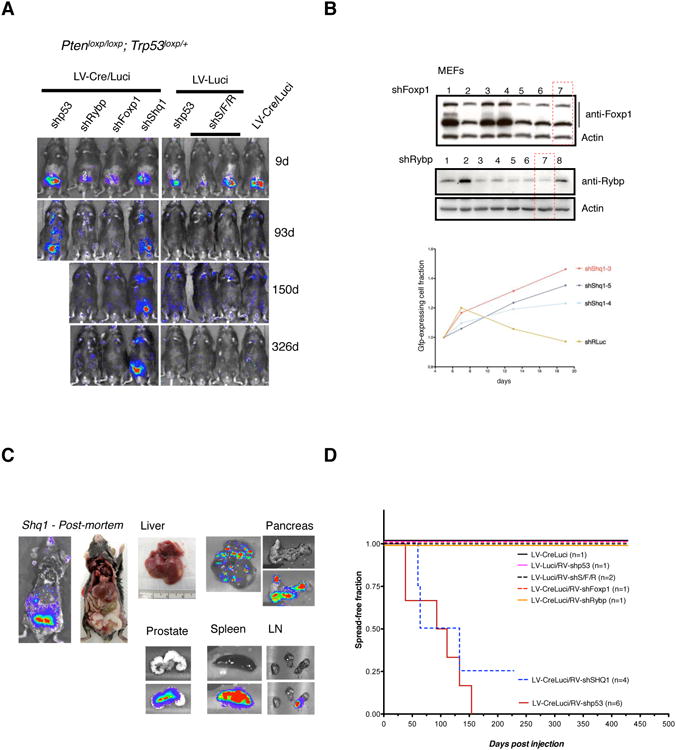
(A) Follow up of RapidCaP mice that were injected with positive control hairpin (shp53), the three candidate gene targeting hairpins alone, or combined (shS/F/R) together with either Cre-positive (LV-Cre/Luci) or -negative control virus (LV-luci). Note that the shp53 positive control animal needed to be sacrificed due to metastatic disease burden in the liver.
(B) Validation and selection of hairpins for best target knockdown as tested by Western blotting analysis (shFoxp1, shRybp) or strongest positive selection in competition with a control hairpin (sh anti-Renilla luciferase).
(C) Post-mortem analysis of a typical shp53 injected sensitized animal as shown in Figure 2A (LV-Cre/Luci + shSHq1) reveals spread of disease to liver, pancreas, spleen and lymph nodes.
(D) Kaplan-Meier analysis of disease onset, progression and outcome for various combinations of hairpin carrying retrovirus with Cre-recombinase and Luciferase marker lentivirus.
2.3. Targeted gene knockdown in vivo
To test the three genes in the candidate locus on chromosome 3p, we generated retrovirus carrying multiple candidate shRNAs against each of them and selected the most potent hairpins as outlined in the Methods section and shown in Figure 2B. The sensitized PtenloxP/loxP; Trp53 +/ loxP RapidCaP mice were then injected with a combination of LV-Cre-Luci to fully delete Pten and one copy of Trp53 together with RV-shRNA-Gfp virus as described above for shp53. The mice were followed up over time using bioluminescence imaging at 2-3 week intervals. As summarized in Figure 2A, suppression of Rybp or Foxp1 showed luciferase signal at 9 days pi, which faded to background levels within 100 days and showed no re-emerging disease even after close to one year post injection. In contrast, suppression of Shq1 showed sustained disease over the entire duration of the experiment. Moreover, these animals presented with disease at secondary sites within 100 days, which persisted throughout the study. This time of onset was similar to the kinetics of disease dissemination in shp53 injected animals, and also comparable to non-sensitized Pten/ Trp53 - homozygous deleted RapidCaP mice [16].
2.4. Isolation and analysis of disease
Post-mortem analysis of shShq1/ Cre/Luci injected RapidCaP animals using luciferase imaging (Figure 2C) confirmed metastasis to lymph nodes, spleen, pancreas and liver in 4 of 4 animals. Furthermore, various negative control injections shown in the Kaplan-Meier analysis (Figure 2C) produced neither primary nor secondary disease. The Kaplan-Meier analysis of the experiments also showed that metastasis induced by Shq1 knockdown had kinetics that were insignificantly different from Trp53 knockdown, the positive control. Collectively, these results showed the feasibility of using the sensitized RapidCaP system for in vivo RNAi-based screening of candidate cancer genes and suggested that SHQ1 can cooperate with the PTEN and TP53 tumor suppressors to block prostate cancer progression to metastasis. Note that based on our experiments it is not possible to exclude a role for the FOXP1 and RYBP genes in disease progression, even though the triple combined targeting was not able to establish stable disease in the absence of Pten/ p53-alteration (Figure 2A, shS/F/R + LV-Luci).
The RNAi-based approach introduced Gfp fluorescence into prostate via the RV-shRNA-Gfp. This allowed us to dissect and collect mutant cells by FACS analysis. We established sorting gates to separate Gfp-negative from -positive cells in prostate and liver tissue and collected cells from each of these gates from both tissues (Figure 3A). After processing of the cells for reverse transcriptase cDNA production we used quantitative PCR to measure the Shq1 mRNA expression levels in Gfp-positive relative to Gfp negative tissue. This analysis revealed greater than 80% knockdown of Shq1 transcription in prostate as well as in the liver metastasis (Figure 3B), compared to the Gfp-negative cells. Importantly, the Gfp-positive cells in liver also expressed the prostate luminal cell marker gene Nkx3.1 at similar levels as the Gfp-positive prostate cells. In contrast, Nkx3.1 expression was virtually absent from the Gfp-negative liver cells. These data were consistent with the prostatic epithelial origin of the Gfp-positive cells in liver. Furthermore, Trp53 genotyping PCR of liver and prostate confirmed the presence of recombined cells in these tissues, specifically in Gfp-positive cells (Figure 3C). Finally, histopathological analysis of the liver confirmed the presence of metastatic nodules (Figure 3D), which contained an admixture of tumor cells, lymphocytes, and entrapped hepatocytes. The tumor cells (see arrows and circles) had dense, amphophilic to eosinophilic cytoplasm and enlarged, hyperchromatic nuclei with irregular nuclear contours. Occasional multinucleated tumor giant cells were noted. Overall, the features of these tumor foci were similar to those identified in the previously published metastatic foci in the lungs [16].
Figure 3. Molecular and histological analysis of Shq1-mutant metastasis.
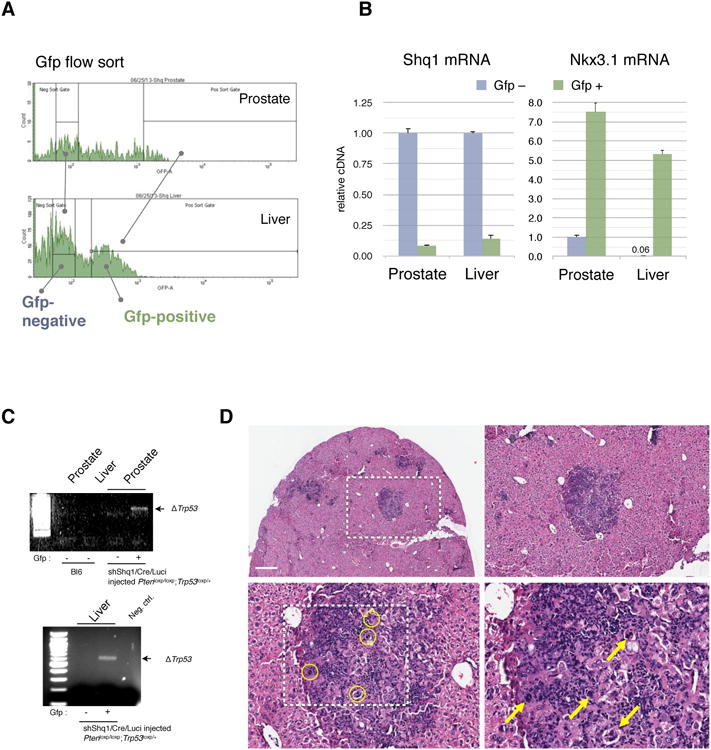
(A) FACS flow sort profile and gates used for separation of Gfp-negative from Gfp-positive cells that were isolated from the LV-shShq1-Gfp positive prostate and liver tissue shown in Figure 2C.
(B) RT-qPCR for Shq1 transcription in the isolated tissue demonstrates greater than 80% knockdown of Shq1 expression in both prostate and liver tissue (left graph). Right graph: Gfp-positive cells from liver show expression of the prostate epithelial marker Nkx3.1, at levels similar to the Gfp-positive cells in from prostate. Error bars show standard deviation of experimental triplicates.
(C) PCR analysis demonstrating recombination in Gfp-positive cells from prostate and liver from the same LV-shShq1-Gfp injected RapidCaP mouse (Figure 2A).
(D) Overview and high magnification H&E analysis of liver from the LV-shShq1-Gfp injected RapidCaP mouse depicted in Figure 2C reveals metastatic nodules that contain tumor cells (circles and arrows) at high magnification. Scale bar, 500μm.
3. Discussion
The above data demonstrate how the viral injection based RapidCaP system can be used to test genes of interest in their contribution to disease. Combination of the knockdown approach with incorporation of fluorescent protein expression for enrichment and identification of mutant cells by FACS analysis facilitates further molecular and genetic analysis of the isolated cells ex vivo. The individual targeting of genes from the common advanced prostate cancer deletion on human chr. 3p14.1-p13 reveals contribution to disease. As discussed in section 2.1, this locus is commonly co-deleted with PTEN and TP53 ([13]), and the RapidCaP results suggest that also in mouse, Shq1 suppression cooperates with deletion of these two tumor suppressors. Note, that while the Rybp and Foxp1 genes did not show cooperation with Pten/ Trp53-loss, their role in prostate cancer progression cannot be excluded at this stage as these negative results require further interrogation of in vivo knockdown efficiency, alone and in combination, also including alteration of Pten/ Trp53. In contrast, the positive result with Shq1 knockdown is demonstrating its potential in growth suppression that is autonomous from the other genes in the locus. SHQ1 functions in ribonucleoprotein (RNP) biogenesis, yet it remains to be seen how it contributes to tumor suppression. Interestingly, several human genomics studies have suggested that SHQ1, also termed GRIM-1 for Gene associated with Retinoid-Interferon-induced Mortality-1, is a tumor suppressor in prostate and other cancer types [13, 23-27].
Collectively, our results show that the RapidCaP system, which is based on Cre/ lox-mediated gene manipulation can be effectively combined with RNA-interference technology to test candidate tumor suppressor genes. We have chosen to recapitulate the Pten-deficient prostate background, as homozygous deletion is a hallmark of Pten-mutant prostate metastasis [11, 28, 29]. In extension, it will be interesting to test if PTEN modifying cancer genes can be identified by using a Pten heterozygous sensitized background, with or without Trp53 alteration. We have previously validated Myc as an oncogene in a RapidCaP system with cDNA over-expression in wt mice. Thus, by using the RapidCaP approach, copy number and expression changes identified in human can be rapidly validated in mouse to pinpoint driver events behind the lethal progression of human prostate cancer.
4. Methods
Human genome copy number analysis
Analysis of the chromosome 3p locus was based on the published and de-identified MSKCC prostate cancer data with 181 primary and 37 metastatic tumors [13] and visualized using the Nexus Copy Number software (v. 7 from BioDiscovery). All analyses for this publication were performed on de-identified patient data and material and thus qualified for exemption from human subjects statements.
Mice
Ptenloxp/loxp; Trp53loxp/+ were used in this study. For genotyping, the following primers were used as previously described [16]: Ptenloxp, primer 1 (5′-TGTTTTTGACCAATTAAAGTAGGCTGTG-3′) and primer 2 (5′-AAAAGTTCCCCTGCTGATGATTTGT-3′). For Trp53loxp, primer 1 (5′-CACAAAAACAGGTTAAACCCAG-3′) and primer 2 (5′-AGCACATAGGAGGCAGAGA C-3′) were used.
All protocols for mouse experiments were in accordance with institutional guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC).
Virus production
Retrovirus was produced by calcium phosphate transfection, 6×106 phoenix cells were plated in 10 cm plates 6 to 12 hours prior to transfection with 15 μg of target construct and 5 μg of ecotropic helper plasmid. Fresh media was added 12 hours after transfection and viral supernatant was collected four times at 24, 36, 48 and 60 hours post-transfection. Viral supernatant was filtered through a 0.45 μm filter, then supplemented with polybrene (4 μg/mL final concentration) for infection of target cells. MEFs for quality control (PC3, HeLa) were split 1:3 from confluent plates 12-24 hours prior to virus collection. The Guava flow cytometer (Millipore, Billerica, MA) was used to determine the infection rate and expression level of RV-shRNA-Gfp virus. Lentivirus was produced by calcium phosphate transfection. 293T cells were plated for transfection at a density of 8×106 cells per 10 cm plate. 10 μg of target plasmid was combined with helper constructs, 8.5 μg of pMD2.G and 3.5 μg of psPAX2, for transfection. Lentiviruses were harvested at 24, 36, 48 and 60 h post-transfection and centrifuged (4500 rpm, 15 min) prior to filtering through 0.45-μm-pore cellulose acetate filters. Viral supernatant was concentrated by ultracentrifugation (2 h at 20,000 × g). For viral injection to prostate, filtered virus was concentrated by ultracentrifugation (2 hours at 50,000 × g), and then an in vitro infection test for each batch of virus was conducted in advance.
RNAi-design and selection
shRNAs against murine Foxp1 (7), Rybp (8) and SHQ1 (5) was designed as recently published [30] and the resulting total of 20 candidates were cloned into the mir30 based microRNA fold using the retroviral backbone plasmid shown in Figure 1B (RV-shRNA-Gfp). Selection of best performing hairpins against Foxp1 and Rybp was done using protein analysis by Western blotting and using stable growth promotion for shShq1 selection due to lack of suitable antibody and growth promoting effects of most designs that were observed as shown in Figure 1B.
Intra-prostate injection
Intra-prostatic injection of virus was carried out as previously described [16]: after exposure to 2% Isoflurane anesthesia, the lower half of the abdomen was shaved and mice were placed in a surgery hood and constantly exposed to Isoflurane via a nose cone. The shaved region was repeatedly cleaned with beta dine and sterile PBS and a 1.5 cm long incision through skin and peritoneum was made along the lower abdominal midline, 5 to 8 mm above the external genitalia. The right seminal vesicle was pulled through the incision, placed on sterile gauze to position the anterior prostate for injection and 30 μl of concentrated virus/ (1:1) admixture was injected into prostate, carefully observing its inflation and the organ was then returned and repositioned below the incision, which was then sutured with a size 4-0 suture. The skin was then stapled shut using 2 to 3 stainless steel EZ Clip wound closures. After the procedure animals were observed for complete recovery from anesthesia and warmed under a heating lamp to regain the ability to maintain sternal recumbence and were given DietGel. The mice were then returned to their respective cages and to the biohazard mouse room.
Bioluminescence Imaging
For in vivo imaging, animals received luciferin at 200 mg/kg by intra-peritoneal injection 5 min prior to imaging. The animals were then anesthetized using 2% isoflurane and were placed onto the warmed stage inside the camera box under continuous exposure to 2% isoflurane to sustain sedation for 3 min of imaging. For quantification, the total signals were measured for each mouse and calls for presence or absence of disease dissemination were based on observations in consecutive images using using a range of 3∼6×104 photons/ sec as background reference.
For ex vivo imaging of tissue, tissues of interest were excised after sacrificing a live imaged animal, placed individually on paraffin film and imaged for 3 min after 3 mg of D-luciferin (200 ul of 15 mg/ml in PBS) were dropped on each organ. Tissues were subsequently either processed, frozen or fixed in 10% neutral-buffered formalin for molecular analysis or standard histopathology evaluation.
Flow cytometry
For flow cytometry analysis, isolated mouse tissues were minced with a sterile scalpel and washed 3 times with Hanks' Balanced Salt Solution (HBSS). Then collagenase was added at 100 units/ml and tissues were incubated at 37 °C for 4 hours. The cell suspension was filtered through a sterile mesh to separate the dispersed cells and tissue fragments from any larger undigested pieces. The cell suspension was then washed 3 times with HBSS and prepared for flow cytometry on a BD FACS-ARIA III cell sorter, where liver and prostate cells from an uninjected control animal were used to set up the Gfp-negative gates (not shown). Using the sorting gates shown in Figure 3A, approximately 100,000 and 400,000 cells were collected for DNA and RNA analyses of prostate and liver, respectively.
RNA Extraction, Reverse Transcription, and Quantitative Polymerase Chain Reaction (RT- qPCR)
Total RNA was extracted from cell lines and tissue samples using the RNeasy kit (Qiagen) or the TRIzol reagent (Life Technologies) following the manufacturer's instructions and 2 μg of RNA were used for first strand synthesis and production of cDNA using random primer and SuperScript II (Invitrogen) or the QuantiTect RT Kit (Qiagen). RNA expression was measured by real-time quantitative reverse transcription- PCR, using the Roche LightCycler 480 (Roche Applied Science) based on the SYBR Green method. Each assay was done in triplicate and the expression level of each gene was calculated relative to expression of β-actin. Quantification was based on a standard curve obtained by serial dilution of the indicated control RT reaction. Primer pair sequences 5′-AGTTCGACGTGTACTTCGAGG-3′ and 5′-TCCAGGCAGAGTTAATCTCAGA-3′ were used for Shq1 amplification and the catalog numbers for the QuantiTect primer assays used are: β-actin: QT00095242; Nkx3.1: QT00102109.
Acknowledgments
We would like to thank the members of the Trotman lab for valuable discussion and help with experimental protocols and reagents, the CSHL Animal Resources team, Lisa Bianco, Lotus Altholtz, Jodi Coblentz, Michael Cahn for help with RapidCaP animal maintenance and trials management, Scott Lowe, Nilgun Tasdemir and Luke Dowe for help with reagents, Aigoul Nourjanova, Raisa Puzis, Denise Hoppe for help with histopathology procedures, and D. Tsang for help with the manuscript.
L.C.T. is a Research Scholar of the American Cancer Society (RSG-14-069-01-TBE) and we are grateful for the support of this work through generous grants from the NIH (CA137050), the Pershing Square Sohn Foundation, the Department of Defense (W81XWH-13-PCRP-IDA), the Robertson Research Fund of Cold Spring Harbor Laboratory and through the NIH Cancer Center Support Grant 5P30CA045508. C.S. is the recipient of a Ruth L. Kirschstein T32 Cancer Research Training Grant from the NIH (5T32CA148056).
Footnotes
Conflict of Interest Statement: The authors declare that they have no competing financial interests.
References
- 1.AC Society. 2014 [Google Scholar]
- 2.Nguyen DX, Bos PD, Massague J. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P, Bischoff J, Harich D, Schlimok G, Riethmuller G, Eils R, Klein CA. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7737–7742. doi: 10.1073/pnas.1331931100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eyles J, Puaux AL, Wang X, Toh B, Prakash C, Hong M, Tan TG, Zheng L, Ong LC, Jin Y, Kato M, Prevost-Blondel A, Chow P, Yang H, Abastado JP. J Clin Invest. 2010;120:2030–2039. doi: 10.1172/JCI42002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, Chen L, Ewing CM, Eisenberger MA, Carducci MA, Nelson WG, Yegnasubramanian S, Luo J, Wang Y, Xu J, Isaacs WB, Visakorpi T, Bova GS. Nat Med. 2009;15:559–565. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, Adejola N, Gurel M, Hicks J, Meeker AK, Halushka MK, Simons JW, Isaacs WB, De Marzo AM, Nelson WG, Yegnasubramanian S. J Clin Invest. 2013;123:4918–4922. doi: 10.1172/JCI70354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nardella C, Carracedo A, Salmena L, Pandolfi PP. Curr Top Microbiol Immunol. 2010;347:135–168. doi: 10.1007/82_2010_62. [DOI] [PubMed] [Google Scholar]
- 9.Frese KK, Tuveson DA. Nat Rev Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- 10.Baca SC, Garraway LA. Front Endocrinol (Lausanne) 2012;3:69. doi: 10.3389/fendo.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, Auclair D, Onofrio RC, Park K, Kitabayashi N, MacDonald TY, Sheikh K, Vuong T, Guiducci C, Cibulskis K, Sivachenko A, Carter SL, Saksena G, Voet D, Hussain WM, Ramos AH, Winckler W, Redman MC, Ardlie K, Tewari AK, Mosquera JM, Rupp N, Wild PJ, Moch H, Morrissey C, Nelson PS, Kantoff PW, Gabriel SB, Golub TR, Meyerson M, Lander ES, Getz G, Rubin MA, Garraway LA. Nature genetics. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, Habegger L, Ambrogio L, Fennell T, Parkin M, Saksena G, Voet D, Ramos AH, Pugh TJ, Wilkinson J, Fisher S, Winckler W, Mahan S, Ardlie K, Baldwin J, Simons JW, Kitabayashi N, MacDonald TY, Kantoff PW, Chin L, Gabriel SB, Gerstein MB, Golub TR, Meyerson M, Tewari A, Lander ES, Getz G, Rubin MA, Garraway LA. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heyer J, Kwong LN, Lowe SW, Chin L. Nature reviews Cancer. 2010;10:470–480. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho H, Herzka T, Zheng W, Qi J, Wilkinson JE, Bradner JE, Robinson BD, Castillo-Martin M, Cordon-Cardo C, Trotman LC. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-13-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O'Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, Murn J, Navin N, Atwal GS, Sander C, Gerald WL, Cordon-Cardo C, Newton AC, Carver BS, Trotman LC. Cancer Cell. 2011;20:173–186. doi: 10.1016/j.ccr.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Science signaling. 2013;6:l1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J, Perry SR, Labrot ES, Wu X, Lis R, Hoshida Y, Hiller D, Hu B, Jiang S, Zheng H, Stegh AH, Scott KL, Signoretti S, Bardeesy N, Wang YA, Hill DE, Golub TR, Stampfer MJ, Wong WH, Loda M, Mucci L, Chin L, DePinho RA. Nature. 2011;470:269–273. doi: 10.1038/nature09677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, Simons BW, Ward JM, Robinson BD, Chu GC, Loda M, Thomas G, Borowsky A, Cardiff RD. Cancer research. 2013;73:2718–2736. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nallar SC, Lin L, Srivastava V, Gade P, Hofmann ER, Ahmed H, Reddy SP, Kalvakolanu DV. PLoS One. 2011;6:e24082. doi: 10.1371/journal.pone.0024082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krohn A, Seidel A, Burkhardt L, Bachmann F, Mader M, Grupp K, Eichenauer T, Becker A, Adam M, Graefen M, Huland H, Kurtz S, Steurer S, Tsourlakis MC, Minner S, Michl U, Schlomm T, Sauter G, Simon R, Sirma H. J Pathol. 2013;231:130–141. doi: 10.1002/path.4223. [DOI] [PubMed] [Google Scholar]
- 25.Lando M, Wilting SM, Snipstad K, Clancy T, Bierkens M, Aarnes EK, Holden M, Stokke T, Sundfor K, Holm R, Kristensen GB, Steenbergen RD, Lyng H. J Pathol. 2013;230:59–69. doi: 10.1002/path.4168. [DOI] [PubMed] [Google Scholar]
- 26.Nallar SC, Kalvakolanu DV. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2013;33:189–198. doi: 10.1089/jir.2012.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu HM, Jiang ZF, Fan XY, Wang T, Ke X, Yan XB, Ma Y, Xiao WH, Liu RY. Hum Pathol. 2014 doi: 10.1016/j.humpath.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Cancer Cell. 18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. Nature. 487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, Park Y, Dickins RA, Hannon GJ, Lowe SW. Nat Protoc. 2012;7:374–393. doi: 10.1038/nprot.2011.446. [DOI] [PMC free article] [PubMed] [Google Scholar]