

Abstract
Bcl-2, the founding member of a family of apoptotic regulators, was initially identified as the protein product of a gene that is translocated and overexpressed in greater than 85% of follicular lymphomas (FLs). Thirty years later we now understand that Bcl-2 modulates the intrinsic apoptotic pathway by binding and neutralizing the mitochondrial permeabilizers Bax and Bak as well as a variety of pro-apoptotic proteins, including the cellular stress sensors Bim, Bid, Puma, Bad, Bmf and, under some conditions, Noxa. Despite extensive investigation of all of these proteins, important questions remain. For example, how Bax and Bak breach the outer mitochondrial membrane remains poorly understood. Likewise, how the functions of anti-apoptotic Bcl-2 family members such as eponymous Bcl-2 are affected by phosphorylation or cancer-associated mutations has been incompletely defined. Finally, whether Bcl-2 family members can be successfully targeted for therapeutic advantage is only now being investigated in the clinic. Here we review recent advances in understanding Bcl-2 family biology and biochemistry that begin to address these questions.
Keywords: Apoptosis, BH3 mimetic, activation-induced cytidine deaminase, mutation, follicular lymphoma
Introduction
Apoptosis is a distinct form of cell death initiated by various physiological and pathological stimuli. Morphologically, apoptosis is characterized by cell shrinkage followed by formation of cell fragments, which are rapidly cleared by phagocytes that recognize “eat me” signals on the plasma membrane [1–3]. Biochemically, apoptosis typically involves the conversion of various signals into caspase-mediated intracellular protease activity [1, 2, 4].
One of the two major pathways leading to apoptosis is the mitochondrial or intrinsic biochemical pathway [5, 6]. The signature biochemical change during activation of this pathway (Fig. 1) is the leakage of cytochrome c into the cytoplasm [7], where it facilitates caspase 9 activation to initiate a caspase cascade [1, 3, 8]. This release of cytochrome c from mitochondria is regulated by members of the Bcl-2 protein family, which include three functionally and structurally distinct subfamilies: i) Pro-apoptotic effector proteins Bax and Bak, which mediate mitochondrial outer membrane permeabilization (MOMP); ii) anti-apoptotic family members, including Bcl-2, Bcl-xL, Mcl-1, Bcl-w and A1, which antagonize MOMP; and iii) pro-apoptotic BH3-only proteins, which promote apoptosis either directly by binding and oligomerizing Bax and Bak or indirectly by neutralizing anti-apoptotic family members [9–12]. When the balance between these Bcl-2 family members tips in favor of cell death, Bax and Bak form oligomers that permeabilize the mitochondrial outer membrane (MOM), resulting in release of cytochrome c and many other mitochondrial intermembrane space proteins [13].
Figure 1. Overview of the mitochondrial pathway.

[1, 2, 4]. The signature event of the mitochondrial or intrinsic death pathway is MOMP, which results in release of cytochrome c and other mitochondrial intermembrane proteins to the cytoplasm [13]. Once in the cytoplasm, cytochrome c facilitates the interaction of the cytoplasmic scaffolding protein Apaf-1 and procaspase 9, activates a caspase cascade leading to cleavage of hundreds to cellular caspase substrates to generate the apoptotic phenotype [3, 4].
As described in the text, Bcl-2 family members regulate this pathway upstream of MOMP. While Bax and Bak are directly responsible for breaching the outer membrane, their oligomerization and activation are modulated by other family members. BH3-only proteins such as Puma, Bim, tBid, and Noxa, which are termed “activators,” transiently bind Bax or Bak [39], facilitating their activation. BH3-only proteins such as Bad, which are unable to directly bind Bax or Bak, are termed “sensitizers” because they bind and neutralize anti-apoptotic Bcl-2 family members.
Apoptosis plays an essential role in development, immune response and tissue homeostasis [14, 15]. Moreover, dysregulation of apoptosis is thought to play a critical role in degenerative diseases and cancer [16]. For example, genomic changes leading to overexpression of the anti-apoptotic proteins Bcl-2, Bcl-xL and Mcl-1 are observed in a variety of neoplasms [17, 18]; and genes encoding pro-apoptotic proteins such as Bax and Bim are mutated or deleted in selected cancers [19, 20]. Accordingly, dysregulation of apoptosis and particularly the intrinsic apoptotic pathway is considered a hallmark of cancer [21].
Although the intrinsic pathway of apoptosis was broadly outlined 20 years ago [1, 3], extensive progress over the past 3–4 years has provided new insight into its critical molecular processes. Structural studies of Bax and Bak have begun to illuminate their oligomerization and function. Moreover, new understanding of anti-apoptotic Bcl-2 family member regulation has emerged. Here we summarize these recent developments, focusing particularly on Bax/Bak activation and Bcl-2 regulation. For more comprehensive reviews, readers are referred to [9–12].
1. Advances in understanding Bax/Bak activation
1.1 Bax/Bak oligomerization: Some answers, more questions
Despite the strong correlation between Bax/Bak oligomerization and MOMP [22, 23], mechanisms of Bax/Bak activation and MOMP remain incompletely understood. Earlier studies showed that homo-oligomerization of Bak and Bax requires their BH3 domains [24]. More detailed mutagenesis and crosslinking assays implicated two interaction interfaces in Bak oligomerization. One is a BH3 domain:BH3 binding groove interaction in which the BH3 domain (α2 helix) of one Bak molecule interacts with the BH3 binding groove (α3-α5 helices) of another [25]. The second interface implicated in oligomerization involves Bak α6 helices [26, 27]. Several models have been advanced to account for Bak oligomerization [26, 27].
First, Bak monomers could form head-to-tail oligomers. Along these lines, an octamer “asymmetric-single-conformer” model of oligomerization has been derived from molecular dynamics simulations [28]. Alternatively, it has been proposed that Bak monomers could form symmetric dimers, with the BH3 domain of one monomer inserted into the BH3 binding groove of its partner and the opposite face of each molecule in the dimer involved in protein-protein interactions leading to higher order oligomerization [10, 26]. A similar model has been proposed for Bax [29]. One potential problem with models based on symmetric dimers is that C-terminal transmembrane (TM) domains of the two Bax or Bak monomers in each homodimer would face in opposite directions, i.e., with one transmembrane domain pointed into the membrane and the other oriented away from it. Nonetheless, recent crystal structures of Bax or Bak fragments fused to EGFP (to facilitate crystallization) provide strong evidence for symmetric dimers [30, 31]. Understanding in greater detail how assembly of these dimers contributes to formation of active Bax or Bak oligomers and MOMP is the next major challenge in this area.
1.2 BH3-only proteins and Bax/Bak activation: Direct activation, indirect activation or both?
There is general agreement that formation of Bax or Bak oligomers requires BH3-only proteins [32]. The exact role of the BH3-only proteins in Bax/Bak oligomerization, however, has been contentious, with three different models proposed.
According to the direct activation model, BH3-only proteins are divided into direct activators and sensitizers [33–35]. Direct activators include Bim, Puma and tBid, which can directly bind to Bax and/or Bak to induce their oligomerization [36–39]. Accordingly, Bid−/−Bim−/−Puma−/− triple knockout mice show some of the same developmental defects as Bax−/−/Bak−/− double knockout mice [40], although other defects appear to be absent [41]. Moreover, Bim−/−Puma−/− double knockout cells [42] and Bid−/−Bim−/−Puma−/− triple knockout cells display extensive, albeit incomplete, resistance to most apoptotic stimuli, suggesting that these proteins play a predominant role in Bax/Bak activation [40]. Although Bid and Bim can both activate Bax and Bak, it has been reported that Bim preferentially activates Bax, while Bid preferentially activates Bak [43].
In this model, the BH3-only protein Bad is a sensitizer, i.e., a protein that neutralizes anti-apoptotic Bcl-2 family members to release activators, which subsequently induce apoptosis [34]. Consensus regarding the role other BH3-only proteins has been more difficult to achieve. For example, while some studies have demonstrated direct activation of Bak by Noxa protein [39, 44], others have reported that Noxa BH3 peptide cannot directly activate Bax or Bak [30, 31, 45]. These different results might reflect, in part, use of full-length protein in some studies versus BH3 domain peptide in others.
Other aspects of BH3-only protein interactions with Bax or Bak also remain unresolved. While transient binding of BH3-only proteins to the canonical BH3 binding grooves of Bax or Bak has been implicated in triggering Bax/Bak activation in some studies [30, 31, 39, 46], a secondary site for triggering Bax activation has been identified in others [37, 47, 48]. Because the peptide derivative used in the latter studies is much more effective than native Bim BH3 peptide in binding and activating Bax [49], detection of the secondary site might reflect unique properties of the ligand.
According to the alternative indirect activation model, the major role of BH3-only proteins is to neutralize anti-apoptotic Bcl-2 family members, causing them to release Bax and Bak, which are then immediately competent to permeabilize the MOM [50, 51]. An earlier argument for this model was the lack of direct evidence for complexes between BH3-only proteins and Bax or Bak in intact cells [50]. However, interactions between BH3-only family members and Bak are now known to be transient [39, 45]; and structural studies have also confirmed interactions between BH3-only proteins and Bax or Bak [30, 31]. While the indirect activation model has fallen out of favor, the possibility that Bax and Bak are present in a preactivated state in cells one of the postulates of this model has not been fully explored and might have important implications for Bax and Bak function.
A more recent, unified model combines elements of both prior models by suggesting that anti-apoptotic Bcl-2 family members inhibit MOMP both by sequestering direct activators and by binding activated Bax and Bak [52]. Consistent with this model, a number of biochemical [38, 53–55], cellular [56–58], and genetic studies [59] now agree that anti-apoptotic Bcl-2 family members have dual functions in inhibiting apoptosis.
1.3 Once activated, how do Bax and Bak permeabilize the MOM?
Once BH3-only proteins trigger oligomerization of Bax and/or Bak, it is still unclear how MOMP occurs. Structural studies of Bcl-xL [60], Bax [61] and Bak [62] have revealed similarity between Bcl-2 family proteins and channel-forming domains of bacterial toxins such as diphtheria toxin [63] and colicin E1 [64], raising the possibility that all of these proteins may utilize similar mechanisms to permeabilize membranes. By analogy to the toxins, which are thought to insert their two hydrophobic core domains as hairpins into membranes to form pore like structures [65], a “hairpin insertion model” has also been proposed for Bax [66]. In particular, Bax α5 and α6, together with α9, are thought to insert into the MOM, while the other α helices from different Bax monomers interact to promote oligomerization at the MOM cytoplasmic surface [66–69].
On the other hand, recent crystal structures of activated Bax and Bak suggest that conformational changes during activation involve separation of α5 and α6 rather than hairpin formation [30, 31]. These observed changes, which are thought to allow Bax and Bak to assume more flexible conformations, may resolve the issue of conflicting TM domain orientation in the dimers described above. However, the observed crystal structures are not consistent with models in which Bax or Bak insert hydrophobic hairpins into the MOM to form pores. Instead, based on further results showing that some of the residues within the putative α5/α6 hairpin label with membrane-impermeable reagents and others do not, it has been suggested that α5 and α6 collapse onto the MOM rather than inserting into it [70]. Whether this leads to MOMP through disruption of membrane curvature [71, 72] or another process still remains to be determined.
1.4 Harnessing BH3-only protein biology to predict chemotherapeutic response
Understanding the mitochondrial pathway has direct implications for cancer therapy. Over the past 20 years, this pathway has been implicated in the cytotoxicity of many chemotherapeutics [73, 74], including DNA damaging agents [75], spindle poisons [76, 77], and kinase inhibitors [78, 79]. Conversely, chemotherapy resistant cancers often contain mitochondrial pathway defects [80, 81].
As described above, BH3-only proteins have been grouped into “direct activators” and “sensitizers” [35, 82]. “Priming of cells for death” was originally defined as a state in which addition of BH3 domains from sensitizer BH3-only proteins to isolated mitochondria (an assay termed “BH3 profiling”) results in robust cytochrome c release [83]. This state was correlated with the presence of Bim bound to anti-apoptotic Bcl-2 family proteins such as Bcl-2 or Mcl-1 as well as simultaneous expression of Bax and/or Bak on the MOM [83]. Moreover, sensitivity to the Bcl-2/Bcl-xL antagonist ABT-737 correlated strongly with release of cytochrome c by certain BH3 domain peptides, notably Bad [83–85].
In further studies, BH3 profiling predicted cancer cell sensitivity to not only BH3 mimetics, but also chemotherapeutic agents, particularly the topoisomerase II inhibitors etoposide, daunorubicin and mitoxantrone [86]. In contrast, this assay was less predictive of sensitivity to the nucleoside analogs cytarabine and clofarabine or the hypomethylating agents azacitidine and decitabine [85–87].
The original cytochrome c release assay employed for BH3 profiling has been replaced by a more efficient cell-based JC-1 dye binding assay [88] that measures loss of mitochondrial membrane potential [85, 89, 90]. This JC-1 assay can be performed by flow cytometry, which enables examination of responses in phenotypically distinct cell types such as leukemic myeloblasts or hematopoietic stem cells (HSC) [86]. Interestingly, this assay demonstrated that leukemic myeloblasts are more highly primed and exhibit a pattern more suggestive of Bcl-2 dependence than HSCs [86, 91], leading to a phase 2 trial of the Bcl-2 antagonist ABT-199. For reasons that are still under investigation, the assay predicted far better clinical activity than was actually observed [92], raising the possibility that it might be better to assay the effects of the BH3 mimetic drugs themselves rather than effects of naked synthetic peptides. Before describing that trial, we first review recent advances in understanding and targeting antiapoptotic Bcl-2 family members.
2. Advances in understanding regulation of anti-apoptotic Bcl-2 family proteins
2.1. Structure and function: Variations on a theme
Since the discovery of Bcl-2 [17, 93, 94] and its ability to promote tumorigenesis by inhibiting cell death [95], five structurally related anti-apoptotic family members, including Bcl- xL, Mcl-1, Bcl-w, BFL1 (A1 in mouse) and Bcl-B (Bcl2L10 in mouse), have been identified [10, 11, 96]. These share four conserved regions [Bcl-2 homology (BH) domains] (Fig. 2). In addition, Bcl-2, Bcl-xL, Mcl-1 Bcl-w and Bcl-B all contain C-terminal TM domains that direct them to intracellular membranes, particularly the MOM. BFL1 lacks a classical TM but is targeted to mitochondria by its C-terminal α-helix [97–99], whereas the mouse homolog Bcl-2A1 appears in some studies to be cytoplasmic. All of these proteins possess a remarkably similar globular structure containing a so-called “Bcl-2 core” [11] consisting of eight α-helices oriented so that helices 3, 4 and 5 form a hydrophobic groove that is capable of binding the BH3 domains of pro-apoptotic family members.
Figure 2. Bcl-2 phosphorylation. (A).
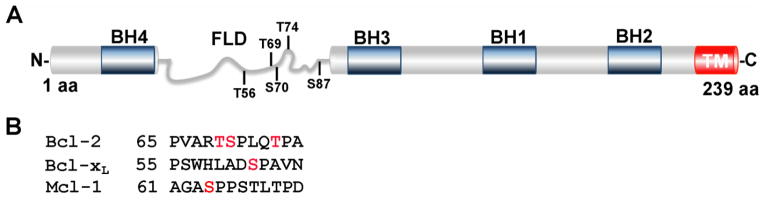
Schematic representation of the Bcl-2 protein with BH domains boxed in blue and flexible loop domain (FLD) labeled in gray. Residues T56, T69, S70, T74 and S87 are located within the FLD and selectively targeted for phosphorylation. S70 (red) is phosphorylated by CDK1 in vivo, inducing a conformation change of the FLD and enhancing the anti-apoptotic function of Bcl-2 [54]. (B). Residues S62 and S64 (labeled in red) within the FLD of Bcl-xL and Mcl-1, respectively, are targeted for phosphorylation by CDK1 [137, 154]. TM, transmembrane domain. (Adapted from [54, 154]).
Despite their similarity, anti-apoptotic Bcl-2 family members exhibit specificity for various pro-apoptotic proteins [51, 100]. Most of the anti-apoptotic proteins bind most of the BH3-only family members, but Bcl-xL and Bcl-w reportedly fail to bind Noxa; Bcl-2 does not bind truncated Bid, Bik, Hrk or Noxa well; Mcl-1 does not interact with Bad, Bik and Hrk; and BFL1/A1 fails to bind Bad and Bmf. Bcl-B, on the other hand, only binds Bim and Bik. Likewise, all of the anti-apoptotic family members bind Bax, but only Bcl-xL, Mcl-1, BFL1 [101, 102] and in some cases Bcl-2 [103] bind Bak.
The selectivity of these interactions, coupled with differences in expression of pro- and anti-apoptotic proteins under divers conditions, are thought to underlie the variable responses of cells to assorted stresses [36, 83, 101]. According to this view, Bcl-2, Bcl-xL and Bcl-w bind to almost all pro-apoptotic proteins and are more potent protectors than Bcl-B, BFL1 and Mcl-1, which bind only a subset of the pro-apoptotic proteins. On the other hand, several observations also argue against this model. First, it has been observed that Mcl1 gene deletion is lethal in a variety of cell lineages [104–109], although this might reflect an obligate role for Mcl-1 in oxidative phosphorylation within mitochondria rather than its anti-apoptotic role on the surface mitochondria [106]. Moreover, recent results suggest that the relative potencies of these proteins might reflect their relative expression levels rather than the range of pro-apoptotic proteins neutralized [110].
2.2. The role of anti-apoptotic Bcl-2 family proteins in tumorigenesis
Apoptosis not only contributes to normal development and tissue homeostasis, but also provides a barrier to cancer development. For example, c-Myc provokes changes that induce not only proliferation, but also apoptosis, limiting its ability to transform cells [81, 111]. As a result, Myc-induced transformation of lymphoid cells in vitro and in vivo is augmented by overexpression of Bcl-2 [95, 112] or Bcl-xL [113]. Similar results in other neoplasms [114] have led to extensive efforts to elucidate apoptotic pathway dysfunction in various cancers.
High Bcl-2 protein levels are detected in a variety of neoplasms, including small cell lung, breast, prostate, colorectal, and bladder cancers, melanoma, and especially human lymphoid malignancies [115–117]. Elevated Bcl-2 expression has also been reported in acute myeloid leukemia (AML), particularly chemotherapy-resistant AML [118, 119], although this has not been universally observed [120].
As summarized in Table 1, many mechanisms contribute to the high Bcl-2 levels observed in various neoplasms. First, the t(14,18) chromosome translocation that places the BCL2 gene next to immunoglobulin heavy chain (IGH) enhancer elements [17, 93, 94] is a major mechanism for elevated Bcl-2 transcription in lymphomas. Second, chromosome deletions and mutations that result in loss of miR-15a and miR-16, which target and repress Bcl-2 mRNA, occur in more than 50% of chronic lymphocytic leukemia (CLL) cases [121]. Third, Bcl-2 levels are regulated by protein ubiquitination [122–124]. The inhibitor of NF-E2-related factor 2 (INrf2) interacts with the BH2 domain of Bcl-2 and directs it to Cul3-Rbx1-mediated ubiquitination. Accordingly, inactivating mutations of INrf2 found in some lung cancer cell lines [124] can stabilize Bcl-2 and confer resistance to apoptotic stimuli. Similar mechanisms, i.e., genomic changes that enhance expression, altered regulation by miRNAs, and diminished ubiquitin-mediated turnover, contribute to upregulation of the other anti-apoptotic Bcl-2 family members in various cancers as well (Table 1).
Table 1.
Mechanisms of Antiapoptotic Bcl-2 Family Member Dysregulation in Cancer
| Bcl-2 family member | Overexpressed in | Genomic alterations | miRNA loss | E3 ubiquitin ligase loss | Other mechanisms |
|---|---|---|---|---|---|
| Bcl-2 | Lymphoid malignancies [115–117] and melanoma [224] as well as small cell lung [21, 225], breast [226], prostate [227], colorectal [228] and bladder cancers [229]. | t(14;18) chromosomal translocation in lymphomas [17, 93, 94] | Loss of miR-15a and miR-16 in CLL [121] Loss of miR- 195, miR-24- 2, miR-365-2 and miR-204 in breast, lung and colon cancer [230, 231] | Loss of INrf2 [124] | |
| Bcl-xL | Hormone-refractory breast cancer [233] prostate cancer [232] and mesenchymal | Gene amplificatio n, often in concert with c-Myc, in multiple solid tumors [18] | Decrease miR-let7c in hepatocellular and lung cancers [234, 235] as well as AML [236] Decreased miR-491-5P in pancreatic [237], colorectal [238], and ovarian cancer [237]. |
Loss of phosphoglycer ate mutase 5, which is required for binding to the E3 ligase INrf2 [124] | Stat5 activation by Bcr/abl in CML [239] |
| Mcl-1 | Relapsed acute leukemia [120], CLL [240], multiple myeloma [241–243] as well as breast [244], ovarian [245], colorectal [246], gastric [247], hepatocellular [248], and non-small cell lung cancers [18]. | Gene amplificatio n in multiple solid tumors [18] | Loss of miR- 29 in many solid tumors [249], miR- 125b in hepatocellular carcinoma [250, 251] or miR-133b in lung cancer [252] | Increased expression of the deubiquitinase USP9X in FL, DLBCL and multiple myeloma [250] Mutation or loss of the E3 ubiquitin ligase FBW7 in a variety of cancers [253, 254] | NFκB- mediated transcription, e.g., in large granular lymphocytic leukemia [255] |
| Bcl-w | Gastric [256, 257] and colorectal cancers [258] as well as glioblastomamultifor me [259, 260]. | Gene amplificatio n in CML, ovarian and colorectal cancers [261, 262] | Downregulati on miR-125b [251], miR- 335 [263, 264], miR- 497 [265], miR-203 [266] or miR- 122 [267] | Not known | Enhanced promoter activity by β-catenin/ TGF4 signaling in colorectal cancer [268]. |
| BFL1 | ALL, CLL and DLBL [269-272]. | Not known | Not known | Regulated by ubiquitination. No E3 ligase has been identified (\[273, 274]. | Transcription al activation by NFκB [275], e.g., downstream of constitutive PI3K or ERK activation [276, 277] that occurs in many solid tumors. |
| Bcl-B | DLBL and breast, prostatic, gastric, colorectal, and small cell lung cancers [278]. High level associates with poor prognosis. | Not known | Not reported | Ubiquitinated protein. No E3 ligase has been identified [279]. | Not known |
2.3. Anti-apoptotic Bcl-2 family member phosphorylation
The anti-apoptotic functions of Bcl-2 and its kin are governed not only by changes in expression, but also by phosphorylation. In particular, Bcl-2 is phosphorylated in response to a number of stimuli, including spindle poisons such as paclitaxel, vincristine and vinblastine, as well as serum starvation [125–128]. As summarized in Fig. 2, several residues within the unstructured flexible loop domain (FLD) of Bcl-2 are phosphorylated, including T56, T69, S70, T74, and S87 [122, 128–130]. Kinases implicated in these phosphorylations include c-Jun N-terminal kinase (JNK) [131, 132], c-Raf [127], protein kinase A [133], p38 MAPK [134], PKCα [135], mTOR [136] and CDK1 [137, 138]. Importantly, Bcl-2 is phosphorylated on several of these residues during mitosis [139, 140], suggesting a possible physiological role for Bcl-2 phosphorylation during cell division.
How phosphorylation affects Bcl-2 function has been somewhat controversial. An S70A Bcl-2 variant protects cells from paclitaxel-induced cell death better than wild type (wt) Bcl-2 does, suggesting that S70 phosphorylation inhibits Bcl-2 function [141]. Likewise, Bcl-2 T69A/S70A/S87A affords enhanced protection from Ca2+-dependent death stimuli [142]. However, phosphomimetic mutants with Glu in place of T69, S70 and/or S87 also exhibit enhanced anti-apoptotic effects [143–145], suggesting that phosphorylation activates Bcl-2.
It was originally suggested that Bcl-2 phosphorylation might alter binding of proteins such as p53 or c-Myc to the Bcl-2 FLD, thereby modulating apoptosis [146, 147]. However, the observation that FLD deletion completely blocks paclitaxel-induced apoptosis [132] raised the possibility that FLD modifications might modulate Bcl-2 function through a process distinct from altering protein-protein interactions involving the FLD. Consistent with this view, we recently found that mutation of T69, S70, T74 or S87 to either Glu or Ala increases the affinity of Bcl-2 for Bak and Bim [54], indicating that FLD modifications modulate Bcl-2 function through a process that does not require introduction of a negative charge. Moreover, we also observed altered protease sensitivity, suggestive of a conformational change, after mutation of Bcl-2 S70 to either Glu or Ala [54]. During paclitaxel-induced mitotic arrest, Bcl-2 was phosphorylated and more tightly bound to Bak and Bim. Collectively, these results suggest that phosphorylation of the Bcl-2 FLD leads to a conformational change in the “Bcl-2 core” that affects binding of pro-apoptotic proteins to the BH3 binding groove.
Interpretation of these results would have been enhanced by structural information about the impact of FLD alterations on overall Bcl-2 conformation. Unfortunately, all published Bcl-2 structures either lack the FLD or replace it with the corresponding region of Bcl-xL [148, 149].
Phosphorylation also modulates the function of other anti-apoptotic Bcl-2 family members. Bcl-xL is phosphorylated on T47 and S62 [150]. Phosphorylation of the latter site by JNK decreases binding of Bcl-xL to Bax, thus diminishing Bcl-xL anti-apoptotic function [151]. Likewise, Mcl-1 phosphorylation at S159 by glycogen synthase kinase-3 (GSK-3) diminishes Mcl-1 anti-apoptotic function, in this case by enhancing Mcl-1 binding to the ubiquitin E3 ligase βTrCP [152] and subsequent turnover [153]. In contrast, CDK- and JNK-mediated Mcl-1 phosphorylation at S64 increases Mcl-1 binding to Noxa, Bak and Bim, thereby potentiating its anti-apoptotic activity [154]. Because phosphorylation of Bcl-2 (on its FLD) and Mcl-1 (at S64) increases the anti-apoptotic effects of these proteins, it has been suggested that these phosphorylations might contribute to chemoresistance [155–157].
3. BCL2 sequence variation
3.1. BCL2 sequence variation in clinical lymphomas
As indicated above, BCL2 translocation to the IGH locus contributes to high level Bcl-2 expression in 90% of FLs and 20% of de novo diffuse large B-cell lymphomas (DLBCLs), mostly of the germinal center B-cell subtype (GCB-DLBCL) [158]. Some of these lymphomas also contain BCL2 mutations. Single nucleotide variants (SNVs), including some that cause amino acid substitutions and others that are silent, were demonstrated soon after BCL2 was cloned [159, 160]. Detection of similar SNVs in untreated FL ruled out chemotherapy as a cause of these BCL2 mutations [159]. In contrast to the impact of other abnormalities involving BCL2, e.g., the extremely poor prognosis of so-called “double hit” lymphomas that harbor translocations involving both the MYC and BCL2 genes [161], the effects of BCL2 SNVs on DLBCL and FL have been less clear.
Recent sequencing of large numbers of primary DLBCL samples and matched germline DNA has demonstrated a high incidence of tumor-associated BCL2 mutations and linked these changes to somatic hypermutation, an activation-induced cytidine deaminase- (AID-) driven mutagenic process that ordinarily generates antibody diversity [162–165]. Because most BCL2 mutations in DLBCL (60–90%) are synonymous [162] and not associated with any impact on chemosensitivity or survival [165], it has been assumed that BCL2 mutations in DLBCL are passenger mutations.
Additional studies have examined BCL2 mutations in FL. More indolent than DLBCL, FL nonetheless carries a 2–3%/year risk of transformation to a more aggressive neoplasm. Transformed FL (tFL) most often resembles DLBCL morphologically [166, 167] but historically has been quite resistant to therapy [167, 168]. This poor therapeutic response of tFL has provided the impetus for recent genomic comparisons of untransformed and transformed FLs [169–171]. These analyses have demonstrated that BCL2 is mutated, at least to some extent, in the majority of FLs and tFLs [169–171]. Analysis of subclonal heterogeneity has suggested that tFL arises from the original mutant FL clone, but not always through linear progression of the clone that predominates during the indolent phase of the disease. Instead, FL transformation commonly reflects divergent evolution from a common mutated ancestor, with independent acquisition of further aberrations in the indolent FL clone and the more aggressive tFL [166, 170].
3.2. Some BCL2 variants exhibit gain of function
Because BCL2 sequence variants inhibit cell death [172] and facilitate lymphomagenesis [173], it has been assumed that BCL2 mutations do not affect Bcl-2 protein function. Our recent studies question this assumption. In particular, we have observed that some variant Bcl-2 proteins from lymphoma cell lines exhibit enhanced activity [103, 174]. For example, the D31H/A60V Bcl-2 variant from the BCL2-translocated lymphoma line RL binds Noxa 20-fold more tightly than wildtype Bcl-2 and provides increased protection from the proteasome inhibitor bortezomib, which upregulates Noxa to kill lymphoma cells [174]. These observations prompted us to examine whether BCL2 mutations in clinical lymphoma might also impact Bcl-2 function.
To address this issue, we compared the incidence, nature, and clinical implications of BCL2 mutations in a number of lymphoid neoplasms [175]. In contrast to DLBCL, where most mutations are silent, a vast excess of amino acid altering (nonsynonymous) variants was observed in FL. Moreover, the presence of BCL2 mutations at diagnosis was an independent prognostic factor that correlated with increased risk of death due to lymphoma [175]. The sequence context of the BCL2 mutations in FL suggested that AID is responsible for these mutations. Consistent with this conclusion, high AID levels in FL were associated with a higher probability of BCL2 mutation at diagnosis and a shorter interval before transformation [175]. In FL, amino acid altering mutations are spread along the Bcl-2 protein [165, 175], with the highest frequency in the FLD (Fig. 3), a region implicated in regulating affinity of Bcl-2 for pro-apoptotic Bcl-2 family members [54] as described above. Consistent with earlier studies in lymphoma cell lines [103, 174], preliminary analysis demonstrated that some variant Bcl-2 proteins identified in FL exhibit increased ability to bind and sequester the pro-apoptotic Bcl-2 family members Bim and Puma [175]. Further analysis of additional variant proteins is needed to determine the prevalence of BCL2 gain of function mutations in this region.
Figure 3. Distribution of Bcl-2 amino acid substitutions in FL. (A).

Schematic representation of the Bcl-2 protein with BH domains boxed in blue and flexible loop domain (FLD) labeled in gray. Somatic variants detected in FL derived from 2 independent cohorts (black and green bars) are distributed throughout the Bcl-2 protein. Color-coded symbols depict distinct types of alterations, with purple for synonymous, white for nonsynonymous with no charge introduction, red for nonsynonymous with negative charge introduction, and blue for nonsynonymous with positive charge introduction. TM, transmembrane domain. (Adapted from Correia et al. [175].) (B). Affinities of Bak BH3 peptide and Bak protein for Bcl-2 variants derived from lymphoid cell lines. (Adapted from Dai et al. [103].) † indicates undetectable binding. (C). Kaplan Meier plots showing impact of BCL2 mutations on death due to lymphoma in a FL cohort (n=128). (Adapted from Correia et al. [175].)
FL-associated mutations also occur in other regions of Bcl-2 (Fig. 3). For example, several mutations alter the BH4 domain, a region partially conserved in Bcl-xL, Bcl-w, BFL1 and Bcl-B [10, 176]. The implications of these BH4 domain mutations are currently unknown. Although multiple studies have reported that deletion of the BH4 domain abrogates Bcl-2 anti-apoptotic function [177–179], the underlying mechanism has been unclear. It has been suggested that BH4 domain deletion abrogates Bcl-2 heterodimerization with Bax [178]. Consistent with this view, binding of a stapled BH4 peptide to unique site on Bax has recently been reported [180]. Other studies, however, have found no impact of BH4 deletion on binding of Bcl-2 to Bax, Bak, Bad, Bik or Bim [177]. Accordingly, further work is required to clarify the function of the Bcl-2 BH4 domain and assess the impact of FL-associated mutations in this region.
Comparison of sequential FL biopsies revealed a 4-fold increase in frequency of overtly detectable BCL2 mutations at transformation compared to the same patients at diagnosis [175], suggesting that mutagenesis is ongoing in FL during the course of disease. Mutations identified at transformation had a lower frequency of classical AID signatures, possibly reflecting AID-induced mutation of cytidines outside the context of preferred AID target sequences [181]. Moreover, some of these mutations were clearly nonfunctional, e.g., a BCL2 variant with a premature stop codon [175], again emphasizing that not all BCL2 mutations enhance Bcl-2 function.
In accord with studies showing that FL transformation is genetically complex and heterogeneous [166, 169–171], BCL2 mutations can suffer several fates during the course of FL. In cases where there is a linear evolution from an original malignant clone, BCL2 mutations would be maintained throughout the course of the disease, perhaps with the acquisition of additional alterations in the same mutant gene over time. In cases where clones that give rise to the indolent FL and tFL follow divergent paths from a common precursor, mutations present in the dominant clone at diagnosis might not predominate at transformation, where new genetic alterations that confer a stronger proliferation or survival advantage could instead emerge. Conversely, in clones that lack BCL2 mutations initially, BCL2 mutations might emerge at transformation, as reported in our cohort [175]. Whether these are driver or passenger mutations remains to be determined.
In comparing recent studies of BCL2 mutations in FL [169–171, 175], several important methodological differences must be kept in mind. First, most of the studies were designed to identify all mutations in the BCL2 locus. These studies not only sequenced the BCL2 promoter and extensive intronic regions in addition to exons, but also counted BCL2 as mutated even when a relatively small percentage of the reads were variant, e.g., as few as three reads out of 85 [169]. In contrast, our recent study, which involved Sanger sequencing of the protein coding exons, was designed to identify mutations that are i) present in predominant clones and ii) capable of altering Bcl-2 protein sequence and function [175].
Second, the studies comprehensively examining genomic changes in FL at transformation relied on the availability of paired FL pathological samples. In contrast, in order to assess the relationship between FL mutations, AID expression and clinical outcome, our recent study relied on a prospectively maintained clinical database and relatively uniform treatment practices at a single institution [175]. To have enough events for survival analysis, however, the vast majority of samples in our study predated the introduction of the anti-CD20 antibody rituximab into clinical FL treatment [182]. Retrospective analyses from the pre-rituximab era have reported a median survival of only 1–2 years after FL transformation [167, 168], whereas recent studies suggest improved survival with tFL in the rituximab era [166, 183], raising the possibility that rituximab might partially counter the adverse impact of genomic changes leading to transformation. Thus, it will be important in the future to assess whether BCL2 mutation status correlates with outcome in FL treated with rituximab-containing chemoimmunotherapy.
4. Targeting Bcl-2 as an anticancer strategy
4.1 Small molecule inhibitors of Bcl-2 family proteins
Because Bcl-2 overexpression contributes to the pathogenesis of various lymphoid neoplasms, particularly FL and CLL [17, 93, 94, 121], and possibly solid tumors [80], there has been substantial effort to develop Bcl-2 inhibitors as therapeutic agents. A combination of nuclear magnetic resonance-based binding assays and fluorescence polarization displacement assays [184, 185] have identified a number of small molecules that interfere with protein-protein interactions involving the Bcl-2, Bcl-xL and/or Mcl-1 BH3 binding grooves [10, 11, 186–189], thus mimicking the effects of sensitizer BH3-only proteins depicted in Fig. 1.
For a small molecule to truly be a BH3 mimetic, its killing must depend on Bax and/or Bak, it must have a high affinity for at least one anti-apoptotic Bcl-2 family member, and it must induce cytotoxicity that correlates with binding to anti-apoptotic protein(s) [186]. Some compounds originally reported to be BH3 mimetics, e.g., BH3I class compounds, HA14-1, antimycin A, and purpurogallin, are no longer thought to kill in this manner because they exhibit poor binding to BH3 binding grooves or Bax/Bak-independent killing [186, 190, 191]. Other agents (Table 2) meet the criteria outlined above and have undergone preclinical and, in some cases, clinical testing. These include AT-101 [the R-(-) enantiomer of gossypol] [192–194]; the gossypol derivatives TW37 [192], apogossypol, and apogossypolone (ApoG2) [195, 196]; and obatoclax (GX15-070) [186, 197–199], although obatoclax also kills Bax/Bak-deficient cells [191, 200], perhaps because it inhibits the pro-survival kinase mTOR [201].
Table 2.
Selective List of BH3 Mimetics
| Agent | Target | BH3 mimetic Statusa | Clinical statusb | References |
|---|---|---|---|---|
| A-124077 | Mcl-1 | Authentic | NAc | [188] |
| ABT-737 | Bcl-xL, Bcl-2, Bcl-w | Authentic | NA | [186, 191, 192, 202, 204] |
| ABT-263 (Navitoclax) | Bcl-xL, Bcl-2, Bcl-w | Authentic | Ph2d | [203] |
| ABT-199 | Bcl-2 | Authentic | Ph3e | [208, 280] |
| GX15-070 (Obatoclax) | Bcl-xL, Bcl-2, Bcl-w, Mcl-1 | Putative/off-target effects | Ph2f | [197] |
| Gossypol | Bcl-2, Bcl-xL, Mcl-1 | Putative/off-target effects | NA | [184, 281] |
| Apogossypolone (ApoG2) | Bcl-xL, Bcl-2, Mcl-1 | Putative/off-target effects | NA | [195] |
| TW37 | Bcl-xL, Bcl-2, Mcl-1 | Putative/off-target effects | NA | [186, 192, 282] |
| AT-101 | Bcl-xL, Bcl-2, Mcl-1 | Putative/off-target effects | Ph2g | [192, 193, 280] |
BH3 mimetic status defined as in [186]
Most advanced clinical testing reported at https://clinicaltrials.gov/
Abbreviations used are: NA, not applicable (no clinical testing); Ph, phase.
Randomized Ph2 study of navitoclax + rituximab vs. rituximab alone in CLL.
Ph3 study of ABT-199 + rituximab vs. bendamustine + rituximab for CLL.
Ph2 studies in acute myelogenous leukemia, small cell lung cancer (with topotecan or carboplatin/etoposide), mantle cell lymphoma (with bortezomib), Hodgkin lymphoma, multiple myeloma (with bortezomib) and non-small cell lung cancer (with docetaxel).
Ph2 studies in chronic lymphocytic leukemia (with rituximab), hormone-resistant prostate cancer (with prednisone/docetaxel), diffuse large B cell lymphoma (with lenalidomide), non-small cell lung cancer (with docetaxel), small cell lung cancer (alone or with topotecan), and glioblastoma (alone or with radiation).
Of particular interest are ABT-737 and its orally bioavailable derivative ABT-263 (navitoclax). These agents bind the BH3 binding grooves of Bcl-xL, Bcl-2, and Bcl-w [186, 191, 192, 202–204] to kill cells in a Bax- or Bak-dependent manner [191, 192]. Interestingly, their major toxicity, thrombocytopenia, also results from Bcl-xL inhibition [205, 206]. To avoid this side effect, the Bcl-2-selective derivative ABT-199 has been developed [207]. Cellular studies have shown that ABT-737, navitoclax and ABT-199 displace Bim from their identified anti-apoptotic binding partner(s), facilitating Bax-mediated MOMP [203, 208, 209]. Priming of Bcl-2 with the activator Bim increases sensitivity to ABT-737, navitoclax and ABT-199, whereas Mcl-1 overexpression favors resistance [56, 191, 203, 208–210]. All of these observations are consistent with the proposed mode of action of these agents.
4.2 Clinical trials of BH3 mimetics
In view of the important role of Bcl-2 in the pathogenesis of lymphoid malignancies (see above), BH3 mimetics have been extensively tested in these disorders (reviewed in [211]). Early clinical testing revealed activity of navitoclax in CLL and non-Hodgkin’s lymphoma (NHL) [212, 213]. With the development of ABT-199, studies of navitoclax continue in solid tumors, where it is being used to target Bcl-xL, whereas studies in Bcl-2-dependent hematological malignancies have focused more extensively on ABT-199.
As of January 2015, 16 clinical trials testing ABT-199 either as a single agent or in combination therapy (www.clinicaltrials.gov) are ongoing. A variety of neoplasms are being studied, including CLL, aggressive NHL, FL, multiple myeloma (MM) and AML (Table 3). In preclinical studies, anti-tumor activity of ABT-199 has also been reported in T-cell ALL, chronic myelogenous leukemia, colorectal and breast cancers [214–217].
Table 3.
Summary of Selected Current ABT-199 Clinical Trials
| Disease | Clinical Characteristics or Eligibility | Regimens Phase | Ref.b | |
|---|---|---|---|---|
| AMLa | Treatment naïve | ABT-199 | Ib | |
| Age ≥65 | + Decitabine or + Azacitidine | |||
| Ineligible for Standard Chemotherapy | ||||
|
| ||||
| AML | Treatment naïve | ABT-199 | I/II | |
| + Cytarabine | ||||
|
| ||||
| AML | NA | ABT-199 | II | [92] |
|
| ||||
| B-Cell NHL, DLBCL | NA | ABT-199 | ||
| + Rituximab or Oblinutuzumab | ||||
| + CHOP | ||||
|
| ||||
| CLL | R/R or Treatment naïve | ABT-199 | Ib | [221] |
| + Oblinutuzumab | ||||
|
| ||||
| CLL | R/R or Treatment naïve | ABT-199 | Ib | [222] |
| + Bendamustine | ||||
| + Rituximab | ||||
|
| ||||
| CLL, NHL | R/R | ABT-199 | I | [219] |
|
| ||||
| CLL, SLL | R/R | ABT-199 | Ib | [220] |
| + Rituximab | ||||
|
| ||||
| CLL | R/R to B-Cell Receptor Inhibitor | ABT-199 | II | |
|
| ||||
| CLL, 17p deletion | R/R | ABT-199 | II | |
|
| ||||
| CLL | R/R | ABT-199 | III | |
| + Rituximab or + Bendamustine and Rituximab | ||||
|
| ||||
| FL | R/R | ABT-199 | II | |
| + Rituximab or + Bendamustine and Rituximab | ||||
|
| ||||
| MM | Treatment with Bortezomib and Dexamethasone | ABT-199 | I | |
|
| ||||
| MM | R/R | ABT-199 | I | |
|
| ||||
| NHL, MM | R/R | ABT-199 | I | |
|
| ||||
| NHL | R/R | ABT-199 | Ib | [223] |
| + Bendamustine | ||||
| + Rituximab | ||||
Abbreviations: AML, acute myeloid leukemia; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; FL, follicular lymphoma; NA, not available; NHL, Non-Hodgkin’s lymphoma; MM, multiple myeloma; R/R, relapsed/refractory; and SLL, small lymphocytic leukemia.
References for ABT-199 trials that have been reported in abstract form.
Early clinical results suggest that ABT-199 has promising activity in relapsed or refractory (R/R) lymphoid malignancies. In particular, ABT-199 has an overall response rate (ORR) of 56% in R/R NHL [218] and 84% (20% complete response) in R/R CLL and small lymphocytic leukemia (SLL) [219]. Indeed, responses in CLL and SLL have been so risk that tumor lysis syndrome has been a major complication of therapy. In contrast, despite tantalizing activity against AML cells ex vivo [91], ABT-199 has shown disappointing activity (12.5% complete remission rate) in R/R AML [92], perhaps because of elevated Mcl-1 in this setting [120].
Additional studies are investigating ABT-199 in combination with other therapies (Table 3). For example, Roberts et al. reported an 86% OR (31% CR) in R/R CLL treated with ABT-199 and the anti-CD20 antibody rituximab [220]. Other studies in R/R or untreated CLL patients combined ABT-199 with Obinutuzumab, a glycoengineered anti-CD20 antibody [221], or in CLL and NHL with Bendamustine/Rituximab [222, 223]. More mature reports of these promising investigations are awaited with interest.
5.0. An agenda for the future
Despite improvements in understanding signaling through the intrinsic apoptotic pathway over the past 2–3 years, important questions remain. Recent studies have clarified how BH3-only proteins induce Bax or Bak activation and provided detailed descriptions of Bax and Bak homodimers, but these results provide limited insight into the mechanism by which Bax and Bak breach the MOM. Further studies of higher order Bax and Bak oligomers, perhaps in the presence of appropriate lipids, are required for progress in this area.
Even though the Bcl-2 protein was identified almost 30 years ago, important questions about its structure and function persist. How phosphorylations in the FLD affect affinity of the BH3 binding groove for its ligands is entirely unclear. While recent studies suggest that changes in the FLD can affect Bcl-2 conformation [54], an NMR or crystal structure of Bcl-2 with its own loop intact is required to better understand this aspect of Bcl-2 regulation.
The consequences of nonsynonymous BCL2 mutations also require further investigation. While these mutations become more prevalent during the course of FL [175], it is unclear whether they are driving FL transformation or are merely bystanders that reflect high AID activity in FLs at highest risk of transformation. Further study is also required to determine which BCL2 mutations affect Bcl-2 protein function and how these amino acid changes, whether in the FLD or elsewhere, alter affinity of the BH3 binding groove.
Finally, a number of questions need to be answered to assure the optimal clinical development of ABT-199. In hematological malignancies, this agent currently appears to be most active in CLL, a disease driven by overexpression of wt Bcl-2. Whether the BCL2 mutations observed in FL will affect ABT-199 sensitivity remains to be determined. Furthermore, it is presently unclear whether ABT-199 will be as active in diseases where Bcl-2 overexpression, while present, plays a less clear-cut role in pathogenesis (e.g., small cell lung cancer). Finally, additional work is required to identify optimal combinations that capitalize on the promising single-agent activity of ABT-199 observed to date.
Given the recent exciting advances, continued investigation of these questions appears warranted.
Highlights.
Here we review recent advances in understanding of Bcl-2 family members.
Recent x-ray crystal structures have provided insight into Bax and Bak activation.
Some lymphoma-associated BCL2 mutations were shown to enhance Bcl-2 function.
The Bcl-2-selective BH3 mimetic ABT-199 has shown promising clinical activity.
Acknowledgments
We gratefully acknowledge provocative discussions with Reuben Harris, David Huang, Greg Gores, Randy Gascoyne and members of the Mayo Clinic Lymphoma Disease-Oriented Group; helpful suggestions of the anonymous reviewers; and editorial assistance of Deb Strauss. Preparation of this review was supported in part by R01 CA166741, F30 CA183507, and predoctoral fellowships from the Mayo Foundation for Education and Research.
Abbreviations
- AID
activation-induced cytidine deaminase
- AML
acute myelogenous leukemia
- BH
Bcl-2 homology
- CLL
chronic lymphocytic leukemia
- CR
complete response
- DLBCL
diffuse large B-cell lymphoma
- FL
follicular lymphoma
- MM
multiple myeloma
- PR
partial response
- R/R
relapsed or refractory
- SNV
single nucleotide variant
- TM
transmembrane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hengartner MO. The Biochemistry of Apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 2.Meng XW, Lee SH, Kaufmann SH. Apoptosis in the treatment of cancer: a promise kept? Current Opin Cell Biol. 2006;18:668–676. doi: 10.1016/j.ceb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 4.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian Caspases: Structure, Activation, Substrates and Functions During Apoptosis. Annual Rev Biochemistry. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 5.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis. A Link Between Cancer Genetics and Chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 6.Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005;26:263–270. doi: 10.1093/carcin/bgh283. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome C. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 8.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annual Rev Biochemistry. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 9.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–3683. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 11.Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem Sci. 2014;39:101–111. doi: 10.1016/j.tibs.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renault TT, Chipuk JE. Death upon a kiss: mitochondrial outer membrane composition and organelle communication govern sensitivity to BAK/BAX-dependent apoptosis. Chem Biol. 2014;21:114–123. doi: 10.1016/j.chembiol.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ekert PG, Vaux DL. The mitochondrial death squad - hardened killers or innocent bystanders? Current Opin Cell Biol. 2005;17:626–630. doi: 10.1016/j.ceb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nature reviews. Neuroscience. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- 16.Thompson CB. Apoptosis in the Pathogenesis and Treatment of Disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 17.Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 18.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M. Somatic Frameshift Mutations in the BAX Gene in Colon Cancers of the Microsatellite Mutator Phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 20.Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, Rosenwald A, Sugimoto KJ, Wheat LM, Karran EL, Garcia JF, Sanchez L, Prosper F, Staudt LM, Pinkel D, Dyer MJ, Martinez-Climent JA. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 21.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Gross A, Waksman G, Korsmeyer SJ. Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol. 1998;18:6083–6089. doi: 10.1128/mcb.18.10.6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, Kluck RM. To trigger apoptosis Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Molecular Cell. 2008;30:369–380. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 26.Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell. 2009;36:696–703. doi: 10.1016/j.molcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Ma S, Hockings C, Anwari K, Kratina T, Fennell S, Lazarou M, Ryan MT, Kluck RM, Dewson G. Assembly of the Bak apoptotic pore: a critical role for the Bak protein alpha6 helix in the multimerization of homodimers during apoptosis. J Biol Chem. 2013;288:26027–26038. doi: 10.1074/jbc.M113.490094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pang YP, Dai H, Smith A, Meng XW, Schneider PA, Kaufmann SH. Bak Conformational Changes Induced by Ligand Binding: Insight into BH3 Domain Binding and Bak Homo-Oligomerization. Sci Rep. 2012;2:257. doi: 10.1038/srep00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M, Boreham D, McFarlane N, Ding J, Johnson AE, Zhang XC, Andrews DW, Lin J. Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem. 2010;285:17614–17627. doi: 10.1074/jbc.M110.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC, Kluck RM, Adams JM, Colman PM. Bax Crystal Structures Reveal How BH3 Domains Activate Bax and Nucleate Its Oligomerization to Induce Apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, Thompson GV, Colman PM, Kluck RM, Czabotar PE. Bak Core and Latch Domains Separate during Activation, and Freed Core Domains Form Symmetric Homodimers. Molecular Cell. 2014;55:938–946. doi: 10.1016/j.molcel.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 32.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Current Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes & development. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- 34.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Molecular Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 35.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 36.Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nature Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 37.Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai H, Pang YP, Ramirez-Alvarado M, Kaufmann SH. Evaluation of the BH3-only protein Puma as a direct Bak activator. J Biol Chem. 2014;289:89–99. doi: 10.1074/jbc.M113.505701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai H, Smith A, Meng XW, Schneider PA, Pang YP, Kaufmann SH. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J Cell Biol. 2011;194:39–48. doi: 10.1083/jcb.201102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villunger A, Labi V, Bouillet P, Adams J, Strasser A. Can the analysis of BH3-only protein knockout mice clarify the issue of 'direct versus indirect' activation of Bax and Bak? Cell Death Differ. 2011;18:1545–1546. doi: 10.1038/cdd.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, Michalak E, Strasser A, Villunger A. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Molecular Cell. 2013;51:751–765. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chm. 2011;286:491–501. doi: 10.1074/jbc.M110.167148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–597. doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 2013;110:E986–995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Molecular Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Molecular Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Molecular Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 50.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Molecular Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 52.Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR. A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Molecular Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, Walensky LD. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol. 2013;20:888–902. doi: 10.1016/j.chembiol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dai H, Ding H, Meng XW, Lee SH, Schneider PA, Kaufmann SH. Contribution of Bcl-2 phosphorylation to Bak binding and drug resistance. Cancer Res. 2013;73:6998–7008. doi: 10.1158/0008-5472.CAN-13-0940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lindner AU, Concannon CG, Boukes GJ, Cannon MD, Llambi F, Ryan D, Boland K, Kehoe J, McNamara DA, Murray F, Kay EW, Hector S, Green DR, Huber HJ, Prehn JH. Systems analysis of BCL2 protein family interactions establishes a model to predict responses to chemotherapy. Cancer Res. 2013;73:519–528. doi: 10.1158/0008-5472.CAN-12-2269. [DOI] [PubMed] [Google Scholar]
- 56.Weber K, Harper N, Schwabe J, Cohen GM. BIM-mediated membrane insertion of the BAK pore domain is an essential requirement for apoptosis. Cell Rep. 2013;5:409–420. doi: 10.1016/j.celrep.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng R, Tong JS, Li H, Yue B, Zou F, Yu J, Zhang L. Targeting Bax interaction sites reveals that only homo-oligomerization sites are essential for its activation. Cell Death Differ. 2013;20:744–754. doi: 10.1038/cdd.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vela L, Gonzalo O, Naval J, Marzo I. Direct interaction of Bax and Bak proteins with Bcl-2 homology domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J Biol Chem. 2013;288:4935–4946. doi: 10.1074/jbc.M112.422204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merino D, Giam M, Hughes PD, Siggs OM, Heger K, O'Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, Bouillet P. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009;186:355–362. doi: 10.1083/jcb.200905153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL, Fesik SW. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 62.Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Molecular Cell. 2006;24:677–688. doi: 10.1016/j.molcel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 63.Choe S, Bennett MJ, Fujii G, Curmi PM, Kantardjieff KA, Collier RJ, Eisenberg D. The crystal structure of diphtheria toxin. Nature. 1992;357:216–222. doi: 10.1038/357216a0. [DOI] [PubMed] [Google Scholar]
- 64.Elkins P, Bunker A, Cramer WA, Stauffacher CV. A mechanism for toxin insertion into membranes is suggested by the crystal structure of the channel-forming domain of colicin E1. Structure. 1997;5:443–458. doi: 10.1016/s0969-2126(97)00200-1. [DOI] [PubMed] [Google Scholar]
- 65.Parker MW, Feil SC. Pore-forming protein toxins: from structure to function. Prog Biophys Mol Biol. 2005;88:91–142. doi: 10.1016/j.pbiomolbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 66.Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuertes G, Garcia-Saez AJ, Esteban-Martin S, Gimenez D, Sanchez-Munoz OL, Schwille P, Salgado J. Pores formed by Baxalpha5 relax to a smaller size and keep at equilibrium. Biophys J. 2010;99:2917–2925. doi: 10.1016/j.bpj.2010.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia-Saez AJ, Coraiola M, Serra MD, Mingarro I, Muller P, Salgado J. Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores. The FEBS J. 2006;273:971–981. doi: 10.1111/j.1742-4658.2006.05123.x. [DOI] [PubMed] [Google Scholar]
- 69.Qian S, Wang W, Yang L, Huang HW. Structure of transmembrane pore induced by Bax-derived peptide: evidence for lipidic pores. Proc Natl Acad Sci U S A. 2008;105:17379–17383. doi: 10.1073/pnas.0807764105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Westphal D, Dewson G, Menard M, Frederick P, Iyer S, Bartolo R, Gibson L, Czabotar PE, Smith BJ, Adams JM, Kluck RM. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc Natl Acad Sci U S A. 2014;111:E4076–4085. doi: 10.1073/pnas.1415142111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Terrones O, Etxebarria A, Landajuela A, Landeta O, Antonsson B, Basanez G. BIM and tBID are not mechanistically equivalent when assisting BAX to permeabilize bilayer membranes. J Biol Chem. 2008;283:7790–7803. doi: 10.1074/jbc.M708814200. [DOI] [PubMed] [Google Scholar]
- 72.Landeta O, Landajuela A, Gil D, Taneva S, Di Primo C, Sot B, Valle M, Frolov VA, Basanez G. Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. The J Biol Chem. 2011;286:8213–8230. doi: 10.1074/jbc.M110.165852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 74.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 75.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 76.Tan TT, Degenhardt K, Nelson DA, Beaudoin B, Nieves-Neira W, Bouillet P, Villunger A, Adams JM, White E. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell. 2005;7:227–238. doi: 10.1016/j.ccr.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 77.Torres K, Horwitz SB. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 1998;58:3620–3626. [PubMed] [Google Scholar]
- 78.Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118:3651–3659. doi: 10.1172/JCI35437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kuroda J, Puthalakath H, Cragg MS, Kelly PN, Bouillet P, Huang DC, Kimura S, Ottmann OG, Druker BJ, Villunger A, Roberts AW, Strasser A. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reed JC. Dysregulation of Apoptosis in Cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 81.Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005;26:263–270. doi: 10.1093/carcin/bgh283. [DOI] [PubMed] [Google Scholar]
- 82.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 83.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, Letai A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 84.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 86.Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, Letai A. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151:344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, del Moore VG, Deng J, Anderson KC, Richardson P, Tai YT, Mitsiades CS, Matulonis UA, Drapkin R, Stone R, Deangelo DJ, McConkey DJ, Sallan SE, Silverman L, Hirsch MS, Carrasco DR, Letai A. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol. 2009;187:429–442. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ryan JA, Brunelle JK, Letai A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci U S A. 2010;107:12895–12900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, Dohner H, Gaidzik VI, Galinsky I, Golfman LS, Haferlach T, Harutyunyan KG, Hu J, Leverson JD, Marcucci G, Muschen M, Newman R, Park E, Ruvolo PP, Ruvolo V, Ryan J, Schindela S, Zweidler-McKay P, Stone RM, Kantarjian H, Andreeff M, Konopleva M, Letai AG. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Konopleva M, Pollyea DA, Potluri J, Chyla BJ, Busman TA, McKeegan E, Salem A, Zhu M, Ricker JL, Blum W, DiNardo CD, Dunbar M, Kirby R, Falotico N, Leverson JD, Humerickhouse RA, Mabry M, Stone RM, Kantarjian HM, Letai A. A Phase 2 Study of ABT-199 (GDC-0199) in Patients with Acute Myelogenous Leukemia (AML) Blood. 2014;124:Abstract 118. [Google Scholar]
- 93.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 94.Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986;47:19–28. doi: 10.1016/0092-8674(86)90362-4. [DOI] [PubMed] [Google Scholar]
- 95.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 96.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 97.Ko JK, Choi KH, Pan Z, Lin P, Weisleder N, Kim CW, Ma J. The tail-anchoring domain of Bfl1 and HCCS1 targets mitochondrial membrane permeability to induce apoptosis. J Cell Sci. 2007;120:2912–2923. doi: 10.1242/jcs.006197. [DOI] [PubMed] [Google Scholar]
- 98.Brien G, Debaud AL, Robert X, Oliver L, Trescol-Biemont MC, Cauquil N, Geneste O, Aghajari N, Vallette FM, Haser R, Bonnefoy-Berard N. C-terminal residues regulate localization and function of the antiapoptotic protein Bfl-1. J Biol Chem. 2009;284:30257–30263. doi: 10.1074/jbc.M109.040824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hind CK, Carter MJ, Harris CL, Chan HT, James S, Cragg MS. Role of the pro-survival molecule Bfl-1 in melanoma. Int J Biochem Cell Biol. 2015;59:94–102. doi: 10.1016/j.biocel.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 100.Rautureau GJ, Yabal M, Yang H, Huang DC, Kvansakul M, Hinds MG. The restricted binding repertoire of Bcl-B leaves Bim as the universal BH3-only prosurvival Bcl-2 protein antagonist. Cell Death Dis. 2012;3:e443. doi: 10.1038/cddis.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simmons MJ, Fan G, Zong WX, Degenhardt K, White E, Gelinas C. Bfl-1/A1 functions, similar to Mcl-1, as a selective tBid and Bak antagonist. Oncogene. 2008;27:1421–1428. doi: 10.1038/sj.onc.1210771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dai H, Meng XW, Lee SH, Schneider PA, Kaufmann SH. Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem. 2009;284:18311–18322. doi: 10.1074/jbc.M109.004770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 105.Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood. 2009;113:2805–2815. doi: 10.1182/blood-2008-05-159145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nature Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Glaser SP, Lee EF, Trounson E, ouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander W, Lowe SW, Robb L, Strasser A. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27:1351–1364. doi: 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sochalska M, Tuzlak S, Egle A, Villunger A. Lessons from gain- and loss-of-function models of pro-survival Bcl2 family proteins: implications for targeted therapy. FEBS J. 2015;282:834–849. doi: 10.1111/febs.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rooswinkel RW, van de Kooij B, de Vries E, Paauwe M, Braster R, Verheij M, Borst J. Antiapoptotic potency of Bcl-2 proteins primarily relies on their stability, not binding selectivity. Blood. 2014;123:2806–2815. doi: 10.1182/blood-2013-08-519470. [DOI] [PubMed] [Google Scholar]
- 111.Lowe SW, Cepro E, Evan G. Intrinsic tumour suppression. Nature. 2004 doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 112.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 113.Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol. 2001;21:5063–5070. doi: 10.1128/MCB.21.15.5063-5070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fanidi A, Harrington EA, Evan GI. Cooperative Interaction Between c-myc and bcl-2 Proto-Oncogenes. Nature. 1992;359:554–556. doi: 10.1038/359554a0. [DOI] [PubMed] [Google Scholar]
- 115.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–6406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 116.Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111:3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Scarfo L, Ghia P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett. 2013;155:36–39. doi: 10.1016/j.imlet.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 118.Maung ZT, MacLean FR, Reid MM, Pearson ADJ, Proctor SJ, Hamilton PJ, Hall AG. The Relationship Between bcl-2 Expression and Response to Chemotherapy in Acute Leukaemia. British J Haematol. 1994;88:105–109. doi: 10.1111/j.1365-2141.1994.tb04984.x. [DOI] [PubMed] [Google Scholar]
- 119.Bincoletto C, Saad ST, da Silva ES, Queiroz ML. Haematopoietic response and bcl-2 expression in patients with acute myeloid leukaemia. Eur J Haematol. 1999;62:38–42. doi: 10.1111/j.1600-0609.1999.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 120.Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, Reed JC. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood. 1998;91:991–1000. [PubMed] [Google Scholar]
- 121.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Breitschopf K, Haendeler J, Malchow P, Zeiher AM, Dimmeler S. Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol Cell Biol. 2000;20:1886–1896. doi: 10.1128/mcb.20.5.1886-1896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Niture SK, Jaiswal AK. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem. 2012;287:9873–9886. doi: 10.1074/jbc.M111.312694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Niture SK, Jaiswal AK. Inhibitor of Nrf2 (INrf2 or Keap1) protein degrades Bcl-xL via phosphoglycerate mutase 5 and controls cellular apoptosis. J Biol Chem. 2011;286:44542–44556. doi: 10.1074/jbc.M111.275073. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 125.Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci U S A. 1995;92:4507–4511. doi: 10.1073/pnas.92.10.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haldar S, Chintapalli J, Croce CM. Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer Res. 1996;56:1253–1255. [PubMed] [Google Scholar]
- 127.Blagosklonny MV, Giannakakou P, el-Deiry WS, Kingston DG, Higgs PI, Neckers L, Fojo T. Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death. Cancer Res. 1997;57:130–135. [PubMed] [Google Scholar]
- 128.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Molecular Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Haldar S, Basu A, Croce CM. Serine-70 is one of the critical sites for drug-induced Bcl2 phosphorylation in cancer cells. Cancer Res. 1998;58:1609–1615. [PubMed] [Google Scholar]
- 130.Furukawa Y, Iwase S, Kikuchi J, Terui Y, Nakamura M, Yamada H, Kano Y, Matsuda M. Phosphorylation of Bcl-2 protein by CDC2 kinase during G2/M phases and its role in cell cycle regulation. J Biol Chem. 2000;275:21661–21667. doi: 10.1074/jbc.M906893199. [DOI] [PubMed] [Google Scholar]
- 131.Maundrell K, Antonsson B, Magnenat E, Camps M, Muda M, Chabert C, Gillieron C, Boschert U, Vial-Knecht E, Martinou JC, Arkinstall S. Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress-activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J Biol Chem. 1997;272:25238–25242. doi: 10.1074/jbc.272.40.25238. [DOI] [PubMed] [Google Scholar]
- 132.Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. 1999;96:3775–3780. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL. Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase. Mol Cell Biol. 1998;18:3509–3517. doi: 10.1128/mcb.18.6.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, Palamara AT, Russo T, Garaci E, Cozzolino F. Bcl-2 Phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–21361. doi: 10.1074/jbc.M511052200. [DOI] [PubMed] [Google Scholar]
- 135.Ruvolo PP, Deng X, Carr BK, May WS. A functional role for mitochondrial protein kinase Calpha in Bcl2 phosphorylation and suppression of apoptosis. J Biol Chem. 1998;273:25436–25442. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- 136.Asnaghi L, Calastretti A, Bevilacqua A, D'Agnano I, Gatti G, Canti G, Delia D, Capaccioli S, Nicolin A. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene. 2004;23:5781–5791. doi: 10.1038/sj.onc.1207698. [DOI] [PubMed] [Google Scholar]
- 137.Terrano DT, Upreti M, Chambers TC. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol Cell Biol. 2010;30:640–656. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sakurikar N, Eichhorn JM, Chambers TC. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J Biol Chem. 2012;287:39193–39204. doi: 10.1074/jbc.M112.391854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ling YH, Tornos C, Perez-Soler R. Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem. 1998;273:18984–18991. doi: 10.1074/jbc.273.30.18984. [DOI] [PubMed] [Google Scholar]
- 140.Scatena CD, Stewart ZA, Mays D, Tang LJ, Keefer CJ, Leach SD, Pietenpol JA. Mitotic phosphorylation of Bcl-2 during normal cell cycle progression and Taxol-induced growth arrest. J Biol Chem. 1998;273:30777–30784. doi: 10.1074/jbc.273.46.30777. [DOI] [PubMed] [Google Scholar]
- 141.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ito T, Deng X, Carr B, May WS. Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem. 1997;272:11671–11673. doi: 10.1074/jbc.272.18.11671. [DOI] [PubMed] [Google Scholar]
- 144.Deng X, Ruvolo P, Carr B, May WS., Jr Survival function of ERK1/2 as IL-3-activated, staurosporine-resistant Bcl2 kinases. Proc Natl Acad Sci U S A. 2000;97:1578–1583. doi: 10.1073/pnas.97.4.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Deng X, Gao F, Flagg T, May WS., Jr Mono- and multisite phosphorylation enhances Bcl2's antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci U S A. 2004;101:153–158. doi: 10.1073/pnas.2533920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Deng X, Gao F, Flagg T, Anderson J, May WS. Bcl2's flexible loop domain regulates p53 binding and survival. Mol Cell Biol. 2006;26:4421–4434. doi: 10.1128/MCB.01647-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jin Z, Gao F, Flagg T, Deng X. Tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone promotes functional cooperation of Bcl2 and c-Myc through phosphorylation in regulating cell survival and proliferation. J Biol Chem. 2004;279:40209–40219. doi: 10.1074/jbc.M404056200. [DOI] [PubMed] [Google Scholar]
- 148.Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T, Fesik SW. Solution structure of the antiapoptotic protein bcl-2. Proc Natl Acad Sci U S A. 2001;98:3012–3017. doi: 10.1073/pnas.041619798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ku B, Liang C, Jung JU, Oh BH. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21:627–641. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schmitt E, Beauchemin M, Bertrand R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) during G2/M cell-cycle checkpoint. Oncogene. 2007;26:5851–5865. doi: 10.1038/sj.onc.1210396. [DOI] [PubMed] [Google Scholar]
- 151.Upreti M, Galitovskaya EN, Chu R, Tackett AJ, Terrano DT, Granell S, Chambers TC. Identification of the major phosphorylation site in Bcl-xL induced by microtubule inhibitors and analysis of its functional significance. J Biol Chem. 2008;283:35517–35525. doi: 10.1074/jbc.M805019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Molecular Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 154.Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW, Craig RW, Kaufmann SH, Gores GJ. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem. 2007;282:18407–18417. doi: 10.1074/jbc.M610010200. [DOI] [PubMed] [Google Scholar]
- 155.Jiffar T, Kurinna S, Suck G, Carlson-Bremer D, Ricciardi MR, Konopleva M, Andreeff M, Ruvolo PP. PKC alpha mediates chemoresistance in acute lymphoblastic leukemia through effects on Bcl2 phosphorylation. Leukemia. 2004;18:505–512. doi: 10.1038/sj.leu.2403275. [DOI] [PubMed] [Google Scholar]
- 156.Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, Chang CJ, Yang Y, Lai CC, Lee DF, Yen CJ, Chen YJ, Hsu JM, Kuo HP, Lin CY, Tsai FJ, Li LY, Tsai CH, Hung MC. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008;68:6109–6117. doi: 10.1158/0008-5472.CAN-08-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Low IC, Loh T, Huang Y, Virshup DM, Pervaiz S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56delta stabilizes its antiapoptotic activity. Blood. 2014;124:2223–2234. doi: 10.1182/blood-2014-03-563296. [DOI] [PubMed] [Google Scholar]
- 158.Visco C, Tzankov A, Xu-Monette ZY, Miranda RN, Tai YC, Li Y, Liu WM, d’Amore ES, Li Y, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Wang HY, Dunphy CH, His ED, Zhao XF, Choi WW, Zhao X, van Krieken JH, Huang Q, Ai W, O'Neill S, Ponzoni M, Ferreri AJ, Kahl BS, Winter JN, Go RS, Dirnhofer S, Piris MA, Moller MB, Wu L, Medeiros LJ, Young KH. Patients with diffuse large B-cell lymphoma of germinal center origin with BCL2 translocations have poor outcome, irrespective of MYC status: a report from an International DLBCL rituximab-CHOP Consortium Program Study. Haematologica. 2013;98:255–263. doi: 10.3324/haematol.2012.066209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tanaka S, Louie DC, Kant JA, Reed JC. Frequent incidence of somatic mutations in translocated BCL2 oncogenes of non-Hodgkin's lymphomas. Blood. 1992;79:229–237. [PubMed] [Google Scholar]
- 160.Seto M, Jaeger U, Hockett RD, Graninger W, Bennett S, Goldman P, Korsmeyer SJ. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988;7:123–131. doi: 10.1002/j.1460-2075.1988.tb02791.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, Kluin PM. Double-hit B-cell lymphomas. Blood. 2011;117:2319–2331. doi: 10.1182/blood-2010-09-297879. [DOI] [PubMed] [Google Scholar]
- 162.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, Saksena G, Stransky N, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Hernandez-Lemus E, Schwarz-Cruz y Celis A, Imaz-Rosshandler I, Ojesina AI, Jung J, Pedamallu CS, Lander ES, Habermann TM, Cerhan JR, Shipp MA, Getz G, Golub TR. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, Grunn A, Messina M, Elliot O, Chan J, Bhagat G, Chadburn A, Gaidano G, Mullighan CG, Rabadan R, Dalla-Favera R. Analysis of the coding genome of diffuse large B-cell lymphoma. Nature Genetics. 2011;43:830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, Boyle M, Slack GW, Marra MA, Connors JM, Brooks-Wilson AR, Gascoyne RD. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012;26:1383–1390. doi: 10.1038/leu.2011.378. [DOI] [PubMed] [Google Scholar]
- 166.Casulo C, Burack WR, Friedberg JW. Transformed follicular non-Hodgkin lymphoma. Blood. 2015;125:40–47. doi: 10.1182/blood-2014-04-516815. [DOI] [PubMed] [Google Scholar]
- 167.Bernstein SH, Burack WR. The incidence, natural history, biology, and treatment of transformed lymphomas. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program. 2009:532–541. doi: 10.1182/asheducation-2009.1.532. [DOI] [PubMed] [Google Scholar]
- 168.Montoto S, Fitzgibbon J. Transformation of indolent B-cell lymphomas. J Clin Oncol. 2011;29:1827–1834. doi: 10.1200/JCO.2010.32.7577. [DOI] [PubMed] [Google Scholar]
- 169.Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, Ouillette P, Trifonov V, Rossi D, Tabbo F, Ponzoni M, Chadburn A, Murty VV, Bhagat G, Gaidano G, Inghirami G, Malek SN, Rabadan R, Dalla-Favera R. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–140. doi: 10.1016/j.celrep.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Green MR, Gentles AJ, Nair RV, Irish JM, Kihira S, Liu CL, Kela I, Hopmans ES, Myklebust JH, Ji H, Plevritis SK, Levy R, Alizadeh AA. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood. 2013;121:1604–1611. doi: 10.1182/blood-2012-09-457283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, Boller S, Cittaro D, Bozek M, Iqbal S, Matthews J, Wrench D, Marzec J, Tawana K, Popov N, O'Riain C, O'Shea D, Carlotti E, Davies A, Lawrie CH, Matolcsy A, Calaminici M, Norton A, Byers RJ, Mein C, Stupka E, Lister TA, Lenz G, Montoto S, Gribben JG, Fan Y, Grosschedl R, Chelala C, Fitzgibbon J. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nature genetics. 2014;46:176–181. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Reed JC, Tanaka S. Somatic point mutations in the translocated bcl-2 genes of non-Hodgkin's lymphoGas and lymphocytic leukemias: implications for mechanisms of tumor progression. Leuk Lymphoma. 1993;10:157–163. doi: 10.3109/10428199309145877. [DOI] [PubMed] [Google Scholar]
- 173.McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18) Nature. 1991;349:254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- 174.Smith AJ, Dai H, Correia C, Takahashi R, Lee SH, Schmitz I, Kaufmann SH. Noxa/Bcl-2 protein interactions contribute to bortezomib resistance in human lymphoid cells. J Biol Chem. 2011;286:17682–17692. doi: 10.1074/jbc.M110.189092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Correia C, Schneider PA, Dai H, Dogan A, Maurer MJ, Church AK, Novak AJ, Feldman AL, Wu X, Ding H, Meng XW, Cerhan JR, Slager SL, Macon WR, Habermann TM, Karp JE, Gore SD, Kay NE, Jelinek DF, Witzig TE, Nowakowski GS, Kaufmann SH. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood. 2014 doi: 10.1182/blood-2014-04-571786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Herman MD, Nyman T, Welin M, Lehtio L, Flodin S, Tresaugues L, Kotenyova T, Flores A, Nordlund P. Completing the family portrait of the anti-apoptotic Bcl-2 proteins: crystal structure of human Bfl-1 in complex with Bim. FEBS letters. 2008;582:3590–3594. doi: 10.1016/j.febslet.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 177.Huang DC, Adams JM, Cory S. The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. EMBO J. 1998;17:1029–1039. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hirotani M, Zhang Y, Fujita N, Naito M, Tsuruo T. NH2-terminal BH4 domain of Bcl-2 is functional for heterodimerization with Bax and inhibition of apoptosis. J Biol Chem. 1999;274:20415–20420. doi: 10.1074/jbc.274.29.20415. [DOI] [PubMed] [Google Scholar]
- 179.Monaco G, Decrock E, Nuyts K, Wagner LE, 2nd, Luyten T, Strelkov SV, Missiaen L, De Borggraeve WM, Leybaert L, Yule DI, De Smedt H, Parys JB, Bultynck G. Alpha-helical destabilization of the Bcl-2-BH4-domain peptide abolishes its ability to inhibit the IP3 receptor. PloS one. 2013;8:e73386. doi: 10.1371/journal.pone.0073386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, Stewart ML, Braun CR, Bird GH, Gavathiotis E, Engen JR, Walensky LD. Inhibition of Pro-Apoptotic BAX by a Noncanonical Interaction Mechanism. Molecular Cell. 2015;57:873–886. doi: 10.1016/j.molcel.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Duquette ML, Huber MD, Maizels N. G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer Res. 2007;67:2586–2594. doi: 10.1158/0008-5472.CAN-06-2419. [DOI] [PubMed] [Google Scholar]
- 182.Friedberg JW. Treatment of follicular non-Hodgkin's lymphoma: the old and the new. Sem Hematol. 2008;45:S2–6. doi: 10.1053/j.seminhematol.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Link BK, Maurer MJ, Nowakowski GS, Ansell SM, Macon WR, Syrbu SI, Slager SL, Thompson CA, Inwards DJ, Johnston PB, Colgan JP, Witzig TE, Habermann TM, Cerhan JR. Rates and outcomes of follicular lymphoma transformation in the immunochemotherapy era: a report from the University of Iowa/MayoClinic Specialized Program of Research Excellence Molecular Epidemiology Resource. J Clin Oncol. 2013;31:3272–3278. doi: 10.1200/JCO.2012.48.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 185.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 186.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 187.Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013;12:1691–1700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 188.Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, Sheppard GS, Wang L, Gierke S, Kategaya L, Anderson DJ, Wong C, Eastham-Anderson J, Ludlam MJ, Sampath D, Fairbrother WJ, Wertz I, Rosenberg SH, Tse C, Elmore SW, Souers AJ. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015;145:76–84. doi: 10.1016/j.pharmthera.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Baell JB, Huang DC. Prospects for targeting the Bcl-2 family of proteins to develop novel cytotoxic drugs. Biochem Pharmacol. 2002;64:851–863. doi: 10.1016/s0006-2952(02)01148-6. [DOI] [PubMed] [Google Scholar]
- 191.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, Shangary S, Qiu S, Gao W, Yang D, Meagher J, Stuckey J, Krajewski K, Jiang S, Roller PP, Abaan HO, Tomita Y, Wang S. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem. 2006;49:6139–6142. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]
- 193.Tang G, Nikolovska-Coleska Z, Qiu S, Yang CY, Guo J, Wang S. Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins. J Med Chem. 2008;51:717–720. doi: 10.1021/jm701358v. [DOI] [PubMed] [Google Scholar]
- 194.Paoluzzi L, Gonen M, Gardner JR, Mastrella J, Yang D, Holmlund J, Sorensen M, Leopold L, Manova K, Marcucci G, Heaney ML, O'Connor OA. Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood. 2008;111:5350–5358. doi: 10.1182/blood-2007-12-129833. [DOI] [PubMed] [Google Scholar]
- 195.Arnold AA, Aboukameel A, Chen J, Yang D, Wang S, Al-Katib A, Mohammad RM. Preclinical studies of Apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in Follicular Small Cleaved Cell Lymphoma model. Mol Cancer. 2008;7:20. doi: 10.1186/1476-4598-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Wei J, Stebbins JL, Kitada S, Dash R, Zhai D, Placzek WJ, Wu B, Rega MF, Zhang Z, Barile E, Yang L, Dahl R, Fisher PB, Reed JC, Pellecchia M. An optically pure apogossypolone derivative as potent pan-active inhibitor of anti-apoptotic bcl-2 family proteins. Front Oncol. 2011;1:28. doi: 10.3389/fonc.2011.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, Goulet D, Viallet J, Belec L, Billot X, Acoca S, Purisima E, Wiegmans A, Cluse L, Johnstone RW, Beauparlant P, Shore GC. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–19517. doi: 10.1073/pnas.0709443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Perez-Galan P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 2007;109:4441–4449. doi: 10.1182/blood-2006-07-034173. [DOI] [PubMed] [Google Scholar]
- 199.Shore GC, Viallet J. Modulating the bcl-2 family of apoptosis suppressors for potential therapeutic benefit in cancer. Am Soc Hematol Educ Program. 2005:226–230. doi: 10.1182/asheducation-2005.1.226. [DOI] [PubMed] [Google Scholar]
- 200.Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009;16:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- 201.Espona-Fiedler M, Soto-Cerrato V, Hosseini A, Lizcano JM, Guallar V, Quesada R, Gao T, Perez-Tomas R. Identification of dual mTORC1 and mTORC2 inhibitors in melanoma cells: prodigiosin vs. obatoclax. Biochem Pharmacol. 2012;83:489–496. doi: 10.1016/j.bcp.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 202.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 203.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 204.Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng SC, Nimmer PM, Oltersdorf T, Park CM, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 205.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, Huang DC, Kile BT. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 206.Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR, White MJ, Kruse EA, Lane RM, Ellis S, Nurden P, Mason KD, O'Reilly LA, Roberts AW, Metcalf D, Huang DC, Kile BT. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med. 2011;208:2017–2031. doi: 10.1084/jem.20110750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 208.Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, Maiga S, Bene MC, Moreau P, Pellat-Deceunynck C, Amiot M. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28:210–212. doi: 10.1038/leu.2013.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 211.Anderson MA, Huang D, Roberts A. Targeting BCL2 for the treatment of lymphoid malignancies. Sem Hematol. 2014;51:219–227. doi: 10.1053/j.seminhematol.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 212.Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, Tulpule A, Dunleavy K, Xiong H, Chiu YL, Cui Y, Busman T, Elmore SW, Rosenberg SH, Krivoshik AP, Enschede SH, Humerickhouse RA. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30:488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, Smyth GK, Christie M, Phillipson LJ, Burns CJ, Mann GB, Visvader JE, Lindeman GJ. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell. 2013;24:120–129. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 215.Peirs S, Matthijssens F, Goossens S, Van de Walle I, Ruggero K, de Bock CE, Degryse S, Cante-Barrett K, Briot D, Clappier E, Lammens T, De Moerloose B, Benoit Y, Poppe B, Meijerink JP, Cools J, Soulier J, Rabbitts TH, Taghon T, Speleman F, Van Vlierberghe P. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014;124:3738–3747. doi: 10.1182/blood-2014-05-574566. [DOI] [PubMed] [Google Scholar]
- 216.Ko TK, Chuah CT, Huang JW, Ng KP, Ong ST. The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors. Oncotarget. 2014;5:9033–9038. doi: 10.18632/oncotarget.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mattoo AR, Zhang J, Espinoza LA, Jessup JM. Inhibition of NANOG/NANOGP8 downregulates MCL-1 in colorectal cancer cells and enhances the therapeutic efficacy of BH3 mimetics. Clinical cancer research : an official journal of the American Association for Cancer Res. 2014;20:5446–5455. doi: 10.1158/1078-0432.CCR-14-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Davids MS, Seymour JF, Gerecitano JF, Kahl BS, Pagel JM, Wierda WG, Anderson M-A, Rudersdorf NK, Gressick LA, Montalvo NP, Yang J, Busman TA, Dunbaar M, Cerri E, Enschede SH, Humerickhouse RA, Roberts AW. The Single-Agent Bcl-2 Inhibitor ABT-199 (GDC-0199) In Patients With Relapsed/Refractory (R/R) Non-Hodgkin Lymphoma (NHL): Responses Observed In All Mantle Cell Lymphoma (MCL) Patients. Blood. 2013;122:Abstract 1789. [Google Scholar]
- 219.Seymour JF, Davids MS, Pagel JM, Kahl BS, Wierda WG, Miller TP, Gerecitano JF, Kipps TJ, Anderson M-A, Huang DCS, Rudersdorf NK, Gressick LA, Montalvo NP, Yang J, Busman TA, Dunbar M, Cerri E, Enschede SH, Humerickhouse RA, Roberts AW. Bcl-2 Inhibitor ABT-199 (GDC-0199) Monotherapy Shows Anti-Tumor Activity Including Complete Remissions In High-Risk Relapsed/Refractory (R/R) Chronic Lymphoma Leukemia (CLL) and Small Lymphocytic Lymphoma (SLL) Blood. 2013;122:Abstract 872. [Google Scholar]
- 220.Roberts AW, Ma S, Brander DM, Kipps TJ, Barrientos JC, Davids MS, Anderson M-A, Tam C, Mason-Bright T, Rudersdorf NK, Gressick LA, Yang J, Munasinghe W, Zhu M, Cerri E, Enschee SH, Humerickhouse RA, Seymour JF. Determination of Recommended Phase 2 Dose of ABT-199 (GDC-0199) Combined with Rituximab ® in Patients with Relapse/Refractory (R/R) Chronic Lymphocytic Leukemia (CLL) Blood. 2014;124:Abstract 325. [Google Scholar]
- 221.Flinn I, Brunvand M, Dyer MJS, Hillman P, Jones J, Lymp J, Elhamy M, Vosganian G, Huang J, Kipps TJ. Preliminary Results of a Phase 1b Study (GP28331) Combining GDC-0199 (ABT-199) and Obinutuzumab in Patients with Relapsed/Refractory or Previously Untreated Chronic Lymphocytic Leukemia. Blood. 2014;124:Abstract 4687. [Google Scholar]
- 222.Salles GA, Boyd TE, Morschhauser F, Wendtner C-M, Lymp J, Hilger J, Vosganian G, Huang J, Stilgenbauer S. Preliminary Results of a Phase 1b Study (GO28440) Combining GDC-0199 (ABT-199) with Bendamustine/Rituximab in Patients with Relapsed/Refractory or Previously Untreated Chronic Lymphocytic Leukemia. Blood. 2014;124:Abstract 3337. [Google Scholar]
- 223.de Vos S, Flowers CR, Wang D, Swinnen LJ, Fowler N, Reid E, Gifford M, D'Amico D, Dunbar M, Zhu M, Yang J, Enschede SH, Ricker JL, Chien D, Humerickhouse RA, Kozloff M. The BCL-2 Inhibitor ABT-199 (GDC-0199) in Combination with Bendamustine and Rituximab in Patients with Relapsed or Refractory Non-Hodgkin's Lymphoma. Blood. 2014;124:Abstract 1722. [Google Scholar]
- 224.Grover R, Wilson GD. Bcl-2 expression in malignant melanoma and its prognostic significance. Eur J Surg Oncol. 1996;22:347–349. doi: 10.1016/s0748-7983(96)90176-6. [DOI] [PubMed] [Google Scholar]
- 225.Jiang SX, Sato Y, Kuwao S, Kameya T. Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas. J Pathol. 1995;177:135–138. doi: 10.1002/path.1711770206. [DOI] [PubMed] [Google Scholar]
- 226.Joensuu H, Pylkkanen L, Toikkanen S. Bcl-2 protein expression and long-term survival in breast cancer. Am J Pathol. 1994;145:1191–1198. [PMC free article] [PubMed] [Google Scholar]
- 227.McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, Tu SM, Campbell ML. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992;52:6940–6944. [PubMed] [Google Scholar]
- 228.Sinicrope FA, Hart J, Michelassi F, Lee JJ. Prognostic value of bcl-2 oncoprotein expression in stage II colon carcinoma. Clin Cancer Res. 1995;1:1103–1110. [PubMed] [Google Scholar]
- 229.Gazzaniga P, Gradilone A, Vercillo R, Gandini O, Silvestri I, Napolitano M, Albonici L, Vincenzoni A, Gallucci M, Frati L, Agliano AM. Bcl-2/bax mRNA expression ratio as prognostic factor in low-grade urinary bladder cancer. Int J Cancer. 1996;69:100–104. doi: 10.1002/(SICI)1097-0215(19960422)69:2<100::AID-IJC5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 230.Singh R, Saini N. Downregulation of BCL2 by miRNAs augments drug-induced apoptosis--a combined computational and experimental approach. J Cell Sci. 2012;125:1568–1578. doi: 10.1242/jcs.095976. [DOI] [PubMed] [Google Scholar]
- 231.Kuwano Y, Nishida K, Kajita K, Satake Y, Akaike Y, Fujita K, Kano S, Masuda K, Rokutan K. Transformer 2beta and miR-204 regulate apoptosis through competitive binding to 3' UTR of BCL2 mRNA. Cell Death Differ. 2014 doi: 10.1038/cdd.2014.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Castilla C, Congregado B, Chinchon D, Torrubia FJ, Japon MA, Saez C. Bcl-xL is overexpressed in hormone-resistant prostate cancer and promotes survival of LNCaP cells via interaction with proapoptotic Bak. Endocrinology. 2006;147:4960–4967. doi: 10.1210/en.2006-0502. [DOI] [PubMed] [Google Scholar]
- 233.Keitel U, Scheel A, Thomale J, Halpape R, Kaulfuss S, Scheel C, Dobbelstein M. Bcl-xL mediates therapeutic resistance of a mesenchymal breast cancer cell subpopulation. Oncotarget. 2014;5:11778–11791. doi: 10.18632/oncotarget.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A, Tatsumi T, Ishida H, Noda T, Nagano H, Doki Y, Mori M, Hayashi N. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. 2010;52:698–704. doi: 10.1016/j.jhep.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 235.Zhan M, Qu Q, Wang G, Zhou H. Let-7c sensitizes acquired cisplatin-resistant A549 cells by targeting ABCC2 and Bcl-XL. Pharmazie. 2013;68:955–961. [PubMed] [Google Scholar]
- 236.Chen Y, Jacamo R, Konopleva M, Garzon R, Croce C, Andreeff M. CXCR4 downregulation of let-7a drives chemoresistance in acute myeloid leukemia. J Clin Invest. 2013;123:2395–2407. doi: 10.1172/JCI66553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Guo R, Wang Y, Shi WY, Liu B, Hou SQ, Liu L. MicroRNA miR-491-5p targeting both TP53 and Bcl-XL induces cell apoptosis in SW1990 pancreatic cancer cells through mitochondria mediated pathway. Molecules. 2012;17:14733–14747. doi: 10.3390/molecules171214733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Nakano H, Miyazawa T, Kinoshita K, Yamada Y, Yoshida T. Functional screening identifies a microRNA, miR-491 that induces apoptosis by targeting Bcl-X(L) in colorectal cancer cells. Int J Cancer. 2010;127:1072–1080. doi: 10.1002/ijc.25143. [DOI] [PubMed] [Google Scholar]
- 239.Gutierrez-Castellanos S, Cruz M, Rabelo L, Godinez R, Reyes-Maldonado E, Riebeling-Navarro C. Differences in BCL-X(L) expression and STAT5 phosphorylation in chronic myeloid leukaemia patients. Eur J Haematol. 2004;72:231–238. doi: 10.1046/j.0902-4441.2003.00201.x. [DOI] [PubMed] [Google Scholar]
- 240.Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, Wang HG, Zhang X, Bullrich F, Croce CM, Rai K, Hines J, Reed JC. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998;91:3379–3389. [PubMed] [Google Scholar]
- 241.Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, Rahmani M, Curiel DT, Dmitriev I, Hedvat M, Wei J, Wu B, Stebbins JL, Reed JC, Pellecchia M, Sarkar D, Fisher PB. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs. 2011;20:1397–1411. doi: 10.1517/13543784.2011.609167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19:1248–1252. doi: 10.1038/sj.leu.2403784. [DOI] [PubMed] [Google Scholar]
- 243.Zhuang L, Lee CS, Scolyer RA, McCarthy SW, Zhang XD, Thompson JF, Hersey P. Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod Pathol. 2007;20:416–426. doi: 10.1038/modpathol.3800750. [DOI] [PubMed] [Google Scholar]
- 244.Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, Lee DF, Yang JY, Xie X, Liu JC, Hung MC. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67:4564–4571. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]
- 245.Shigemasa K, Katoh O, Shiroyama Y, Mihara S, Mukai K, Nagai N, Ohama K. Increased MCL-1 expression is associated with poor prognosis in ovarian carcinomas. Jpn J Cancer Res. 2002;93:542–550. doi: 10.1111/j.1349-7006.2002.tb01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Backus HH, van Riel JM, van Groeningen CJ, Vos W, Dukers DF, Bloemena E, Wouters D, Pinedo HM, Peters GJ. Rb, mcl-1 and p53 expression correlate with clinical outcome in patients with liver metastases from colorectal cancer. Ann Oncol. 2001;12:779–785. doi: 10.1023/a:1011112227044. [DOI] [PubMed] [Google Scholar]
- 247.Likui W, Qun L, Wanqing Z, Haifeng S, Fangqiu L, Xiaojun L. Prognostic role of myeloid cell leukemia-1 protein (Mcl-1) expression in human gastric cancer. J Surg Oncol. 2009;100:396–400. doi: 10.1002/jso.21344. [DOI] [PubMed] [Google Scholar]
- 248.Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia BP, Peck-Radosavljevic M, Wacheck V. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol. 2006;44:151–157. doi: 10.1016/j.jhep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 249.Steele R, Mott JL, Ray RB. MBP-1 upregulates miR-29b that represses Mcl-1, collagens, and matrix-metalloproteinase-2 in prostate cancer cells. Genes Cancer. 2010;1:381–387. doi: 10.1177/1947601910371978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 251.Gong J, Zhang JP, Li B, Zeng C, You K, Chen MX, Yuan Y, Zhuang SM. MicroRNA-125b promotes apoptosis by regulating the expression of Mcl-1, Bcl-w and IL-6R. Oncogene. 2013;32:3071–3079. doi: 10.1038/onc.2012.318. [DOI] [PubMed] [Google Scholar]
- 252.Crawford M, Batte K, Yu L, Wu X, Nuovo GJ, Marsh CB, Otterson GA, Nana-Sinkam SP. MicroRNA 133B targets pro-survival molecules MCL-1 and BCL2L2 in lung cancer. Biochem Biophys Res Commun. 2009;388:483–489. doi: 10.1016/j.bbrc.2009.07.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O'Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 254.Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. Mcl-1 ubiquitination and destruction. Oncotarget. 2011;2:239–244. doi: 10.18632/oncotarget.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, Wang J-M, Yang-Yen H-F, Karras J, Jove R, Loughran TP. Inhibition of STAT3 Signaling Leads to Apoptosis of Leukemic Large Granular Lymphocytes and Decreased Mcl-1 Expression. J Clin Invest. 2001;107:351–362. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lee HW, Lee SS, Lee SJ, Um HD. Bcl-w is expressed in a majority of infiltrative gastric adenocarcinomas and suppresses the cancer cell death by blocking stress-activated protein kinase/c-Jun NH2-terminal kinase activation. Cancer Res. 2003;63:1093–1100. [PubMed] [Google Scholar]
- 257.Pritchard DM, Print C, O'Reilly L, Adams JM, Potten CS, Hickman JA. Bcl-w is an important determinant of damage-induced apoptosis in epithelia of small and large intestine. Oncogene. 2000;19:3955–3959. doi: 10.1038/sj.onc.1203729. [DOI] [PubMed] [Google Scholar]
- 258.Wilson JW, Nostro MC, Balzi M, Faraoni P, Cianchi F, Becciolini A, Potten CS. Bcl-w expression in colorectal adenocarcinoma. Br J Cancer. 2000;82:178–185. doi: 10.1054/bjoc.1999.0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough WS, Sloan A, Coons SW, Berens ME. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia. 2005;7:7–16. doi: 10.1593/neo.04535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Lee WS, Woo EY, Kwon J, Park MJ, Lee JS, Han YH, Bae IH. Bcl-w Enhances Mesenchymal Changes and Invasiveness of Glioblastoma Cells by Inducing Nuclear Accumulation of beta-Catenin. PLoS One. 2013;8:e68030. doi: 10.1371/journal.pone.0068030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Yasui K, Mihara S, Zhao C, Okamoto H, Saito-Ohara F, Tomida A, Funato T, Yokomizo A, Naito S, Imoto I, Tsuruo T, Inazawa J. Alteration in copy numbers of genes as a mechanism for acquired drug resistance. Cancer Res. 2004;64:1403–1410. doi: 10.1158/0008-5472.can-3263-2. [DOI] [PubMed] [Google Scholar]
- 262.Valdez BC, Murray D, Ramdas L, de Lima M, Jones R, Kornblau S, Betancourt D, Li Y, Champlin RE, Andersson BS. Altered gene expression in busulfan-resistant human myeloid leukemia. Leuk Res. 2008;32:1684–1697. doi: 10.1016/j.leukres.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Cao J, Cai J, Huang D, Han Q, Yang Q, Li T, Ding H, Wang Z. miR-335 represents an invasion suppressor gene in ovarian cancer by targeting Bcl-w. Oncol Rep. 2013;30:701–706. doi: 10.3892/or.2013.2482. [DOI] [PubMed] [Google Scholar]
- 264.Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang X, Jiang L, Sun Z, Miao Z, Xu H. MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene. 2012;31:1398–1407. doi: 10.1038/onc.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Shen L, Li J, Xu L, Ma J, Li H, Xiao X, Zhao J, Fang L. miR-497 induces apoptosis of breast cancer cells by targeting Bcl-w. Exp Ther Med. 2012;3:475–480. doi: 10.3892/etm.2011.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Bo J, Yang G, Huo K, Jiang H, Zhang L, Liu D, Huang Y. microRNA-203 suppresses bladder cancer development by repressing bcl-w expression. FEBS J. 2011;278:786–792. doi: 10.1111/j.1742-4658.2010.07997.x. [DOI] [PubMed] [Google Scholar]
- 267.Lin CJ, Gong HY, Tseng HC, Wang WL, Wu JL. miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem Biophys Res Commun. 2008;375:315–320. doi: 10.1016/j.bbrc.2008.07.154. [DOI] [PubMed] [Google Scholar]
- 268.Lapham A, Adams JE, Paterson A, Lee M, Brimmell M, Packham G. The Bcl-w promoter is activated by beta-catenin/TCF4 in human colorectal carcinoma cells. Gene. 2009;432:112–117. doi: 10.1016/j.gene.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 269.Nagy B, Lundan T, Larramendy ML, Aalto Y, Zhu Y, Niini T, Edgren H, Ferrer A, Vilpo J, Elonen E, Vettenranta K, Franssila K, Knuutila S. Abnormal expression of apoptosis-related genes in haematological malignancies: overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br J Haematol. 2003;120:434–441. doi: 10.1046/j.1365-2141.2003.04121.x. [DOI] [PubMed] [Google Scholar]
- 270.Mahadevan D, Spier C, Della Croce K, Miller S, George B, Riley C, Warner S, Grogan TM, Miller TP. Transcript profiling in peripheral T-cell lymphoma, not otherwise specified, and diffuse large B-cell lymphoma identifies distinct tumor profile signatures. Mol Cancer Ther. 2005;4:1867–1879. doi: 10.1158/1535-7163.MCT-05-0146. [DOI] [PubMed] [Google Scholar]
- 271.Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F, Costa G, Ruggeri BA, Cheng M, Chiarle R, Palestro G, Neri A, Inghirami G. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCL2A1 as critical target genes. J Clin Invest. 2006;116:3171–3182. doi: 10.1172/JCI29401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, Osorio LM. High expression of bfl-1 contributes to the apoptosis resistant phenotype in B-cell chronic lymphocytic leukemia. Int J Cancer. 2005;113:730–737. doi: 10.1002/ijc.20614. [DOI] [PubMed] [Google Scholar]
- 273.Kucharczak JF, Simmons MJ, Duckett CS, Gelinas C. Constitutive proteasome-mediated turnover of Bfl-1/A1 and its processing in response to TNF receptor activation in FL5.12 pro-B cells convert it into a prodeath factor. Cell Death Differ. 2005;12:1225–1239. doi: 10.1038/sj.cdd.4401684. [DOI] [PubMed] [Google Scholar]
- 274.Herold MJ, Zeitz J, Pelzer C, Kraus C, Peters A, Wohlleben G, Berberich I. The stability and anti-apoptotic function of A1 are controlled by its C terminus. J Biol Chem. 2006;281:13663–13671. doi: 10.1074/jbc.M600266200. [DOI] [PubMed] [Google Scholar]
- 275.Zong WX, Edelstein LC, Chen C, Bash J, Gelinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999;13:382–387. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Morgan RK, Kingham PJ, Walsh MT, Curran DR, Durcan N, McLean WG, Costello RW. Eosinophil adhesion to cholinergic IMR-32 cells protects against induced neuronal apoptosis. J Immunol. 2004;173:5963–5970. doi: 10.4049/jimmunol.173.10.5963. [DOI] [PubMed] [Google Scholar]
- 277.Vogler M. BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 2012;19:67–74. doi: 10.1038/cdd.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Krajewska M, Kitada S, Winter JN, Variakojis D, Lichtenstein A, Zhai D, Cuddy M, Huang X, Luciano F, Baker CH, Kim H, Shin E, Kennedy S, Olson AH, Badzio A, Jassem J, Meinhold-Heerlein I, Duffy MJ, Schimmer AD, Tsao M, Brown E, Sawyers A, Andreeff M, Mercola D, Krajewski S, Reed JC. Bcl-B expression in human epithelial and nonepithelial malignancies. Clin Cancer Res. 2008;14:3011–3021. doi: 10.1158/1078-0432.CCR-07-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.van de Kooij B, Rooswinkel RW, Kok F, Herrebout M, de Vries E, Paauwe M, Janssen GM, van Veelen PA, Borst J. Polyubiquitination and proteasomal turnover controls the anti-apoptotic activity of Bcl-B. Oncogene. 2013;32:5439–5448. doi: 10.1038/onc.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 281.Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, Kipps TJ, Reed JC, Pellecchia M. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L) Chem Biol. 2004;11:389–395. doi: 10.1016/j.chembiol.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 282.Zeitlin BD, Joo E, Dong Z, Warner K, Wang G, Nikolovska-Coleska Z, Wang S, Nor JE. Antiangiogenic effect of TW37, a small-molecule inhibitor of Bcl-2. Cancer Res. 2006;66:8698–8706. doi: 10.1158/0008-5472.CAN-05-3691. [DOI] [PubMed] [Google Scholar]