

Abstract
The impact of statin therapy on plasma asymmetric dimethylarginine (ADMA) levels has not been conclusively studied. Therefore the aim of the meta-analysis was to assess the effect of statins on circulating ADMA levels. We searched selected databases (up to August 2014) to identify randomized controlled trials (RCTs) that investigate the effect of statins on plasma ADMA concentrations. A weighted meta-regression (WMD) using unrestricted maximum likelihood model was performed to assess the impact of statin dose, duration of statin therapy and baseline ADMA concentrations as potential variables on the WMD between statin and placebo group. In total, 1134 participants in 9 selected RCTs were randomized; 568 were allocated to statin treatment and 566 were controls. There was a significant reduction in plasma ADMA concentrations following statin therapy compared with placebo (WMD: − 0.104 μM, 95% confidence interval: − 0.131 to − 0.077, Z = − 7.577, p < 0.0001). Subgroups analysis has shown a significant impact of hydrophilic statins (WMD: − 0.207 μM, 95%CI: − 0.427 to + 0.013, Z = − 7.250, p < .0001) and a non-significant effect of hydrophobic statins (WMD: − 0.101 μM, 95%CI: − 0.128 to − 0.074, Z = − 1.845, p = 0.065). In conclusion, this meta-analysis of available RCTs showed a significant reduction in plasma ADMA concentrations following therapy with hydrophilic statins.
Endothelial dysfunction is an early event in atherogenesis characterized by decreased availability of nitric oxide (NO), which diffuses towards the vascular smooth muscle tissues (VSMCs), triggers a rise of intracellular cyclic guanosine monophosphate (cGMP), leading to vasorelaxation1. Endothelial dysfunction may be associated with increased circulating asymmetric dimethylarginine (ADMA) levels - an L-arginine analogue, which inhibits NO formation2. ADMA is a pan-inhibitor of all 3 NO synthases (NOS) isoforms (potent noncompetitive inhibitor of neuronal NOS and week inhibitor of inducible and endothelial NOS)3 and its plasma concentrations in the general population is 0.4–0.7 μM4.
The first study that showed that middle-aged smoking men in the highest quartile of ADMA levels were at an almost 4-fold risk for acute coronary events was conducted in 20015. Since then, it has been shown that higher ADMA levels are related to increased mortality and adverse clinical outcomes in patients with coronary artery disease (CAD), diabetes, renal disease and ischemic stroke6,7,8,9. Moreover, in the Coronary Artery Risk Determination investigating the Influence of ADMA Concentration (CARDIAC) study, ADMA was shown to be a risk factor for CAD, independently of traditional predictors10.
Formation of NO is regulated by both substrate availability (L-arginine) and the presence of the inhibitor (ADMA), which in turn may be represented by their ratio11. However, the application of L-arginine/ADMA ratio is much limitted due to the fact that L-arginine varies much stronger that ADMA levels in the circulation, and therefore the ratio need not reflect the intracellular situation10. The Hoorn Study showed that systemic inflammation was associated with decreased arginine and increased ADMA plasma levels resulting in an unfavorable NOS substrate-to-inhibitor ratio12.
The interplay of inflammation, endothelial dysfunction, and oxidative stress might play a crucial role in ADMA pathophysiology, and reduction of ADMA levels might be a significant target for preventing endothelial dysfunction13. Statins may provide an effective response to reverse endothelial dysfunction via reduction of ADMA levels; however, the available evidence is not conclusive. Therefore, the aim of this systematic review and meta-analysis was to assess the impact of statins on circulating ADMA levels.
Methods
Data Sources
This study was designed in conformity to the guidelines of the 2009 Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) statement14. Our search included PubMed, Web of Science, Cochrane Library, Scopus and EMBASE databases and was limited to randomized controlled trials (RCTs) carried out from January 1, 1970 to August 1, 2014, investigating the potential effects of statins on circulating ADMA levels. The references of relevant publications were searched and articles of interest were retrieved. The databases were searched using the following search terms in titles and abstracts (also in combination with MESH terms): (rosuvastatin OR pravastatin OR fluvastatin OR simvastatin OR atorvastatin OR pitavastatin OR lovastatin OR cerivastatin OR “statin therapy” OR statins) AND (ADMA OR “asymmetric dimethylarginine”). The wild-card term ‘‘*’’ was used to increase the sensitivity of the search strategy. Two reviewers (CS and AS) evaluated each article separately. Disagreements were resolved by agreement and discussion with a third party (MB). Uncontrolled studies or those with results that did not consider the main objectives of the meta-analysis were omitted.
Study selection
Inclusion criteria
Study design had to meet the following criteria: (1) randomized, placebo-controlled parallel or cross-over trial, (2) population enrolled: adults ≥ 18 years, and, (3) plasma ADMA levels at baseline and after statin administration were available.
Exclusion criteria
The studies were excluded if: (1) had a non-randomized or uncontrolled design, (2) the study was not conducted in statin-treated subjects, (3) no numerical values were presented concerning plasma ADMA levels at baseline and at the end of the study, (3) had duplicate data on ADMA concentrations, (4) we were unable to obtain adequate details of study methodology or results from the article or the investigators, and, (5) the study was an ongoing trial.
Quality assessment
The quality of involved studies in this meta-analysis was evaluated using Jadad scale15. This scale includes randomization (0–2 points), blinding (0–2 points), and dropouts and withdrawals (0–1 point). The overall score of a study in accordance with this scale varies among 0-5, with greater scores as a measure of better quality16. Studies with Jadad scale of ≤ 2 and ≥ 3 were considered as low- and high-quality, respectively17.
Quantitative Data Synthesis
Meta-analysis was conducted using Comprehensive Meta-Analysis (CMA) V2 software (Biostat, NJ). Since all studies used the same methods for the measurement of ADMA levels (plasma levels measured in μM), weighted raw mean difference and 95% confidence interval (CI) was used as summary statistic. Weighting of results was performed using the inverse variance method (Borenstein M, et al. Comprehensive meta-analysis version 2. Engelwood, NJ: Biostat, 2005). Mean difference in measurements was calculated as follows: (measure at end of follow-up in the statin group − measure at baseline in the statin group) − (measure at end of follow-up in the placebo group − measure at baseline in the placebo group). Standard deviations (SDs) of the mean difference were calculated using the following formula: SD = square root [(SDpre-treatment)2 + (SDpost-treatment)2 − (2R × SDpre-treatment × SDpost-treatment)], assuming a correlation coefficient (R) = 0.518. A random-effect model and the generic inverse variance method were used for quantitative data synthesis in order to address the inter-study variations in time of statin type, statin dose and duration of treatment. Pooled effect size was expressed as weighted mean difference (WMD) with 95%CI. In order to evaluate the influence of each study on the overall effect size, sensitivity analysis was conducted using the one-study remove (leave-one-out) approach19,20. In case the values were only presented as graph, the software GetData Graph Digitizer 2.24 (http://getdata-graph-digitizer.com/) was applied to digitize and extract the data; otherwise the authors of the article were contacted to provide numerical values of ADMA concentrations in statin and/or placebo group.
A weighted meta-regression using unrestricted maximum likelihood model was performed to assess the impact of statin dose, duration of statin therapy and baseline ADMA concentrations as potential moderator variables on the WMD in ADMA concentrations between statin and placebo group19,21.
Presence of publication bias was explored graphically using funnel plots of precision (1/standard error) by study effect size (mean difference). Asymmetric funnel plot was further assessed for publication bias using Duval & Tweedie trim-and-fill and classic “fail-safe N” methods, as well as Begg’s rank correlation and Egger’s weighted regression tests18,19.
Results
Search results and trial flow
A summary of the study selection process is shown in Fig. 1. The initial screening for potential relevance excluded articles whose titles and/or abstracts were clearly irrelevant. After removing the trials not assessing the effects of statins in reducing plasma ADMA concentrations, only 17 RCTs met the inclusion criteria and the full-texts were obtained. After assessment 9 articles met the inclusion criteria and were selected for the final meta-analysis.
Figure 1.

Flow chart of number of studies identified and included into the meta-analysis.
Description of studies
In total, 1134 participants in the 9 selected RCTs were randomized; 568 were allocated to statin treatment and 566 were controls. The number of participants in these trials ranged from 53 to 650. The included studies were published between 2003 and 2012, and were conducted in Norway, Taiwan, the Netherlands, Bulgaria, Italy Turkey, New Zealand, China and Finland22,23,24,25,26,27,28,29,30. Statins (pravastatin, rosuvastatin, simvastatin, fluvastatin, and atorvastatin) were administered at doses from 10 to 80 mg/day. Duration of trials ranged between 6 weeks and 24 months. Six trials were designed as parallel-group studies22,25,27,29-31 and 2 trials as cross-over studies23,28. One study26 was a prospective follow-up trial conducted in 3 stages. Demographic and baseline parameters of the included studies are shown in Table 1.
Table 1. Demographic characteristics of the included studies.
| Panichi et al.[22] | Eid et al. [23] | Lu et al.[24] | Nanayakkara et al.[25] | Vladimira-Kitova et al[26] | Oguz et al.[27] | Young et al.[28] | Xia et al. [29] | Janatuinen et al. [30] | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Year | 2008 | 2003 | 2004 | 2009 | 2012 | 2008 | 2008 | 2009 | 2003 | ||
| Jadad score | 3 | 4 | 3 | 3 | 3 | 3 | 3 | 4 | 3 | ||
| Location | Italy | Norway | Taiwan | Netherlands | Bulgaria | Turkey | New Zealand | China | Finland | ||
| Design | Randomized double-blinded placebo-controlled parallel trial | Double blinded, placebo-controlled cross-over trial | Multicenter, randomized, double-blinded, placebo-controlled parallel trial | Secondary analysis of a randomized double-blind placebo-controlled parallel trial | Prospective follow-up randomized controlled trial conducted in three stages | Randomized controlled parallel trial | Randomized double-blinded placebo-controlled cross-over trial | Randomized controlled parallel trial | Randomized double-blinded placebo-controlled parallel trial | ||
| Duration of trial | 6 months | 8 weeks | 6 weeks | 24 months | 3 months | 6 weeks | 6 weeks | 3 months | 6 months | ||
| Inclusion criteria | Patients with chronic kidney diseases (creatinine clearance ranging from 15 to 60 ml/min/1.73 m2) and LDL cholesterol > 100 mg/dL | Men with untreated hypercholesterolemia | Patients with hypercholesterolemia with fasting plasma LDL cholesterol > 160 mg/dl and triglyceride levels < 350 mg/dl after an initial 6 weeks of diet control | Patients with creatinine clearance of 15 to 70 mL/min/1.73 m2 (according to the Cockcroft-Gault equation) | Patients over 16 years of age with severe hypercholesterolemia defined as fasting total cholesterol level ≥ 7.5 mmol/l and LDL-C level of ≥ 4.9 mmol/l and a family history of premature atherosclerosis. | Patients over 20 years of age with diagnosis of metabolic syndrome, a LDL cholesterol level between 100-160 mg/dL, and a triglyceride level lower than 400 mg/dL. | Patients with symptomatic heart failure (ejection fraction < 40%, New York Heart Association Functional Classes II and III) | Patients consecutively subjected to elective electrical cardioversion to treat persistent atrial fibrillation ( > 48 h). | Men aged 25–40 years; total cholesterol levels 5.5–9.0 mmol/l measured previously at routine controls provided by employers; otherwise healthy; no continuous medication or use of antioxidant vitamins. | ||
| Statin intervention | Simvastatin 40 mg/day | Pravastatin 40 mg/day | Rosuvastatin 10 mg/day | Pravastatin 40 mg/day | Simvastatin 40 mg/day | Simvastatin 80 mg/day | Fluvastatin 80 mg/day | Atorvastatin 40 mg/day | Rosuvastatin 10 mg/day | Pravastatin 40 mg/day | |
| Participants | Treatment | 20 | 32 | 23 | 46 | 325 | 120 | 42 | 23 | 32 | 25 |
| Control | 15 | 32 | 23 | 47 | 325 | 120 | 43 | 23 | 32 | 26 | |
| Age (years) | Treatment | 60 ± 12 | 33-71 | 62.8 ± 11.2 | 54 ± 11 | 46 ± 4 | 46 ± 3 | 55.50 ± 10.46 | 60.7 ± 10.4 | 62.28 ± 8.55 | 35.7 ± 3.6 |
| Control | 58 ± 11 | 33-71 | 59.8 ± 11.8 | 52 ± 13 | 46 ± 2 | 46 ± 2 | 56.16 ± 7.56 | 60.7 ± 10.4 | 60.72 ± 8.21 | 34.6 ± 4.3 | |
| Male (%) | Treatment | 70.0 | 100.0 | 43.5 | 52.1 | 51.1 | 46.7 | 38.1 | NS | 68.7 | 100.0 |
| Control | 60.0 | 100.0 | 73.9 | 61.7 | 52.3 | 49.2 | 37.2 | NS | 62.5 | 100.0 | |
| BMI (kg/m2) | Treatment | 25.1 ± 3.0 | NS | 25.3 ± 2.7 | 27 ± 5 | 25 ± 2 | 24 ± 4 | NS | NS | 23.74 ± 2.26 | 25.3 ± 2.8 |
| Control | 24.9 ± 2.3 | NS | 24.8 ± 2.9 | 26 ± 4 | 25 ± 3 | 25 ± 2 | NS | NS | 23.49 ± 2.20 | 24.6 ± 1.8 | |
| Baseline plasma ADMA concentration (μM) | Treatment | 0.90 ± 0.10 | 1.50 (1.18, 1.75)* | 0.60 ± 0.19 | 0.53 ± 0.06 | 1.17 ± 0.15 | 1.26 ± 0.38 | 1.57 ± 1.07 | NS | 1.60 ± 0.41 | 0.38 ± 0.18 |
| Control | 0.74 ± 0.12 | 1.64 (1.24, 1.75)* | 0.54 ± 0.14 | 0.53 ± 0.09 | 1.16 ± 0.17 | 1.25 ± 0.21 | 1.17 ± 1.41 | NS | 1.58 ± 0.40 | 0.42 ± 0.15 |
Values are expressed as mean ± SD. *Median values and 25, 75 percentiles are given;
ABBREVIATIONS: BMI: body mass index; LDL-C: low-density lipoprotein cholesterol; NA: not applicable, NS: not stated.
Quantitative data synthesis
Combining results of retrieved RCTs indicated a significant reduction in plasma ADMA concentrations following treatment with statins compared with placebo (WMD: − 0.104 μM, 95%CI: − 0.131 to − 0.077, Z = − 7.577, p < 0.0001). Forest plots detailing the meta-analysis of RCTs assessing the impact of statin therapy on plasma ADMA levels is illustrated in Fig. 2. Subgroup analysis revealed a significant impact of hydrophilic statins (rosuvastatin, pravastatin and fluvastatin; n = 403; WMD: − 0.207 μM, 95%CI: − 0.427 to + 0.013, Z = − 7.250, p < 0.0001), and a non-significant effect of hydrophobic statins (simvastatin and atorvastatin; n = 321; WMD: − 0.101 μM, 95%CI: − 0.128 to − 0.074, Z = − 1.845, p = 0.065) (Fig. 3).
Figure 2.
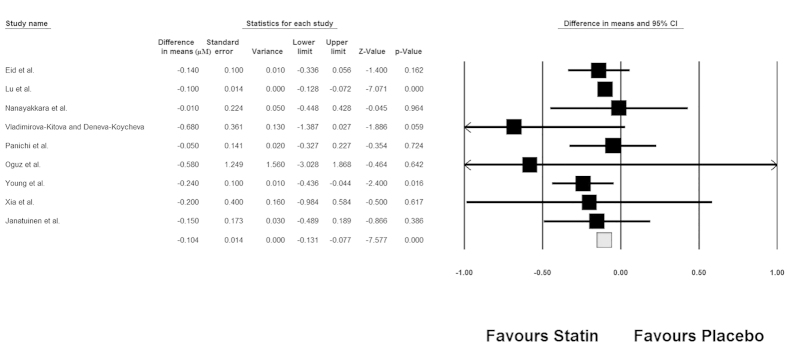
Forest plot detailing weighted mean difference and 95% confidence intervals for the impact of statin therapy on plasma concentrations of ADMA. Meta-analysis was performed using a random-effect model with inverse variance weighting.
Figure 3.

Forest plot detailing weighted mean difference and 95%Cl for the impact of hydrophilic (left) and hydrophobic (right) statins on plasma concentrations of ADMA. Meta-analysis was performed using a random-effect model with inverse variance weighting.
The strength of the pooled estimate was robust and did not significantly differ according to the characteristics of individual studies in the leave-one-out sensitivity analysis. The results of sensitivity analysis are summarized in Fig. 4.
Figure 4.
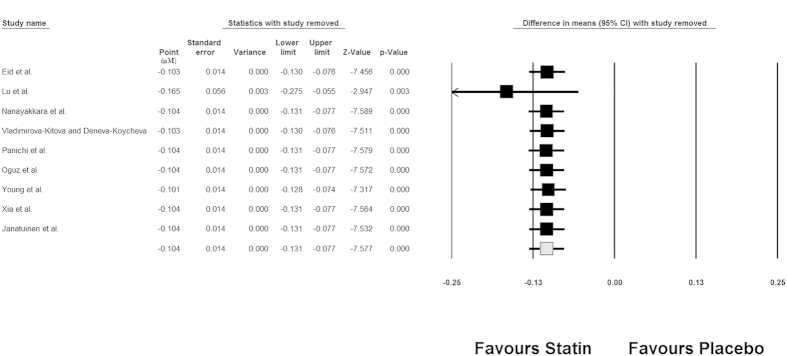
Leave-one-out sensitivity analysis for the impact of statin therapy on plasma concentrations of ADMA.
Meta-regression
Weighted unrestricted maximum likelihood meta-regression analysis was performed to assess the impact of potential moderators on the pooled effect size. None of the moderator parameters i.e. statin dose (slope: − 0.003; 95%CI: − 0.006 to 0.001; p = 0.164), duration of statin therapy (slope: 0.001; 95%CI: − 0.008 to 0.011; p = 0.774) and baseline ADMA concentrations (slope: − 0.083; − 0.275 to 0.109; p = 0.399) was significantly associated with the pooled estimate of the statin effect on plasma ADMA concentrations. Meta-regression bubble plots are illustrated in Supplementary Figure S1.
Publication bias
Visual inspection of funnel plot asymmetry suggested potential publication bias for the effects of statin therapy on plasma ADMA concentrations (Supplementary Figure S2). Imputation of theoretically missed studies using Duval and Tweedie’s trim-and-fill method added 3 studies, leading to an imputed WMD of − 0.100 (95%CI: − 0.127 to − 0.074) which was still significant (Supplementary Figure S2). The “fail safe N” test showed that 50 theoretically missing studies would be needed to add to the analysis of the effect of statin therapy on plasma ADMA concentrations in order to yield a statistically non − significant overall effect. Likewise, Begg’s rank correlation test (Kendall’s Tau with continuity correction = − 0.167, Z = 0.626, two-tailed p = 0.532) and Egger’s linear regression tests suggested no evidence of publication bias (intercept = − 0.478, 95%CI = − 1.150 to 0.195, t = 1.679, df = 7.00, two-tailed p = 0.137).
Discussion
To our knowledge this meta-analysis is the first that assessed the effects of statin therapy on plasma levels of ADMA. The findings provide a thorough synthesis of results from available RCTs and showed a significant reduction in plasma ADMA concentrations. Additionally, statin therapy was examined by class: hydrophobic (simvastatin and atorvastatin), that might be dispersed at low levels throughout human tissues and hydrophilic (pravastatin, rosuvastatin and fluvastatin) that functions mainly in the liver and are present in the circulation31,32. In our meta-analysis hydrophilic statins (rosuvastatin, pravastatin and fluvastatin) had a significant impact on ADMA levels while hydrophobic statins (simvastatin and atorvastatin) non-significantly reduced ADMA levels.
Currently ADMA is considered a prognostic marker of cardiovascular disease and mortality6. The available data also suggests that ADMA has been involved in systemic vascular inflammation through induction of reactive oxygen species (ROS) in endothelial cells32. In patients undergoing coronary bypass surgery, it was observed that ADMA levels were correlated with elevated NOS-derived generation of ROS33. Furthermore, it has been shown that ROS upregulate ADMA synthesis and protein arginine N-methyltransferase expression34. In cell culture studies, it has been shown that pro-oxidant and pro-inflammatory stimulants inhibit dimethylarginine dimethylaminohydrolase (DDAH) activity35. Decreased DDAH, the enzyme responsible for ADMA degeneration, is generally followed by the consecutive decrease of NOS activity, increase of ADMA concentrations and development of atherosclerosis36,37. However, it should also be mentioned that there are some doubts on the ADMA/DDAH association – e.g. DDAH activity is not associated with oxidative stress in the elderly patients with peripheral arterial occlusive disease38,39. In human monocytic cells, ADMA induces tumor necrosis factor (TNF)-α production via the inhibitory effect of reinioside C and ROS/nuclear factor (NF)–κB dependent pathways40. Since both ROS and systemic inflammation are responsible for increased ADMA levels, and statins are recognized as anti-inflammatory and antioxidant agents41, the hypothesis was that statin therapy might decrease ADMA levels. Indeed, several smaller studies have shown that statin therapy reduces ADMA levels 25,42, however other studies with high dose statins (e.g. simvastatin 80 mg/day or atorvastatin 40 mg/day) did not decrease plasma ADMA levels43. It seems that our meta-analysis provides the answer to the question on the role of statins on ADMA levels (mainly hydrophilic), irrespective of the statins doses and therapy duration. These results also show the marginal or lack of effect of simvastatin and atorvastatin (hydrophobic statins) on plasma ADMA levels43.
There are few hypotheses on how statins influence ADMA levels. One of them concerns the inhibition of ADMA-induced inflammatory reaction, modulated by mitogen-activated protein kinase (MAPK) pathway in human endothelial cells44. Statins also activate the transcription factor sterol response element binding protein (SREBP) through decreasing content of the cholesterol in the membrane45. SREBP specifically enhances the expression of more than 30 genes associated with the synthesis and uptake of fatty acids, phospholipids, cholesterol and triglycerides46. One of its isoforms - nuclear SREBP-2 increases the transcription of proprotein convertase subtilisin/kexin type 9 (PCSK9)47. It has been shown that statins upregulate both PCSK9 mRNA levels and LDLR via activation of sterol-mediated SREBP-2, an important activator of DDAH transcription and activity48. Since reduced DDAH activity is linked to endothelial dysfunction, we speculate that statin therapy might decrease ADMA levels through multiple mechanisms such as activation of sterol-mediated SREBP-2, increasing of transcription of PCSK9 or by decreasing ADMA-induced inflammatory reaction, modulated by MAPK49,50.
This meta-analysis has several limitations. Most importantly, the eligible RCTs usually had small populations and short follow-up (up to 6 months in 8/9 included studies). The included studies were also heterogeneous with regards to population characteristics (there were patients with hyperlipidemia, renal failure or atrial fibrillation), study design, and statin preparation and dose. In order to cover these variabilities we used a more conservative random-effects model and performed the sensitivity analysis. The meta-regression analysis also revealed that none of the moderator parameters i.e. statin dose, duration of statin therapy and baseline ADMA concentrations were significantly associated with the pooled estimate of statin effect on plasma ADMA concentrations. Finally, the smoking status, an important determinant of ADMA levels (as well as other variables, such as: hyperhomocysteinemia, hypertension, coronary artery disease, heart failure, and administration of the following drugs: antioxidants, estrogen, vitamin A, angiotensin converting enzyme inhibitors, angiotensin AT1 receptor antagonists, and beta-adrenoreceptor blocking drugs), could not be considered in this meta-analysis due to lack of data.
In conclusion, this meta-analysis of RCTs showed a significant reduction in plasma ADMA concentrations following hydrophilic statin therapy. These results might reveal an additional benefit of statins, which might contribute to the observed reduction of cardiovascular risk. Larger, well-designed studies involving smoking status are needed to validate our findings.
Author Contributions
CS - designed the study, made the literature search, drafted the manuscript, prepared the revised version; AS - designed the study, made the statistical analysis, corrected the draft of the paper; SU - made the statistical analysis, drafted the manuscript; DPM, MR, GYHL, GKH, JJPK, LK, JR - corrected the draft of the paper and the revised version; MB - designed the study, made the literature search, drafted the manuscript, prepared the revised version, submitted the paper.
Additional Information
How to cite this article: Serban, C. et al. A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations Sci. Rep. 5, 09902; doi: 10.1038/srep09902 (2015).
Supplementary Material
Supplementary Figures
Acknowledgments
The meta-analysis has been prepared within Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group (www.lbpmcgroup.umed.pl)
References
- Margaritis M., Channon K. M. & Antoniades C. Statins as regulators of redox state in the vascular endothelium: beyond lipid lowering. Antioxidants & redox signaling 20, 1198–1215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger R. H. & Ron E. S. L-Arginine improves vascular function by overcoming deleterious effects of ADMA, a novel cardiovascular risk factor. Alternative medicine review : a journal of clinical therapeutic 10, 14–23 (2005). [PubMed] [Google Scholar]
- Kielstein A., Tsikas D., Galloway G. P. & Mendelson J. E. Asymmetric dimethylarginine (ADMA)—A modulator of nociception in opiate tolerance and addiction? Nitric oxide: biology and chemistry/official journal of the Nitric Oxide Society 17, 55–59 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekarova M. et al. Asymmetric dimethylarginine regulates the lipopolysaccharide-induced nitric oxide production in macrophages by suppressing the activation of NF-kappaB and iNOS expression. European journal of pharmacology 713, 68–77, doi: 10.1016/j.ejphar.2013.05.001 (2013). [DOI] [PubMed] [Google Scholar]
- Valkonen V.-P. et al. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. The Lancet 358, 2127–2128 (2001). [DOI] [PubMed] [Google Scholar]
- Böger R. H., Maas R., Schulze F. & Schwedhelm E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—an update on patient populations with a wide range of cardiovascular risk. Pharmacological Research 60, 481–487 (2009). [DOI] [PubMed] [Google Scholar]
- Kielstein J. T., Frolich J. C., Haller H. & Fliser D. ADMA (asymmetric dimethylarginine): an atherosclerotic disease mediating agent in patients with renal disease? Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association 16, 1742–1745 (2001). [DOI] [PubMed] [Google Scholar]
- Siegerink B. et al. Asymmetric and symmetric dimethylarginine and risk of secondary cardiovascular disease events and mortality in patients with stable coronary heart disease: the KAROLA follow-up study. Clinical research in cardiology : official journal of the German Cardiac Society 102, 193–-202., doi: 10.1007/s00392-012-0515-4 (2013). [DOI] [PubMed] [Google Scholar]
- Anderssohn M., Schwedhelm E., Luneburg N., Vasan R. S. & Boger R. H. Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diabetes & vascular disease research : official journal of the International Society of Diabetes and Vascular Disease 7, 105-118 , doi: 10.1177/1479164110366053 (2010). [DOI] [PubMed] [Google Scholar]
- Schulze F. et al. Asymmetric dimethylarginine is an independent risk factor for coronary heart disease: results from the multicenter Coronary Artery Risk Determination investigating the Influence of ADMA Concentration (CARDIAC) study. American heart journal 152, 493. e491-493. e498 (2006). [DOI] [PubMed] [Google Scholar]
- Bode-Böger S. M., Scalera F. & Ignarro L. J. The L-arginine paradox: importance of the L-arginine/asymmetrical dimethylarginine ratio. Pharmacology & therapeutics 114, 295–306 (2007). [DOI] [PubMed] [Google Scholar]
- van der Zwan L. et al. Systemic inflammation is linked to low arginine and high ADMA plasma levels resulting in an unfavourable NOS substrate-to-inhibitor ratio: the Hoorn Study. Clinical science 121, 71–78 (2011). [DOI] [PubMed] [Google Scholar]
- Landim M. B. P., Casella Filho A. & Chagas A. C. P. Asymmetric dimethylarginine (ADMA) and endothelial dysfunction: implications for atherogenesis. Clinics 64, 471–478 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D., Liberati A., Tetzlaff J., Altman D. G. & Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. International journal of surgery 8, 336–341, 10.1016/j.ijsu.2010.02.007 (2010). [DOI] [PubMed] [Google Scholar]
- Jadad A. R. et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary? Control Clin Trials 17, 1–12 (1996). [DOI] [PubMed] [Google Scholar]
- Jadad A. R. The merits of measuring the quality of clinical trials: is it becoming a Byzantine discussion? Transplant international : official journal of the European Society for Organ Transplantation 22, 1028, 10.1111/j.1432-2277.2009.00919.x (2009). [DOI] [PubMed] [Google Scholar]
- Moher D. et al. Assessing the quality of reports of randomised trials: implications for the conduct of meta-analyses. Health technology assessment (Winchester, England) 3, i-iv, 1–98 (1998). [PubMed] [Google Scholar]
- Sahebkar A. Effects of resveratrol supplementation on plasma lipids: a systematic review and meta-analysis of randomized controlled trials. Nutrition reviews 71, 822–835, doi: 10.1111/nure.12081 (2013). [DOI] [PubMed] [Google Scholar]
- Higgins J. P. & Green S. Cochrane handbook for systematic reviews of interventions . Vol. 5 Wiley Online Library 2008). [Google Scholar]
- Sahebkar A. Are curcuminoids effective C-reactive protein-lowering agents in clinical practice? Evidence from a meta-analysis. Phytotherapy research : PTR 28, 633–642, 10.1002/ptr.5045 (2014). [DOI] [PubMed] [Google Scholar]
- Sahebkar A. Does PPARgamma2 gene Pro12Ala polymorphism affect nonalcoholic fatty liver disease risk? Evidence from a meta-analysis. DNA and cell biology 32, 188–198, doi: 10.1089/dna.2012.1947 (2013). [DOI] [PubMed] [Google Scholar]
- Panichi V. et al. Effect of simvastatin on plasma asymmetric dimethylarginine concentration in patients with chronic kidney disease. Journal of nephrology 21, 38–44 (2008). [PubMed] [Google Scholar]
- Eid H. M., Eritsland J., Larsen J., Arnesen H. & Seljeflot I. Increased levels of asymmetric dimethylarginine in populations at risk for atherosclerotic disease. Effects of pravastatin. Atherosclerosis 166, 279–284 (2003). [DOI] [PubMed] [Google Scholar]
- Lu T. M. et al. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. The American journal of cardiology 94, 157–161, doi: 10.1016/j.amjcard.2004.03.052 (2004). [DOI] [PubMed] [Google Scholar]
- Nanayakkara P. W. et al. Randomized placebo-controlled trial assessing a treatment strategy consisting of pravastatin, vitamin E, and homocysteine lowering on plasma asymmetric dimethylarginine concentration in mild to moderate CKD. American journal of kidney diseases : the official journal of the National Kidney Foundation 53, 41–50, 10.1053/j.ajkd.2008.06.016 (2009). [DOI] [PubMed] [Google Scholar]
- Vladimirova-Kitova L. G. & Deneva-Koycheva T. I. The effect of simvastatin on asymmetric dimethylarginine and flow-mediated vasodilation after optimizing the LDL level: a randomized, placebo-controlled study. Vascular pharmacology 56, 122–130, doi: 10.1016/j.vph.2011.10.004 (2012). [DOI] [PubMed] [Google Scholar]
- Oguz A. & Uzunlulu M. Short term fluvastatin treatment lowers serum asymmetric dimethylarginine levels in patients with metabolic syndrome. International heart journal 49, 303–311 (2008). [DOI] [PubMed] [Google Scholar]
- Young J. M. et al. Effect of atorvastatin on plasma levels of asymmetric dimethylarginine in patients with non-ischaemic heart failure. European journal of heart failure 10, 463–466, doi: 10.1016/j.ejheart.2008.03.010 (2008). [DOI] [PubMed] [Google Scholar]
- Xia W., Yin Z., Li J., Song Y. & Qu X. Effects of rosuvastatin on asymmetric dimethylarginine levels and early atrial fibrillation recurrence after electrical cardioversion. Pacing and clinical electrophysiology :PACE 32, 1562–1566, 10.1111/j.1540-8159.2009.02554.x (2009). [DOI] [PubMed] [Google Scholar]
- Janatuinen T. et al. Plasma asymmetric dimethylarginine modifies the effect of pravastatin on myocardial blood flow in young adults. Vascular medicine 8, 185–189 (2003). [DOI] [PubMed] [Google Scholar]
- Lu T.-M. et al. Effect of<i>rosuvastatin</i>on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. The American journal of cardiology 94, 157–161 (2004). [DOI] [PubMed] [Google Scholar]
- Mangoni A. A. The emerging role of symmetric dimethylarginine in vascular disease. Advances in clinical chemistry 48, 73–94 (2009). [DOI] [PubMed] [Google Scholar]
- Antoniades C. et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. European heart journal 30, 1142-1150 , doi: 10.1093/eurheartj/ehp061 (2009). [DOI] [PubMed] [Google Scholar]
- Antoniades C. et al. Role of asymmetrical dimethylarginine in inflammation-induced endothelial dysfunction in human atherosclerosis. Hypertension 58, 93–98, doi: 10.1161/HYPERTENSIONAHA.110.168245 (2011). [DOI] [PubMed] [Google Scholar]
- Tran C. T., Leiper J. M. & Vallance P. The DDAH/ADMA/NOS pathway. Atherosclerosis. Supplements 4, 33–40 (2003). [DOI] [PubMed] [Google Scholar]
- Ito A. et al. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation 99, 3092–3095 (1999). [DOI] [PubMed] [Google Scholar]
- Wadham C. & Mangoni A. A. Dimethylarginine dimethylaminohydrolase regulation: a novel therapeutic target in cardiovascular disease. Expert opinion on drug metabolism & toxicology 5, 303–319, doi: 10.1517/17425250902785172 (2009). [DOI] [PubMed] [Google Scholar]
- Schneider J. Y., Pham V. V., Froelich J. C. & Tsikas D. DDAH activity is not associated with oxidative stress in elderly patients with peripheral arterial occlusive disease. Experimental gerontology 55, 159 (2014). [DOI] [PubMed] [Google Scholar]
- Tsikas D. & Chobanyan K. Pitfalls in the measurement of tissue DDAH activity: is DDAH sensitive to nitrosative and oxidative stress&quest. Kidney international 74, 969–969 (2008). [DOI] [PubMed] [Google Scholar]
- Zhang G. G. et al. Asymmetric dimethylarginine induces TNF-alpha production via ROS/NF-kappaB dependent pathway in human monocytic cells and the inhibitory effect of reinioside C. Vascular pharmacology 48, 115–121, doi: 10.1016/j.vph.2008.01.004 (2008). [DOI] [PubMed] [Google Scholar]
- Aydin U. et al. Effects of atorvastatin on vascular intimal hyperplasia: an experimental rodent model. Angiology 60, 370–377, doi: 10.1177/0003319708321102 (2009). [DOI] [PubMed] [Google Scholar]
- Grosso A. F. et al. Synergistic anti-inflammatory effect: simvastatin and pioglitazone reduce inflammatory markers of plasma and epicardial adipose tissue of coronary patients with metabolic syndrome. Diabetology & metabolic syndrome 6, 47, doi: 10.1186/1758-5996-6-47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkonen V. P. et al. Asymmetrical dimethylarginine (ADMA) and risk of acute coronary events. Does statin treatment influence plasma ADMA levels ? Atherosclerosis. Supplements 4, 19–22 (2003). [DOI] [PubMed] [Google Scholar]
- Jiang J. L. et al. The inhibitory effect of simvastatin on the ADMA-induced inflammatory reaction is mediated by MAPK pathways in endothelial cells. Biochemistry and cell biology=Biochimie et biologie cellulaire 85, 66–77, doi: 10.1139/o06-146 (2007). [DOI] [PubMed] [Google Scholar]
- Ivashchenko C. Y. et al. Regulation of the ADMA-DDAH system in endothelial cells: a novel mechanism for the sterol response element binding proteins, SREBP1c and -2. American journal of physiology. Heart and circulatory physiology 298, H251–258, 10.1152/ajpheart.00195.2009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120, 261–273, 10.1016/j.cell.2004.11.043 (2005). [DOI] [PubMed] [Google Scholar]
- Dragan S., Serban M. C. & Banach M. Proprotein Convertase Subtilisin/Kexin 9 Inhibitors: An Emerging Lipid-Lowering Therapy? Journal of cardiovascular pharmacology and therapeutics , doi: 10.1177/1074248414539562 (2014). [DOI] [PubMed] [Google Scholar]
- Rashid S. et al. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proceedings of the National Academy of Sciences of the United States of America 102, 5374–5379, doi: 10.1073/pnas.0501652102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkacz A. et al. Plasma asymmetric dimethylarginine predicts restenosis after coronary angioplasty. Archives of medical science : AMS 7, 444–448, 10.5114/aoms.2011.23410 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goch A., Banach M., Mikhailidis D. P., Rysz J. & Goch J. H. Endothelial dysfunction in patients with noncomplicated and complicated hypertension. Clinical and experimental hypertension 31, 20–30, doi: 10.1080/10641960802409846 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures