

Abstract
Cell-targeted therapies (smart drugs), which selectively control cancer cell progression with limited toxicity to normal cells, have been developed to effectively treat some cancers. However, many cancers such as metastatic prostate cancer (PC) have yet to be treated with current smart drug technology. Here, we describe the thorough preclinical characterization of an RNA aptamer (A9g) that functions as a smart drug for PC by inhibiting the enzymatic activity of prostate-specific membrane antigen (PSMA). Treatment of PC cells with A9g results in reduced cell migration/invasion in culture and metastatic disease in vivo. Importantly, A9g is safe in vivo and is not immunogenic in human cells. Pharmacokinetic and biodistribution studies in mice confirm target specificity and absence of non-specific on/off-target effects. In conclusion, these studies provide new and important insights into the role of PSMA in driving carcinogenesis and demonstrate critical endpoints for the translation of a novel RNA smart drug for advanced stage PC.
Introduction
Upon initial diagnosis of locally advanced prostate cancer (PC), many patients currently receive endocrine therapy (e.g., androgen ablation/castration). However, in most cases, the cancer will only respond to this treatment for a limited time and most patients will eventually develop metastatic castration-resistant prostate cancer (mCRPC).1 mCRPC is invariably fatal with a mean survival time of 30 months following diagnosis.1 Patients who progress following medical castration are offered standard chemotherapy (e.g., docetaxel or cabazitaxel) and/or novel treatment options directed against the androgen receptor (AR) signaling pathway which was recently shown to continue to play an essential role in mCRPC.1 The new AR signaling pathway inhibitors include abiraterone (an inhibitor of the hepatic drug-metabolizing enzyme CYP2D6 involved in androgen synthesis) and enzalutamide (an inhibitor of the AR signaling pathway). Although these drugs result in significant survival prolongation, they are not yet curative therapies as their median duration of efficacy is around 6–12 months each.1 Furthermore, these drugs are not cell-specific (e.g., do not selectively kill or inhibit cancer cells) and thus often result in toxicity to normal tissues (e.g., hepatotoxicity, hypertension, uneven heart rate, hypokalemia, peripheral edema, muscle pain, diarrhea, and respiratory infections).1,2,3 Considering this current landscape, new therapeutic options that selectively target mCRPC are needed to increase the efficacy and safety benefits to patients with advanced stage disease.
A promising molecular marker for targeted therapy of PC is prostate-specific membrane antigen (PSMA). PSMA is a glycosylated type-II membrane protein which is primarily expressed in prostatic epithelial cells, though low levels of expression can also be observed in kidney, proximal small intestine, salivary gland, and brain.4,5 Predominantly, normal prostatic epithelial cells express an alternative spliced variant of PSMA that lacks the transmembrane domain (PSM') and is found in the cytoplasm. However, during malignant transformation, PC cells greatly upregulate the expression of the transmembrane isoform, resulting in abnormally high levels of PSMA on the cell surface.6,7,8 PSMA cell-surface expression has also been associated with more aggressive disease and with circulating tumor cells, increasing its attractiveness as a target for imaging and therapy in late-stage mCRPC patients.4,6,9,10,11 Of note, elevated PSMA expression has also been reported in the neovasculature of many solid tumors expanding its therapeutic profile.12,13 Given PSMA's potential for targeted therapy, we and others have sought to develop therapies and diagnostics that selectively target PC via PSMA. Examples of PSMA-targeted approaches include delivery of radionuclides,4,14 small molecule drugs/toxins,15 and therapeutic siRNAs16,17,18,19 to PSMA-expressing PC cells with antibodies, small molecule inhibitors, and RNA aptamers.
A less explored aspect of PSMA as a target for PC therapy is its enzymatic activity.20,21 Although several studies have suggested a role for PSMA's enzymatic activity in cancer progression, PSMA inhibitors have not been evaluated as potential therapeutics for PC.22,23,24 Thus, we sought to explore whether inhibition of PSMA's enzymatic activity would result in inhibition of PC metastases. We previously described the clinical optimization of a PSMA inhibitory RNA aptamer, A9g.25 This aptamer is a derivative of the A9 aptamer26 that was rationally truncated to enhance its target specificity (and minimize off-target effects) and enable large-scale chemical synthesis for therapeutic applications.25 In recent years, aptamers with similar chemical compositions have proven valuable for a variety of clinical applications, highlighting their potential as a therapeutic platform.27
Here, we show that A9g inhibits PC cell migration and invasion in vitro and exhibits potent antimetastatic activity in a mouse model of metastatic PC when administered intravenously. Importantly, A9g is selective for PC cells expressing PSMA and has no observed toxicity in normal mouse tissues or in human peripheral blood mononuclear cells (PBMCs). In addition, we present thorough pharmacokinetic (PK) and biodistribution data toward understanding the in vivo disposition of this new class of RNA-based smart drugs prior to testing in humans. As a whole, these preclinical studies provide critical endpoints for the clinical translation of a novel cell-targeted RNA smart drug for managing mCRPC. To the best of our knowledge, this is the first demonstration that an inhibitor of PSMA's enzymatic activity has therapeutic efficacy against advanced stage PC.
Results
Effect of PSMA aptamers on cancer cell growth, migration/invasion, and survival in vitro
Given PSMA's proposed role in driving carcinogenesis,22,23,28 we assessed the effect of PSMA expression on proliferation, migration, invasion, and survival of cancer cells in culture (Figure 1 and Supplementary Figures S1 and S2). Cell proliferation was evaluated over a 5-day period using an MTS (3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium) assay. Human PC cells lacking PSMA expression (PC-3(PSMA−)) were compared to PC-3 cells engineered to stably express PSMA (PC-3(PSMA+)) (Figure 1a). PC-3(PSMA+) cells had a clear growth advantage over cells lacking PSMA with a 1.8-fold increase in the rate of proliferation (Figure 1a, see inset). These data suggest that PSMA expression confers PC cells a growth advantage.
Figure 1.
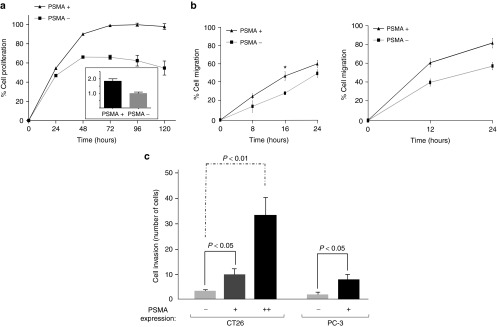
PSMA expression drives cancer cell proliferation, migration and invasion. (a) Proliferation of PC-3(PSMA-) and PC-3(PSMA+) cells grown in culture determined by MTS assay. (Inset) Fold increase in the rate of proliferation from 24 to 48 hours (b) (Left panel) Migration of PC-3(PSMA-) and PC-3(PSMA+) cells determined by scratch-wound assay. (Right panel) Migration of CT26(PSMA−) and CT26(PSMA+) cells determined by scratch-wound assay. *P < 0.05, **P < 0.001 (c) Invasion of CT26 and PC3 cells, expressing varying amounts of PSMA on the cell surface, was determined by Matrigel invasion assay. (−) = no PSMA expression, (+) = low PSMA expression, (++) = high PSMA expression as determined by flow cytometry analysis MTS, 3-(4 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium.
Next, we determined the effect of PSMA expression on cell migration using a scratch-wound assay (Figure 1b, left panel). Cells were synchronized via serum-starvation, and wound closure was assessed over a 24-hour period to minimize artifacts from cellular proliferation. Our results demonstrate a significant increase in the migratory ability of PSMA+ cells compared to cells lacking PSMA (PSMA−). The effects of PSMA on migration were corroborated in PSMA-negative CT26 cells (mouse colon carcinoma cells), which were engineered to stably express human PSMA (Figure 1b, right panel). We next evaluated the ability of PSMA+ cells to invade through an artificial basement membrane (i.e., Matrigel) (Figure 1c). Of note, the extent of cell invasion was proportional to the levels of PSMA expression on the cell surface (Figure 1c and Supplementary Figure S3). In contrast, PSMA expression had no effect on cell survival in response to various insults such as γ-irradiation, docetaxel, or ultra-violet (UV) radiation (Supplementary Figure S1 and Supplementary Materials and Methods).
As mentioned above, PSMA's enzymatic activity has been directly implicated in promoting cell growth and migration.22,23,24 We sought to explore inhibition of PSMA enzymatic activity on cancer cell proliferation, migration and invasion, using previously described RNA aptamers to PSMA (A10-3.218 and A9g)25 (Figure 2a). First, we assessed the effect of these aptamers on PSMA's enzymatic activity (NAALADase) (Figure 2b). Aptamers were incubated with membrane extracts of PC cells expressing PSMA. Aptamer A9g inhibited PSMA enzymatic activity in a dose-dependent manner. In contrast, a different PSMA aptamer (A10-3.2) and a point mutant aptamer version of A9g, which does not bind PSMA (A9g.625), had no significant effect on PSMA enzymatic activity under the same experimental conditions.
Figure 2.
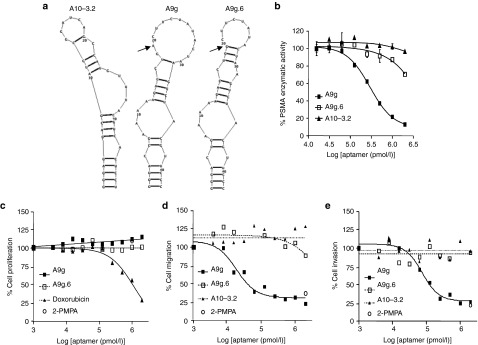
Functional characterization of PSMA RNA aptamers. (a) Predicted secondary structures of PSMA-specific aptamers A10-3.218 and A9g,25 as well as, control non-binding aptamer (A9g.6). Arrows indicate the nucleotide that was mutated in A9g to generate the non-binding, control aptamer A9g.6. Structural predictions were generated using RNA structure version 5.03. (b) Effect of PSMA aptamers on PSMA enzymatic activity determined by NAALADase Assay. (c) Effect of A9g on PSMA-mediated cell proliferation determined by MTS Assay. Doxorubicin: antiproliferative chemotherapeutic drug; 2-PMPA: small molecule inhibitor of PSMA enzymatic activity. (d) Effect of A9g on PSMA-mediated cell migration determined by transwell migration assay. (e) Effect of A9g on PSMA-mediated cell invasion determined by Matrigel invasion assay MTS, 3-(4 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2h-tetrazolium.
We next evaluated the effect of A9g on PSMA-mediated cell proliferation (Figure 2c). PC-3(PSMA+) cells were incubated with either increasing amounts of A9g, the non-binding aptamer (A9g.6), or a standard antiproliferative chemotherapeutic (doxorubicin). A single, high concentration of the small molecule inhibitor of PSMA (2-PMPA) was used as a positive control for inhibition of PSMA enzymatic activity. Interestingly, we observed no change in cellular proliferation when PSMA enzymatic activity was inhibited with either A9g or 2-PMPA. In contrast, doxorubicin significantly inhibited cell proliferation under the same experimental conditions. These results suggest that PSMA enzymatic activity is not required for cell proliferation.
Although A9g had no effect on cell proliferation, increasing amounts of A9g strongly inhibited migration (Figure 2d) and invasion (Figure 2e) of PC cells in culture. The inhibitory effects on cell migration and invasion were confirmed with 2-PMPA. In contrast, the inert PSMA RNA aptamer (A10-3.2) and the non-binding mutant aptamer (A9g.6) had no effect on either cell migration or invasion under the same experimental conditions. Collectively, these data suggest that PSMA's enzymatic activity is necessary for promoting migration and invasion, but not proliferation, of PC cells in vitro.
In vivo efficacy of A9g PSMA aptamer in mouse model of metastatic PC
The underlying cause of PC mortality in humans is disseminated bone disease (bony mets).1 To better evaluate the effect of A9g on migration and invasion of PC cells in vivo, we developed a mouse model of metastatic PC characterized by high osteotropism. For this model, PSMA+, luciferase-expressing PC cells (22Rv1(1.7)) were injected into the left ventricle of the mouse heart. Images were acquired weekly for 4 weeks using the AMI 1000 instrument, which features bioluminescence as well as X-ray computed tomography (X-ray CT) capabilities. Similar to the human disease, we show that 80–90% of the metastases in these mice are localized to the bone (Supplementary Figure S4, top panel).
To determine the effect of the A9g PSMA aptamer on disseminated disease, male SCID mice were divided into three treatment groups: vehicle control (n = 10), A9g (n = 18), and A9g.6 (n = 16). On day 0, 22Rv1(1.7) cells were incubated for 30 minutes with the above treatments prior to intracardiac injection into mice. Systemic administration (via tail vein) of the RNA reagents (1 nmol or approximately 0.63 mg/kg) was continued every 24 hours until day 4, after which, administration was reduced to once a week for 4 additional weeks. Representative images of mice from each treatment group (at week 4) can be seen in Figure 3a. All data were collected and plotted as percentage of mice with metastases (Figure 3b, left panel) or number of metastases per mouse (Figure 3b, right panel). As shown in Figure 3b, A9g significantly reduced the spread of PC cells in this mouse model. Only 2 out of 18 mice in the A9g treatment group developed any visible metastases (Figure 3b, left panel) and the number of metastases in these mice was greatly reduced compared to the vehicle or A9g.6 control groups (Figure 3b, right panel). In contrast, 8 out of 10 mice in the vehicle group and 13 out of 16 mice in the A9g.6 group developed metastases (Figure 3b, left panel). The number of metastases per mouse, observed in the vehicle and A9g treated groups, varied considerably, but averaged about three metastases per mouse. In order to confirm the presence of PC cells in bone, luciferase-positive (by bioluminescence imaging (BLI)) bone tissue was excised and processed for histological examination (Figure 3c). Hematoxylin and Eosin (H&E) staining confirmed the presence of tumor cells focally effacing approximately 25% of the marrow in most bone tissues sampled (representative image of leg tumor shown).
Figure 3.

In vivo efficacy and safety of A9g PSMA aptamer. (a) Representative images of vehicle (n = 10), A9g (n = 18) and A9g.6 (n = 16) treated SCID mice following intra-cardiac injection of luciferase (Luc+) expressing PSMA+ prostate cancer cells, 22Rv1(1.7). Images were acquired on the AMI 1000 Instrument (Spectral Instruments Imaging). (b) (Left panel) Percentage of mice with metastases from the three treatment groups (vehicle, A9g or A9g.6) 4 weeks after intra-cardiac injection of 22rv1(1.7) cells (*P < 0.0001, Fisher's exact test comparing A9g-treated group to either vehicle or A9g.6-treated groups). (Right panel) Number of bioluminescent (Luc+) foci (metastatic foci) per mouse, per group was quantified. (**P < 0.001, Student t-test comparing A9g-treated group to either vehicle or A9g.6-treated groups) (c) Representative histological section of bone with disseminated disease. Hematoxylin and Eosin (H&E) staining of bone section at 100× and 400× shown * = indicates bone metastases. (d) Effect of A9g aptamer on mouse weight during course of treatment. Normalized weight: weights normalized to pretreatment values. (e) Effect of A9g on blood cells after a complete treatment course. CBC is reported for white blood cells (WBC), red blood cells (RBC), platelets (PLT), neutrophils (NEU), lymphocytes (LYM), and monocytes (MON). (f) Assessment of potential immune stimulatory effect of A9g in immune-competent mice. Poly I:C: positive control for immune stimulation. Spleens (top panels) and livers (bottom panels) of treated mice were collected at the indicated time points and processed for total RNA. Expression levels of several immune responsive genes were determined by RT-qPCR. OAS-1: 2′-5′ oligoadenylate synthetase 1A, IFN-β: interferon beta 1, IFN-γ: interferon gamma, IL-6: interleukin 6, and IFIT1: interferon-induced protein with tetratricopeptide repeats 1.
In vivo safety evaluation of A9g PSMA aptamer in mice
An important consideration for clinical translation of a drug is safety.29 To determine potential toxicity of the RNA aptamer drug, we monitored general appearance (increase/decrease activity, hypo/hyperthermia, pallor, dermal/ocular/respiratory abnormalities), behavior (righting reflex, prostration, catatonia, ataxia, tremors, convulsions), and mouse weights over the course of the 4-week treatment. No apparent changes in appearance or behavior were observed (data not shown). Similarly, no significant decreases in mouse weights were observed for any of the treatment groups (Figure 3d). Because changes in hematological parameters provide important information regarding the mechanism of toxicity,29 blood was collected from each mouse at the end of the 4-week treatment and processed to determine any abnormal deviations in blood cell counts (Figure 3e). As seen in Figure 3e, treatment of mice with A9g had no significant effect on any of the blood cell types evaluated. This was also true for the control RNA aptamer, confirming the overall safety of these chemically modified RNA reagents. Finally, an abbreviated pathology assessment of major organs (heart, kidneys, liver, lung, spleen, stomach, and testes) was conducted at termination of treatment. No gross organ abnormalities were observed in all treatment groups (data not shown).
Unmodified nucleic acids (e.g., siRNA, RNA aptamers) are potent activators of the mammalian innate immune system. The induction of innate immunity by these RNAs is dependent on the RNA sequence/structure, method of delivery, and cell-type.30,31 To assess any potential immune activation induced by the systemic administration of our RNA reagent, we performed in-depth immune stimulation studies using immune-competent mice (Figure 3f). For these studies, 1 nmol (~0.63 mg/kg) of A9g or A9g.6 was injected in the tail veins of C57/BL6 male mice. Spleens and livers from treated mice were collected at early (0.5 hours), intermediate (6 hours), and late (24 hours) time points following injection of the RNA and processed for total RNA. Levels of inflammatory cytokines (IL-6), type I (IFN-β) and type II (IFN-γ) interferons, and viral RNA recognition genes (OAS-1, IFIT1) were quantified using RT-qPCR. As a positive control for immunostimulation, 100 µg of polyinosinic:polycytidylic acid (Poly I:C) was injected into mice and spleens and livers from these mice processed as above. As seen in Figure 3f, treatment of A9g did not result in stimulation of inflammatory cytokines, interferons, or viral recognition genes. In contrast, Poly I:C was a potent inducer of IL-6, OAS-1, and IFIT1 (see 6-hour time point). Of note, a slight increase in both type I and type II interferon response genes (IFN-β/γ) was observed in the spleens of mice treated with the non-binding, point mutant aptamer (A9g.6) (Figure 3f, top panels). The reason for this is unclear although, it is unlikely to be due to differences in sequence (A9g.6 varies from A9g by only one nucleotide) or chemistry (both RNAs are 2′-F modified) of the RNAs. It is possible that the increase in immune-stimulatory genes is due to secondary/tertiary structural differences of these RNAs (see Figure 2a for predicted secondary structures). Together, these data suggest that A9g is safe for in vivo applications. Furthermore, the immune stimulatory studies highlight the need for careful and thorough assessment of these RNA-based drugs prior to use in humans.
Pharmacokinetics and biodistribution of A9g PSMA aptamer
Although biodistribution and PK of some aptamers have been described, these basic properties can depend on the structure and binding characteristics of each individual aptamer.27 To determine the biodistribution and PK of our RNA smart drug, we performed non-invasive imaging with fluorescent near-infrared (NIR)-labeled material. Briefly, A9g and A9g.6 were conjugated to an NIR fluorophore (800 CW) through amine coupling.32 Labeling efficiency (Supplementary Figure S5a) and overall purity (Supplementary Figure S5b) of the NIR-RNAs were assessed prior to the start of our studies. Activity of the NIR-labeled A9g was confirmed using the PSMA enzymatic assay (Supplementary Figure S5c).
NIR-labeled aptamers were injected into the tail veins of mice bearing PC-3(PSMA+) tumors in their right flanks (Figure 4a). Mice were imaged over 72 hours using BLI. RNA aptamers were evenly distributed throughout the mouse's body 5 minutes after administration. Both RNAs were cleared from circulation as early as 8 hours following administration. Importantly, NIR-A9g was retained in the PSMA+ tumor up to 72 hours after administration (Figure 4a, top panels). In contrast, no NIR-A9g.6 was detected in the tumor or elsewhere after 8 hours (Figure 4a, bottom panels). Tumor xenografts were excised at 96 hours following administration of the NIR-labeled RNAs and imaged ex vivo (Figure 4a, inset). We observed intense fluorescence in the tumors that were isolated from NIR-A9g treated mice, while no fluorescence was observed in control (NIR-A9g.6) treated mice. These results clearly demonstrate selective targeting of A9g to PSMA+ tumors in vivo.
Figure 4.

PK and biodistribution of PSMA aptamer, A9g. (a) Near NIR labeled aptamers NIR-A9g and NIRA9g.6 were injected tail vein in mice bearing PSMA+ tumors (implanted subcutaneously) and tracked over time with the IVIS 200 imaging system (PerkinElmer). Ex-vivo: tumors were excised at day 4 (96 hours) post-injection. (b) PK and biodistribution of NIR-A9g in a control, non-tumor-bearing mouse. (c) Specific targeting of PSMA positive tumor by A9g. NIR-A9g (top row) or NIR-A9g.6 (bottom row) were injected tail vein in SCID mice bearing subcutaneous PSMA+ (right flank) or PSMA− (left flank) prostate cancer tumors. Images were acquired with the IVIS 200 imaging system (PerkinElmer) at the indicated time points following injection of the labeled RNAs.
We then sought to determine the PK and biodistribution of A9g in a control, non-tumor-bearing mouse (Figure 4b). NIR-A9g was injected in the tail veins of male SCID mice and imaged over a period of 24 hours. As expected, the in vivo profile of A9g in non-tumor-bearing mice was similar to that observed for the control non-binding aptamer (A9g.6) in Figure 4a. Together, these data suggest that the longer in vivo retention time of A9g observed in tumor-bearing mice is due to tumor-specific targeting by this RNA. To confirm tumor-target specificity and rule out any potential enhanced permeability and retention (EPR) effect of our RNA smart drug, we performed PK and biodistribution studies in mice bearing both PSMA+ (right flank) and PSMA− (left flank) tumors implanted subcutaneously (Figure 4c). EPR is a process by which macromolecules (e.g., liposomes, nanoparticles, and macromolecular drugs) tend to accumulate in tumor tissue much more than they do in normal tissues.33 NIR-A9g rapidly localized to the PSMA+ tumor. NIR signal in the PSMA+ tumor persisted for 24 hours in this experiment. This signal was specific to tumors with PSMA expression, as no NIR-A9g was detected in PSMA− tumors. In contrast, NIR-A9g.6 did not accumulate in either the PSMA+ or PSMA− tumor (Figure 4c, bottom panel). Together, these data suggest that uptake of NIR-A9g into PMSA+ tumors is specific to PSMA surface expression and not due to non-targeted RNA uptake (e.g., potential EPR effect).
Stability of A9g PSMA aptamer in human serum
Preclinical stability and safety studies performed in human samples are critical to the successful translation of potential future RNA smart drugs. Serum stability of RNA is limited by endonuclease cleavage (which can be overcome by the 2′-F-pyrimidines or similar modifications)27 and serum exonuclease cleavage of the 3′-end (which can be avoided using terminal modifications such as PEGylation).18 To determine the stability of A9g, we incubated the RNA in human serum for a total of 1 week. Serum-RNA samples at the indicated time-points (0, 1, 2, 4, 8, 24, 48, 72, and 168 hours) were resolved on a denaturing polyacrylamide gel electrophoresis (PAGE) (Figure 5a, left panel) and RNA bands quantified using ImageJ software (Figure 5a, right panel). To evaluate the effect of additional modifications on stability, we modified the A9g sequence to incorporate 2′-O-methyls (O′Me) in a subset of purines (data shown for modification of three purines (A9g +3)) or modification of all purines (A9g + 21). A9g and A9g +3 have a T1/2 of ~70 hours in human serum. The modifications of the three purines did not confer additional stability to the RNA. In contrast, 2′-O′Me modifications of all purines within the A9g sequence resulted in a significant increase in serum stability (T1/2 > 1 w). However, modification of all purines post-selection resulted in a nonfunctional A9g RNA as determined by PSMA enzymatic activity (Figure 5b). This was not the case for A9g +3, which still retained functionality. These studies suggest caution when optimizing RNA aptamers post-selection for clinical applications.
Figure 5.
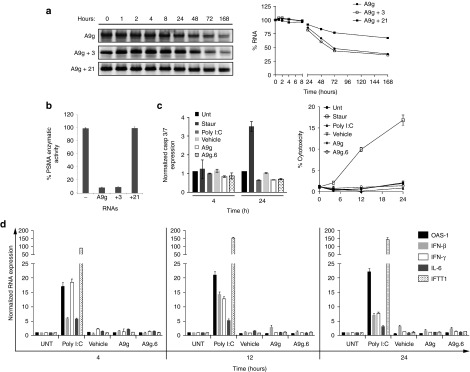
Stability and safety assessment of A9g aptamer in human serum and cells. (a) Effect of chemical modifications on stability of A9g aptamer in human serum. (Left panel) Denaturing PAGE gel of RNA aptamers following incubation with 100% human serum over a 1-week period. (Right panel) Band intensity was quantified using Image J version 1.47 and plotted relative to input/time 0 RNA. A9g = All pyrimidines modified with 2′-fluoro chemistry, A9g + 3 = All pyrimidines modified with 2′-fluoro chemistry and three purines modified with 2′-O methyl chemistry; A9g + 21 = All pyrimidines modified with 2′-fluoro chemistry and all 21 purines modified with 2′-O methyl chemistry. (b) Functional characterization of chemically modified A9g aptamers using NAALADAse Assay. Percentage of PSMA enzymatic activity is reported. (c) Assessment of potential toxicity of A9g in human peripheral blood mononuclear cells (hPBMC) from healthy adult volunteers. Staurosporine: positive control for apoptosis; Poly I:C: positive control for immune stimulation. Vehicle: Binding Buffer or a non-binding, control aptamer (A9g.6) for the indicated time points. (Left panel) Apoptosis assessed by caspase 3/7 activation. (Right panel) Cytotoxicity assessed by lactate dehydrogenase activity. (d) Assessment of potential immune stimulatory effect of A9g in hPBMCs as in Figure 3f for above.
Safety assessment of A9g PSMA aptamer in human PBMCs
Next, we assessed potential cytotoxicity and immune-stimulatory effects of A9g in human PBMCs. Importantly, incubation of A9g with human PBMCs did not result in an increase in caspase 3/7 activity (two markers of cellular apoptosis) at 4 hours or 24 hours following incubation with the RNA. In contrast, an increase in caspase 3/7 activity was observed following a 24-hour incubation of PBMCs with staurosporine (positive control for caspase-dependent and independent apoptotic pathways) (Figure 5c, left panel). Interestingly, poly I:C did not induce caspase 3/7 activation. As an independent measure of cytotoxicity, we determined the release of lactate dehydrogenase (LDH) from human PBMCs treated with the RNA smart drug (Figure 5c, right panel). LDH is a soluble cytosolic enzyme that is released into the culture medium following loss of membrane integrity resulting from either apoptosis or necrosis. LDH activity, therefore, can be used as an indicator of cell membrane integrity and serves as a general means to assess cytotoxicity resulting from chemical compounds or environmental toxic factors. Importantly, A9g did not induce LDH. In contrast, a robust induction of LDH was observed when human PBMCs were incubated with staurosporine (Figure 5c). As a final measure of toxicity, we assessed any potential immune stimulation due to A9g in human PBMCs (Figure 5d). Human PBMCs were incubated with either A9g, A9g.6, or Poly I:C as a positive control. RNA from treated cells was isolated at early (4 hours), intermediate (12 hours), and late (24 hours) time points as described above for the mouse safety studies. Upregulation of specific immune responsive genes was assessed as in Figure 3f. No increase in inflammatory cytokines (IL-6), type I (IFN-β) and II (IFN-γ) interferons, or viral RNA recognition genes (OAS-1, IFIT1) was observed. Together, these studies verify that A9g does not elicit non-specific immune stimulation in human PBMCs and suggest that it is safe for clinical applications.
Discussion
In this preclinical study, we describe a cell-targeted RNA-based smart drug for managing mCRPC that is both safe and effective in vivo when delivered systemically and is amenable to chemical synthesis for large-scale production. The RNA smart drug (A9g) was previously shown by us to bind to PSMA on the surface of PC cells and to act as a potent inhibitor of PSMA enzymatic activity in vitro.25 We now show that A9g inhibits PSMA-dependent cell migration and invasion in cancer cells in culture (Figure 2d,e) and in vivo in a mouse model of metastatic PC (Figure 3). Interestingly, A9g has no effect on PSMA-dependent cell proliferation (Figure 2c) or survival (Supplementary Figure S1), two cellular processes also thought to be linked to PSMA's enzymatic activity.22,23,24 Based on these findings, we believe that A9g is inhibiting specific downstream effectors (e.g., p130cas) of the focal adhesion kinase pathway or implicated in cell motility.24,34 Alternatively, A9g could be blocking the ability of PSMA to interact with Filanim A, a cytoskeletal protein which plays a central role in cell motility and metastasis.24 Importantly, we demonstrate that A9g is safe when administered to either tumor-bearing (immune-compromised) or to immune-competent mice (Figure 3d–f). Similarly, in studies performed in human PBMCs, A9g was not cytotoxic (Figure 5c) and did not induce high levels of inflammatory cytokines and interferons (Figure 5d). PK and biodistribution studies performed with NIR-labeled A9g (Figure 4) confirm its target specificity and suggest the absence of any non-specific on-target or off-target effects of the RNA drug.
The majority of cancer drugs are non-specific cytotoxic agents that poorly discriminate between cancer cells and normal dividing cells.1 These drugs often present with severe side-effects including liver toxicity, bone marrow suppression, severe edema, and cardiotoxicity.1 Cell-targeted approaches, which promise improved efficacy and safety profiles when compared to conventional non-targeted drugs, have long been recognized as the Holy Grail of cancer therapeutics. Recently, several targeted cancer drugs (smart drugs) have been described. These include: (1) trastuzumab (Herceptin), an antibody to HER2 used to treat HER2+ breast cancers and other solid cancers with elevated HER2 expression.35 (2) Lapatinib (Tykerb), a small molecule tyrosine kinase inhibitor (TKI) used to treat cancers which are driven by HER2 and epidermal growth factor receptor (EGFR).36 (3) Imatinib (Gleevec), a TKI specific for Abl, c-kit and PDGFR, used to treat certain leukemias (e.g., CML) and gastrointestinal stromal tumors whose growth and survival are driven by the elevated expression or constitutive activation of these protein kinases.37 Despite the impressive successes obtained with this class of targeted drugs, which demonstrate the utility of selectively controlling cancer cell progression while limiting toxicity to normal cells, many cancers such as mCRPC are currently not being treated with smart drug technology. The work described herein is of importance since we demonstrate potent and specific inhibition of metastatic PC foci spread with a low therapeutic dose (nine treatments at ~0.63 mg/kg) of an RNA aptamer inhibitor of PSMA.
Given the potential of aptamers for targeted cell therapies, much emphasis over the past several years has been on optimizing current selection methodologies for establishing a platform technology that would facilitate the identification of aptamers to cell-surface receptors. Indeed, until recently, only a handful of aptamers to cell-surface receptors had been described, due primarily to difficulties in obtaining purified preparations of recombinant membrane proteins for selection. Also, since complex glycosylation patterns and structural conformations of membrane proteins tend not to be replicated in recombinant protein preparations, aptamers selected against recombinant targets have often failed post-selection to bind to their targets on the surface of cells.38,39 Recent progress made in cell-based selection technologies have favored the isolation of aptamers specific for cell-surface proteins within their native milieu (i.e., the cell membrane).40 These selections have demonstrated clear advantages over selections performed using purified recombinant proteins. To date, several aptamers against receptors expressed on the surface of cancer cells have been identified using the cell-based selection approach.41 These aptamers have been used to inhibit the function of the target proteins as well as for selective delivery of RNAi modulators (e.g., siRNAs/miRNAs) to cells.41 Importantly, given the role of these aptamer targets in cancer and other diseases, these aptamers may turn out to be ideal candidates for the development of future smart drugs.
The amenability of aptamers to being modified with chemistry facilitates conjugation to various molecules including fluorophores, radiopharmaceuticals, small molecule drugs/toxins, and other therapeutic oligonucleotides.41 Here, we demonstrate the use of a noninvasive approach, based on NIR imaging technology, for tracking the RNA drug in preclinical mouse models of disease. Similar approaches, which incorporate radiopharmaceuticals into the RNA drug, will facilitate in vivo tracking studies in larger preclinical animal models as well as in humans.42 Progress made toward developing methods to noninvasively image delivery and actions of these nucleic acid-based drugs will be important in a clinical setting where understanding the safety profile and PK of the drug is crucial.43,44
In addition to in vivo imaging applications, cell-targeted aptamers have also shown promise in selective drug delivery.41 Toward this end, we previously described the conjugation of an inert (non-inhibitory) PSMA aptamer (A10-3.2) to cytotoxic siRNA duplexes.18 The siRNAs were designed to target cancer-specific genes such as polo-like kinase -1 and B-cell lymphoma-2. Although the aptamer itself had no effect on PC progression, the therapeutic potential of this dual-targeted RNA drug stems from its exquisite selectivity: the A10-3.2 aptamer targets PSMA on PC cells while delivering a cancer-specific siRNA cargo. Since the initial description of this targeted approach, various derivatives of the A10-3.2 aptamer have been linked to other therapeutic siRNAs.17,19 Importantly, all these studies have reported efficacy, following systemic administration of the aptamer-siRNA drug at doses under 1 mg/kg in preclinical mouse models of PC.
While the use of inert aptamers for selective drug delivery is a major achievement on its own, conjugation of therapeutic aptamers (such as A9g) with other cytotoxic drugs for combinatorial therapy is of particular interest in the setting of complex diseases such as mCRPC. Indeed, in the last year, strong evidence points to PC as a highly heterogenous disease driven by multiple mechanisms of cell survival, which are thought to be responsible for the development and resistance to standard monotherapy. Attempts to establish individualized therapy in order to better tailor the treatment to the individual patient have reached significant advance. In addition, clinical trials exploring combination of therapies (e.g., endocrine therapy with chemotherapy) are ongoing.1 As combination therapy becomes the new frontier for the clinical management of PC, future studies, which focus on evaluating the in vivo efficacy and safety of therapeutic PC-specific aptamers coupled to existing small molecule inhibitors/toxins or therapeutic siRNAs (e.g., siRNAs directed against molecular pathways recently implicated in driving the progression of mCRPC), will be critical. Indeed, while the current study focused on evaluating the effect of A9g on preventing metastatic colonization, it will be valuable to evaluate an aptamer combination smart drug where A9g targets the colonization of new metastatic foci and a cytotoxic drug load targets the growth/survival of established metastases.
In conclusion, we describe a novel RNA aptamer-based smart drug for managing mCRPC that can be developed as an adjuvant to current cytotoxic therapies or conjugated to existing/novel drugs for the development of targeted, single agent, combination therapies. This would significantly expand the current repertoire of drugs used to treat PC patients with advanced disease. Importantly, the aptamer-targeted approach also lends itself to the development of individualized therapy where the drug cargo can be modified to better tailor the treatment to the individual patient. Finally, while our proof-of-concept, preclinical study focused on the effect of A9g on migration and invasion of PC cells, given PSMA's expression in the neovasculature of many sarcomas and carcinomas,12,13 targeting neoangiogenesis via PSMA may also serve as a viable therapeutic option in other solid cancers.
Materials and Methods
Cell culture. CT26 and CT26(PSMA+) mouse colorectal carcinoma cells were a generous gift from E. Gilboa (University of Miami). CT26(PSMA++) was derived from CT26(PSMA+) by sorting for cells with high PSMA cell-surface expression. PC-3 and PC-3(PSMA+) human prostate adenocarcinoma cells were developed as previously described.13 PC-3(PSMA++) cells were derived from PC-3(PSMA+) by sorting for cells with high PSMA cell-surface expression. All cells were maintained in NuAire water-jacketed CO2 incubators at 37 °C and 5% CO2 (Plymouth, MN). CT26 cell lines were maintained in RPMI1640 media supplemented with 10% fetal bovine serum (FBS). All PC-3 cells were grown in F12K media supplemented with 10% FBS. The human, PSMA+, luciferase (Luc)+, prostate carcinoma cell line, 22Rv1(1.7)18 was grown in RPMI 1640 medium (Gibco, Grand Island, NY) supplemented with 10% FBS (Hyclone), 1 mmol/l nonessential amino acids (GIBCO) and 100 μg/ml G-418.
DNA templates and primers. All in vitro assays were performed using in vitro transcribed RNA unless otherwise noted. All in vivo assays were performed using chemically synthesized RNA (TriLink Biotechnologies, San Diego, CA) modified with 2′-fluoro pyrimidines, 2′-hydroxyl purines and a C12-NH2 5′ terminal modification.
A9g. DNA Template: 5′-GGGACCGAAAAAGACCTGACTTCTATACTAAGTCTACGTTCCC-3′
5′ Primer: 5′-TAATACGACTCACTATAGGGACCGAAAAAGACC-3′
3′ Primer: 5′-GGGAACGTAGACTTAG-3′
A9g.6. DNA Template: 5′-GGGACCGAAAAAGACCTGGCTTCTATACTAAGTCTACGTTCCC-3′
5′ Primer: 5′-TAATACGACTCACTATAGGGACCGAAAAAGACC-3′
3′ Primer: 5′-GGGAACGTAGACTTAG-3′
A10-3.2. DNA Template: 5′-GGGAGGACGATGCGGATCAGCCATGTTTACGTCACTCCTTGTCAATCCTCATCGGCAGACGACTCGCCCGA-3′
5′ Primer: 5′-TAATACGACTCACTATAGGGAGGACGATGCGGA-3′
3′ Primer: 5′-AGGAGTGACGTAAACATG-3′
RNA transcription/purification. RNA aptamers were transcribed as previously described.16 Briefly, template DNAs and primers were ordered from Integrated DNA Technologies (IDT; Coralville, IA). Double-stranded DNA templates for transcription were generated via PCR. DNA templates were purified with Qiagen DNA purification columns (Cat# 27106) and used for in vitro transcription reactions to make individual RNA aptamers. An Y639F mutant T7 RNA polymerase45 was used to incorporate 2′-fluoro pyrimidines in order to generate RNA aptamers resistant to serum nucleases. The in vitro transcribed RNAs were resolved on denaturing PAGE gels (10% polyacrylamide/7 M urea), and visualized using UV shadowing. The RNA was excised from the gel, eluted in 4 ml of TE buffer (0.1 mmol/l ethylenediaminetetraacetic (EDTA)), washed twice with 4 ml of TE buffer and concentrated with an Amicon 10,000 MW-cutoff spin filter (Cat# UFC801024).
Proliferation assay. PC-3 and PC-3(PSMA+) cells were plated onto 96-well tissue culture plates (Corning Costar) in triplicate at a concentration of 2.5 × 105 cells/ml in a volume of 100 μl/well. At indicated time point, 20μl of Cell Titer 96 AQueous One Solution (Promega Catalog #G3581) was added directly to cells in 96-well plates. Cells and solution were incubated at 37 °C for 1 hour. Absorbance at 490 nm was then assessed on a Molecular Devices Thermo Max Microplate Reader (GMI Inc., MN).
Assessing effect of PSMA aptamers on cell proliferation. Chemically synthesized A9g and A9g.6 RNAs were allowed to fold at 4 μmol/l in 1.5 ml total volume by heating to 98 °C for 10 minutes, followed by incubation at 65 °C for 10 minutes and finally incubation at 37 °C for 10minutes. Stock solutions of 2-PMPA (Enzo Life Sciences Catalog #ALX-550-358-M001) and Doxorubicin (Dox; Sigma Catalog #D1515-10MG) were diluted to 4 μmol/l per manufacturer recommendation. All reagents were then serial diluted 1:2 in cell culture media for a total of 10 concentrations. PC3(PSMA++) cells were plated at a concentration of 5 × 105 cells/ml in a volume of 50μl/well. Fifty microliters of A9g, A9g.6, 2-PMPA, and Dox at all concentrations were added to cells in triplicate. Cell proliferation was assessed as above.
Migration/invasion assays
Scratch-wound assay. Cells were starved in serum-free, folate-free RPMI 1640 medium for 24 hours. Cells were then trypsinized and counted using Trypan Blue to exclude dead cells. 2.5 × 105 viable cells were added to each well in a 6-well cell culture plate and allowed to adhere for 24 hours. A linear scratch was made in the cell monolayer using a sterile pipette tip and cells were washed 2× with DPBS to remove scraped/dead cells before returning to folate-free RPMI 1640. Three to four images of each scratch were taken consistently from the time of scratch induction and every 8 hours until scratch closure. Measurements of images were performed at three locations perpendicular to the scratch in a blinded fashion and expressed as the percentage of wound closure relative to the initial scratch.
Transwell migration assay. CT26 or PC-3 cells were serum starved in serum-free, folate-free RPMI 1640 medium for 24 hours. Following starvation, 3.5 × 104 cells with or without PSMA expression were seeded on uncoated transwell filters (BD Biosciences, San Jose, CA) in folate-free, serum-free RPMI 1640 media with or without aptamer or PSMA small molecule inhibitor (2-PMPA) and allowed to migrate for 20 hours toward the lower chamber containing 10% FBS or serum-free media. Cells migrating through the membrane were fixed with methanol followed by mounting and staining with VECTASHIELD containing DAPI (Vector Laboratories, Burlingame, CA). Cell migration is expressed as the fraction of total cells that migrated through the filter relative to untreated cells. Each assay was set up in duplicate, and each experiment was conducted a minimum of three times and represents three random fields per condition. Images were acquired with an Olympus IX71 inverted microscope equipped with a 10× objective an EXFO X-Cite Q xenon light source and a cooled CCD digital camera.
Matrigel invasion assay. CT26 or PC-3 cells were serum starved in serum-free, folate-free RPMI 1640 medium for 24 hours prior to initiation of experiment. Following starvation, 3.5 × 104 cells with or without the expression of PSMA were seeded in folate-free, serum-free RPMI 1640 media with or without aptamer or PSMA small molecule inhibitor (2-PMPA) on Matrigel-coated filters (BD Biosciences) and allowed to invade for 22–24 hours toward the lower chamber containing 10% FBS or serum-free media. Cells invading through the Matrigel layer were fixed with methanol followed by mounting and staining with VECTASHIELD containing DAPI (Vector Laboratories, Burlingame, CA). Cell invasion is expressed as the fraction of total cells that invaded through the filter relative to untreated cells. Each assay was set up in duplicate, and each experiment was conducted a minimum of three times and represents three random fields per condition. Images were acquired with an Olympus IX71 inverted microscope (Olympus, Tokyo, Japan) equipped with a 10× objective an EXFO X-Cite Q xenon light source and a cooled CCD digital camera.
Cell membrane extract preparation. Cell membrane fractions were prepared as described previously.46 Briefly, cell membranes from PC-3 cells, with and without stable expression of human PSMA, were grown at 90–95% confluent in T75 flasks (Costar). Because phosphates are known to inhibit PSMA NAALADase activity at micromolar amounts,46 media was aspirated and cells were washed with ice-cold HEPES-buffered saline instead of DBPS. Cells were detached from plates using cell scrapers and pelleted at 1,000 rpm for 4 minutes in a LABNET Hermle Z400K benchtop centrifuge. Supernatants were decanted. Cell pellets were resuspended in 500 µl of ultrapure H2O and pellets were sonicated four times at 10 seconds each on maximum tone using a Fisher Model 100 sonicator (Fisher Scientific). Cells were spun for 30 minutes at 100,000×g using a TLA 100.2 rotor in a Beckman TL-100 Ultracentrifuge (Beckman Coulter). The supernatant was discarded and membrane pellets were resuspended and homogenized by sonication in 250 µl of pre-warmed 50 mmol/l Tris–Cl, pH 7.4.
NAALADase assay. The PSMA NAALADase activity assay was performed as previously described46 with minor modifications. Briefly, the final reaction volume was modified to 200 µl. Double-distilled H2O (ddH2O) was used for all solutions. RNA aptamers were refolded in 1× Binding Buffer (1×BB: 20 mmol/l HEPES, 150 mmol/l NaCl, 2 mmol/l CaCl2) at 1 µmol/l concentration. RNA was refolded by heating for 10 minutes at 98 °C, 15 minutes at 65 °C, 15 minutes at 37 °C, and cooled for at least 25 minutes at room temperature. Aptamers were concentrated through an Amicon 10,000MW-cutoff spin filter (Amicon Cat# UFC801024). The remaining procedures were performed on ice. Ten microliters of refolded RNA in 1×BB were combined with 50 µl of 200 mmol/l Tris buffer, pH 7.5, and 20 µl 10 mmol/l CoCl2. Recombinant PSMA was prepared by diluting 2 µg recombinant human PSMA (4234-ZN-010) from R&D Systems (Minneapolis, MN) in 500 µl of 50 mmol/l pH 7.5 Tris buffer. Either 10 µl of the recombinant PSMA solution (40 ng PSMA) or 10 µl of cell membrane preparations described above was added to the reaction mixture, and the reaction was incubated for 15 minutes at 37ºC to promote RNA–PSMA interaction. Ten microliters of a working solution containing 30 nmol/l NAAG having a specific activity of 2.5 µCi/µl of (glutamate-3,4-3H)-NAAG from PerkinElmer (Waltham, MA; Cat# NET1082250UC) were added to the reaction mixture. The reaction was allowed to proceed for 15 minutes, mixing once by gentle vortexing at 7.5 minutes. Reactions were terminated by adding an equal volume (200 µl) of ice-cold 0.1 M Na2HPO4 buffer. AG 1-X8 formate resin (200–400 mesh) from Bio-Rad Laboratories (Hercules, CA Cat# 140–1454) was used in column chromatography to isolate the [3H]-glutamate reaction product. Before use, the columns were equilibrated with 2 ml of ddH2O. Half of the final reaction volume (200 µl) was added to a column. The columns were eluted with 4 ml of 1 M formic acid and added to 10 ml of Bio-Safe II scintillation fluid (Research Products International Corp., Mt. Prospect, IL). Activity was counted using a Beckman-Coulter liquid scintillation counter (Beckman Coulter), and was normalized to the amount of activity obtained in the reaction with no RNA added.
In vivo efficacy studies. Animals in this study will be utilized in accordance with all PHS policies and the Guide for the Care and Use of Laboratory Animals, National Institutes of Health Publication No. 85–223, revised 1996 (http://oacu.od.nih.gov/regs/guide/guidex.htm).
Intracardiac injections of 22Rv1(1.7) cells. Intracardiac injections were performed as previously described.47 Briefly, 22Rv1(1.7) cells were grown to 80% confluence in a T75 flask (Sigma-Aldrich, St Louis, MO CLS3375), washed once with Dulbecco's phosphate buffered saline (DPBS), trypsinized for 5 minutes with 0.05% trypsin containing EDTA (Invitrogen, Grand Island, NY), resuspended in 10 ml RPMI 1640 medium supplemented with 10% FBS and spun at 1,000 rpm for 5 minutes to pellet the cells. Next, cells were re-suspended in DPBS, pipeted several times to disperse the clumps and passed through a 70 µm Nylon cell strainer (BD Biosciences) into a 50 ml conical tube. Cells were further assessed for viability with Trypan Blue stain and brought to a final concentration of 2 × 106 cells/ml in RPMI medium in 1.5-ml Eppendorf tube. Shortly before intracardiac injections, cells were pelleted and resuspended in 1×BB (vehicle), A9g aptamer (10 µmol/l in 1×BB) or A9g.6 aptamer (10 µmol/l in 1×BB). Before incubating with the cells, A9g and A9g.6 aptamers were folded by heating for 10 minutes at 98 °C, 15 minutes at 65 °C, 15 minutes at 37 °C, and cooled for at least 25 minutes at room temperature. The effect of A9g and A9g.6 aptamers on PSMA enzymatic activity was confirmed by NAALADase assay, as described above. Cell suspensions were kept on ice until injections were performed.
Aptamer treatment. Mice were randomized into three groups: control vehicle (1× BB) group, A9g (PSMA aptamer), and A9g.6 (non-binding, control aptamer). Briefly, 5- to 8-week-old male SCID mice (NCI) were anesthetized in a chamber with 3% isoflurane (Abbott Laboratories, North Chicago, IL). After the mice were anesthetized, they were placed in a ventral presentation for injections where they were maintained on 2.5% isoflurane delivered through nose cones on a manifold. Hundred microliters of cell suspension (2 × 105 cells final) were injected over a period of 30 seconds into the left ventricle of the heart, using a 30-gauge needle. The needle was placed to the left of the xiphoid process and through the diaphragm into the heart. The presence of a rapid pulsatile flow of bright red blood was indicative of correct needle placement. Following intracardiac injections, 100 µl of 10 µmol/l (1 nmol) A9g, A9g.6 or vehicle (1 × BB) was systemically administered (via tail vein) to mice every day for 4 consecutive days and then weekly for 5 weeks.
BLI. All small animal imaging experiments were performed at the Central Microscopy Research Facility (University of Iowa; http://cmrf.research.uiowa.edu/). At weeks 2, 3, 4, and 5 following intracardiac injections, mice were imaged (ventral and dorsal presentations) using an IVIS 200 imaging system (PerkinElmer) with a 1-minute exposure time. Images were acquired within 10–15 minutes after administration (intra-peritoneal) of 100 µl of 15 mg/ml luciferin (Gold Biotechnology, Inc St. Louis, MO). To confirm metastases localized to the bone, final images (weeks 4–5) were acquired using the SPECTRAL AMI 1000 system with X-ray CT capabilities (25 kV 1-minute exposure). The number of mice per group that had at least one foci of bioluminescence signal by weeks 4 and 5 was plotted as percentage of the total mice assigned per group. Total number of metastatic foci, as assessed by BLI, were counted for each mouse (for ventral and dorsal positions) and plotted as number of metastases per mouse for each respective treatment group. Statistical analysis was performed using GraphPad Prism 5 Software. The percentage of mice with metastases per group was analyzed using Fisher's exact test. For all other analyses, student t-test analysis was used.
Histology. Histology was performed at the Comparative Pathology and Histology Research Laboratories (University of Iowa; http://www.medicine.uiowa.edu/pathology/research/dcp/). At 5 weeks, mice were euthanatized with ketamine/xylazine. Luciferase-positive tissues as determined by BLI (e.g., limb, spine, mandibula, lungs, kidneys, spleen, heart, liver, adrenals) were excised and fixed in 10% neutral buffered formalin for at least 48 hours at room temperature. Bone tissue samples were washed three times in DPBS for 5 minutes and subsequently decalcified in 10% EDTA for 7 days, replacing EDTA daily. After decalcification, the samples were placed in 70% ethanol. All fixed tissues were processed in a series of alcohol and xylene baths, paraffin-embedded, and 7 μm sections were stained with H&E. A veterinary pathologist (David K Meyerholz) examined all tissue sections. High-resolution digital images were acquired with a DP71 camera (Olympus) mounted on a BX51 microscope (Olympus) with MicroSuite Pathology Edition Software (Olympus).
Mouse safety studies
Weights. Mouse weights were recorded prior to intracardiac injections, as well as, prior to every aptamer treatment using a digital laboratory balance (Denver Instrument MXX-601, Bohemia, NY). Weights were normalized to pretreatment values and plotted using GraphPad Prism 5 Software.
Blood counts. At 5 weeks post-intracardiac injection, mice were anesthetized with ketamine/xylazine. Approximately, 150 µl of blood was collected from each mouse via submandibular venous puncture into an EDTA tripotassium salt multivette (Multivette 600 K3E; Sarstedt, Germany). Complete blood counts (CBC) were measured from undiluted blood samples within 30 minutes of collection using a Sysmex XT-200i Automated Hematology Analyzer (Sysmex America Inc, Mundelein, IL).
Immune stimulation studies in immune-competent mice. Hundred microliters of A9g (10 µmol/l), A9g.6 (10 µmol/l) or Poly I:C (1 mg/ml) were injected in the tail veins of immune-competent C57BL/6 mice. Both A9g and A9g.6 RNAs were folded as previously described above. At indicated time points, mice were euthanatized with ketamine/xylazine. Spleens and livers were excised and immediately flash frozen. Total cellular RNA was isolated from tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA). The purity and concentration of isolated RNA was determined using a Nano-Drop 2000c spectrophotometer (Thermo Scientific, Waltham, MA). RT-qPCR was used to determine the expression level of indicated immune responsive genes. This was carried out using the iScript One-Step RT-PCR Kit with SYBR Green (Bio-Rad) following the manufacturer's recommendations. Briefly, 50 ng total RNA was used per 25 µl reaction, with a final primer concentration of 300 nmol/l. PCR cycle program: 10 minutes at 50 °C, 5 minutes at 95 °C, (30 seconds at 95 °C, 30 seconds at 60 °C, 15 seconds at 72 °C (45 cycles)), 1 minute at 95 °C, 1 minute at 55 °C, melt curve 10 seconds from 55 °C to 95 °C. Each reaction was carried out in triplicate and gene expression data normalized to that of beta-actin expression. Mouse-specific primers used in study: β-Actin (Forward: CGGTTCCGATGCCCTGAGGCTCTT; Reverse: CGTCACACTTCATGATGGAATTGA) OAS-1 (Forward: ACCGTCTTGGAACTGGTCAC; Reverse: ATGTTCCTTGTTGGGTCAGC) IFN-β (Forward: CTGGCTTCCATCATGAACAA; Reverse: CATTTCCGAATGTTCGTCCT), IFN-γ (Forward: AGCGGCTGACTGAACTCAGATTGTAG; Reverse: GTCACAGTTTTCAGCTGTATAGGG), IL-6 (Forward: GAGACTTCCATCCAGTTGCC; Reverse: AAGTGCATCATCGTTGTTCATACA), IFIT1 (Forward: CAGAAGCACACATTGAAGAA; Reverse: TGTAAGTAGCCAGAGGAAGG).
PK and biodistribution studies
Conjugation of RNA aptamers to NIR dye (IRDye 800CW). Chemically synthesized A9g and A9g.6 aptamers were incubated with IRDye 800CW-NHS ester (LI-COR, cat# 929–70021), at a ratio of 1:40 (RNA:dye) for 2 hours at 25 °C in 1 mol/l potassium phosphate buffer (pH 9) under slow mixing conditions. Unconjugated free dye was removed by successive spin filtrations using an Amicon 10,000 MW-cutoff centrifugal filter (Cat# UFC801024). Briefly, RNA was added to filter and brought up to 4 ml total volume using PBS+/+ (GIBCO). Filters were spun at 3,500 rpm for 15 minutes and subsequently brought back up to 4 ml total volume with PBS+/+. This process was repeated for a total of five times. Aptamer labeling with IRDye 800CW was confirmed by electrospray ionization (ESI) spectrometry and ultraviolet-visible (UV–Vis) spectroscopy (IDT, IA). Functionality (binding) of the NIR-aptamer conjugates was verified using the PSMA NAALADase activity assay as described above.
In vivo disposition of NIR-labeled aptamers. All small animal imaging experiments were performed at the Central Microscopy Research Facility (University of Iowa; http://cmrf.research.uiowa.edu/.). Six- to eight-week-old male SCID mice (NCI) were injected subcutaneously (s.c.) with PC3 (PSMA++) cells 1 × 106 mixed with 100 µl of Matrigel (BD Biosciences, Bedford, MA) into the forward right flank. Tumor-bearing mice were imaged when the xenografts reached 5–9 mm in diameter, using the IVIS 200 imaging system (PerkinElmer) and images analyzed using the Living Image software (Caliper Life Sciences, Hopkinton, MA). The mice were placed on a warm (37 °C) stage inside a light-tight camera box with continuous exposure to 2% isoflurane. At least three tumor-bearing mice or non-tumor-bearing mice were injected (via tail vein) with 2 nmol (in 100 µl) of either A9g-NIR or A9g.6-NIR aptamers. Images were acquired before injection (to establish background fluorescence signal) and at various time points post-injection. For aptamer specificity studies, mice were injected subcutaneously with 1 × 106 PC3 (PSMA−) (left flank) or PC3 (PSMA++) (right flank) cells in 100 µl of Matrigel. Images were acquired with the ICG excitation filter passband (710–760) and ICG emission filter passband (810–875). The acquisition time for each image was 1 second.
RNA stability in human serum. The A9g aptamer was modified with additional 2′-O methyl chemistry to increase stability in human serum. A9g = original aptamer where all pyrimidines are modified with 2′-fluoro chemistry. A9g + 3 = A9g aptamer containing three purines modified with 2′-O methyl chemistry. A9g + 21 = A9g aptamer containing all 21 purines modified with 2′-O methyl chemistry. RNAs were incubated with 100% human serum (Sigma) in DPBS at 37 °C. A 20-µl aliquot was taken out at each time point and immediately mixed with 20 µl of gel loading buffer II (Denaturing PAGE, Ambion, Grand Island, NY) and stored at −80 °C. RNA from all time points was resolved with PAGE (12% polyacrylamide with 8 M urea). The gel was stained with SYBR Gold (Invitrogen) to visualize the RNA bands. Quantification of band intensities was performed using Image J software and plotted utilizing GraphPad Prism 5 software.
Assessment of aptamer safety in human PBMCs
Human PBMC isolation. Heparinized venous blood was collected from healthy adult volunteers using a protocol approved by the Institutional Review Board of the University of Iowa, and with donor informed consent. PBMCs were isolated by Ficoll-Hypaque density gradient centrifugation. PBMCs were washed twice in HEPES (N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid))-buffered, endotoxin-free RPMI-1640 medium supplemented with l-glutamine (both from Lonza, Walkersville, MD). Human serum was prepared by incubation of non-heparinized whole blood at 37 °C for 30 minutes followed by 30 minutes on ice. The clotted blood was manually disrupted and centrifuged at 1,100×g for 15 minutes at 4 °C. Serum was sterile filtered before use. Where indicated, pooled human serum was prepared from a minimum of 14 different donors.
PBMC cytotoxicity/apoptosis studies. PBMCs were diluted in RPMI containing 2.5% human serum to a final concentration of 2 × 106 PBMCs in 1 ml total volume. Samples were placed in twelve 75 mm polypropylene round-bottom tubes (BD Biosciences, Franklin Lakes, NJ) and were allowed to rest for 2 hours at 37 °C, 5% CO2. After this, an unstimulated sample was processed and is represented as time 0. RNA-aptamer constructs were added to a final concentration of 150 nmol/l and control samples were stimulated with poly(I:C) at 100 µg/ml or 1 µM staurosporine. At 4, 12, and 24 hours post-stimulation, 10 µl aliquots of the PBMCs were taken and mixed with Caspase-Glo 3/7 Reagent (Promega, Madison, WI) for detection of caspase activity according to the manufacturer's protocols. The remaining cells were pelleted by centrifugation at 900×g, for 15 minutes at 4 °C. Supernatants were removed and the cell pellets were re-suspended in 1 ml of TRIzol Reagent (Life Technologies, Carlsbad, CA) and stored at −80 °C until RNA isolation was performed (methods below). Cytotoxicity was determined by measuring LDH release from the supernatants with the CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega) per manufacturer's recommendation. Percentage cytotoxicity was calculated by comparison of each sample to the maximum LDH release achieved by addition of 9% Triton X-100 to an untreated sample at each time point.
PBMC immune stimulation studies. Total cellular RNA was isolated from treated hPBMCs resuspended in TRIzol (see above) using the PureLink RNA Mini Kit (Ambion). The purity and concentration of isolated RNA was determined using a Nano-Drop 2000c spectrophotometer (Thermo Scientific). RT-qPCR was used to determine the expression level of indicated immune responsive genes according to the methods described above for the mouse immune stimulation studies. Each reaction was carried out in triplicate and results were normalized to GAPDH expression. Human-specific primers were as follows: GAPDH (Forward: AGCCACATCGCTCAGACAC; Reverse: GCCCAATACGACCAAATCC) OAS-1 (Forward: GGTGGAGTTCGATGTGCTG; Reverse: AGGTTTATAGCCGCCAGTCA) IFN-β (Forward: CAGAAGGAGGACGCCGCATTGAC; Reverse: CCAGGCACAGTGACTGTACTCC), IFN-γ (Forward: CCAACGCAAAGCAATACATGA; Reverse: CCTTTTTCGCTTCCCTGTTTTA), IL-6 (Forward: GGTACATCCTCGACGGCATCT; Reverse: GTGCCTCTTTGCTGCTTTCAC), IFIT1 (Forward: GCCTCCTTGGGTTCGTCTACAA; Reverse: CTCAGGGCCCGCTCATAGTA).
SUPPLEMENTARY MATERIAL Figure S1. Functional characterization of PSMA RNA aptamers. Figure S2. PSMA expression drives cancer cell migration. Figure S3. PSMA expression in cancer cell lines used in this study. Figure S4. In vivo efficacy of A9g PSMA aptamer. Figure S5. NIR-RNA conjugation chemistry. Materials and Methods
Acknowledgments
We thank Rui Sousa (University of Texas, San Antonio) for his generous gift of the mutant (Y639F) T7 RNAP. We thank Michael Henry (Department of Molecular Physiology and Biophysics, University of Iowa) for supplying PC-3 and 22Rv1(1.7) luciferase-positive cells. We thank James McNamara (University of Iowa), David Lubaroff (University of Iowa), Eli Gilboa (University of Miami), and George Weiner (University of Iowa) for helpful discussions. J.P.D. is supported by a postdoctoral training grant from the National Institutes of Health (T32HL07344). M.E.W., L.A.A., and D.K.M. are supported by the National Institutes of Health. This work was supported by grants to PHG from the National Institutes of Health (R01CA138503 and R21DE019953), Mary Kay Foundation (9033-12 and 001-09), Elsa U Pardee Foundation (E2766), and the Roy J Carver Charitable Trust (RJCCT 01-224). Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA086862.
J.P.D., L.I.H., G.S.T., M.E.L., W.M.R., C.A.H., Y.C., F.J.H., X.Y.L., and P.H.G. performed the research. J.P.D., L.I.H., G.S.T., M.E.L., W.M.R., C.A.H., and D.K.M. analyzed the data. D.K.M., M.E.W., D.A.V., and L.A.A. provided the expertise. J.P.D., L.I.H., G.S.T., and P.H.G. designed the research. P.H.G. coordinated the research. J.P.D. and P.H.G. wrote the manuscript.
Supplementary Material
Functional characterization of PSMA RNA aptamers.
PSMA expression drives cancer cell migration.
PSMA expression in cancer cell lines used in this study.
In vivo efficacy of A9g PSMA aptamer.
NIR-RNA conjugation chemistry.
References
- Cereda V, Formica V, Massimiani G, Tosetto L, Roselli M. Targeting metastatic castration-resistant prostate cancer: mechanisms of progression and novel early therapeutic approaches. Expert Opin Investig Drugs. 2014;23:469–487. doi: 10.1517/13543784.2014.885950. [DOI] [PubMed] [Google Scholar]
- Pal SK, Twardowski P, Sartor O. Critical appraisal of cabazitaxel in the management of advanced prostate cancer. Clin Interv Aging. 2010;5:395–402. doi: 10.2147/CIA.S14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N, Dizon DS. New-generation platinum agents for solid tumors. Future Oncol. 2009;5:33–42. doi: 10.2217/14796694.5.1.33. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Heston WD. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J Cell Biochem. 2004;91:528–539. doi: 10.1002/jcb.10661. [DOI] [PubMed] [Google Scholar]
- Israeli RS, Powell CT, Corr JG, Fair WR, Heston WD. Expression of the prostate-specific membrane antigen. Cancer Res. 1994;54:1807–1811. [PubMed] [Google Scholar]
- Sweat SD, Pacelli A, Murphy GP, Bostwick DG. Prostate-specific membrane antigen expression is greatest in prostate adenocarcinoma and lymph node metastases. Urology. 1998;52:637–640. doi: 10.1016/s0090-4295(98)00278-7. [DOI] [PubMed] [Google Scholar]
- Mlcochová P, Barinka C, Tykvart J, Sácha P, Konvalinka J. Prostate-specific membrane antigen and its truncated form PSM'. Prostate. 2009;69:471–479. doi: 10.1002/pros.20894. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Teske S, Vessella RL, True LD, Zakrajsek BA. Expression of prostate specific membrane antigen and three alternatively spliced variants of PSMA in prostate cancer patients. Int J Cancer. 2003;107:323–329. doi: 10.1002/ijc.11402. [DOI] [PubMed] [Google Scholar]
- Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res. 1997;3:81–85. [PubMed] [Google Scholar]
- Wright GL, Jr, Grob BM, Haley C, Grossman K, Newhall K, Petrylak D, et al. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48:326–334. doi: 10.1016/s0090-4295(96)00184-7. [DOI] [PubMed] [Google Scholar]
- Yates DR, Rouprêt M, Drouin SJ, Comperat E, Ricci S, Lacave R, et al. Quantitative RT-PCR analysis of PSA and prostate-specific membrane antigen mRNA to detect circulating tumor cells improves recurrence-free survival nomogram prediction after radical prostatectomy. Prostate. 2012;72:1382–1388. doi: 10.1002/pros.22488. [DOI] [PubMed] [Google Scholar]
- Chang SS, O'Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin Cancer Res. 1999;5:2674–2681. [PubMed] [Google Scholar]
- Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59:3192–3198. [PubMed] [Google Scholar]
- Elsässer-Beile U, Bühler P, Wolf P. Targeted therapies for prostate cancer against the prostate specific membrane antigen. Curr Drug Targets. 2009;10:118–125. doi: 10.2174/138945009787354601. [DOI] [PubMed] [Google Scholar]
- Olson WC, Israel RJ. Antibody-drug conjugates targeting prostate-specific membrane antigen. Front Biosci (Landmark Ed) 2014;19:12–33. doi: 10.2741/4193. [DOI] [PubMed] [Google Scholar]
- McNamara JO, 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, et al. Cell type-specific delivery of sirnas with aptamer-sirna chimeras. Nat Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- Pastor F, Kolonias D, Giangrande PH, Gilboa E. Induction of tumour immunity by targeted inhibition of nonsense-mediated mRNA decay. Nature. 2010;465:227–230. doi: 10.1038/nature08999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, et al. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Zhang Y, Ribas J, Chowdhury WH, Castanares M, Zhang Z, et al. Prostate-targeted radiosensitization via aptamer-shRNA chimeras in human tumor xenografts. J Clin Invest. 2011;121:2383–2390. doi: 10.1172/JCI45109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto JT, Suffoletto BP, Berzin TM, Qiao CH, Lin S, Tong WP, et al. Prostate-specific membrane antigen: a novel folate hydrolase in human prostatic carcinoma cells. Clin Cancer Res. 1996;2:1445–1451. [PubMed] [Google Scholar]
- Carter RE, Feldman AR, Coyle JT. Prostate-specific membrane antigen is a hydrolase with substrate and pharmacologic characteristics of a neuropeptidase. Proc Natl Acad Sci USA. 1996;93:749–753. doi: 10.1073/pnas.93.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidus RG, Tiffany CW, Isaacs JT, Slusher BS. Prostate-specific membrane antigen (PSMA) enzyme activity is elevated in prostate cancer cells. Prostate. 2000;45:350–354. doi: 10.1002/1097-0045(20001201)45:4<350::aid-pros10>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Yao V, Berkman CE, Choi JK, O'Keefe DS, Bacich DJ. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate. 2010;70:305–316. doi: 10.1002/pros.21065. [DOI] [PubMed] [Google Scholar]
- Conway RE, Petrovic N, Li Z, Heston W, Wu D, Shapiro LH. Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol Cell Biol. 2006;26:5310–5324. doi: 10.1128/MCB.00084-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey WM, Hernandez FJ, Huang SY, Cao S, Howell CA, Thomas GS, et al. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther. 2011;21:299–314. doi: 10.1089/nat.2011.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. 2010;9:537–550. doi: 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao V, Parwani A, Maier C, Heston WD, Bacich DJ. Moderate expression of prostate-specific membrane antigen, a tissue differentiation antigen and folate hydrolase, facilitates prostate carcinogenesis. Cancer Res. 2008;68:9070–9077. doi: 10.1158/0008-5472.CAN-08-2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlich JA, Taggart P, Proctor J, Stahle P, Colis R, Hall L, et al. The non-GLP toleration/dose range finding study: design and methodology used in an early toxicology screening program. Proc West Pharmacol Soc. 2009;52:94–98. [PubMed] [Google Scholar]
- Robbins M, Judge A, MacLachlan I. siRNA and innate immunity. Oligonucleotides. 2009;19:89–102. doi: 10.1089/oli.2009.0180. [DOI] [PubMed] [Google Scholar]
- Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther. 2006;13:644–670. doi: 10.1016/j.ymthe.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams KE, Ke S, Kwon S, Liang F, Fan Z, Lu Y, et al. Comparison of visible and near-infrared wavelength-excitable fluorescent dyes for molecular imaging of cancer. J Biomed Opt. 2007;12:024017. doi: 10.1117/1.2717137. [DOI] [PubMed] [Google Scholar]
- Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11:812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- Dienstmann R, Markman B, Tabernero J. Application of monoclonal antibodies as cancer therapy in solid tumors. Curr Clin Pharmacol. 2012;7:137–145. doi: 10.2174/157488412800228929. [DOI] [PubMed] [Google Scholar]
- Larsen PB, Kümler I, Nielsen DL. A systematic review of trastuzumab and lapatinib in the treatment of women with brain metastases from HER2-positive breast cancer. Cancer Treat Rev. 2013;39:720–727. doi: 10.1016/j.ctrv.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Thanopoulou E, Judson I. The safety profile of imatinib in CML and GIST: long-term considerations. Arch Toxicol. 2012;86:1–12. doi: 10.1007/s00204-011-0729-7. [DOI] [PubMed] [Google Scholar]
- Thiel KW, Giangrande PH. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides. 2009;19:209–222. doi: 10.1089/oli.2009.0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kuan CT, Mi J, Zhang X, Clary BM, Bigner DD, et al. Aptamers selected against the unglycosylated EGFRvIII ectodomain and delivered intracellularly reduce membrane-bound EGFRvIII and induce apoptosis. Biol Chem. 2009;390:137–144. doi: 10.1515/BC.2009.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Du L, Li M. Advances and perspectives in cell-specific aptamers. Curr Pharm Des. 2011;17:80–91. doi: 10.2174/138161211795049769. [DOI] [PubMed] [Google Scholar]
- Dassie JP, Giangrande PH. Current progress on aptamer-targeted oligonucleotide therapeutics. Ther Deliv. 2013;4:1527–1546. doi: 10.4155/tde.13.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey WM, Huang L, Kloepping KC, Baumhover NJ, Giangrande PH, Schultz MK. Synthesis and radiolabeling of chelator-RNA aptamer bioconjugates with copper-64 for targeted molecular imaging. Bioorg Med Chem. 2011;19:4080–4090. doi: 10.1016/j.bmc.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth S, Johns R, Templin MV. Delivery and biodistribution of siRNA for cancer therapy: challenges and future prospects. Ther Deliv. 2012;3:245–261. doi: 10.4155/tde.11.155. [DOI] [PubMed] [Google Scholar]
- Ifediba MA, Moore A. In vivo imaging of the systemic delivery of small interfering RNA. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4:428–437. doi: 10.1002/wnan.1158. [DOI] [PubMed] [Google Scholar]
- Huang Y, Eckstein F, Padilla R, Sousa R. Mechanism of ribose 2'-group discrimination by an RNA polymerase. Biochemistry. 1997;36:8231–8242. doi: 10.1021/bi962674l. [DOI] [PubMed] [Google Scholar]
- Tiffany CW, Slusher BS. Measurement of glutamate carboxypeptidase II (NAALADase) enzyme activity by the hydrolysis of [³H]-N-acetylaspartylglutamate (NAAG) Curr Protoc Pharmacol. 2002;Chapter 3:Unit3.10. doi: 10.1002/0471141755.ph0310s15. [DOI] [PubMed] [Google Scholar]
- Drake JM, Gabriel CL, Henry MD. Assessing tumor growth and distribution in a model of prostate cancer metastasis using bioluminescence imaging. Clin Exp Metastasis. 2005;22:674–684. doi: 10.1007/s10585-006-9011-4. [DOI] [PubMed] [Google Scholar]
- Freshney RI. Culture of Animal Cells: A Manual of Basic Technique. Wiley-Liss; New York; 1994. [Google Scholar]
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78:1039–1049. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Functional characterization of PSMA RNA aptamers.
PSMA expression drives cancer cell migration.
PSMA expression in cancer cell lines used in this study.
In vivo efficacy of A9g PSMA aptamer.
NIR-RNA conjugation chemistry.