

Abstract
Prostaglandin (PG) E2 and PGI2 are essential to hyperalgesia in inflammatory tissues. These prostaglandins are produced from arachidonic acid, which is cleaved from membrane phospholipids by the action of phospholipase A2 (PLA2). Which isozyme of PLA2 is responsible for the cleavage of arachidonic acid and the production of prostaglandins essential to inflammation-induced hyperalgesia is not clear. In this study, we examined the effects of two PLA2 isozyme-specific inhibitors on carrageenan-induced production of PGE2 and PGI2 in rat hind paw and behavioral nociceptive response to radiant heat. Local administration of bromoenol lactone (BEL), an inhibitor of calcium-independent PLA2 (iPLA2), significantly reduced carrageenan-induced elevation of prostaglandins in the inflamed foot pad 3 h after injection. It also ameliorated the hyperalgesic response between 1 h and 3 h after carrageenan injection. On the other hand, AACOCF3, an inhibitor of cytosolic PLA2, suppressed neither prostaglandin production nor the hyperalgesic response. BEL did not suppress the mRNA levels of iPLA2 β, iPLA2 γ, cyclooxygenase-2, microsomal prostaglandin E synthase, prostaglandin I synthase, or proinflammatory cytokines in the inflamed foot pad, indicating that BEL did not suppress inflammation itself. These results suggest that iPLA2 is involved in the production of prostaglandins and hyperalgesia at the inflammatory loci.
1. Introduction
Acute inflammation produces a condition known as hyperalgesia, which is characterized by enhanced pain sensation and reduced pain threshold. This abnormal sensory state is brought about, at least in part, by sensitization of peripheral nociceptors. Accumulating evidence indicates that inflammatory mediators including prostaglandin E2 (PGE2) and PGI2 are responsible for the sensitization of nociceptors [1, 2]. Recent studies further clarified the molecular mechanism of sensitization: PGE2 and PGI2 enhance or sensitize the activity of the capsaicin receptor transient potential receptor V1 (TRPV1) through the activation of PGE2 EP1 and PGI2 receptors, respectively [3]. Protein kinase C and protein kinase A are involved in the activation of TRPV1 by these prostaglandins. In contrast, cyclopentenone prostaglandins, including 15-d-PGJ2, PGA2, and PGA1, which are metabolites of PGD2, PGE2, and PGE1, respectively, directly activate the irritant receptor TRPA1 [4]. For pharmacological control of inflammatory hyperalgesia, it is important to understand how these prostaglandins are produced in inflamed tissues.
Prostaglandins are generated through the arachidonic acid cascade [5], which involves three enzymatic steps: first phospholipase A2 (PLA2) cleaves membrane phospholipids and releases arachidonic acid; second, cyclooxygenase (COX) converts arachidonic acid into PGH2; and, third, various types of prostaglandin isomerases convert PGH2 to bioactive prostaglandins, including PGE2 and PGI2. Each enzymatic reaction can involve multiple isozymes. Recent studies have shown that COX-2, an inducible isozyme of COX, and microsomal prostaglandin synthase-1 (mPGES-1), an inducible isozyme of PGE synthase, are responsible for the generation of PGE2 in inflamed tissues [6, 7]. In these studies, administration of a COX-2 inhibitor to rats [7] or disruption of the mPGES-1 gene in mice lowered PGE2 content in inflamed tissues and eased pain-related behaviors [6]. Thus, COX-2 and mPGES-1 could be pharmacological targets for the treatment of inflammatory pain.
However, it is not clear which isozyme of PLA2 is responsible for the production of prostaglandins and development of hyperalgesia in inflamed tissues. PLA2 has over ten isozymes which are classified into three categories based on their structural and functional similarities [8, 9]. First, cytosolic PLA2s (cPLA2 or group IV PLA2) are located in the cytoplasm and are activated by low calcium ion concentrations (μM levels). Second, secretory PLA2s (sPLA2 or group IB, II, V, or X PLA2) are released into the extracellular space where they are activated by high concentrations (mM levels) of calcium ions. Third, calcium-independent PLA2s (iPLA2 or group VI PLA2) are present in the cytoplasm and do not require calcium ions for their enzymatic activity, although the precise mechanisms for activation are unclear. Among these PLA2s, a significant role for iPLA2 in the production of prostaglandins in carrageenan-induced pleuritis in rats was demonstrated [10]. It is also reported that, in the spinal cord, cPLA2 seems to be involved in inflammation-induced hyperalgesia [11]. In the present study, we examined if iPLA2 and/or cPLA2 are responsible for the production of prostaglandins and development of hyperalgesia in carrageenan-induced inflammation in the rat hind paw.
2. Materials and Methods
2.1. Materials
Male Sprague-Dawley rats (nine weeks old, 300–320 g) were purchased from Charles River Laboratories (Yokohama, Japan). All chemicals were obtained from commercial suppliers: bromoenol lactone (BEL; iPLA2 inhibitor), arachidonyl trifluoromethyl ketone (AACOCF3; cPLA2 inhibitor), PGE2 enzyme immunoassay (EIA) kit, and 6-keto-PGF1α EIA kit from Cayman Chemical (Ann Arbor, MI); Isogen (RNA extraction solution) from Nippon Gene (Tokyo, Japan); reverse transcription Kit from Invitrogen (Carlsbad, CA); PCR Sybr Green master mix, LightCycler TaqMan Master, and TaqMan Probes from Roche Diagnostics (Indianapolis, IN); and RNAlater (RNA stabilization solution) from Ambion (Austin, TX).
2.2. Animals
All experiments were carried out according to protocols approved by the Institutional Animal Care Committee of Kyoto Prefectural University of Medicine. Rats were housed four per cage and maintained on a 12 h light/dark cycle (light on 8:00–20:00) with controlled temperature (25 ± 3°C) and humidity (55 ± 15%). Animals were allowed free access to food and water at all times.
2.3. Pharmacological Treatment
The plantar surface of the left paw received a subcutaneous injection of either 3 mg type λ carrageenan (Sigma-Aldrich, St. Louis, MO) dissolved in 100 μL saline or saline alone. Just after carrageenan injection, one of the two PLA2 inhibitors (30 nmol) or vehicle (100 μL of 0.1% dimethyl sulfoxide (DMSO) in saline) was injected into the same site. The inhibitor dose was determined according to the study by Gilroy et al. [10]. For biochemical analyses, each rat was anesthetized with pentobarbital (60 mg/kg, intraperitoneal injection) 3 h after subcutaneous injection, and the left hind leg was cut at the knee, quickly frozen in dry ice powder, and kept at −80°C until further processing. Rats were killed by decapitation. This time point was determined based on the following nociceptive behavior study, in which a PLA2 inhibitor was effective between 1 h and 3 h after injection.
For the assessment of nociceptive behavior response, rats were injected with the carrageenan/saline and PLA2 inhibitors/vehicle in the manner described above. Thermally evoked paw-withdrawal response was assessed using a device developed in Yaksh's lab [12]. The paw-withdrawal latency was measured for the left hind paw before and every 60 minutes after the subcutaneous injection for a total of 360 minutes. For each time point, the latencies were measured 3 times in each rat and averaged. Paw-withdrawal latencies were expressed as ratios to the baseline value of each rat.
2.4. Biochemical Assay
For analyses of PGE2 and 6-keto-PGF1α (a metabolite of PGI2), the hind paws were coronally cut into 50 μm thick frozen sections in a cryostat at −20°C. Twenty of these sections were collected in a plastic tube containing precooled ethanol (1 mL) and indomethacin (10 μg) which prevents the synthesis of prostaglandins during tissue processing. After measuring wet tissue weight, the sections were homogenized with polytron for 30 s followed by sonication for 20 s. The homogenates were centrifuged (15000 rpm for 20 min at 4°C) and the supernatant was collected. Ethanol was evaporated from the supernatant using a vacuum centrifuge. Prostaglandins, which were retained in the tube, were dissolved in EIA buffer. PGE2 and 6-keto-PGF1α were measured using EIA kits according to the manufacturer's instructions. Tissue pellet remaining in the plastic tube was heated in a heat block to completely evaporate the ethanol. The weight of dried pellet was considered to be the dry tissue weight of the paw from which the prostaglandins were extracted.
2.5. Real-Time RT-PCR
Frozen paw sections were prepared as described above. Twenty of these sections were placed into a vial containing RNA later (1 mL) and stored at −30°C until further processing. For RNA extraction, the samples were homogenized in 1 mL phenol-based RNA extraction solution (Isogen) with polytron for 30 s followed by sonication for 20 s. Total RNA was isolated according to the manufacturer's instructions. cDNA was prepared from total RNA using M-MLV reverse transcriptase and random hexamer as the primer. The reverse-transcribed cDNA was amplified using a light cycler (Roche Diagnostics). mRNAs of COX-2, mPGES-1, iPLA2 β, iPLA2 γ, and GAPDH were quantified with the Sybr Green protocol. For the quantification of mRNAs of prostaglandin I synthase (PGIS), interleukin-1β (IL1β), and interleukin-6 (IL6), the TaqMan Probe protocol was used. Primer pairs used for the PCR reaction were as follows:
-
COX-2: 5′-ctcactttgagtcattc-3′, 5′-gattagtactgtagggttaatg-3′,
-
mPGES-1: 5′-aatgaacccacgcattcgct-3′, 5′-cagccttcatggctccgtct-3′,
-
iPLA2 β: 5′-caaggaactgggcaagatgg-3′, 5′-agagggcgttgaccagcact-3′,
-
iPLA2 γ: 5′-gaataccacaacatacacga-3′, 5′-acctaaaatacgtgtcagca-3′,
-
GAPDH: 5′-tgaacgggaagctcactgg-3′, 5′-tccaccaccctgttgctgta-3′,
-
PGIS: 5′-atgccatcaacagcatcaaa-3′, 5′-gctccaggtcgaaatgagtc-3′, TaqMan Probe (UPL #18),
-
IL1β: 5′-tgtgatgaaagacggcacac-3′, 5′-cttcttctttgggtattgtttgg-3′, TaqMan Probe (UPL #78),
-
IL6: 5′-cccttcaggaacagctatgaa-3′, 5′-acaacatcagtcccaagaagg-3′, TaqMan Probe (UPL #20),
-
GAPDH: 5′-agctggtcatcaatgggaaa-3′, 5′-atttgatgttagcgggatcg-3′, TaqMan Probe (UPL #9).
Relative expression levels of each gene were calculated with the following formula:
| (1) |
Specificity of the PCR products was checked by the melting curve and by agarose gel electrophoresis in the Sybr Green protocol.
2.6. Statistics
The significance of differences between two groups was determined by t-test. The significance of differences among multiple groups was determined by multiple t-tests with Bonferroni correction. Differences were considered significant at P < 0.05. Data are presented as mean ± SEM.
3. Results
We examined the effects of PLA2 inhibitors on PGE2 and 6-keto-PGF1α (a metabolite of PGI2) levels in inflamed foot pad. Carrageenan and PLA2 inhibitors/vehicle were injected into the right foot pad at the same time. Three hours after the injection, carrageenan significantly elevated PGE2 and 6-keto-PGF1α levels compared to injection of saline alone in vehicle-, BEL- and AACOCF3-coinjected groups (N = 4 in each group, P = 0.0002–0.014) (Figure 1). BEL, an iPLA2 inhibitor, significantly suppressed carrageenan-induced increases in PGE2 by 57% (P = 0.009) and 6-keto-PGF1α by 49% (P = 0.017) compared to vehicle. On the other hand, AACOCF3, a cPLA2 inhibitor and less potent iPLA2 inhibitor, did not suppress the prostaglandin levels compared to the vehicle-treated rats. The two inhibitors did not exert significant effects on the prostaglandin levels in the saline-injected foot pad.
Figure 1.

Contents of PGE2 (a) and 6-keto-PGF1α (b) in rat hind paw. Carrageenan (Car) injection (filled bars) significantly elevated both prostaglandin levels compared to saline (Sal) injection (open bars) in vehicle-, BEL-, and AACOCF3- (AACO-) treated groups (P = 0.0002–0.014, t-test, N = 4 in each group). BEL but not AACOCF3 significantly suppressed carrageenan-induced increases in PGE2 (a) and 6-keto-PGF1α (b) compared to vehicle (P = 0.009 for PGE2 and P = 0.017 for 6-keto-PGF1α, multiple t-tests with Bonferroni correction, 2 comparisons among 3 groups; vehicle versus BEL and vehicle versus AACOCF3). Asterisks indicate the statistical significance of the difference between vehicle + carrageenan group and BEL + carrageenan group.
Real-time RT-PCR studies revealed that mRNAs of both iPLA2 isozymes, that is, iPLA2 β and iPLA2 γ, were present in the noninflamed foot pad, and their levels did not change following carrageenan injection (Figure 2).
Figure 2.
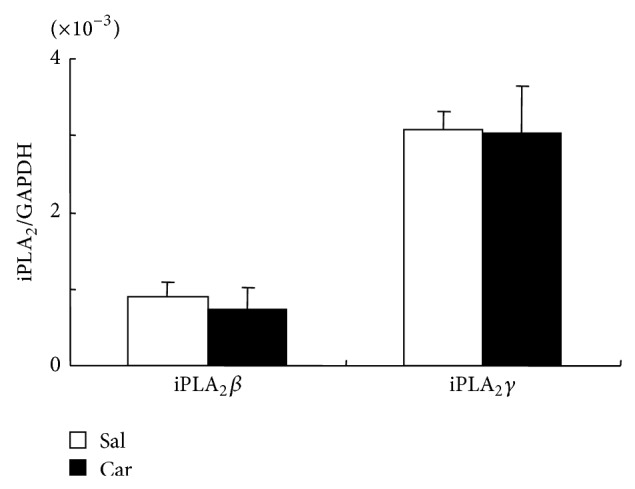
Levels of mRNA of iPLA2 β and iPLA2 γ relative to that of an internal control gene (GAPDH) in rat hind paw. Their relative levels were not influenced by carrageenan-induced inflammation. Open bars and filled bars represent results from saline (Sal)-injected group and carrageenan (Car)-injected group, respectively. N = 4 in each group.
We then asked if BEL influences the induction of COX-2, mPGES-1, and PGIS, which are possibly involved in carrageenan-induced prostaglandin synthesis. COX-2 mRNA, mPGES-1 mRNA, and PGIS mRNA were increased by carrageenan injection though the increases did not reach the statistically significant level except for COX-2 mRNA and PGIS mRNA in carrageenan + BEL group (Figure 3). There was no significant effect of BEL on the COX-2 mRNA, mPGES-1 mRNA, or PGIS mRNA levels (Figure 3). We also examined the effects of BEL on carrageenan-induced proinflammatory cytokine mRNAs (Figure 4). Carrageenan significantly elevated the mRNA levels of IL1β and IL6. BEL slightly but significantly elevated carrageenan-induced IL1β mRNA and did not change carrageenan-induced IL6 mRNA. Therefore, BEL seemed to act solely on the enzyme activity of iPLA2s resulting in suppression of prostaglandin levels in inflamed tissue.
Figure 3.

Relative expression of COX-2 mRNA (a), mPGES-1 mRNA (b), and PGIS mRNA (c) in rat hind paw. Carrageenan (Car) injection (filled bars) elevated COX-2 mRNA and mPGES-1 mRNA levels compared to saline (Sal) injection (open bars) though the elevations were not statistically significant by t-test except for COX-2 and PGIS in BEL-treated groups. BEL did not significantly influence COX-2 mRNA, mPGES-1 mRNA, or PGIS mRNA levels.
Figure 4.
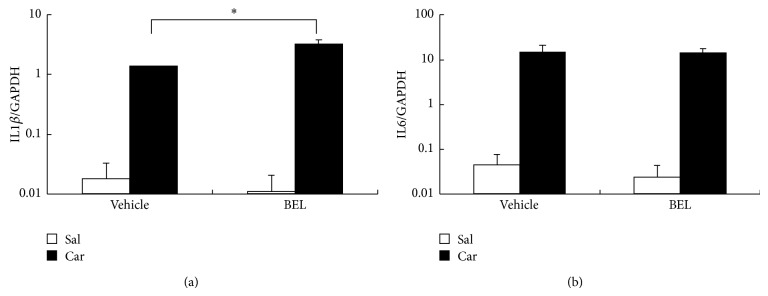
Relative expression of IL1β mRNA (a) and IL6 mRNA (b) in rat hind paw. Carrageenan (Car) injection (filled bars) significantly elevated IL1β mRNA and IL6 mRNA levels compared to saline (Sal) injection (open bars) (P < 0.05). BEL slightly but significantly elevated carrageenan-induced IL1β mRNA (P < 0.05) and did not affect carrageenan-induced IL6 mRNA.
Lastly, we studied the effects of PLA2 inhibitors on carrageenan-induced thermal hyperalgesia. When carrageenan was injected with vehicle, foot withdrawal latency to radiant heat was reduced by approximately 70% compared to the preinjection level (at time point 0), indicating the induction of thermal hyperalgesia (Figure 5(a)). The thermal hyperalgesia was statistically significant throughout the observation period of 6 h (N = 5, P < 0.005). Carrageenan-induced thermal hyperalgesia was ameliorated by BEL by 44% at 1 h, 61% at 2 h, and 46% at 3 h after injection compared to vehicle-treated group. At these 3 time points, the difference between carrageenan + BEL group (N = 6) and carrageenan + vehicle group (N = 5) was statistically significant (P = 0.026, 0.014, and 0.015, resp.). On the other hand, AACOCF3 did not influence the thermal hyperalgesia (Figure 5(a)). Injection of saline did not induce thermal hyperalgesia (Figure 5(b)). Both BEL and AACOCF3 did not show significant effect in saline-injected foot pad.
Figure 5.

Paw-withdrawal latencies to radiant heat. (a) The latencies were reduced by approximately 70% after carrageenan (Car) injection in vehicle-treated group. The reduction was statistically significant throughout the observation period of 6 h (N = 5, P < 0.005, multiple t-tests with Bonferroni correction, 6 comparisons among 7 groups; time point 0 versus time points 1–6). Treatment with BEL but not with AACOCF3 prolonged the latencies at 1 h, 2 h, and 3 h after injection. At these time points, the effect of BEL was statistically significant (P = 0.026, 0.014, and 0.015, resp., multiple t-tests with Bonferroni correction, 2 comparisons among 3 groups; vehicle versus BEL and vehicle versus AACOCF3 in each time point). Asterisks indicate the statistical significance of the difference between vehicle + carrageenan group and BEL + carrageenan group. (b) Saline (Sal) injection had little effect on paw-withdrawal latencies. PLA2 inhibitors did not influence the latencies in saline-injected groups.
4. Discussion
PGE2 and PGI2 are known to sensitize nociceptors and develop hyperalgesia during inflammation [1, 2]. However, which isoform of PLA2 is involved in the synthesis of these prostaglandins in inflammatory tissues was unclear. In the present study, we showed that BEL, a potent inhibitor of iPLA2, lowered these prostaglandins by approximately 50% in rat foot pads that were inflamed with carrageenan. The inhibitor also halved carrageenan-induced thermal hyperalgesia during the first 3 h after injection. We confirmed that mRNAs of iPLA2 β and iPLA2 γ were constitutively expressed in the rat foot pad. This result is in line with the study by Gilroy et al. [10], who demonstrated that BEL reduced PGE2 content in the cell-free inflammatory exudates of the pleural cavity 3 h after carrageenan injection.
It should be noted that BEL is also a potent inhibitor of phosphatidic acid phosphohydrolase. Grkovich et al. [13] reported that BEL suppressed COX-2 expression in LPS-activated macrophages through the inhibition of phosphatidic acid phosphohydrolase-1. Therefore, we wondered if BEL might have reduced prostaglandin levels by suppressing COX-2 induction. However, in the present study, carrageenan-induced COX-2 mRNA was not affected by BEL. This was also the case for mRNAs of mPGES-1 and PGIS, the final enzymes in PGE2 and PGI2 synthesis, respectively. In line with these results, BEL did not reduce carrageenan-induced proinflammatory cytokines, which are strong inducers of COX-2 and other enzymes in the arachidonic acid cascade. These results strongly suggest that BEL did not significantly suppress inflammation but inhibited iPLA2 enzyme activity to reduce carrageenan-induced PGE2 and PGI2 synthesis and, consequently, thermal hyperalgesia. Therefore, we conclude that iPLA2 is responsible for the synthesis of prostaglandins in carrageenan-induced tissue inflammation.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for the treatment of inflammatory pain. NSAIDs potently inhibit COX but do not suppress the supply of its substrate, that is, arachidonic acid. Thus, free arachidonic acid may enter lipoxygenase or epoxygenase pathways to be various potent eicosanoids such as leukotrienes. This may result in side effects of NSAIDs. On the other hand, since inhibitors of PLA2, such as BEL, reduce the supply of free arachidonic acid, PLA2 inhibitors may be more ideal in the treatment of symptoms induced by eicosanoids than NSAIDs.
The inhibition of prostaglandin synthesis by BEL was partial—approximately 50% of the control (vehicle alone). This fact suggests that other PLA2 isoforms, that is, cPLA2 or sPLA2s, also participated in carrageenan-induced prostaglandin synthesis. Traditionally, cPLA2 has been considered the major PLA2 that cleaves arachidonic acid from the sn-2 position of membrane phospholipids during inflammation. In the present study injection of AACOCF3, a cPLA2 inhibitor, into the foot pad neither reduced PGE2 and PGI2 nor suppressed thermal hyperalgesia, suggesting that cPLA2 does not play a significant role in these responses. This result is consistent with those of two previous studies, the first of which by Gilroy et al. [10] showed no effect of AACOCF3 on the PGE2 content of cell-free inflammatory exudates of the pleural cavity 3 h after carrageenan injection. The second study by Lucas et al. [11] reported that intraplantar injection of AACOCF3 did not suppress carrageenan-induced thermal hyperalgesia even with a dose approximately 20 times higher than that used in the present study. On the other hand, it is reported that a new type of cPLA2 inhibitor reduced carrageenan-induced PGE2 production in the rat air pouch [14]. The reason for the discrepancy is unclear at present, and we cannot completely exclude the possibility that cPLA2 is partly involved in PGE2 and PGI2 production in inflammatory tissues.
PGE2 and possibly PGI2 augment nociceptive responses by acting in multiple sites along the pain processing neural pathway. In each site, distinct isoforms of PLA2 seem to be involved in prostaglandin synthesis. In the peripheral inflammatory site, these prostaglandins are produced by the action of iPLA2 and sensitize nociceptors, as shown in this study. In the spinal cord, PGE2 is produced in response to nociceptive neural inputs from the primary sensory neurons and possibly enhances the pain signal transmission [15]. In this site, cPLA2 and sPLA2 but not iPLA2 are reported to play major roles in PGE2 synthesis and hyperalgesic response [11, 16]. Furthermore, in the spinal cord and brain, peripheral inflammation induces prostaglandin synthesis through the action of circulating inflammatory cytokines such as interleukin-6 [17]. This is evidenced by an elevation of PGE2 in the cerebrospinal fluid and induction of COX-2 in endothelial cells throughout the brain and spinal cord after intraplantar injection of carrageenan [18]. Antiserum to interleukin-6 suppressed these responses [17]. However, little is known about which isoform of PLA2 is activated upstream of COX-2. Further studies are necessary to thoroughly determine which PLA2 isoforms are involved in prostaglandin synthesis in the central nervous system under inflammatory conditions.
5. Conclusion
The present study has indicated the importance of iPLA2 in prostaglandin synthesis at the inflammatory site and suggested that iPLA2 is a possible target for the treatment of inflammatory hyperalgesia.
Acknowledgments
The authors thank Dr. Aiko Hori and Ms. Tomoko Yamamoto for helping in the measurements of prostaglandins. This work was supported by KAKENHI (25460322 and 24592354).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Ferreira S. H. Peripheral analgesia: mechanism of the analgesic action of aspirin like drugs and opiate-antagonists. British Journal of Clinical Pharmacology. 1980;10(2):237S–245S. doi: 10.1111/j.1365-2125.1980.tb01806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meves H. The action of prostaglandins on ion channels. Current Neuropharmacology. 2006;4(1):41–57. doi: 10.2174/157015906775203048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moriyama T., Higashi T., Togashi K., et al. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Molecular Pain. 2005;1, article 3 doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Materazzi S., Nassini R., Andrè E., et al. Cox-dependent fatty acid metabolites cause pain through activation of the irritant receptor TRPA1. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(33):12045–12050. doi: 10.1073/pnas.0802354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith W. L., Marnett L. J., Dewitt D. L. Prostaglandin and thromboxane biosynthesis. Pharmacology and Therapeutics. 1991;49(3):153–179. doi: 10.1016/0163-7258(91)90054-P. [DOI] [PubMed] [Google Scholar]
- 6.Kamei D., Yamakawa K., Takegoshi Y., et al. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin E synthase-1. The Journal of Biological Chemistry. 2004;279(32):33684–33695. doi: 10.1074/jbc.m400199200. [DOI] [PubMed] [Google Scholar]
- 7.Seibert K., Zhang Y., Leahy K., et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(25):12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kudo I., Murakami M. Phospholipase A2 enzymes. Prostaglandins & Other Lipid Mediators. 2002;68-69:3–58. doi: 10.1016/s0090-6980(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 9.Schaloske R. H., Dennis E. A. The phospholipase A2 superfamily and its group numbering system. Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids. 2006;1761(11):1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Gilroy D. W., Newson J., Sawmynaden P., Willoughby D. A., Croxtall J. D. A novel role for phospholipase A2 isoforms in the checkpoint control of acute inflammation. The FASEB Journal. 2004;18(3):489–498. doi: 10.1096/fj.03-0837com. [DOI] [PubMed] [Google Scholar]
- 11.Lucas K. K., Svensson C. I., Hua X. Y., Yaksh T. L., Dennis E. A. Spinal phospholipase A2 in inflammatory hyperalgesia: role of group IVA cPLA 2. British Journal of Pharmacology. 2005;144(7):940–952. doi: 10.1038/sj.bjp.0706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dirig D. M., Salami A., Rathbun M. L., Ozaki G. T., Yaksh T. L. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. Journal of Neuroscience Methods. 1997;76(2):183–191. doi: 10.1016/S0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- 13.Grkovich A., Johnson C. A., Buczynski M. W., Dennis E. A. Lipopolysaccharide-induced cyclooxygenase-2 expression in human U937 macrophages is phosphatidic acid phosphohydrolase-1-dependent. The Journal of Biological Chemistry. 2006;281(44):32978–32987. doi: 10.1074/jbc.m605935200. [DOI] [PubMed] [Google Scholar]
- 14.McKew J. C., Lee K. L., Shen M. W., Thakker P., Mckew J. C., Shen M. Indole cytosolic phospholipase A2 alpha inhibitors: discovery and in vitro and in vivo characterization of 4-{3-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(dipheny lmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. Journal of Medicinal Chemistry. 2008;51(12):3388–3413. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- 15.Yaksh T. L., Hua X.-Y., Kalcheva I., Nozaki-Taguchi N., Marsala M. The spinal biology in humans and animals of pain states generated by persistent small afferent input. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(14):7680–7686. doi: 10.1073/pnas.96.14.7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Svensson C. I., Lucas K. K., Hua X. Y., Powell H. C., Dennis E. A., Yaksh T. L. Spinal phospholipase A2 in inflammatory hyperalgesia: role of the small, secretory phospholipase A2. Neuroscience. 2005;133(2):543–553. doi: 10.1016/j.neuroscience.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 17.Oka Y., Ibuki T., Matsumura K., et al. Interleukin-6 is a candidate molecule that transmits inflammatory information to the CNS. Neuroscience. 2007;145(2):530–538. doi: 10.1016/j.neuroscience.2006.10.055. [DOI] [PubMed] [Google Scholar]
- 18.Ibuki T., Matsumura K., Yamazaki Y., Nozaki T., Tanaka Y., Kobayashi S. Cyclooxygenase-2 is induced in the endothelial cells throughout the central nervous system during carrageenan-induced hind paw inflammation; its possible role in hyperalgesia. Journal of Neurochemistry. 2003;86(2):318–328. doi: 10.1046/j.1471-4159.2003.01848.x. [DOI] [PubMed] [Google Scholar]