

Abstract
The antileprosy drug clofazimine has shown potential for shortening tuberculosis treatment; however, the current dosing of the drug is not evidence based, and the optimal dosing is unknown. Our objective was to conduct a preclinical evaluation of the pharmacokinetics and pharmacodynamics of clofazimine in the mouse model of tuberculosis, with the goal of providing useful information on dosing for future studies. Pharmacokinetic parameters were evaluated in infected and uninfected BALB/c mice. Pharmacodynamic parameters were evaluated in Mycobacterium tuberculosis-infected mice that were treated for 12 weeks with one of six different clofazimine dosing regimens, i.e., doses of 6.25, 12.5, and 25 mg/kg of body weight/day and 3 regimens with loading doses. Clofazimine progressively accumulated in the lungs, livers, and spleens of the mice, reaching levels of greater than 50 μg/g in all tissues by 4 weeks of administration, while serum drug levels remained low at 1 to 2 μg/ml. Elimination of clofazimine was extremely slow, and the half-life was dependent on the duration of drug administration. Clofazimine exhibited dose-dependent tissue and serum concentrations. At any dose, clofazimine did not have bactericidal activity during the first 2 weeks of administration but subsequently demonstrated potent, dose-independent bactericidal activity. The antituberculosis activity of clofazimine was dependent on neither the dose administered nor the drug concentrations in the tissues, suggesting that much lower doses could be effectively used for tuberculosis treatment.
INTRODUCTION
Clofazimine (CFZ) is a phenazine dye developed in the 1950s for the treatment of tuberculosis (TB) (1, 2). Despite promising activity against Mycobacterium tuberculosis both in vitro and in vivo, clofazimine was not advanced as a TB drug but instead became incorporated decades later into the multidrug treatment of leprosy, where it remains a key drug (3, 4). Interest in clofazimine for TB treatment has been revitalized following the 2010 report by Van Deun and colleagues that a clofazimine-containing regimen not only was highly effective for the treatment of multidrug-resistant (MDR) TB, but was also associated with a significantly decreased duration of therapy (5); 9 months of a clofazimine-containing regimen resulted in 87.9% relapse-free cure, which is in sharp contrast to the limited efficacy (less than 50% relapse-free cure) of the World Health Organization (WHO)-recommended, at least 20-month-long MDR-TB treatment regimen (6, 7).
This report was complemented by experimental chemotherapy studies in which the inclusion of clofazimine in first- and second-line regimens significantly shortened the duration of treatment needed for relapse-free cure in mouse models of drug-susceptible and MDR TB, respectively (8, 9). These clinical and preclinical data suggest that clofazimine has great potential for TB treatment.
Because clofazimine was abandoned as an option for TB treatment shortly after its discovery, and also because the use of clofazimine for leprosy is not based on extensive experimental studies (10), the pharmacokinetics (PK) and pharmacodynamics (PD) of the drug are poorly understood, and the dosing for both TB and leprosy patients is not evidence based. It is well known that, both in people and in mice, clofazimine accumulates to high levels in the tissues and has a long half-life in vivo (9, 11, 12). A better understanding of the PK/PD of clofazimine is essential to maximize its therapeutic potential, and experimental chemotherapy in the mouse model is fundamental for all preclinical studies of new antibiotics and new multidrug regimens for TB (13). Optimizing the use of drugs in this model is critical for conducting meaningful, hypothesis-driven preclinical experiments. The objective of this study was to characterize the PK/PD properties of clofazimine in the mouse model of TB.
MATERIALS AND METHODS
All experimental work was performed at the KwaZulu-Natal Research Institute for Tuberculosis and HIV, Durban, South Africa, and the Biomedical Resources Unit, College of Health Sciences, University of KwaZulu-Natal, Westville, South Africa. Work with live bacteria and infected animals was performed in biosafety level 3 facilities. Animal procedures were approved by the Animal Ethics Sub-Committee of the University of KwaZulu-Natal (reference numbers 068/13/Animal and 025/14/Animal).
Animals.
Female BALB/c mice (6 to 8 weeks old; 18 to 20 g) from in-house breeding were used in all experiments except the serum protein binding experiment, where male mice were used. Mice were sacrificed by cardiac puncture under anesthesia; blood, lungs, spleen, and liver were collected.
Drug preparation and administration.
All drugs were purchased from Sigma and were prepared and administered by oral gavage as previously described (9). Clofazimine was prepared as a suspension in 0.05% (wt/vol) agarose, and isoniazid and rifampin were prepared in distilled water. All drugs were prepared to deliver the indicated dose in a volume of 0.2 ml. Drugs were prepared weekly and stored at 4°C.
PK experiments.
For the single-dose experiment, 27 mice received one dose of clofazimine at 25 mg/kg of body weight, and groups of 3 mice were sacrificed 0.5, 1, 2, 4, 7, 24, 48, 72, and 96 h later. In the multiple-dose experiment, 111 mice received clofazimine at 25 mg/kg daily (5 days/week) for up to 20 weeks, and groups of 3 mice were sacrificed after 1, 2, 4, 8, 12, 16, and 20 weeks of administration. The sacrifices occurred 24 h after the last dose or 72 h after if the previous dose was on Friday. Three control mice that did not receive clofazimine were also sacrificed. Administration was discontinued for subgroups of mice after 4, 8, 12, 16, and 20 weeks of daily clofazimine, and 3 mice from each subgroup were sacrificed 1, 2, 4, 8, 16, and 24 weeks after discontinuation.
Dose-ranging PK/PD experiment.
Two-hundred sixty mice were aerosol infected with M. tuberculosis H37Rv (ATCC 27294) in three infection runs using a Glas-Col Inhalation Exposure System as previously described (14). The day after infection, 2 mice from each infection run were sacrificed to determine implantation. Six weeks after infection, on the day of treatment initiation, 3 mice from each infection run were sacrificed to determine baseline pretreatment CFU counts. The treatment regimens administered are outlined in Table 1. After treatment for 1 day; 2 days; and 1, 2, 4, 8, and 12 weeks, groups of 5 mice per treatment group were sacrificed to determine CFU counts. The mice were sacrificed 24 h after the previous dose of clofazimine or 72 h after if the previous dose was on a Friday. Treatment efficacy was assessed by lung and spleen CFU counts, performed as previously described, including the use of charcoal-containing plates to detect and limit drug carryover (8). Raw CFU count data are presented in Tables S1 (lungs) and S2 (spleens) in the supplemental material.
TABLE 1.
Clofazimine dosing schemes used in the dose-ranging PK/PD experiment
| Drug regimen | Descriptiona | Total clofazimine dosed (mg)b after administration duration of: |
|
|---|---|---|---|
| 2 wk | 12 wk | ||
| Untreated | No drug administered | 0 | 0 |
| Clofazimine A | Day 1 to wk 12, 25 mg/kg daily | 5 | 30 |
| Clofazimine B | Day 1 to wk 12, 12.5 mg/kg daily | 2.5 | 15 |
| Clofazimine C | Day 1 to wk 12, 6.25 mg/kg daily | 1.25 | 7.5 |
| Clofazimine D | Day 1, 200 mg/kg; day 2, 100 mg/kg; days 3–14, 75 mg/kg daily; wk 2–12, 25 mg/kg, 3 days/wk | 18 | 33 |
| Clofazimine E | Day 1, 100 mg/kg; day 2, 75 mg/kg; days 3–14, 50 mg/kg daily; wk 2–12, 25 mg/kg, 3 days/wk | 11.5 | 26.5 |
| Clofazimine F | Day 1, 50 mg/kg; days 2–14, 25 mg/kg daily; wk 2–12, 25 mg/kg, 3 days/wk | 5.5 | 20.5 |
Daily dosing indicates administration 5 days per week (Monday-Friday); dosing 3 days/week indicates administration on Monday, Wednesday, and Friday.
The total amounts of clofazimine administered for the first 2 weeks and all 12 weeks of treatment were calculated based on a mouse mass of 20 g.
Serum protein binding assay.
Groups of 10 mice received a single dose of clofazimine (25 or 50 mg/kg), isoniazid (10 mg/kg; negative control) (15), or rifampin (10 mg/kg; positive control) (16). Mice were sacrificed 1 and 2 h after administration of isoniazid and rifampin, respectively (the time points were based on the half-lives of the drugs in mouse serum) (13), and 24 h after administration of clofazimine. Serum protein binding was determined using the Single-Use Rapid Equilibrium Dialysis Plate (Thermo Scientific, Rockford, IL, USA) according to the manufacturer's instructions. Serum samples were equilibrated for 4 h.
LC/MS.
Drug concentrations in samples were determined by liquid chromatography/mass spectrometry (LC/MS) as previously described (9).
Statistical analyses.
PK parameters were analyzed using Phoenix WinNonlin version 6.3. All other analyses were performed with GraphPad Prism version 6.05. Nonlinear regression analysis of clofazimine accumulation and elimination was performed using one-phase association (constrained to fit accumulation/elimination intersects). Clofazimine concentrations during treatment of infected mice were compared by 2-way analysis of variance (ANOVA) with Tukey's multiple-comparison test. Treatment efficacy analyses were performed as previously described (8).
RESULTS
PK of clofazimine in uninfected BALB/c mice.
To assess the PK of clofazimine in uninfected BALB/c mice, clofazimine was administered at 25 mg/kg, a dose comparable to the customary 100-mg daily dose for MDR-TB patients (17, 18).
PK of clofazimine after a single dose.
The PK parameters in the serum, lungs, liver, and spleen following a single oral dose of clofazimine are presented in Table 2. The peak drug concentrations were reached 4 and 7 h after administration in the serum and tissues, respectively. Clofazimine levels declined slowly in the serum and organs, with half-life values all greater than 45 h.
TABLE 2.
PK parameters of clofazimine in serum and tissues of uninfected mice after a single oral 25-mg/kg dosea
| Sample | t½ (h) | Cmax (μg/unit)b | tmax (h) | AUC0–24 (μg · h/unit)b | AUC0–∞ (μg · h/unit)b |
|---|---|---|---|---|---|
| Serum | 87.46 | 0.43 | 1–4 | 19.13 | 38.22 |
| Liver | 45.25 | 2.78 | 2–7 | 68.59 | 88.72 |
| Lung | 66.94 | 0.76 | 2–7 | 38.30 | 56.36 |
| Spleen | 77.96 | 1.73 | 2–7 | 82.64 | 132.47 |
t½, half-life; Cmax, maximum concentration; tmax, time to maximum concentration (range); AUC, area under the concentration curve.
Unit, milliliters for serum and grams for liver, lung, and spleen tissue.
Accumulation of clofazimine.
After 1 week of daily clofazimine, the adipose tissue in the mice was slightly orange in color, and this coloration increased in intensity during the entire 20 weeks of clofazimine administration (Fig. 1). The lungs became red-orange in color, while the spleens became almost black. Clofazimine levels in the lungs and liver increased during the first 8 weeks of administration and then plateaued at about 35 and 80 μg/g tissue, respectively (Fig. 2A and B; see Tables S3 and S4 in the supplemental material). The concentration of clofazimine in the serum, however, remained comparatively low throughout the course of administration, reaching around 1 μg/ml at 4 weeks and then increasing and plateauing at about 2 μg/ml after 8 weeks (Fig. 2C; see Table S5 in the supplemental material). In the spleen, clofazimine accumulated during the first 12 weeks of administration and then plateaued at about 5,000 μg/g (Fig. 3; see Table S6 in the supplemental material). Thus, in the serum and tissues, the concentration of clofazimine by the first week of administration was already well above the MIC for M. tuberculosis, 0.25 μg/ml (5).
FIG 1.

Clofazimine-induced discoloration in uninfected BALB/c mice. Photographs of the lungs, spleen, and subcutaneous adipose tissue were taken at each time point during the 20 weeks of clofazimine administration and during the 24 weeks after stopping administration. Postadministration photographs of the mice that received clofazimine for 4, 8, 12, and 16 weeks are presented in Fig. S1 in the supplemental material.
FIG 2.
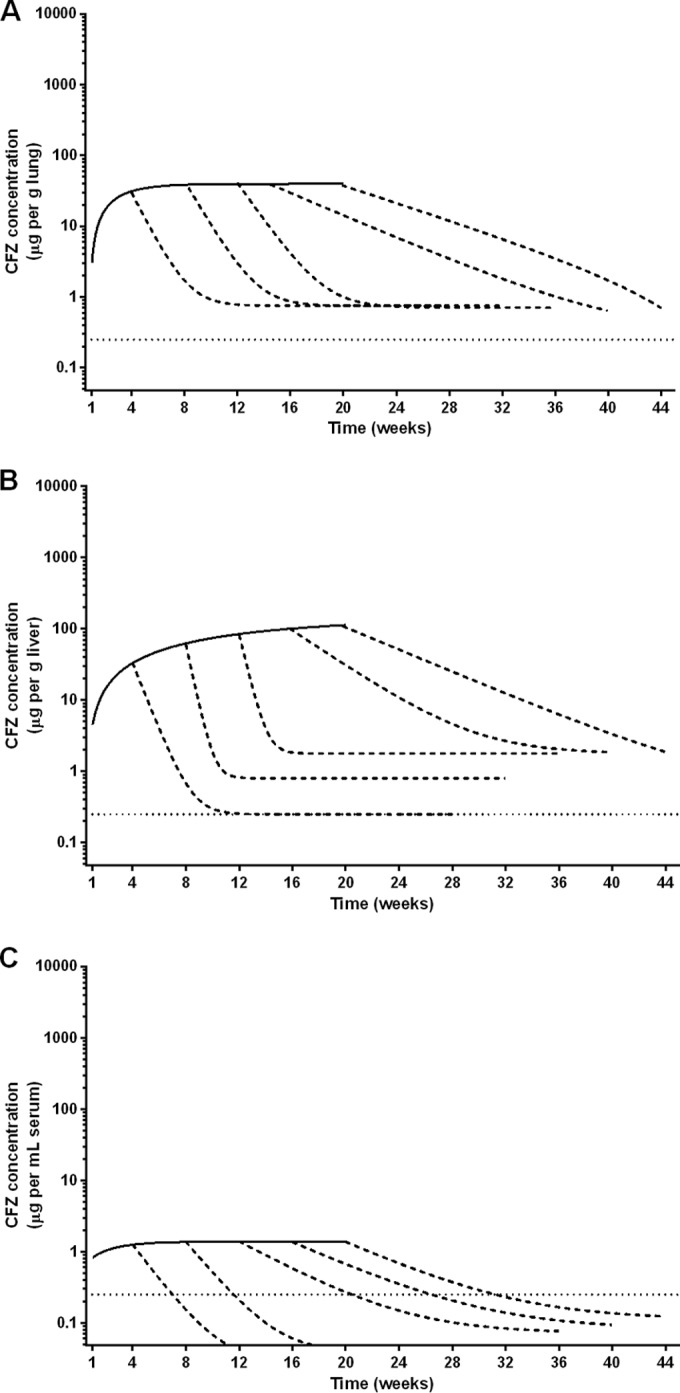
Nonlinear regression of clofazimine accumulation and elimination in the lungs (A), liver (B), and serum (C) of uninfected BALB/c mice. Elimination curves are included for each group of mice that discontinued clofazimine administration after 4, 8, 12, 16, and 20 weeks. Clofazimine concentration data for the lungs, liver, and serum are presented in Tables S3, S4, and S5 in the supplemental material, respectively. The dotted lines represent the MIC of clofazimine for M. tuberculosis.
FIG 3.
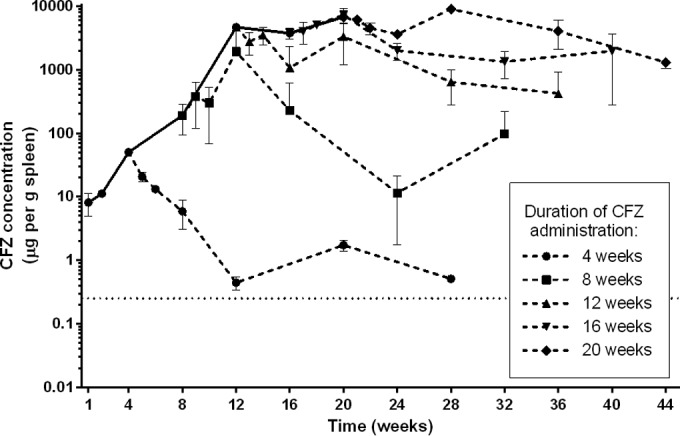
Clofazimine accumulation and elimination in the spleens of uninfected BALB/c mice. The concentrations of clofazimine measured after stopping administration are represented by the dashed lines. The error bars represent standard deviations (3 mice per group per time point). Clofazimine concentration values are presented in Table S6 in the supplemental material. The dotted line represents the MIC of clofazimine for M. tuberculosis.
Elimination of clofazimine.
During 20 weeks of clofazimine administration, subgroups of mice stopped receiving the drug at 4-week intervals, and elimination of clofazimine from the blood and tissues was monitored for an additional 24 weeks postadministration. The intense red-orange coloration noticeably faded from the adipose tissue during this period; however, the coloration of the lungs and spleens did not return to preadministration levels within the 24-week follow-up (Fig. 1; see Fig. S1 in the supplemental material). The half-life values for clofazimine varied by serum/tissue site and also by duration of administration (Fig. 2 and 3 and Table 3). In mice receiving clofazimine for at least 8 weeks, the concentration in the spleen continued to increase for several weeks after discontinuation of the drug (making the elimination of clofazimine from the organ difficult to model). In the lungs, liver, and spleen, the clofazimine concentration remained above the MIC for M. tuberculosis throughout the follow-up period, regardless of the duration of administration (Fig. 2 and 3; see Tables S3 to S6 in the supplemental material).
TABLE 3.
PK parameters of clofazimine in uninfected mice after daily administration for up to 20 weeks
| Sample | No. of wk of CFZ administration | t½ (wk) | C (μg/unit)a,b | AUC0–∞ (μg · wk/unit)b |
|---|---|---|---|---|
| Serum | 4 | 1.45 | 1.33 | 7.15 |
| 8 | 1.91 | 2.25 | 14.97 | |
| 12 | 6.49 | 2.25 | 25.72 | |
| 16 | 6.29 | 2.25 | 30.18 | |
| 20 | 8.19 | 2.25 | 35.18 | |
| Liver | 4 | 3.09 | 33.26 | 107.96 |
| 8 | 5.57 | 59.97 | 312.19 | |
| 12 | 4.10 | 82.37 | 714.66 | |
| 16 | 3.32 | 111.29 | 1,302.99 | |
| 20 | 2.82 | 156.91 | 2,514.44 | |
| Lung | 4 | 4.33 | 29.98 | 105.27 |
| 8 | 4.27 | 52.59 | 387.34 | |
| 12 | 3.55 | 52.59 | 557.31 | |
| 16 | 4.17 | 52.59 | 739.39 | |
| 20 | 3.36 | 106.11 | 1,188.01 | |
| Spleen | 4 | 4.96 | 50.43 | 180.95 |
| 8 | 1.65 | 1,936.85 | 9,376.44 | |
| 12 | 5.38 | 4,684.68 | 54,082.57 | |
| 16 | 7.18 | 7,297.53 | 113,878.1 | |
| 20 | 13.46 | 9,020.41 | 192,658.5 |
C, highest mean concentration observed.
Unit, milliliters for serum and grams for liver, lung, and spleen tissue.
Serum protein binding of clofazimine.
Twenty-four hours after administration of a single dose of clofazimine at 25 or 50 mg/kg, the drug was extremely highly bound in the serum. For both of the samples, the amount of free clofazimine that diffused into the buffer dialysis chamber was below the lower limit of quantitation (0.048 μg/ml). Using the lower limit of quantitation as the nominator, we calculated that the percentage of free clofazimine must be less than 15% (Table 4). However, the concentration of clofazimine in the serum dialysis well was essentially equal to the total serum clofazimine concentration; therefore, it is likely that nearly all of the clofazimine is protein bound, which is in agreement with what has previously been reported (19). As expected, rifampin (2 h after a single 10-mg/kg dose) was also highly protein bound in the serum (2.4% free), which is higher protein binding than has been reported in human plasma (about 15% free drug) (16). Also as expected, isoniazid (1 h after a single 10-mg/kg dose) was almost entirely non-protein bound (93.4% free).
TABLE 4.
Mouse serum protein binding for isoniazid (negative control), rifampin (positive control), and clofazimine
| Drug administered (dose [mg/kg]) | Time pointa (h) | Drug concnb (SD) (μg/ml) |
% free drugc | ||
|---|---|---|---|---|---|
| Total drug | Buffer dialysis well | Serum dialysis well | |||
| Isoniazid (10) | 1 | 0.985 (0.063) | 0.576 (0.023) | 0.617 (0.032) | 93.4 |
| Rifampin (10) | 2 | 10.7 (0.141) | 0.237 (0.005) | 9.68 (0.085) | 2.4 |
| Clofazimine (25) | 24 | 0.319 (0.003) | Below LLOQ | 0.321 (0.012) | <15.0 |
| Clofazimine (50) | 24 | 0.296 (0.008) | Below LLOQ | 0.317 (0.031) | <15.1 |
Time point, the time post-drug administration when blood was collected from the mice by cardiac puncture.
Total drug, the total amount of drug measured in the serum, processed for analysis prior to setting up the protein binding assay. The drug concentrations in the serum and buffer dialysis wells were determined after 4 h of equilibration. The lower limit of quantitation (LLOQ) was 0.048 μg/ml.
The percent free drug was calculated as follows: (concentration in buffer chamber/concentration in serum sample chamber) × 100%.
PK/PD of clofazimine in infected BALB/c mice.
The massive tissue accumulation and slow elimination of clofazimine in the tissues of uninfected mice raised several key questions. As clofazimine is known to be sequestered in macrophages (20–22), would administration of clofazimine to M. tuberculosis-infected mice affect the drug distribution in tissues? Would administration of lower doses of clofazimine lead to different levels of tissue accumulation, and how would this impact the antimycobacterial activity of clofazimine? The last question was of particular interest, because the dosing of clofazimine for human TB patients is not evidence based. To address these questions and to carefully examine the PK/PD relationship, we performed a dose-ranging experiment with clofazimine in M. tuberculosis-infected mice (Table 1 shows the dosing scheme).
Establishment of infection.
Aerosol infection with M. tuberculosis (5.92 log10 CFU/ml suspension) resulted in a mean implantation of 2.17 (standard deviation [SD], 0.28) log10 CFU/lung. Six weeks later, at the start of treatment, the mean log10 CFU count had increased to 6.49 (SD, 0.30) in the lungs and to 4.66 (SD, 0.21) in the spleens.
Clofazimine in serum.
In mice treated with constant daily doses of 25, 12.5, and 6.25 mg/kg (regimens A, B, and C, respectively), the clofazimine concentration in the serum increased in a steady and dose-dependent manner during the first 8 weeks of treatment and then plateaued (Fig. 4). Differences in serum concentrations in the three groups became statistically significant by 4 weeks of administration. After the second dose, the serum clofazimine concentrations in mice receiving 12.5 and 25 mg/kg were greater than the drug's MIC for M. tuberculosis. In mice treated with the loading doses (regimens D, E, and F), the serum clofazimine concentration was already higher than the MIC for M. tuberculosis 24 h after the first dose (Fig. 4A). A significant difference in serum drug concentrations between mice receiving the two higher loading doses and mice receiving the lowest loading dose was maintained for the first 2 weeks, but by 4 weeks of administration (by which time dosing was consistent for these regimens), the serum clofazimine concentrations did not differ between the groups (Fig. 4B).
FIG 4.
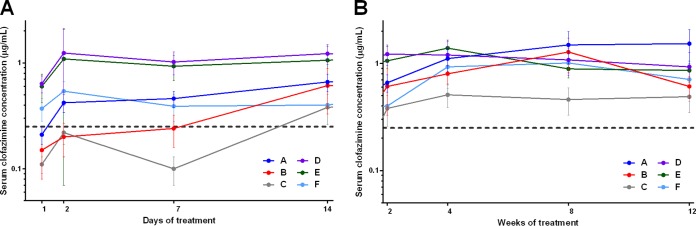
Mean clofazimine concentrations in the sera of M. tuberculosis-infected mice during the first 2 weeks (A) or weeks 2 to 12 (B) of treatment. The dashed lines represent the MIC of clofazimine for M. tuberculosis. The error bars represent standard deviations (5 mice per group per time point). The drug regimens are described in Table 1.
Clofazimine in tissues.
The clofazimine concentrations in the lungs, liver, and spleen were dose dependent and loading dose dependent, but accumulation of clofazimine occurred during the first 2 weeks of treatment and then stabilized or even slightly decreased (Fig. 5). In all tissues, by 24 h after the first dose, the clofazimine concentration was greater than the MIC for M. tuberculosis. After 1 and 2 weeks of clofazimine administration, mice treated with the highest loading dose regimens (regimens D and E, which represent total clofazimine doses of 15 and 11.5 mg, respectively, during the first 2 weeks [Table 1]) had significantly higher concentrations in all tissues than mice that did not receive loading doses, but these differences were no longer apparent by week 4. From day 7 through week 12, at almost every time point in every tissue, the clofazimine concentrations in mice treated with 6.25 mg/kg/day (regimen C) were significantly lower than those in mice receiving the other clofazimine regimens.
FIG 5.

Mean clofazimine concentrations in the lungs (A and B), liver (C and D), and spleen (E and F) of M. tuberculosis-infected mice during the first 2 weeks (A, C, and E) or weeks 2 to 12 (B, D, and F) of treatment. The dashed lines represent the MIC of clofazimine for M. tuberculosis. The error bars represent standard deviations (5 mice per group per time point). The drug regimens are described in Table 1.
PD data. (i) Early bactericidal activity.
During the first week of administration, clofazimine at any dose had no impact on the CFU counts in the mouse lungs, with no statistically significant differences between mice receiving any regimen and the untreated controls (Fig. 6A; see Table S7 in the supplemental material). Thus, despite the high serum and tissue drug concentrations, none of the clofazimine-containing regimens exhibited early bactericidal activity (EBA) (a decline in log10 CFU per lung per day) (Table 5). By day 14, however, bactericidal activity was apparent in all treatment groups, although only mice that received the highest loading dose regimen (regimen D, which delivered the largest total amount of clofazimine during the first 2 weeks) had lung CFU counts that were statistically significantly different from those of the untreated controls (P ≤ 0.01). In the spleens, there was similarly no statistically significant difference in CFU counts during the first week of treatment, regardless of the regimen administered. However, by day 14, the CFU counts in the spleens of all treated mice were significantly different than those of the untreated controls (P ≤ 0.01 for regimen D; P ≤ 0.0001 for all other regimens) (Fig. 6B; see Table S7 in the supplemental material). As in the lungs, neither the regimen nor the total dose of clofazimine had any impact on the decline in CFU counts.
FIG 6.

Mean log10 CFU counts in the lungs (A and C) and spleens (B and D) of M. tuberculosis-infected mice during the first 2 weeks (A and B) and weeks 2 to 12 (C and D) of treatment. The error bars represent standard deviations (5 mice per group per time point). The drug regimens are described in Table 1.
TABLE 5.
Fourteen-day extended EBAa of clofazimine-containing drug regimens in M. tuberculosis-infected mice
| Clofazimine regimen | EBA0–2 | EBA2–14 | EBA0–7 | EBA7–14 | EBA0–14 |
|---|---|---|---|---|---|
| Untreated | −0.010 | 0.002 | 0.003 | −0.001 | 0.001 |
| A | −0.050 | 0.078 | 0.040 | 0.079 | 0.059 |
| B | 0.035 | 0.074 | 0.069 | 0.069 | 0.069 |
| C | 0.170 | 0.054 | 0.009 | 0.133 | 0.071 |
| D | 0.030 | 0.091 | 0.089 | 0.076 | 0.082 |
| E | 0.020 | 0.056 | 0.047 | 0.054 | 0.051 |
| F | 0.025 | 0.086 | 0.037 | 0.117 | 0.077 |
EBA is defined as the decline in log10 CFU per lung per day of treatment, as indicated by the subscript. The clofazimine regimens are described in Table 1.
(ii) Weeks 2 to 12 of treatment.
From weeks 4 to 12 of treatment, all mice receiving any dose of clofazimine had significantly lower CFU counts in the lungs (Fig. 6C; see Table S7 in the supplemental material) and the spleens (Fig. 6D; see Table S7 in the supplemental material) than the untreated control mice (P ≤ 0.0001 for all regimens). However, at each time point in both organs, there was no statistically significant difference in CFU counts between any of the treatment regimens, and in both organs, there were no visible differences in the gross pathology across any of the treatment groups (see Fig. S2 in the supplemental material).
Drug carryover.
To ensure that the decline in CFU counts observed in the clofazimine-treated mice was not due to growth inhibition on agar plates caused by drug carryover from homogenized tissues, the lung and spleen homogenates were systematically plated on selective 7H11 agar with and without a supplement of 0.4% activated charcoal, which can help to adsorb the released drug (23). As early as day 2 of treatment, drug carryover was observed; bacterial growth was totally inhibited on the charcoal-free agar plates that were inoculated with neat (undiluted) lung homogenate from the mice treated with the loading dose regimens, while confluent bacterial lawns were observed on the charcoal-containing plates inoculated with the same neat homogenates (see Table S1 in the supplemental material). Similar effects of carryover were observed with the spleen homogenates (see Table S2 in the supplemental material). However, even at the latest time point (day 84/week 12), growth inhibition was rarely observed on plates inoculated with the 10−1 dilutions of homogenates and no inhibition was observed on plates inoculated with the 10−2 dilutions of the homogenates, a finding that we considered when counting colonies.
DISCUSSION
Despite being an essential component of multidrug leprosy treatment, clofazimine has never been very well understood, and uncertainty regarding how the drug works and the implications of its unusual PK properties has limited official interest in clofazimine for TB treatment. The work presented here raises a number of key issues that simultaneously contribute both to understanding the PK/PD of clofazimine for TB and to highlighting how much more there is to learn about this enigmatic drug.
The first and most striking finding from this work is that dose-dependent concentrations of clofazimine in the blood and tissues did not translate into dose-dependent antimicrobial activity. In M. tuberculosis-infected mice, no differences in bacterial killing were observed between any of the clofazimine doses administered; the lowest dose was as effective as the highest dose. At face value, our data indicate that lower doses in patients might be effectively used, and if such lower doses of clofazimine produce the same treatment-shortening effect that has been observed with the standard dosing, then there is real promise for the use of low doses of the drug in TB patients. Beyond their face value, our data raise interesting questions. What is the real meaning of such dose-independent antimicrobial activity? How does clofazimine really work in vivo? Most likely, the antimicrobial activity occurs when a threshold has been reached, and when that occurs, an increase in the amount of drug does not increase activity. In other words, when clofazimine reaches levels lethal to the bacteria, it cannot become “more lethal.” If, as has been proposed, clofazimine acts by causing the release of reactive oxygen species (2, 24), perhaps this is an on/off or all-or-nothing effect.
The second key finding from this work is that the PK profiles of clofazimine were similar in uninfected and M. tuberculosis-infected mice, with the exception of drug levels in the spleen and lungs. Our data confirmed the well-known effects of tissue and skin discoloration (Fig. 1; see Fig. S1 and S2 in the supplemental material) and the associated massive accumulation of clofazimine in the tissues (Fig. 2, 3, and 5) during administration. In uninfected mice receiving 25 mg/kg/day of clofazimine, the drug concentration steadily increased with the duration of administration, leveling out at 50 μg/g tissue in the lungs (Fig. 2A); in the spleen, clofazimine levels increased to more than 6,500 μg/g after 20 weeks of administration (Fig. 3). These findings are in agreement with recent work by Baik and colleagues, who reported that 8 weeks of administration of approximately 10 mg/kg/day of clofazimine in the diet to uninfected BALB/c mice resulted in extremely high accumulation in the spleen, although they found much higher accumulation in their study (more than 10 mg/g spleen tissue), and that approximately 90% of the drug was sequestered in intracellular crystal-like drug inclusions (25). Interestingly, in our study, when clofazimine was administered at 25 mg/kg/day to infected mice, the concentration of the drug in the lungs and spleen stabilized at approximately 100 μg/g tissue after 4 to 8 weeks of administration (Fig. 5). Thus, the concentration in the lungs of infected mice was higher than in uninfected mice, while in the spleens of infected mice, the clofazimine concentration was considerably lower than in the uninfected mice. The difference in tissue concentrations in infected versus uninfected mice could be due to the redistribution of the drug in relation to the movement of macrophages or other cells containing accumulated crystals or liquid crystals of clofazimine (20–22, 25) from the spleen to the lungs during infection and treatment and/or the natural change in the biodistribution of the drug, as has been demonstrated by Baik and colleagues (25).
Another intriguing aspect of clofazimine, and our third key finding, is the high levels of drug maintained in the tissues after stopping administration. The constant release of clofazimine from the tissues may explain the increase in the half-life of clofazimine in the serum from 1.45 to 8.19 weeks when the duration of administration increased from 4 to 20 weeks (Table 3), because more accumulated clofazimine is released into the serum as the obligate route of elimination from the tissues.
The fourth key finding is that, despite the extremely high tissue concentrations of clofazimine, the levels in the serum remained comparatively low and stable in both infected and uninfected mice at about 1 to 2 μg/ml, which is well above the 0.25-μg/ml MIC for M. tuberculosis. In the dose-ranging PK/PD experiment, the serum clofazimine concentration was dose and loading dose dependent during the first 2 weeks, after which the serum concentration stabilized at the 1- to 2-μg/ml level in all mice except those receiving the lowest dose of clofazimine (6.25 mg/kg/day; regimen C), in which the concentration was slightly lower (Fig. 4). In mice receiving the two highest loading dose regimens (regimens D and E), the serum concentration reached 1 to 2 μg/ml after the first dose of the drug but did not increase further. Our findings are in agreement with a previous report by Banerjee and colleagues, who demonstrated progressively increasing high tissue concentrations of clofazimine (reaching up to 15-mg/g levels in the spleens of uninfected mice) coupled with a low, steady serum concentration of about 1.3 μg/ml in mice given clofazimine as part of their diet (17).
Very few studies have been done to characterize the PK of clofazimine in humans. Our single- and multiple-dose PK studies in uninfected mice suggest that the 25-mg/kg dose that we administered to the mice resulted in PK properties that were similar to those reported by Williams et al., who administered clofazimine at 20 mg/kg to uninfected BALB/c mice (18), and are also similar to what was reported for the 100-mg daily dose in human leprosy patients (17). However, Diacon and colleagues found much lower plasma clofazimine concentrations after 14 days of administration; TB patients who received 300 mg/day of clofazimine for 3 days (i.e., loading doses) followed by 100 mg daily for the following 11 days had a mean plasma drug concentration of 0.2315 μg/ml (26). Thus, the PK of clofazimine in humans also needs to be better understood.
Our data also show that the serum clofazimine level is maintained above the MIC for weeks after stopping administration (3 to 12 weeks after receiving clofazimine for 4 to 20 weeks, respectively) (Fig. 2C). When clofazimine was developed in the 1950s, Barry and colleagues suggested that the tissue accumulation of the drug could contribute to sustained antimicrobial activity after cessation of administration (2, 27). Our data support this hypothesis; if the sustained serum concentration of clofazimine translates into sustained antimicrobial activity, this could be a reason for the drastic treatment-shortening effect observed when clofazimine is incorporated into first- and second-line drug regimens for TB treatment (5, 8, 9). Thus, the tissue accumulation and long half-life may indeed be truly beneficial properties of the drug.
Taken together, these results suggest that high tissue concentrations of clofazimine do not reflect free/active drug but rather clofazimine that is bound or in crystal deposits and thus devoid of antimicrobial activity. This idea is further supported by the fact that drug carryover in lung and spleen homogenates inhibited M. tuberculosis growth on charcoal-free agar in a concentration-associated manner (see Tables S1 and S2 in the supplemental material). Thus, it may be that it is the drug levels in the serum that are the key to clofazimine's antimicrobial activity. We found that in the serum, clofazimine is also likely highly protein bound (Table 4), which is in agreement with what has been reported by Irwin and colleagues (19). If total serum clofazimine levels are maintained at around 1 to 2 μg/ml, then the amount of free clofazimine could be expected to be well below the 0.25-μg/ml MIC for M. tuberculosis. This raises the question of what concentration of clofazimine is relevant to antimicrobial activity. This remains to be answered.
The fifth key finding is the lack of significant bactericidal activity of clofazimine during the first 2 weeks of administration to infected mice, despite the fact that the concentration of clofazimine in the blood and tissues was strongly dose dependent and well above the MIC for M. tuberculosis during this time. This lack of EBA, especially during the first week of administration, has been previously reported with clofazimine when administered in combination with first-line drugs in the mouse model (9). Recently, Diacon and colleagues reported no bactericidal activity of clofazimine when administered alone for 2 weeks in an EBA study in human TB patients (26). It is possible that the delay in antimicrobial activity is due to the tissue and protein binding of clofazimine and that a level of saturation must be achieved before there is enough free drug to exert activity. However, if this were the case, then accelerating the saturation by the use of loading doses should have resulted in earlier response to treatment, which was not the case in this study. Similarly, in the human EBA study reported by Diacon and colleagues, TB patients were given clofazimine at a loading dose of 300 mg/day for the first 3 days of treatment, followed by the usual dose of 100 mg/day for days 4 to 14, but no decrease in sputum CFU counts was observed in these patients during the 2-week study (26). The results from our mouse study indicate that the antimicrobial activity cannot be accelerated by administering loading doses of clofazimine. Thus, the question of the cause of clofazimine's delayed bactericidal activity remains. It is possible that it may be due to the mechanism of action of the drug, which, although it is not well understood, seems to involve the respiratory chain (24). In this case, perhaps bacterial energy stores need to be depleted before the effect of the drug can be observed, or it takes some time for reactive oxygen species to induce lethal DNA damage in the bacteria. Another possibility is that clofazimine, which is highly lipophilic, needs to accumulate to a certain level in the lipid-rich cell wall of M. tuberculosis before it is able to reach the cell membrane, where it impacts the respiratory chain. Thus, plenty of work remains to be done to understand the biological meaning of our findings and to decipher how clofazimine exerts its activity.
Recently, Irwin and colleagues published data from an elegant series of experiments assessing the antimicrobial activity of clofazimine when administered to M. tuberculosis (strain Erdman)-infected BALB/c and C3HeB/FeJ mice (19). They found that clofazimine (administered at 20 mg/kg/day) was highly active in BALB/c mice, resulting in reductions of 2.5 and 4.0 log10 CFU in the lungs after 4 and 8 weeks of treatment, respectively. This bactericidal activity is similar to, although slightly more potent than, what we observed in our study at all clofazimine doses (Fig. 6A and C). Likewise, we also observed bactericidal activity in the spleens of clofazimine-treated BALB/c mice similar to that reported by Irwin et al. Most interestingly, Irwin and colleagues found that clofazimine did not exhibit bactericidal activity in the lungs of M. tuberculosis-infected C3HeB/FeJ mice, which, in contrast to BALB/c mice, are hypersusceptible to M. tuberculosis infection and form hypoxic, caseous lung lesions in response to the infection (28, 29). However, bacterial killing was observed in the spleens of the treated mice, suggesting that this discrepant activity of clofazimine was associated with the pathology in the lungs and spleens. The implication of these findings as they relate to dosing of clofazimine across mouse models and in human TB remains an open question.
Finally it should be noted that the PK/PD properties of clofazimine are strikingly similar to those of the newest anti-TB drug, bedaquiline. Bedaquiline similarly accumulates to very high levels in the tissues in both people (30) and mice (31), and the accumulated drug in tissue homogenates results in carryover and growth inhibition of M. tuberculosis on non-charcoal-containing agar (23). Despite the extremely high tissue concentrations, bedaquiline is maintained at a relatively low and steady concentration in serum, which is thought to be due to steady release of the drug from the tissues (30); and it has also been suggested that it is the serum drug levels, rather than the tissue drug levels, that represent active drug (31). Like clofazimine, the half-life of bedaquiline is extremely long (days to weeks) and is dependent on the duration of administration (31, 32), and bedaquiline is also highly protein bound in the serum (30, 31), with free drug concentration estimates well below the MIC of the drug for M. tuberculosis. Bedaquiline also exhibits delayed bactericidal activity against M. tuberculosis, both in vitro (33, 34) and in vivo (32, 35), and it has been hypothesized that this delay is related to bedaquiline's mechanism of action targeting ATP synthase (36), with bactericidal activity evident only after bacterial energy stores have been depleted (33). Finally, low-level, efflux-driven cross-resistance to bedaquiline and clofazimine has been reported in M. tuberculosis (37). Comparative studies between clofazimine and bedaquiline may contribute to a better understanding of how to optimally use both drugs in TB treatment.
Clofazimine is known to be safe for long-term administration in people, as has been demonstrated by the successful use of the drug in millions of leprosy patients (3), but it is associated with the cosmetically significant adverse effect of reversible skin discoloration. This effect could perhaps be greatly minimized in TB treatment if lower doses can be effectively used and the duration of treatment can be shortened. Based on this work and previous studies (5, 8, 9), a reasonable next step could be an assessment of clofazimine activity for the treatment of human TB.
Supplementary Material
ACKNOWLEDGMENTS
The work was funded by the KwaZulu-Natal Research Institute for Tuberculosis and HIV, the Howard Hughes Medical Institute, and the University of KwaZulu-Natal.
We thank T. Govender at the University of KwaZulu-Natal, Westville, South Africa, for the donation of the dialysis plate.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00260-15.
REFERENCES
- 1.Barry VC, Conalty ML, Gaffney EE. 1956. Antituberculosis activity in the phenazine series; isomeric pigments obtained by oxidation of o-phenylenediamine derivatives. J Pharm Pharmacol 8:1089–1096. doi: 10.1111/j.2042-7158.1956.tb12238.x. [DOI] [PubMed] [Google Scholar]
- 2.Barry VC, Belton JG, Conalty ML, Denneny JM, Edward DW, O'Sullivan JF, Twomey D, Winder F. 1957. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 179:1013–1015. doi: 10.1038/1791013a0. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2004. Multidrug therapy against leprosy: development and implementation over the past 25 years. WHO, Geneva, Switzerland. [Google Scholar]
- 4.World Health Organization. 2012. WHO Expert Committee on Leprosy. World Health Organ Tech Rep Ser 968:1–61. [PubMed] [Google Scholar]
- 5.Van Deun A, Maug AK, Salim MA, Das PK, Sarker MR, Daru P, Rieder HL. 2010. Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med 182:684–692. doi: 10.1164/rccm.201001-0077OC. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. 2013. Shorter treatment regimens for multi-drug-resistant tuberculosis (MDR-TB). WHO, Geneva, Switzerland. [Google Scholar]
- 7.World Health Organization. 2011. Guidelines for the programmatic management of drug-resistant tuberculosis. 2011 update. WHO, Geneva, Switzerland. [PubMed] [Google Scholar]
- 8.Grosset JH, Tyagi S, Almeida DV, Converse PJ, Li SY, Ammerman NC, Bishai WR, Enarson D, Trébucq A. 2013. Assessment of clofazimine activity in a second-line regimen for tuberculosis in mice. Am J Respir Crit Care Med 188:608–612. doi: 10.1164/rccm.201304-0753OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tyagi S, Ammerman NC, Li SY, Adamson J, Converse PJ, Swanson RV, Almeida DV, Grosset JH. 2015. Clofazimine shortens the duration of the first-line treatment regimen for experimental chemotherapy of tuberculosis. Proc Natl Acad Sci U S A 112:869–874. doi: 10.1073/pnas.1416951112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang YT. 1955. Chemotherapy of murine leprosy. IV. The effects of amithiozone (TB1/698), p-aminosalicylic acid (PAS), B 283 (a phenazine pigment), five antibiotics and three diphenylthiourea compounds on mouse leprosy. Int J Lepr 23:167–180. [PubMed] [Google Scholar]
- 11.Levy L. 1974. Pharmacologic studies of clofazimine. Am J Trop Med Hyg 23:1097–1109. [DOI] [PubMed] [Google Scholar]
- 12.Mansfield RE. 1974. Tissue concentrations of clofazimine (B663) in man. Am J Trop Med Hyg 23:1116–1119. [DOI] [PubMed] [Google Scholar]
- 13.Grosset J, Ji B. 1998. Experimental chemotherapy of mycobacterial diseases, p 51–97. In Gangadharam PRJ, Jenkins PA (ed), Mycobacteria, vol II: chemotherapy Chapman & Hall, New York, NY. [Google Scholar]
- 14.Grosset J, Almeida D, Converse PJ, Tyagi S, Li SY, Ammerman NC, Pym AS, Wallengren K, Hafner R, Lalloo U, Swindells S, Bishai WR. 2012. Modeling early bactericidal activity in murine tuberculosis provides insights into the activity of isoniazid and pyrazinamide. Proc Natl Acad Sci U S A 109:15001–15005. doi: 10.1073/pnas.1203636109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrera AM, Scott DO, Lunte CE. 1990. Microdialysis sampling for determination of plasma protein binding of drugs. Pharm Res 7:1077–1081. doi: 10.1023/A:1015955503708. [DOI] [PubMed] [Google Scholar]
- 16.Burman WJ, Gallicano K, Peloquin C. 2001. Comparative pharmacokinetics and pharmacodynamics of the rifamycin antibacterials. Clin Pharmacokinet 40:327–341. doi: 10.2165/00003088-200140050-00002. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee DK, Ellard GA, Gammon PT, Waters MF. 1974. Some observations on the pharmacology of clofazimine (B663). Am J Trop Med Hyg 23:1110–1115. [DOI] [PubMed] [Google Scholar]
- 18.Williams K, Minkowski A, Amoabeng O, Peloquin CA, Taylor D, Andries K, Wallis RS, Mdluli KE, Nuermberger EL. 2012. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother 56:3114–3120. doi: 10.1128/AAC.00384-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Irwin SM, Gruppo V, Brooks E, Gilliland J, Scherman M, Reichlen MJ, Leistikow R, Kramnik I, Nuermberger EL, Voskuil MI, Lenaerts AJ. 2014. Limited activity of clofazimine as a single drug in a mouse model of tuberculosis exhibiting caseous necrotic granulomas. Antimicrob Agents Chemother 58:4026–4034. doi: 10.1128/AAC.02565-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baik J, Rosania GR. 2012. Macrophages sequester clofazimine in an intracellular liquid crystal-like supramolecular organization. PLoS One 7:e47494. doi: 10.1371/journal.pone.0047494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDougall AC. 1974. Electron microscope studies of the antileprosy drug B663 (clofazimine; Lamprene). Int J Lepr Other Mycobact Dis 42:1–12. [PubMed] [Google Scholar]
- 22.Conalty ML, Barry VC, Jina A. 1971. The antileprosy agent B.663 (Clofazimine) and the reticuloendothelial system. Int J Lepr Other Mycobact Dis 39:479–492. [PubMed] [Google Scholar]
- 23.Tasneen R, Li SY, Peloquin CA, Taylor D, Williams KN, Andries K, Mdluli KE, Nuermberger EL. 2011. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 55:5485–5492. doi: 10.1128/AAC.05293-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yano T, Kassovska-Bratinova S, Teh JS, Winkler J, Sullivan K, Isaacs A, Schechter NM, Rubin H. 2011. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem 286:10276–10287. doi: 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baik J, Stringer KA, Mane G, Rosania GR. 2013. Multiscale distribution and bioaccumulation analysis of clofazimine reveals a massive immune system-mediated xenobiotic sequestration response. Antimicrob Agents Chemother 57:1218–1230. doi: 10.1128/AAC.01731-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, van Niekerk C, Everitt D, Hutchings J, Burger DA, Schall R, Mendel CM. 26 January 2015. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am J Respir Crit Care Med doi: 10.1164/rccm.201410-1801OC. [DOI] [PubMed] [Google Scholar]
- 27.Barry VC. 1969. Synthetic phenazine derivatives and mycobacterial disease: a twenty year investigation. Boyle Medal Lecture no. 16. Sci Proc R Dublin Soc Ser A 3:153–170. [Google Scholar]
- 28.Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, Bishai WR, Pomper MG, Nuermberger EL, Jain SK. 2012. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis 205:595–602. doi: 10.1093/infdis/jir786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I. 2005. Ipr1 gene mediates innate immunity to tuberculosis. Nature 434:767–772. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janssen Therapeutics Division, Janssen Products LP. 2013. Sirturo (bedaquiline) 100 mg tablets full prescribing information. Reference ID 3644668. Revised 2014 Janssen Therapeutics, Titusville, NJ. [Google Scholar]
- 31.Rouan MC, Lounis N, Gevers T, Dillen L, Gilissen R, Raoof A, Andries K. 2012. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis. Antimicrob Agents Chemother 56:1444–1451. doi: 10.1128/AAC.00720-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rustomjee R, Diacon AH, Allen J, Venter A, Reddy C, Patientia RF, Mthiyane TC, De Marez T, van Heeswijk R, Kerstens R, Koul A, De Beule K, Donald PR, McNeeley DF. 2008. Early bactericidal activity and pharmacokinetics of the diarylquinoline TMC207 in treatment of pulmonary tuberculosis. Antimicrob Agents Chemother 52:2831–2835. doi: 10.1128/AAC.01204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koul A, Vranckx L, Dhar N, Göhlmann HWH, Özdemir E, Neefs JM, Schulz M, Lu P, Mørtz E, McKinney JD, Andries K, Bald D. 2014. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat Commun 5:3369. doi: 10.1038/ncomms4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhillon J, Andries K, Phillips PP, Mitchison DA. 2010. Bactericidal activity of the diarylquinoline TMC207 against Mycobacterium tuberculosis outside and within cells. Tuberculosis 90:301–305. doi: 10.1016/j.tube.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Diacon AH, Dawson R, Von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, Conradie A, Erondu N, Ginsberg AM, Egizi E, Winter H, Becker P, Mendel CM. 2013. Randomized dose-ranging study of the 14-day early bactericidal activity of bedaquiline (TMC207) in patients with sputum microscopy smear-positive pulmonary tuberculosis. Antimicrob Agents Chemother 57:2199–2203. doi: 10.1128/AAC.02243-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 37.Hartkoorn RC, Uplekar S, Cole ST. 2014. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2979–2981. doi: 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.